Introduction
Breast cancer is the most common cancer in women in
the western world with the majority of tumors being classified as
estrogen receptor α (ER) positive. Tamoxifen treatment is the
standard first-line therapy for premenopausal women with ER
positive breast cancer; however, resistance to treatment is a major
clinical problem, and about one third of patients receiving
adjuvant treatment and almost all patients with advanced disease
develop resistance (1). The
mechanisms behind tamoxifen resistance are poorly understood.
Tamoxifen is a selective ER modulator that can exert both
antagonistic and agonistic effects. In estrogen responsive breast
cancer cells, binding of tamoxifen to ER antagonizes the growth
stimulation from estrogen (2). We
have previously shown that tamoxifen-resistant cell lines rely on
ER for growth and that tamoxifen acts as an agonist (3). ER co-regulators play important roles
for determining whether tamoxifen functions as an antagonist or
agonist (4). Activated ER is
associated to the chromatin through actions of pioneer factors,
e.g., the forkhead protein FOXA1, which ensures the opening of the
chromatin allowing binding of other proteins to the DNA (5). When estrogen binds to ER, the AF2
domain of the receptor is activated, leading to binding of
co-activators and initiation of transcription. One such
co-activator is amplified in breast cancer 1 (AIB1), which is a
member of the p160 steroid receptor co-activator family (6). AIB1 has been shown to be
phosphorylated by mitogen activated protein kinase (MAPK), thereby
enhancing its transactivation potential (7). Other co-activators can bind to the
AF1 domain of ER, mediated by ligand-independent activation of the
receptor (8).
Ligand-independent activation of ER can occur
through phosphorylation by receptor tyrosine kinase (RTK) pathways
(9,10). RTKs are high affinity transmembrane
receptors for many growth factors, hormones and cytokines.
Activation of RTKs leads to the activation of different signaling
pathways, including the MAPK and phosphoinositide-3-kinase (PI3K)
pathways, which play important roles in cell growth (11). Therefore, treatment with kinase
inhibitors may be a way to target tumor growth and prevent the
ligand-independent activation of ER. Sorafenib is an inhibitor of
vascular endothelial growth factor receptor (VEGFR), platelet
derived growth factor receptor (PDGFR), rearranged during
transformation (RET) and Raf (12), where especially the kinase RET has
been associated with tamoxifen resistance (13). Sorafenib is approved for treatment
of advanced kidney and liver cancer (14,15)
and is currently in clinical trials for treatment of advanced
breast cancer (16,17). Studies in mice have shown that
treatment with sorafenib inhibits breast tumor growth and
angiogenesis (18). Nilotinib is
an inhibitor of PDGFR, c-Kit and the fusion protein Bcr/Abl, and
was designed as a high affinity molecule against Bcr/Abl to treat
chronic myeloid leukemia (CML) after resistance to imatinib
(19,20). Nilotinib is now used as front line
treatment for newly diagnosed CML patients (21). Preclinical studies show that
nilotinib has a growth inhibitory effect on long-term estrogen
deprived (LTED) MCF-7 breast cancer cells via ER (22).
In this study, we tested sorafenib and nilotinib as
potential new treatments for tamoxifen-resistant breast cancer. We
show that the compounds preferentially inhibit the growth of
tamoxifen-resistant cells compared with parental MCF-7 cells and
resensitize the resistant cells to tamoxifen. We also show that
sorafenib and nilotinib downregulate total and phosphorylated ER as
well as FOXA1 and AIB1, and that sorafenib and nilotinib have no
effect in presence of estradiol supporting a mechanism of action
via ER.
Materials and methods
Cell lines and culture conditions
The MCF-7 cell line was originally obtained from the
Human Cell Culture Bank (Mason Research Institute, Rockville, MD,
USA) and adapted to grow in medium with low estrogen (MCF-7/S0.5;
hereafter called MCF-7) (23). The
tamoxifen resistant cell lines; MCF-7/TAMR-1,
MCF-7/TAMR-4, MCF-7/TAMR-7 and
MCF-7/TAMR-8 (hereafter called TAMR-1,
TAMR-4, TAMR-7, and TAMR-8,
respectively) were established from the MCF-7 cell line as
previously described (3,24). All cell lines were maintained at
37°C in humidified air with 5% CO2 in phenol-red-free
DMEM/F12 medium (Invitrogen, Carlsbad, CA, USA) containing 1% FBS
(Invitrogen), 2.5 mM L-glutamax (Invitrogen) and 6 ng/ml insulin
(Sigma-Aldrich, St. Louis, MO, USA). Growth medium for
tamoxifen-resistant cell lines was supplemented with 1 μM tamoxifen
(Sigma-Aldrich). The MCF-7 cell line used in this study was
authenticated in January, 2014 by DNA profiling using short tandem
repeat loci performed by Leibniz-Institut DSMZ (Braunschweig,
Germany) and found to be matching the genetic profile reported for
the MCF-7 cell line (DSMZ ACC 115).
Growth experiments
For tamoxifen dose-response experiments, and the
experiments where cells were grown without tamoxifen, tamoxifen was
withdrawn from the growth medium of resistant cells one week prior
to treatment. For dose-response experiments with inhibitors, cells
were grown in standard growth medium, i.e., containing 1 μM
tamoxifen for resistant cell lines. Cells were seeded two days
prior to treatment with the indicated concentrations of tamoxifen
(Sigma-Aldrich), 4-OH tamoxifen (Sigma-Aldrich), fulvestrant
(ICI182,780; Tocris Bioscience, Bristol, UK), estradiol
(Sigma-Aldrich), sorafenib (Selleck, Houston, TX, USA) or nilotinib
(Selleck). Control cells received similar amount of vehicle as the
treated cultures; i.e., 0.1% ethanol or 0.1% DMSO. Treatment medium
was renewed after three days and cell number was determined after 5
days of treatment using a crystal violet stain colorimetric assay
as previously described (25). All
growth experiments were performed with triplicate samples or more
and repeated at least twice with similar results.
Western blot analysis
MCF-7, TAMR-1 and TAMR-4 cells
were seeded in their standard medium and treated for 1–72 h with
the indicated compounds. Cells were harvested in RIPA buffer and
western blot analysis was performed as previously described
(26). Immunostaining was
performed overnight with primary antibodies directed against the
following proteins: Akt (9272), phospho-Ser473-Akt (9271),
phospho-Ser118-ERα (2511), ERK1/2 (9102) and
phospho-Thr202/Tyr204-ERK1/2 (4377) from Cell Signaling Technology
(Danvers, MA, USA); ERα (RM-9101), Hsp70 (MS-482-PO) from
Neomarkers (Fremont, CA, USA); β-actin (A5441) from Sigma-Aldrich;
PARP (511024) and AIB1 (611105) from BD Biosciences (San Jose, CA,
USA) and FOXA1 (23738) from Abcam (Cambridge, MA, USA). Western
blot analyses were done on at least two independent sets of lysates
with similar results.
LDH assay
MCF-7 and TAMR-4 cells were seeded in
96-well plates and treated with the indicated concentrations of
sorafenib or nilotinib. To investigate inhibition of cell death,
cells were treated with 5 μM z-Val-Ala-dl-Asp-fluoromethylketone
(zVAD-fmk) (Bachem, Torrance, CA, USA) or 85 μM
z-Phe-Ala-fluoromethylketone (zFA-fmk) (Calbiochem, Darmstadt,
Germany). Cytotoxicity was measured using lactate dehydrogenase
(LDH) cytotoxicity assay (Roche, Basel, Switzerland) according to
the manufacturer’s instructions. All experiments were repeated at
least twice with similar results.
Statistical analysis
Statistical analyses were performed on results from
cell growth assays and LDH assays in order to determine significant
differences between groups. Data from the representative
experiments are shown and expressed as mean ± SD as a percentage of
controls or as cytotoxicity values ± SD. Group comparisons were
done using a two-tailed t-test with Bonferroni adjusted p-values
for multiple testing. The level of statistical significance was set
to p<0.05 and indicated by asterisks in the figures.
Results
Treatment with sorafenib or nilotinib
preferentially inhibits the growth of the tamoxifen-resistant cell
lines compared with MCF-7
The effect of sorafenib and nilotinib on
tamoxifen-resistant cell growth was explored using four different
tamoxifen-resistant breast cancer cell lines, derived by long-term
treatment of MCF-7 cells with tamoxifen. Both the parental MCF-7
cell line and the four tamoxifen-resistant cell lines were growth
inhibited in a dose-dependent manner by treatment for 5 days with
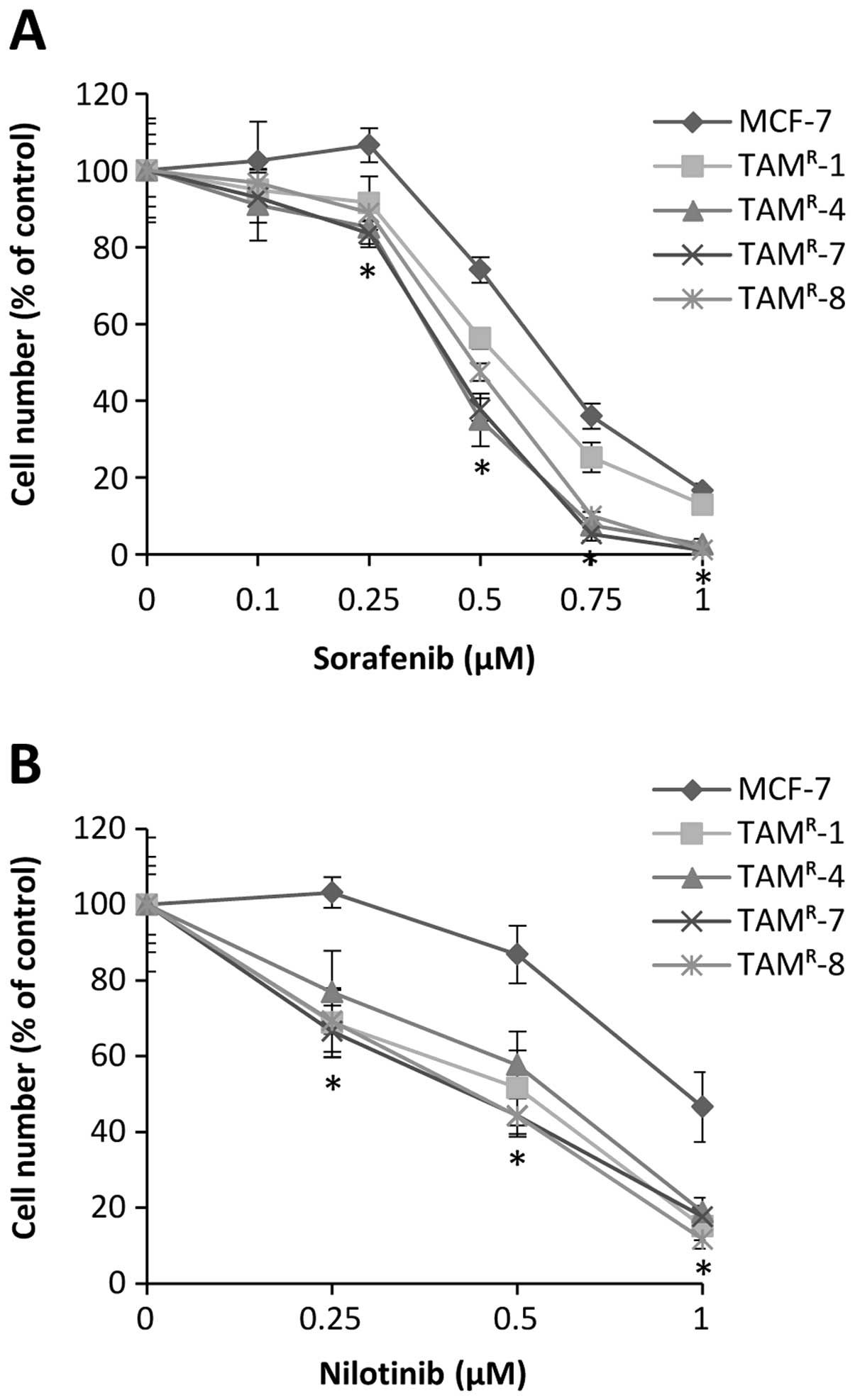
sorafenib or nilotinib (Fig. 1).
The tamoxifen-resistant cell lines TAMR-4,
TAMR-7 and TAMR-8 were significantly more
growth inhibited by treatment with 0.25, 0.5, 0.75 and 1 μM
sorafenib when compared with MCF-7 cells (Fig. 1A). Cell growth of TAMR-1
cells was significantly more reduced using 0.5 and 0.75 μM
sorafenib compared with MCF-7 cells (Fig. 1A). Preferential growth inhibition
was achieved with 0.25 and 0.5 μM nilotinib in TAMR-1,
TAMR-7 and TAMR-8, and with 1 μM nilotinib,
the growth of all four tamoxifen-resistant cell lines was
significantly more inhibited compared with MCF-7 (Fig. 1B).
Treatment with sorafenib or nilotinib
renders the resistant cells sensitive to tamoxifen inhibition
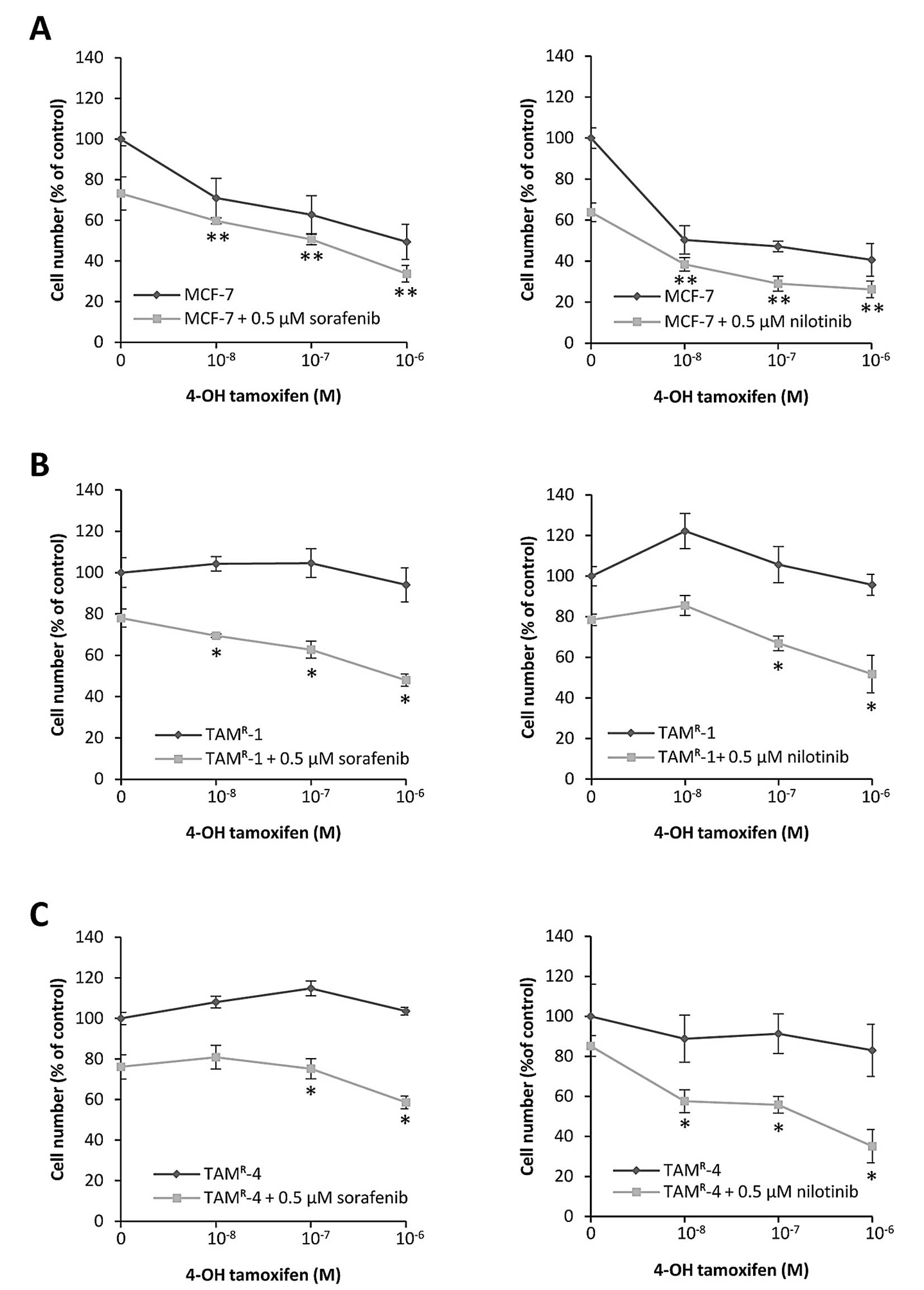
Next, we investigated whether sorafenib and
nilotinib rendered the tamoxifen-resistant cell lines sensitive to
tamoxifen treatment. Growth assays showed that the
tamoxifen-sensitive, parental MCF-7 cells, grown without sorafenib
or nilotinib, were growth inhibited in a dose-dependent manner by 5
days of treatment with 4-OH-tamoxifen (Fig. 2A). Adding 0.5 μM sorafenib or
nilotinib had only modest effect on the 4-OH-tamoxifen response in
MCF-7 cells (Fig. 2A). As
expected, TAMR-1 and TAMR-4 cells, grown
without sorafenib or nilotinib, were resistant to 4-OH-tamoxifen
treatment and a small agonistic effect was observed (Fig. 2B and C), in agreement with previous
findings (3). However, when grown
in the presence of 0.5 μM sorafenib or nilotinib, which resulted in
about 20% growth inhibition, addition of 4-OH-tamoxifen exerted a
further dose-dependent growth inhibition (Fig. 2B and C). The combined treatment
inhibited growth of the resistant cell lines to almost the same
level as tamoxifen treated MCF-7 cells, demonstrating that
sorafenib and nilotinib resensitize the resistant cell lines to
tamoxifen treatment. Furthermore, TAMR-4 cells could not
be propagated continuously in presence of 1 μM tamoxifen plus 0.5
μM sorafenib or nilotinib (data not shown), suggesting that the
combined treatment effectively prevents resistant cell growth.
Sorafenib and nilotinib induce ERK/Akt
phosphorylation and downregulate total and S118-phosphorylated
ER
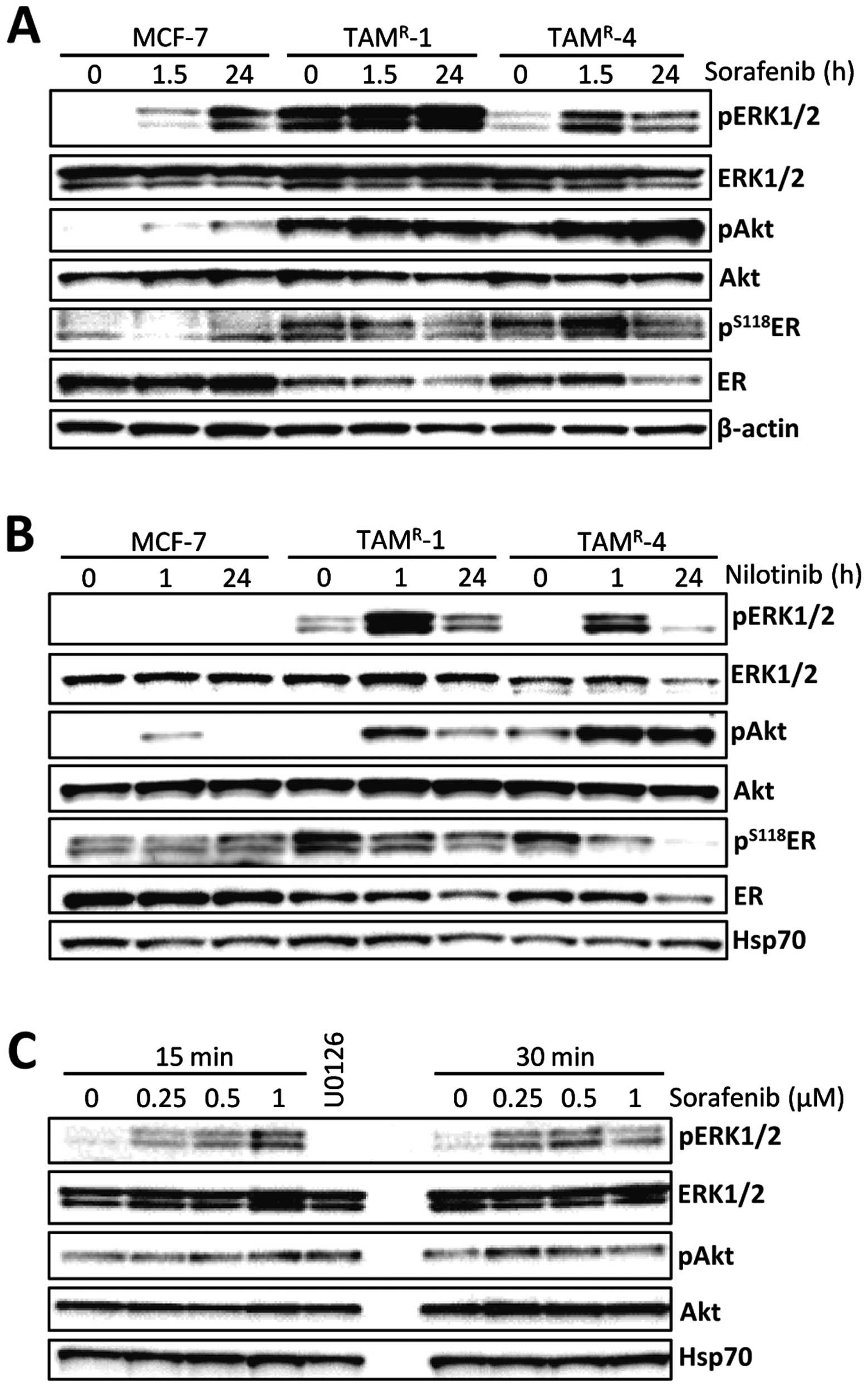
The effect of sorafenib and nilotinib treatment on
downstream signaling was analyzed in MCF-7 and the two
tamoxifen-resistant cell lines TAMR-1 and
TAMR-4 (Fig. 3). The
cells were short-term (1–1.5 h) and long-term (24 h) treated with 1
μM sorafenib or 1 μM nilotinib. Since sorafenib and nilotinib are
inhibitors of several RTKs, a decreased activity of MAPK and PI3K
pathways would be expected. However, both in MCF-7 and
tamoxifen-resistant cell lines, phosphorylation of ERK and Akt was
increased upon treatment with sorafenib and nilotinib (Fig. 3A and B). The increase of
phosphorylated ERK was observed after only 15 min using
concentrations of sorafenib down to 0.25 μM in TAMR-4
cells (Fig. 3C) and a decline in
the level of phosphorylated ERK was seen in most experiments after
24 h. Furthermore, in our cell model, we were unable to detect any
decrease in phosphorylation of the primary targets RET and PDGFR
(data not shown), indicating that sorafenib and nilotinib may act
through other targets. We have previously shown that the
tamoxifen-resistant cell lines rely on ER for growth (3). ER has several phosphorylation sites,
the most well studied being serine 118 (S118) which is an ERK
phosphorylation site (27)
associated with tamoxifen resistance (28). Interestingly, the levels of both
S118-phosphorylated and total ER were decreased upon treatment with
sorafenib or nilotinib in the resistant cell lines but not in MCF-7
cells (Fig. 3A and B). Treatment
with nilotinib caused a lower level of phosphorylated ER already
after 1 h of treatment. After 24 h of treatment with sorafenib or
nilotinib, ER was clearly reduced in both tamoxifen-resistant cell
lines, indicating that sorafenib and nilotinib may act through
similar mechanisms, involving reduction of phosphorylated ER and of
total ER protein.
Sorafenib and nilotinib downregulate ER
independent of tamoxifen
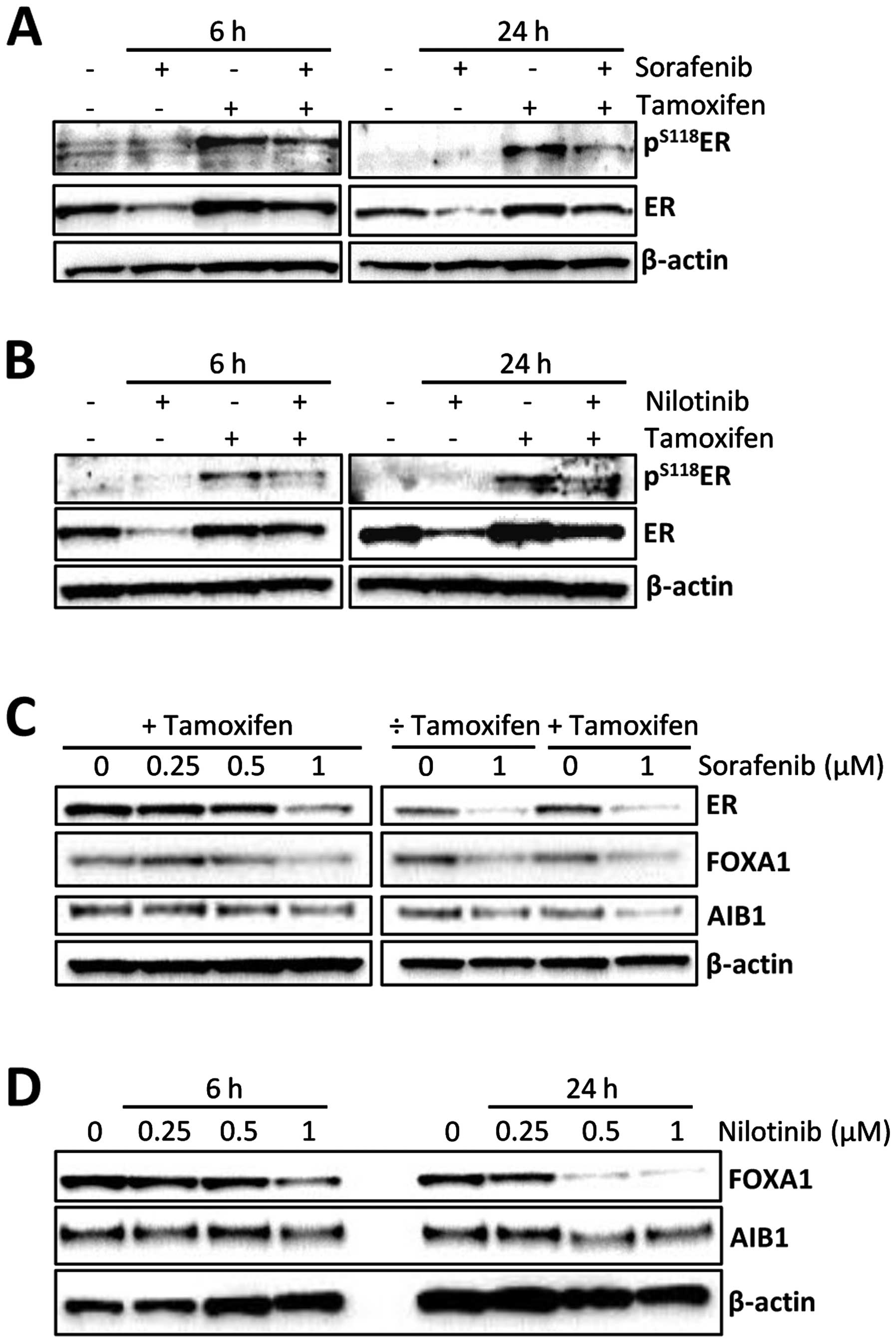
The observed resensitization of the resistant cell
lines to tamoxifen (Fig. 2) and
the decreased level of ER seen upon treatment with sorafenib and
nilotinib (Fig. 3) could indicate
that ER plays a role for the effect of the two kinase inhibitors in
tamoxifen-resistant cells. We and others have previously shown that
treatment with tamoxifen causes an increase in the level of
phosphorylated ER, presumably due to a ligand-dependent mechanism
caused by tamoxifen (3,29). This tamoxifen-induced increase in
S118-phosphorylated ER is seen in Fig.
4A and B, where stabilization of the ER protein, mediated by
tamoxifen (30), is also evident.
In this experiment, 6 h of treatment with the kinase inhibitors was
included to investigate an intermediate time point between 1 and 24
h. In presence of tamoxifen, sorafenib and nilotinib caused a
decrease in S118-phosphorylation of ER after both 6 and 24 h of
treatment (Fig. 4A and B), similar
to the results shown in Fig. 3
after 24 h of treatment. The level of total ER protein was reduced
upon 6 and 24 h of treatment with sorafenib or nilotinib, both in
the presence and absence of tamoxifen (Fig. 4A and B), indicating that the
sorafenib- and nilotinib-induced ER downregulation occurs
independently of tamoxifen. To further investigate the effect of
the kinase inhibitors on ER and ER co-regulators, TAMR-4
cells were treated for 24 h with increasing concentrations of
sorafenib or 1 μM sorafenib with and without tamoxifen. A reduction
in ER protein was observed with 0.5 and 1 μM sorafenib (Fig. 4C). The levels of ER pioneer factor
FOXA1 and the co-activator AIB1 were also decreased upon treatment
with 1 μM sorafenib, though the decrease in AIB1 protein level was
less than for ER and FOXA1. In TAMR-4 cells treated with
1 μM sorafenib with or without tamoxifen, FOXA1 was decreased by
sorafenib in a tamoxifen independent manner (Fig. 4C). Concerning AIB1, it seemed that
the decrease in AIB1 protein after sorafenib treatment was more
pronounced in the presence than in the absence of tamoxifen
(Fig. 4C). A substantial decrease
in the level of FOXA1 and a minor reduction in AIB1 were also
observed in TAMR-4 cells upon 24 h of treatment with 0.5
and 1 μM nilotinib (Fig. 4D).
ER is required for the growth inhibiting
effect of sorafenib and nilotinib
To further study the importance of ER for inhibition
of tamoxifen-resistant cell growth by sorafenib and nilotinib,
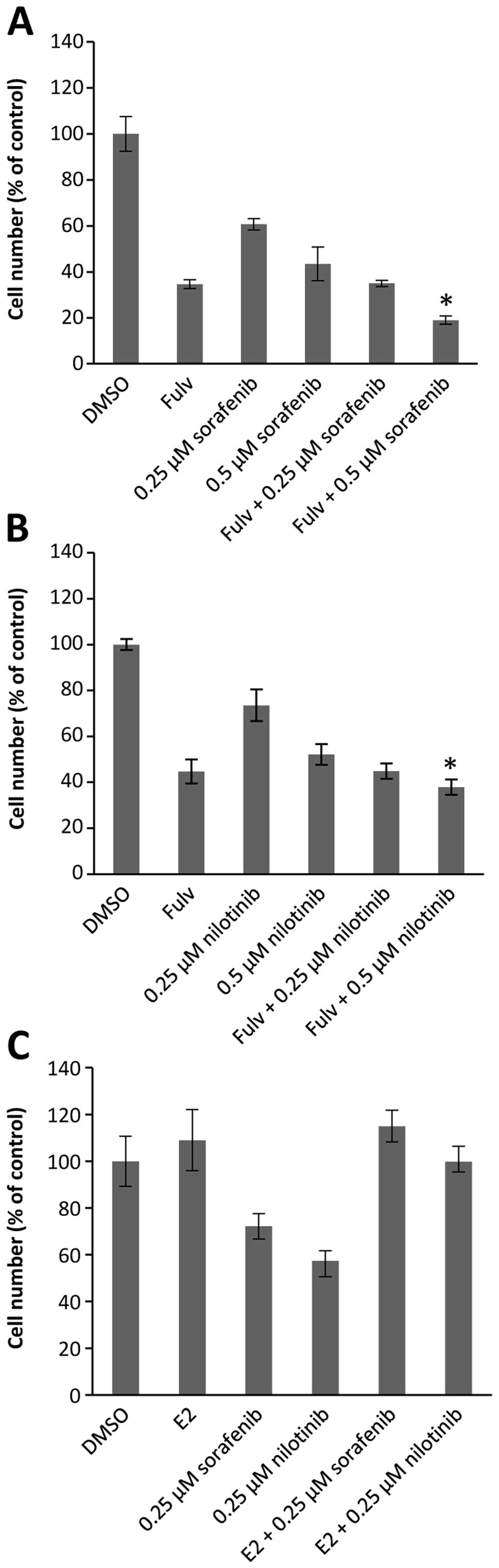
TAMR-4 cells were treated with fulvestrant or estradiol
(E2) in combination with the kinase inhibitors (Fig. 5). Fulvestrant binds competitively
to ER resulting in degradation of the receptor (31). As previously demonstrated (24), fulvestrant inhibited growth of the
tamoxifen-resistant cell line TAMR-4 (Fig. 5A and B). Addition of 0.25 μM
sorafenib (Fig. 5A) or nilotinib
(Fig. 5B) to fulvestrant did not
result in further growth inhibition, whereas 0.5 μM sorafenib or
nilotinib in combination with fulvestrant exerted significantly
more growth inhibition than fulvestrant alone. Noteworthy, in
combination with estradiol neither 0.25 μM sorafenib nor 0.25 μM
nilotinib affected cell growth (Fig.
5C). Together, these results demonstrate that at low
concentrations the growth inhibiting effect of the two kinase
inhibitors involves ER. At higher kinase inhibitor concentrations,
ER-independent mechanisms may also be involved in the growth
inhibitory effect.
High concentrations of sorafenib induce
caspase and cathepsin-mediated cell death
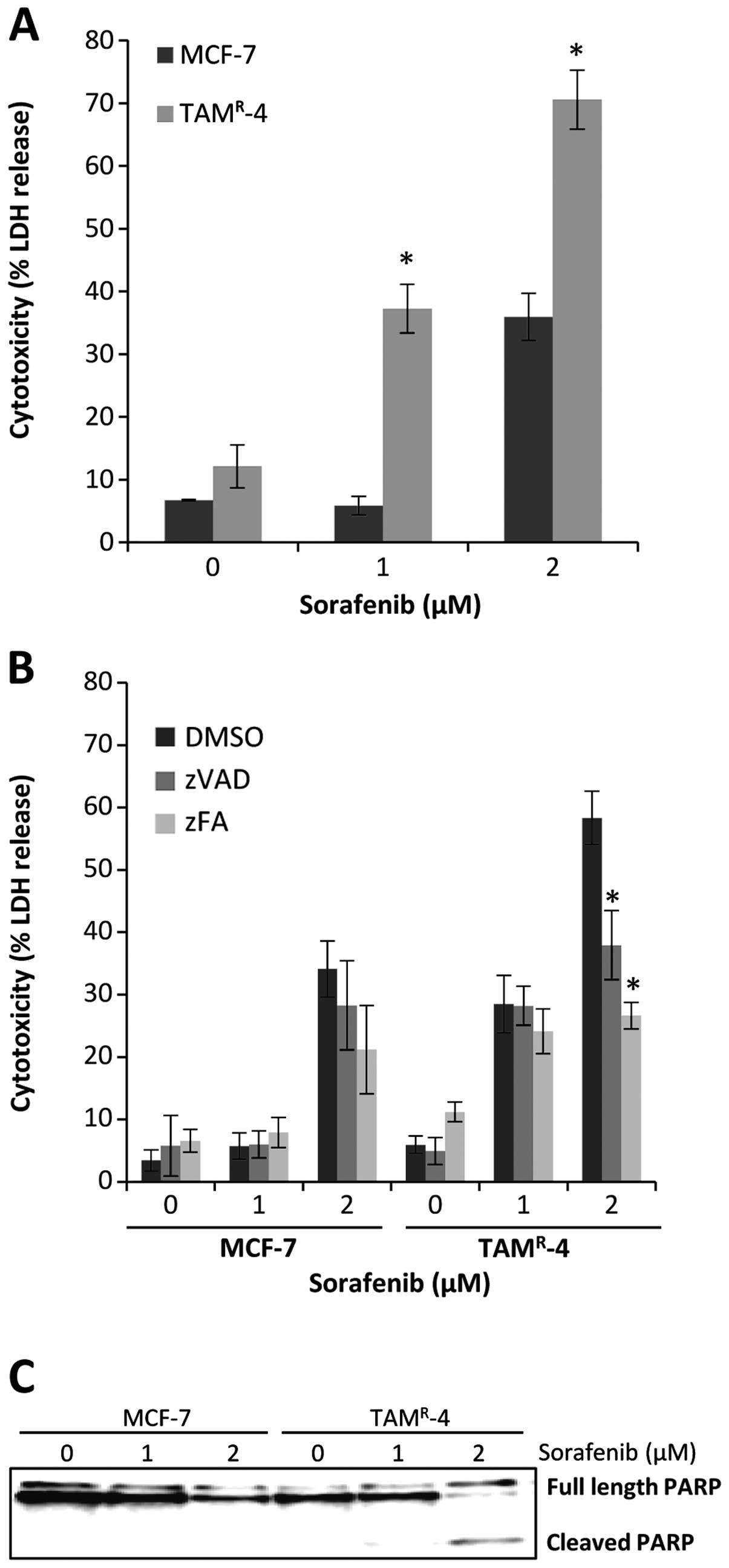
To investigate if the observed ER-independent growth
inhibition at higher drug concentrations could be due to cell
death, an LDH assay was performed on MCF-7 and TAMR-4
after treatment with 1 or 2 μM sorafenib for 72 h. Both cell lines
showed increased cell death with increasing sorafenib
concentrations and the cell death was significantly higher in
TAMR-4 when compared to MCF-7 cells (Fig. 6A). The broad-range caspase
inhibitor z-Val-Ala-dl-Asp-fluoromethylketone (zVAD-fmk) was used
to investigate if the sorafenib-induced cell death was due to
caspase-mediated apoptosis and the cathepsin inhibitor
z-Phe-Ala-fluoromethylketone (zFA-fmk) was used to investigate if
the sorafenib-induced cell death was mediated through cathepsins,
indicative of cell death by lysosomal membrane permeabilization
(32). MCF-7 and TAMR-4
cells were treated with 1 or 2 μM sorafenib in combination with
zVAD-fmk or zFA-fmk. Cell death was induced by 2 μM sorafenib in
MCF-7 and could not be significantly prevented by the two
inhibitors (Fig. 6B). For
TAMR-4 cells, cell death was induced at 1 μM. However,
the cell death could not be inhibited until cytotoxicity was
reached with 2 μM sorafenib. At this point the cell death of
TAMR-4 cells could be inhibited by both the caspase
inhibitor zVAD-fmk and the cathepsin inhibitor zFA-fmk (Fig. 6B). Furthermore, western blot
analyses revealed PARP cleavage in TAMR-4 cells after
treatment with 2 μM sorafenib for 72 h, confirming induction of
apoptotic cell death (Fig.
6C).
Discussion
Resistance to endocrine therapy is a major clinical
problem in the treatment of ER-positive breast cancer. Therefore,
identification of drugs that target tamoxifen-resistant cells is
important in order to find new treatment options. RTKs play
important roles in the activation of pathways involved in cell
proliferation, cell survival and ligand-independent activation of
ER and may be important for circumventing the growth inhibitory
effect of tamoxifen. Therefore, inhibitors of RTKs may have
potential as treatment for tamoxifen-resistant breast cancer.
Sorafenib and nilotinib are both multitargeting RTK inhibitors
targeting a broad range of kinases important for cellular
functions. We show here that both sorafenib and nilotinib
preferentially inhibit the growth of tamoxifen-resistant cell lines
compared with their parental tamoxifen-sensitive cell line MCF-7.
Furthermore, the two inhibitors render the resistant cell lines
sensitive to tamoxifen inhibition. As shown previously (3,33,34),
and also here, tamoxifen can stimulate growth of
tamoxifen-resistant cells presumably by an agonistic effect on ER.
Together these results indicate that in tamoxifen-resistant cells,
sorafenib and nilotinib may switch the effect of tamoxifen from
being agonistic to antagonistic, resulting in growth inhibition by
tamoxifen. These results also suggest that sorafenib or nilotinib
should be administered together with tamoxifen as a potential
combined treatment of tamoxifen-resistant breast cancer.
Sorafenib and nilotinib are inhibitors of kinases
that may signal through the MAPK or PI3K pathways. However, we did
not observe inhibition but rather activation of these pathways,
indicating that sorafenib and nilotinib have other mechanisms of
action. Our findings that treatment with the two inhibitors lowered
both the total and the phosphorylated level of ER and that
estradiol completely abrogated the growth inhibitory effect of
sorafenib and nilotinib demonstrated that the kinase inhibitors
function by targeting ER activity in tamoxifen-resistant cells.
Tamoxifen induces S118 phosphorylation of ER, which could
contribute to the agonistic effect of tamoxifen in the resistant
cells (3,35). The observed lowered levels of
S118-phosphorylated ER upon treatment with sorafenib or nilotinib
may therefore cause growth inhibition. The tamoxifen-resistant cell
lines are highly dependent on ER for growth as the ER downmodulator
fulvestrant exerted severe growth inhibition of the resistant
cells. Thus, the observed decrease in ER would cause growth
inhibition of the tamoxifen-resistant cells. In support of our
data, Weigel et al have also observed a decrease in the
level of total ER and growth inhibition upon treatment of LTED
cells with nilotinib (22).
We found that downregulation of ER by sorafenib and
nilotinib is independent of tamoxifen, suggesting that the
resensitization to tamoxifen by the inhibitors involves mechanisms,
such as changes in ER co-regulators, causing a switch from
agonistic to antagonistic effect of tamoxifen. Notably, we show
here a decrease in the level of the two ER co-regulators AIB1 and
FOXA1 upon treatment with sorafenib and nilotinib. The co-activator
AIB1 is, like ER, phosphorylated by MAPKs (7) and therefore high levels of activated
AIB1 could reduce the antagonistic effects of tamoxifen and cause
tamoxifen resistance. In support of this, high levels of AIB1 in
tumors from tamoxifen treated patients were associated with poor
disease-free survival (36).
Additionally, knockdown of AIB1 in the tamoxifen-resistant breast
cancer cell line BT474 restored its sensitivity to tamoxifen
(37). Studies have also shown
that high levels of AIB1 enhance the agonistic properties of
tamoxifen (38,39). Thus, the reduced level of AIB1 in
sorafenib or nilotinib treated tamoxifen-resistant cells could
cause a shift of tamoxifen from agonist to antagonist. FOXA1 is
required for both the agonistic and antagonistic function of
tamoxifen bound ER. Studies on a tamoxifen-resistant MCF-7-based
cell line showed that silencing of FOXA1 suppressed
ligand-independent ER binding to chromatin, inhibiting cell growth
(5). This demonstrates that FOXA1
is essential for ligand-independent growth of tamoxifen resistant
cells. The decrease in FOXA1 protein induced by sorafenib and
nilotinib may therefore reduce cell growth by inhibiting
ligand-independent ER activity.
We observed that the concentration of sorafenib and
nilotinib used to treat the tamoxifen-resistant cell lines was
crucial for the mechanism of action of the inhibitors. At low
concentration (0.25 μM), only ER-dependent mechanisms were
observed, while at higher concentrations (>0.5 μM), a
combination of both ER dependent and independent mechanisms, e.g.,
cytotoxic effects, were present. This is in agreement with other
studies showing that sorafenib and nilotinib induce cell death at
concentrations >1 μM (40–42).
In conclusion, we have shown that sorafenib and
nilotinib in combination with tamoxifen inhibit growth of
tamoxifen-resistant breast cancer cells. The mechanisms of action
are complex and both reduced total ER and phosphorylated ER,
reduced ligand-independent ER activation due to lowered FOXA1
level, and a switch in the effect of tamoxifen from agonistic to
antagonistic via reduced AIB1 appears to contribute to growth
inhibition. Collectively, this suggests that sorafenib or nilotinib
together with tamoxifen have potential as a combined treatment of
tamoxifen-resistant breast cancer patients.
Acknowledgements
We thank Jane Lind Christensen and Birgit Reiter for
excellent technical assistance. This study was supported by Astrid
Thaysen’s grant (ATL12/01), A.P. Møller Foundation for the
Advancement of Medical Science (12-374), Danish Cancer Research
Foundation, Danish Cancer Society, Danske Bank Foundation, Leo
Nielsen’s grant (LN12/07) and Sigvald and Edith Rasmussen’s
grant.
References
|
1
|
Davies C, Godwin J, Gray R, et al:
Relevance of breast cancer hormone receptors and other factors to
the efficacy of adjuvant tamoxifen: patient-level meta-analysis of
randomised trials. Lancet. 378:771–784. 2011. View Article : Google Scholar
|
|
2
|
Dutertre M and Smith CL: Molecular
mechanisms of selective estrogen receptor modulator (SERM) action.
J Pharmacol Exp Ther. 295:431–437. 2000.PubMed/NCBI
|
|
3
|
Thrane S, Lykkesfeldt AE, Larsen MS,
Sorensen BS and Yde CW: Estrogen receptor alpha is the major
driving factor for growth in tamoxifen-resistant breast cancer and
supported by HER/ERK signaling. Breast Cancer Res Treat. 139:71–80.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lavinsky RM, Jepsen K, Heinzel T, et al:
Diverse signaling pathways modulate nuclear receptor recruitment of
N-CoR and SMRT complexes. Proc Natl Acad Sci USA. 95:2920–2925.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hurtado A, Holmes KA, Ross-Innes CS,
Schmidt D and Carroll JS: FOXA1 is a key determinant of estrogen
receptor function and endocrine response. Nat Genet. 43:27–33.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu J and Li Q: Review of the in vivo
functions of the p160 steroid receptor coactivator family. Mol
Endocrinol. 17:1681–1692. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Font de Mora J and Brown M: AIB1 is a
conduit for kinase-mediated growth factor signaling to the estrogen
receptor. Mol Cell Biol. 20:5041–5047. 2000.PubMed/NCBI
|
|
8
|
Bunone G, Briand PA, Miksicek RJ and
Picard D: Activation of the unliganded estrogen receptor by EGF
involves the MAP kinase pathway and direct phosphorylation. EMBO J.
15:2174–2183. 1996.PubMed/NCBI
|
|
9
|
Kato S, Endoh H, Masuhiro Y, et al:
Activation of the estrogen receptor through phosphorylation by
mitogen-activated protein kinase. Science. 270:1491–1494. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Campbell RA, Bhat-Nakshatri P, Patel NM,
Constantinidou D, Ali S and Nakshatri H: Phosphatidylinositol
3-kinase/ AKT-mediated activation of estrogen receptor alpha: a new
model for anti-estrogen resistance. J Biol Chem. 276:9817–9824.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zwick E, Bange J and Ullrich A: Receptor
tyrosine kinase signalling as a target for cancer intervention
strategies. Endocr Relat Cancer. 8:161–173. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wilhelm SM, Adnane L, Newell P, Villanueva
A, Llovet JM and Lynch M: Preclinical overview of sorafenib, a
multikinase inhibitor that targets both Raf and VEGF and PDGF
receptor tyrosine kinase signaling. Mol Cancer Ther. 7:3129–3140.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Plaza-Menacho I, Morandi A, Robertson D,
et al: Targeting the receptor tyrosine kinase RET sensitizes breast
cancer cells to tamoxifen treatment and reveals a role for RET in
endocrine resistance. Oncogene. 29:4648–4657. 2010. View Article : Google Scholar
|
|
14
|
Escudier B, Eisen T, Stadler WM, et al:
Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J
Med. 356:125–134. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Llovet JM, Ricci S, Mazzaferro V, et al:
Sorafenib in advanced hepatocellular carcinoma. N Engl J Med.
359:378–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Luu T, Frankel P, Chung C, et al: Phase
I/II trial of vinorelbine and sorafenib in metastatic breast
cancer. Clin Breast Cancer. 14:94–100. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baselga J, Costa F, Gomez H, et al: A
phase 3 trial comparing capecitabine in combination with sorafenib
or placebo for treatment of locally advanced or metastatic
HER2-negative breast cancer (the RESILIENCE study): study protocol
for a randomized controlled trial. Trials. 14:2282013. View Article : Google Scholar
|
|
18
|
Wilhelm SM, Carter C, Tang L, et al: BAY
43-9006 exhibits broad spectrum oral antitumor activity and targets
the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in
tumor progression and angiogenesis. Cancer Res. 64:7099–7109. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weisberg E, Manley P, Mestan J,
Cowan-Jacob S, Ray A and Griffin JD: AMN107 (nilotinib): a novel
and selective inhibitor of BCR-ABL. Br J Cancer. 94:1765–1769.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
O’Hare T, Walters DK, Deininger MW and
Druker BJ: AMN107: tightening the grip of imatinib. Cancer Cell.
7:117–119. 2005.PubMed/NCBI
|
|
21
|
Piccaluga PP, Paolini S, Bertuzzi C, De
Leo A and Rosti G: First-line treatment of chronic myeloid leukemia
with nilotinib: critical evaluation. J Blood Med. 3:151–156. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Weigel MT, Ghazoui Z, Dunbier A, Pancholi
S, Dowsett M and Martin LA: Preclinical and clinical studies of
estrogen deprivation support the PDGF/Abl pathway as a novel
therapeutic target for overcoming endocrine resistance in breast
cancer. Breast Cancer Res. 14:R782012. View
Article : Google Scholar
|
|
23
|
Briand P and Lykkesfeldt AE: Effect of
estrogen and antiestrogen on the human breast cancer cell line
MCF-7 adapted to growth at low serum concentration. Cancer Res.
44:1114–1119. 1984.PubMed/NCBI
|
|
24
|
Lykkesfeldt AE, Madsen MW and Briand P:
Altered expression of estrogen-regulated genes in a
tamoxifen-resistant and ICI 164,384 and ICI 182,780 sensitive human
breast cancer cell line, MCF-7/TAMR-1. Cancer Res.
54:1587–1595. 1994.PubMed/NCBI
|
|
25
|
Lundholt BK, Briand P and Lykkesfeldt AE:
Growth inhibition and growth stimulation by estradiol of estrogen
receptor transfected human breast epithelial cell lines involve
different pathways. Breast Cancer Res Treat. 67:199–214. 2001.
View Article : Google Scholar
|
|
26
|
Larsen MS, Yde CW, Christensen IJ and
Lykkesfeldt AE: Carboplatin treatment of antiestrogen-resistant
breast cancer cells. Int J Oncol. 41:1863–1870. 2012.
|
|
27
|
Arnold SF, Obourn JD, Jaffe H and Notides
AC: Phosphorylation of the human estrogen receptor by
mitogen-activated protein kinase and casein kinase II: consequence
on DNA binding. J Steroid Biochem Mol Biol. 55:163–172. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
De Leeuw R, Neefjes J and Michalides R: A
role for estrogen receptor phosphorylation in the resistance to
tamoxifen. Int J Breast Cancer. 2011:2324352011.PubMed/NCBI
|
|
29
|
Chen D, Riedl T, Washbrook E, et al:
Activation of estrogen receptor alpha by S118 phosphorylation
involves a ligand-dependent interaction with TFIIH and
participation of CDK7. Mol Cell. 6:127–137. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wijayaratne AL and McDonnell DP: The human
estrogen receptor-alpha is a ubiquitinated protein whose stability
is affected differentially by agonists, antagonists, and selective
estrogen receptor modulators. J Biol Chem. 276:35684–35692. 2001.
View Article : Google Scholar
|
|
31
|
Osborne CK, Wakeling A and Nicholson RI:
Fulvestrant: an oestrogen receptor antagonist with a novel
mechanism of action. Br J Cancer. 90(Suppl 1): S2–S6. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kroemer G and Jaattela M: Lysosomes and
autophagy in cell death control. Nat Rev Cancer. 5:886–897. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shou J, Massarweh S, Osborne CK, et al:
Mechanisms of tamoxifen resistance: increased estrogen
receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J
Natl Cancer Inst. 96:926–935. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hodges LC, Cook JD, Lobenhofer EK, et al:
Tamoxifen functions as a molecular agonist inducing cell
cycle-associated genes in breast cancer cells. Mol Cancer Res.
1:300–311. 2003.PubMed/NCBI
|
|
35
|
Vendrell JA, Bieche I, Desmetz C, et al:
Molecular changes associated with the agonist activity of
hydroxy-tamoxifen and the hyper-response to estradiol in
hydroxy-tamoxifen-resistant breast cancer cell lines. Endocr Relat
Cancer. 12:75–92. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Osborne CK, Bardou V, Hopp TA, et al: Role
of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in
tamoxifen resistance in breast cancer. J Natl Cancer Inst.
95:353–361. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Su Q, Hu S, Gao H, et al: Role of AIB1 for
tamoxifen resistance in estrogen receptor-positive breast cancer
cells. Oncology. 75:159–168. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Smith CL, Nawaz Z and O’Malley BW:
Coactivator and corepressor regulation of the agonist/antagonist
activity of the mixed antiestrogen, 4-hydroxytamoxifen. Mol
Endocrinol. 11:657–666. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Reiter R, Oh AS, Wellstein A and Riegel
AT: Impact of the nuclear receptor coactivator AIB1 isoform
AIB1-Delta3 on estrogenic ligands with different intrinsic
activity. Oncogene. 23:403–409. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rahmani M, Davis EM, Crabtree TR, et al:
The kinase inhibitor sorafenib induces cell death through a process
involving induction of endoplasmic reticulum stress. Mol Cell Biol.
27:5499–5513. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu L, Cao Y, Chen C, et al: Sorafenib
blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and
induces tumor cell apoptosis in hepatocellular carcinoma model
PLC/PRF/5. Cancer Res. 66:11851–11858. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shaker ME, Ghani A, Shiha GE, Ibrahim TM
and Mehal WZ: Nilotinib induces apoptosis and autophagic cell death
of activated hepatic stellate cells via inhibition of histone
deacetylases. Biochim Biophys Acta. 1833:1992–2003. 2013.
View Article : Google Scholar : PubMed/NCBI
|