Introduction
Pancreatic cancer is one of the most aggressive
types of cancer. With a 5-year survival rate of <5%, it is the
fourth most common cause of cancer-related deaths in the developed
world (1). The reasons for the
extremely poor prognosis are the late diagnosis, resistance to
conventional chemotherapies, and high immunosuppression (2). Immunotherapy approaches designed to
target tumor-associated antigens (TAAs) are promising treatments
for pancreatic cancer. The major goal of immunotherapy is to
activate CD8+ cytotoxic T lymphocytes (CTLs).
Tumor-specific CTLs, activated by immunotherapy, are the effector
cells most capable of directly recognizing and lysing cancer cells.
However, immunotherapy alone is limited by the number of CTLs that
can penetrate a large and established pancreatic tumor. To identify
more efficient immunotherapies for pancreatic cancer, it is
important to have an understanding of the following basic issues:
i) the identity of tumor antigens and means to evaluate the immune
response in pancreatic cancer; ii) mechanisms used by tumors to
escape the immune system and strategies to overcome them; and iii)
development of efficient immune interventions to eliminate
pancreatic cancer cells. In particular, the identification of
appropriate pancreatic cancer TAAs remains critical to the
development of effective immunotherapy strategies and the
assessment of tumor-specific CTL responses. Pancreatic cancer
immunotherapies have targeted a few known proteins that were either
the products of oncogenes (e.g., mutated Kras) (3) or differentially expressed
glycoproteins such as MUC1, CEA (4), and mesothelin (5). However, vaccination against a single
antigen has some disadvantages because it is unknown which of the
identified antigens have the potential to induce an effective
anti-tumor immune response. Furthermore, immunity against a single
antigen may be ineffective in tumors with heterogeneous cell
populations. In addition, the cellular environment in pancreatic
cancer consists of not only cancer cells but also immune
suppressive cells such as cancer-associated fibroblasts (CAFs),
tolerogenic dendritic cells, myeloid-derived suppressor cells
(MDCSs), immunosuppressive tumor-associated macrophages (TAMs), and
T regulatory cells (6). These
immunosuppressive cells inhibit the antitumor immunity induced by
pancreatic cancer vaccines. The accumulation of these
immunosuppressive cells in pancreatic cancer might be closely
related to the extent of disease and fails to provide clinically
relevant benefits (7).
Anti-Gal is the most abundant natural antibody in
human sera from both normal subjects and patients with
malignancies, and constitutes ~1% of serum IgG (8). This antibody interacts specifically
with the α-gal epitopes on glycolipids and glycoproteins (8). Anti-Gal is produced primarily by
anti-Gal B cells (i.e., B cells that can produce anti-Gal) present
along the gastrointestinal tract due to continuous stimulation by
bacteria of the natural flora (8).
The α-gal epitope is absent in humans but is synthesized by the
glycosylation enzyme, α1,3GT, in very large amounts in cells from
non-primate mammals, prosimians and New World monkeys (8). The α1,3GT gene was inactivated as a
pseudogene in ancestral Old World primates (8); thus, humans, apes, and Old World
monkeys all lack α-gal epitopes and instead produce anti-Gal in
large amounts (8,9). Introduction of cancer cells, or
molecules such as TAAs and tumor lysates expressing α-gal epitopes,
into humans results in the binding of anti-Gal to these epitopes
in vivo. This interaction is evident in xenotransplantation,
in which in vivo binding of anti-Gal to α-gal epitopes on
transplanted pig hearts or kidneys is the main cause of hyperacute
rejection of such grafts (9–11).
This in situ interaction between anti-Gal/α-gal epitopes may
be exploited for targeting cancer vaccines expressing α-gal
epitopes to antigen presenting cells (APCs).
In a recent study, we investigated the in
vitro and in vivo effects of whole cell vaccination with
α-gal epitope-expressing pancreatic cancer cells (12). However, the effect was some-what
weak because melanoma cells transplanted in athymic mice formed
tumors despite vaccination with α-gal epitopes expressing
pancreatic cancer cells. To further develop an effective
immunotherapy for pancreatic cancer, we hypothesized that tumor
lysate is a more suitable source of TAAs because it contains
several known and unknown antigens in cancer cells and stromal
cells that can elicit a broad spectrum anti-tumor immune response.
Moreover, the primary tumor of pancreatic adenocarcinoma contains a
subset of pancreatic cancer cells with stem cell properties (i.e.,
pancreatic cancer stem cells: pancreatic CSCs) (13,14).
These pancreatic CSCs, whose phenotypic identification is still a
matter of debate, could have different biologically important
characteristics, such as the capacity to self-renew and divide
asymmetrically (13,14). In pancreatic cancer, recent data
suggest that the presence of these putative CSCs in primary tumors
is associated with shorter overall survival, resistance to the
standard cytotoxic agent gemcitabine and enhanced metastatic
potential (13,14). However, it is noteworthy that the
induction of the immune response against pancreatic CSCs by
standard vaccination with tumor lysate, as described above, is
often difficult because the CSCs constitute only 1% of all cancer
cells (13,14). Accordingly, it is desirable to
prepare a vaccine from lysates of tumors engineered to express
α-gal epitopes to increase the immunogenicity of the broad-spectrum
of TAAs present in both differentiated pancreatic cancer cells and
pancreatic CSCs.
In the present study, we investigated the effects of
vaccination with lysate from α-gal epitope-expressing tumors, using
adoptive transfer mouse models. The tumor growth of pancreatic
cancer cells, which include differentiated pancreatic cancer cells
and pancreatic CSCs, in NOD/SCID mice was examined as well as the
survival of recipients. Furthermore, the immunoresponses of both B
and T cells were investigated in details.
Materials and methods
Ethics statement
All animals were bred and maintained as specific
pathogen-free condition (SPF) at the Institute of Experimental
Animal Sciences, Osaka University Medical School. All animal care
and procedures described in the present study were approved by the
Ethics Review Committee for Animal Experimentation of Osaka
University (experimental number 20-055-0), and animal wellbeing was
taken into consideration in the study design. All animal
experiments were performed in accordance with the Guidelines for
proper conduct for animal experiments from Scientific Council of
Japan.
Mice
Mice used in the present study had disrupted
α1,3-galactosyltransferase (α1,3GT) genes and are referred as
α1,3GT knockout (KO) mice. The α1,3GT KO mice were generated on a
C57BL/6×BALB/c genetic background (H-2bxd) (15,16).
Prior to the experimental procedure, anti-Gal antibody (Ab)
production was elicited in 6- to 8-week-old α1,3GT KO mice by four
weekly intraperitoneal (i.p.) injections with 100 mg of pig kidney
membrane homogenate (9). The
amount (titer) of anti-Gal Ab was confirmed to be similar to that
observed in humans (1:400–1:2,000, designated as high anti-Gal KO
mice) by enzyme-linked immunosorbent assay (ELISA) with synthetic
α-gal epitopes linked to bovine serum albumin (BSA) (Dextra
Laboratories Ltd., Berkshire, UK) as the solid phase antigen
(9,12,15).
Preparation of tumor lysate vaccines
expressing α-gal epitopes
The human pancreatic cancer cell line, PANC1 (ATCC,
Manassas, VA, USA), which intrinsically expressed the Mucin1 (MUC1)
molecule, was employed (12,17).
We established stable PANC1-transfected cells, expressing α-gal
epitopes, by mouse α1,3GT gene transfection (called α-gal PANC1) as
previously described (12). To
generate PANC1 tumors, 2×106 live cells (either parental
or α-gal PANC1) were injected subcutaneously into the back of
non-obese diabetic severe combined immunodeficiency (NOD/SCID) mice
(NOD. CB17-Prkdcscid/J mice; Charles River, Tokyo
Japan). The grown PANC1 tumors were enucleated and homogenized
under sterile conditions, washed with 200 ml of PBS and centrifuged
at 30,000 × g. The tumor membranes were resuspended at 100 mg/ml
(weight/volume) in saline, and were subsequently irradiated with 50
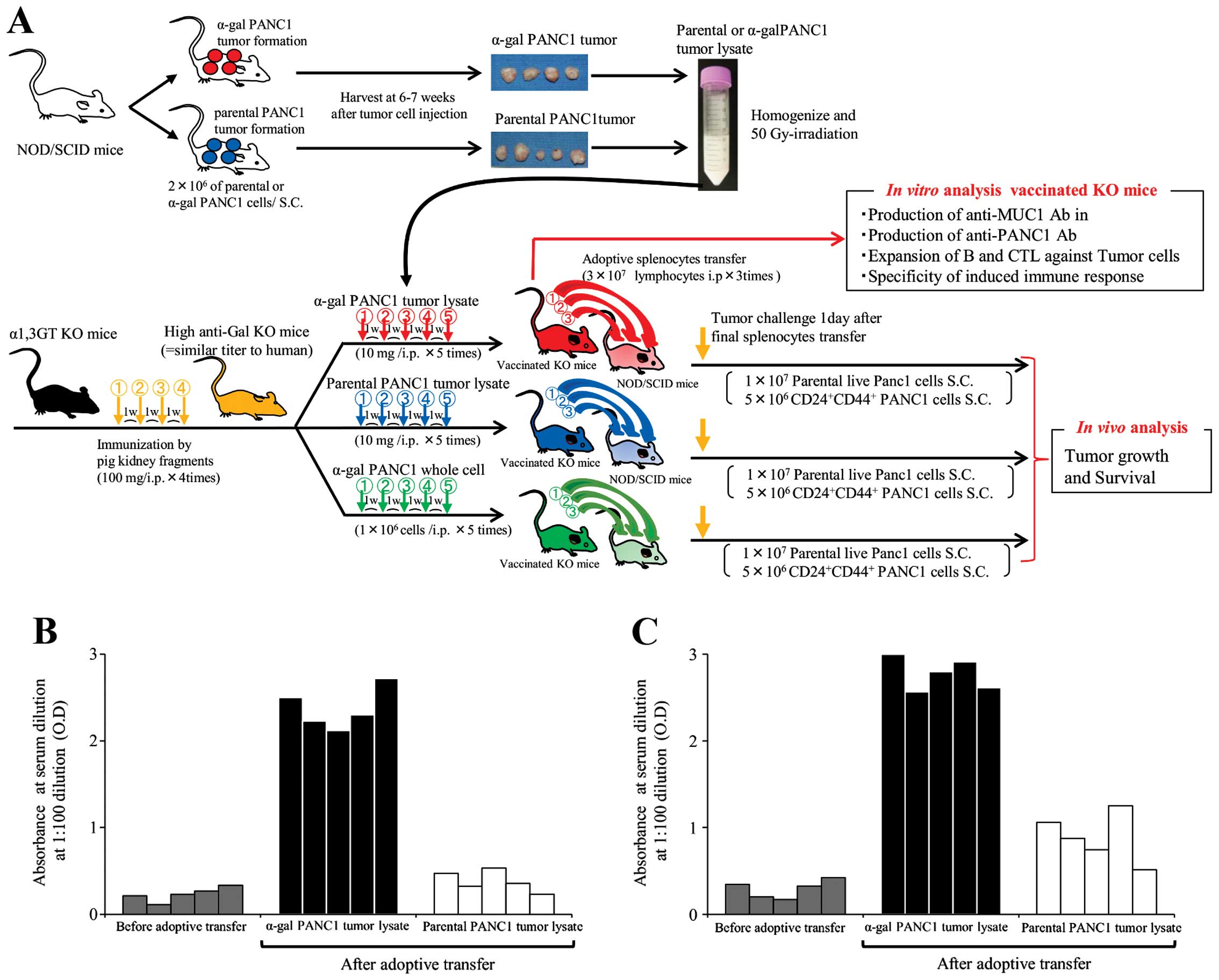
Gy and frozen until needed (Fig.
1A).
Tumor lysate vaccination
The high anti-Gal KO mice were vaccinated by i.p.
injection five times at 1-week intervals with 10 mg of
50-Gy-irradiated parental or α-gal PANC1 tumor lysates (abbreviated
here as pt-lysate or α-gal-t-lysate, respectively). One week after
the 5th vaccination, the mice were assessed for immune response
induced by tumor lysate vaccination as described below (Fig. 1A). To compare the effectiveness of
the α-gal PANC1 whole cell (abbreviated here as α-gal-whole-c)
vaccine with that of α-gal-t-lysate vaccine, the mice received five
i.p. injections of 1×106 cells of 50 Gy-irradiated α-gal
PANC1 whole cell vaccine in a manner similar to the tumor lysate
vaccine (Fig. 1A) (12).
Enzyme-linked immunosorbent assay
(ELISA)
To determine whether the studied tumor lysates
expressed α-gal epitopes, the tumor homogenates were assayed by
ELISA using the monoclonal anti-Gal IgM Ab, M86, as previously
described (17–19). The expression level of MUC1 in
tumor lysates was assessed by ELISA using anti-MUC1 monoclonal
antibody (mAb) (clone VU4H5; Santa Cruz Biotechnology, Santa Cruz,
CA, USA; cat. no. sc-7313, lot no. B1611). Anti-MUC1 IgG
production, was detected by ELISA using MUC1-BSA as the solid phase
antigen, as previously described (12). Anti-PANC1 IgG production was
detected by ELISA using dried-up PANC1 cells as the solid phase
antigen, as previously described (20).
Enzyme-linked immunospot analysis
(ELISPOT)
An enzyme-linked immunospot (ELISPOT) assay was used
to identify the expansion of anti-MUC1 secreting B cells and
MUC1-specific activated T cells (i.e., IFN-γ secreting T cells),
using a previously described method (12).
Immunohistochemical analysis and
immunofluorescence microscopy
Parental PANC1 and α-gal PANC1 tumor specimens,
generated in NOD/SCID mice, were cut into small blocks, fixed in
formalin and then embedded in paraffin. Tissue sections (4 μm
thick) were incubated with either mouse anti-human MUC1 mAb (1:100;
Santa Cruz Biotechnology; clone VU4H5, cat. no. sc-7313, lot. no.
B1611) or M86 anti-Gal mAb (1:2) (21) in PBS Tween-20 (0.05% w/v) for 16 h
at 4°C. The sections were then incubated with appropriate
antibodies (for anti-MUC1 Ab, HRP-conjugated goat anti-mouse IgG,
dilution 1:1,000; for M86 mAb, HRP-conjugated goat anti-mouse IgM,
dilution 1:1,000). Immunostaining was visualized with 0.02%
diaminobenzidine (DAB; Sigma-Aldrich) as the chromogen. The
specificity of the primary Abs was verified using control sections
prepared as described above but without the use of the primary
Abs.
To evaluate the expression of CD44 and CD24, which
are CSC markers of pancreatic cancer, on parental and α-gal PANC1
tumors, tissue sections were incubated with either rabbit
anti-human CD44 mAb (dilution 1:100; Abcam, Cambridge, MA, USA;
cat. no. ab97478) or rabbit anti-human CD24 mAb (dilution 1:100,
Santa Cruz Biotechnology; cat. no. FL-80, sc-11406), respectively,
followed by incubation with Alexa Fluor 555 goat anti-rabbit IgG Ab
(A21429, dilution 1:1,000; Invitrogen). Fluorescence signals were
observed with a Biozero fluorescence microscope (Keyence
Corporation of America, Elmwood Park, NJ, USA). The α-gal epitopes
in PANC1 tumors were detected by incubating the sections with M86
anti-Gal mAb (1:2 dilution) (21)
in PBS Tween-20 for 16 h at 4°C, followed by incubation with Alexa
Fluor 488 goat anti-mouse IgM Ab (A21042; dilution 1:1,000;
Invitrogen). Fluorescence signals were assessed by fluorescence
microscopy.
Flow cytometric analysis
To investigate Ab production against differentiated
pancreatic cancer cells (isolated differentiated cancer cells from
PANC1 cells; i.e., CD44−CD24− PANC1 cells)
and pancreatic CSCs (isolated cancer stem cells from PANC1 cells;
i.e., CD44+CD24+ PANC1 cells), cells were
stained with sera from KO mice vaccinated with pt-lysate,
α-gal-t-lysate, or α-gal-whole-c, as previously described (12). To determine whether or not
splenocytes from the vaccinated α1,3GT KO mice can be specifically
stimulated by MUC1 peptide, PANC1 cells or PANC1 tumor lysate, a
carboxyfluorescein diacetate succinimidyl ester (CFSE; Invitrogen,
Carlsbad, CA, USA; CellTrace™ CFSE Cell Proliferation kit, cat. no.
C34554) assay was performed according to the manufacturer’s
recommended protocol. Human embryonic kidney HEK293 cells were also
employed as stimulatory cells. The CFSE labeled mouse splenocytes
were cultured in 96-well round bottom plates (cat. no 3870-096;
Iwaki, Japan) at 2×105 cells/well with 1×104
stimulatory cells (irradiated PANC1 or HEK293 cells), and 10
μg/well of MUC1 peptide, 10 mg/well of PANC1 tumor lysate or 3
μg/ml of ConA. The stimulated cells were cultured for 72 h.
Proliferation of either CD4+ or CD8+
responder T-cells was measured with a FACSCalibur and analyzed with
CellQuest software (BD Biosciences).
In vivo studies of the tumor lysate
vaccine
As shown in Fig.
1A, high anti-Gal KO mice (n=90) were generated by immunization
with pig kidney fragments, then vaccinated with pt-lysate (n=30),
α-gal-t-lysate (n=30) or α-gal-whole-c (n=30). One week after the
last vaccination, splenocytes were prepared from successfully
vaccinated donor KO mice and then suspended in warm (37°C), sterile
RPMI complete medium containing 50 μM of 2-mercaptoethanol. For
adoptive transfer, these isolated splenocytes were transferred by
i.p. injection into NOD/SCID mice three times at 3-day intervals
(75-150×106 cells/vaccinated KO mouse). Splenocytes
obtained from pt-lysate-, α-gal-t-lysate- or
α-gal-whole-c-vaccinated KO mice were injected in equal amounts
into NOD/SCID recipient mice (in total, 90×106
splenocytes were transferred; each group, n=10 transferred NOD/SCID
mice). One day after adoptive transfer, all NOD/SCID mice were
challenged with subcutaneous injection of either 10×106
live PANC1 cells or 5×106
CD44+CD24+ PANC1 cells (i.e., the pancreatic
CSC fraction of PANC1 cells) (14). Subsequently, these mice were
examined for both tumor growth and survival. All mice were
monitored every day after injection to detect the changes of
general signs. Mice were sacrificed at the humane endpoints defined
as following changes: i) physical appearance (self-injury, soiling
of hair with urine of faces, bleeding, severe body weight loss
defined by >20% loss in maximal body weight and loss of
appetite); ii) clinical physiology (tachypnea and low body
temperature). When remarkable increase of tumor size, defined by
>10% increase in body weight was observed, mice were humanely
sacrificed. The mice were induced deep anesthesia by isoflurane and
subsequently sacrificed by cervical dislocation.
Statistical analysis
Data were collected from at least five independent
experiments. Quantitative data were expressed as the mean ± SD.
Statistical analysis was performed using the Student’s t-test.
Kaplan-Meier curves of estimated survival were generated, and
comparisons between parental PANC1 tumor lysate, α-gal PANC1 tumor
lysate, and α-gal PANC1 whole cell vaccine groups were performed
using a two-sided log rank test. A P-value <0.05 was considered
significant.
Results
Generation of a tumor lysate vaccine
expressing α-gal epitopes
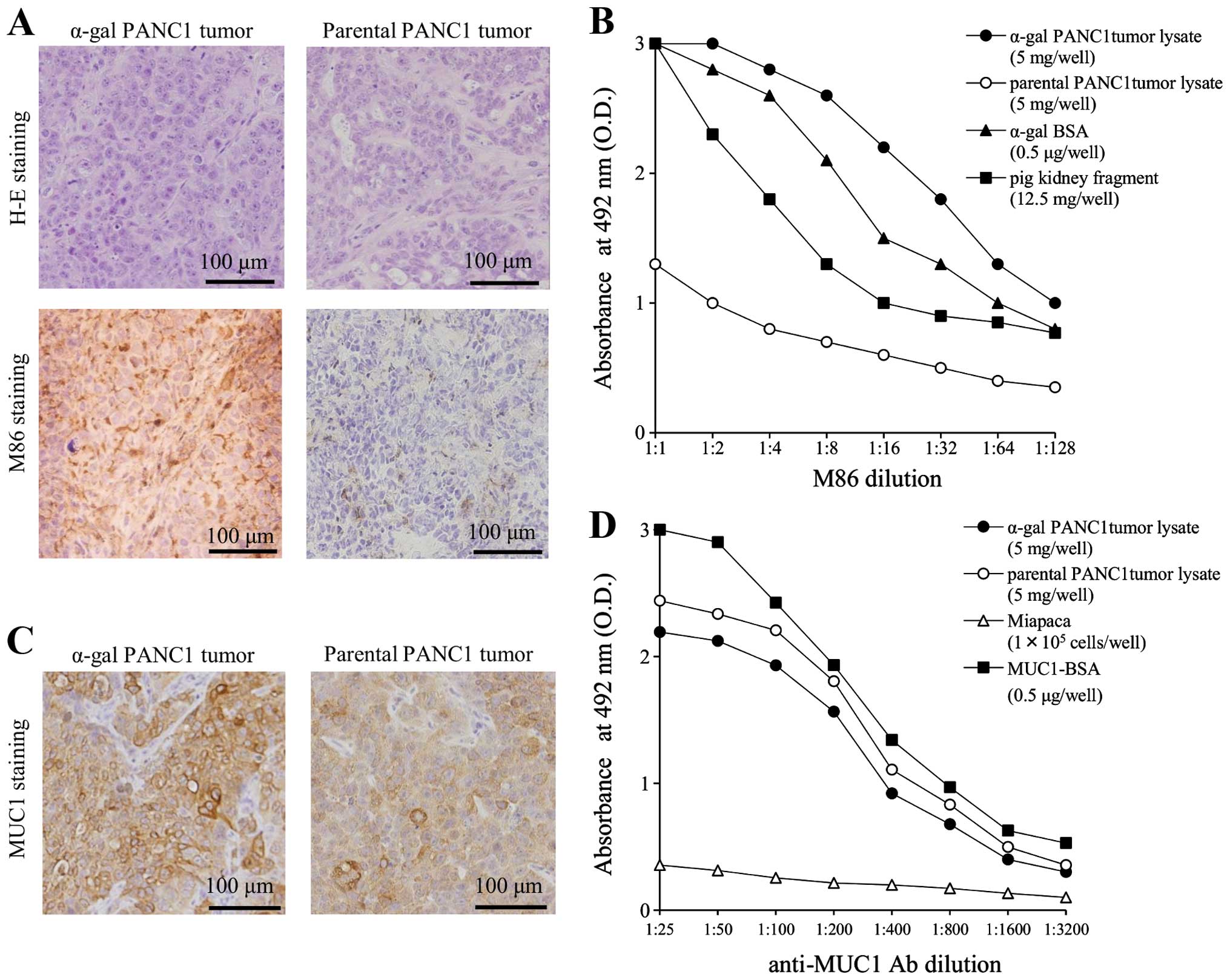
The histological findings of the PANC1 tumors
originating from parental and α-gal PANC1 cells were compatible
with those of human pancreatic cancer (Fig. 2A). Low expression levels of α-gal
epitopes were observed in the parental PANC1 tumor, whereas high
expression levels were detected on the cell surface of α-gal PANC1
tumors (Fig. 2A). The low
expression of α-gal epitopes in parental PANC1 tumor was likely
dependent on the migration of stromal tissues, including vascular
and fibrous cells that originated from recipient NOD/SCID mice.
We previously reported the expression of
5×1013 α-gal epitopes/mg-lysate in pig kidney fragments
(9,19). ELISA determined that approximately
2×1014 α-gal epitopes/mg-lysate were expressed in
α-gal-t-lysate (Fig. 2B). For
α-gal-whole-c, similar levels of α-gal epitope expression were
detected (~2×109 α-gal epitopes/cell). A BCA protein
assay was performed to assess the accurate protein concentration of
tumor lysates or α-gal-whole-c. The protein concentration was
approximately 1 mg/ml for both tumor lysates and α-gal-whole-c.
Therefore, 100 mg of glycoprotein/i.p. injection, expressing
2×1015 α-gal epitopes, was contained in either the 10 mg
α-gal-t-lysate or 1×106 α-gal-whole-c vaccination given
to high anti-Gal KO mice. Similar levels of MUC1 expression were
observed in parental and α-gal PANC1 tumors by both
immunohistochemical staining and ELISA (Fig. 2C and D). Similar levels of MUC1
expression were also observed in α-gal-whole-c (data not
shown).
Vaccination with α-gal PANC1 tumor lysate
induces an effective antitumor immune response of both B- and
T-cells
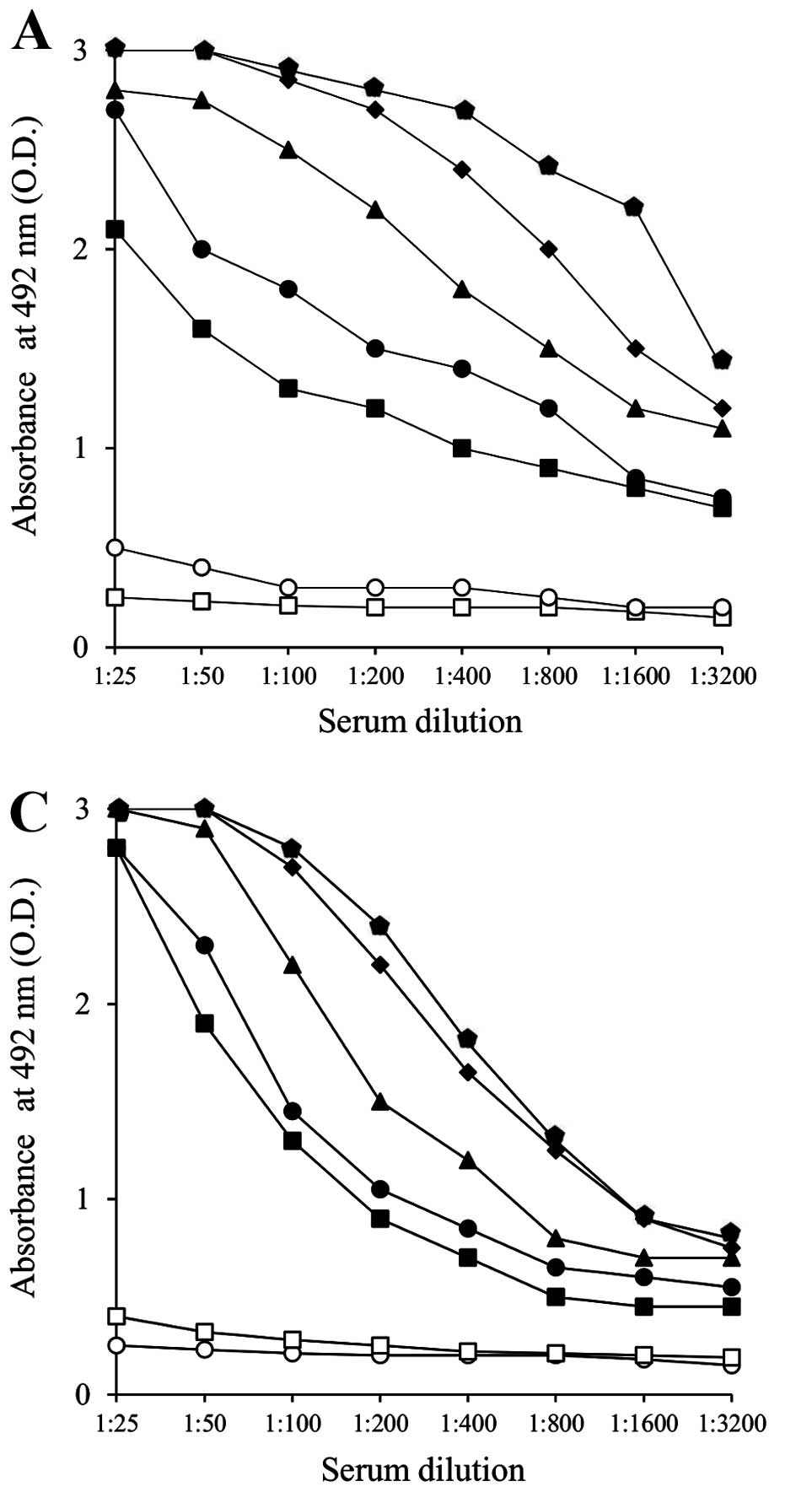
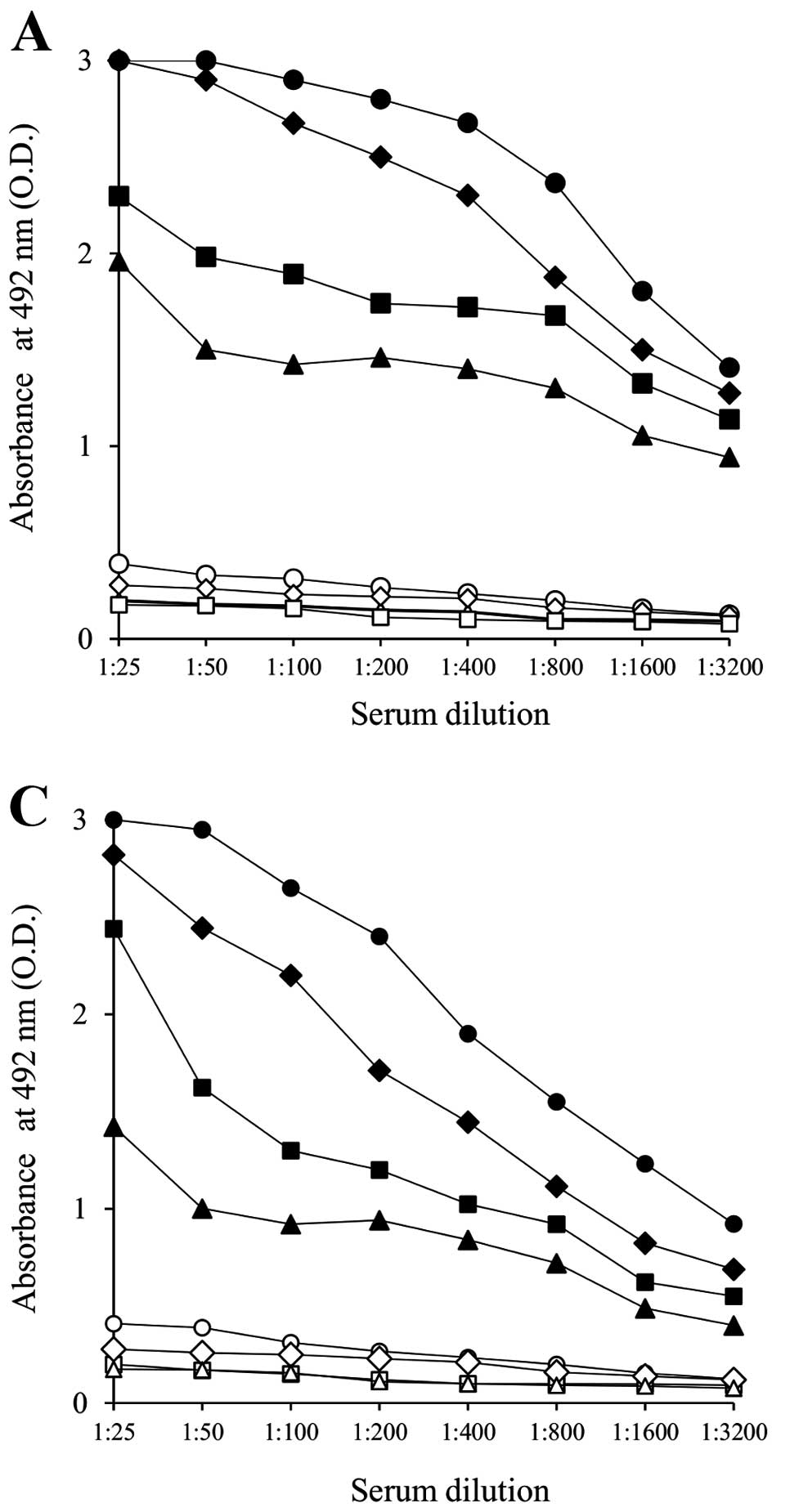
As shown in Fig. 3
(A and B; anti-PANC1 IgG response, C and D; anti-MUC1 IgG
response), repeated vaccinations (five times) with 10 mg/i.p.
injection of α-gal-t-lysate elicited strong responses of anti-PANC1
IgG and anti-MUC1 IgG. Vaccinations with pt-lysate did not induce
these Ab responses. Vaccination with the α-gal-t-lysate elicited an
~16-fold increase in both anti-PANC1 IgG and anti-MUC1 IgG
production, compared with the pt-lysate vaccination. There was
~2–4-fold higher production of anti-PANC1 IgG observed in sera from
α-gal-t-lysate vaccinated KO mice than detected after vaccination
with α-gal-whole-c; however, there were no differences in anti-MUC1
IgG production (data not shown).
To further investigate the subclass of
immunoglobulin reactivity of either anti-PANC1 IgG or anti-MUC1
IgG, we performed an ELISA using HRP-conjugated goat anti-mouse
IgG1, IgG2a, IgG2b and IgG3 mAbs as secondary antibodies. All
secondary antibodies were purchased from Bethyl Laboratories Inc.
Sera from α-gal-t-lysate-vaccinated KO mice showed large amounts of
all IgG subclasses, including IgG1, IgG2b, IgG2a and IgG3. The IgG1
subclass of both anti-PANC1 and anti-MUC1 IgG was especially
expressed, and induces strong antitumoral cytolysis through
antibody-dependent cell-mediated cytotoxicity and
complement-dependent cytotoxicity (Fig. 4A and C). On the other hand, sera
from pt-lysate-vaccinated KO mice produced only a small amount of
IgG1 and did not produce the IgG2a, IgG2b and IgG3 subclasses
(Fig. 4B and D). With the
α-gal-whole-c vaccination, there was no production of the IgG2a
subclass of either anti-PANC1 or anti-MUC1 IgG in sera despite
production of large amounts of the other IgG subclasses (data not
shown) (12).
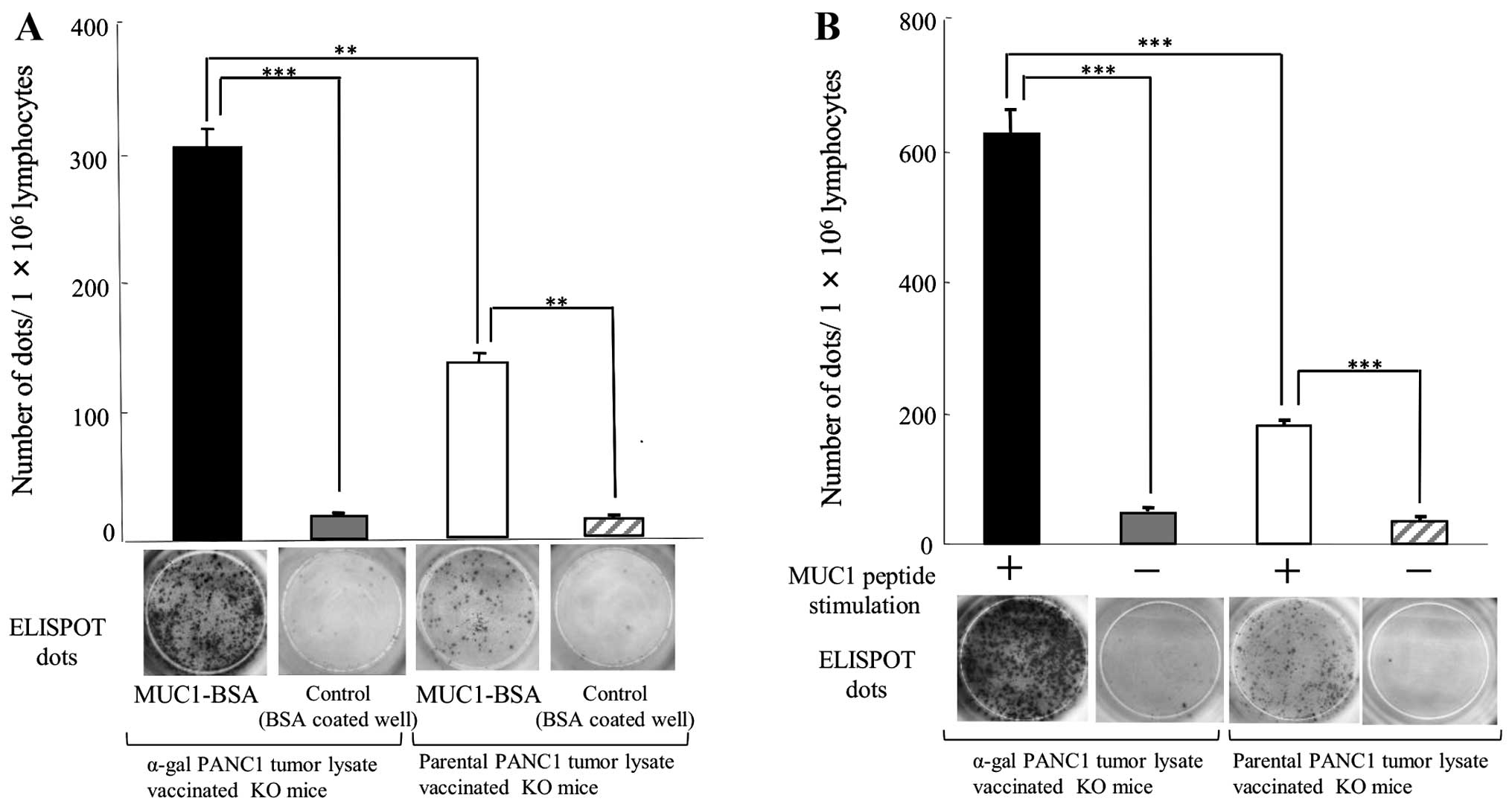
As shown in Fig.
5A, splenocytes isolated from pt-lysate-vaccinated KO mice
displayed 136.7±13.2 spots/1×106 splenocytes of
anti-MUC1-secreting B cells. In contrast, α-gal-t-lysate-vaccinated
KO mice had 305.3±44.0 spots/1×106 splenocytes
(P=0.0071). In pt-lysate-vaccinated KO mice, we detected 181.7±27.5
and 44.3±6.5 spots of IFN-γ secreting T cells in the presence and
absence of the MUC1 stimulator peptide, respectively; thus, a
significant increase in the number of spots was observed with MUC1
peptide stimulation (P=0.0011; Fig.
5B). In α-gal-t-lysate-vaccinated α1,3GT KO mice, 626.7±118.6
and 76.3±12.9 spots were detected with or without MUC1 peptide
stimulation, respectively, and the difference in the number of
spots was also significant (P=0.0013; Fig. 5B). Furthermore, the number of spots
in the presence of the MUC1 peptide was significantly higher in the
α-gal-t-lysate-vaccinated group than in the pt-lysate group
(P=0.0032; Fig. 5B).
Immune response in α-gal PANC1 tumor
lysate-vaccinated α1,3GT KO mice is specific against MUC1 peptide,
PANC1 cells and PANC1 tumor lysate
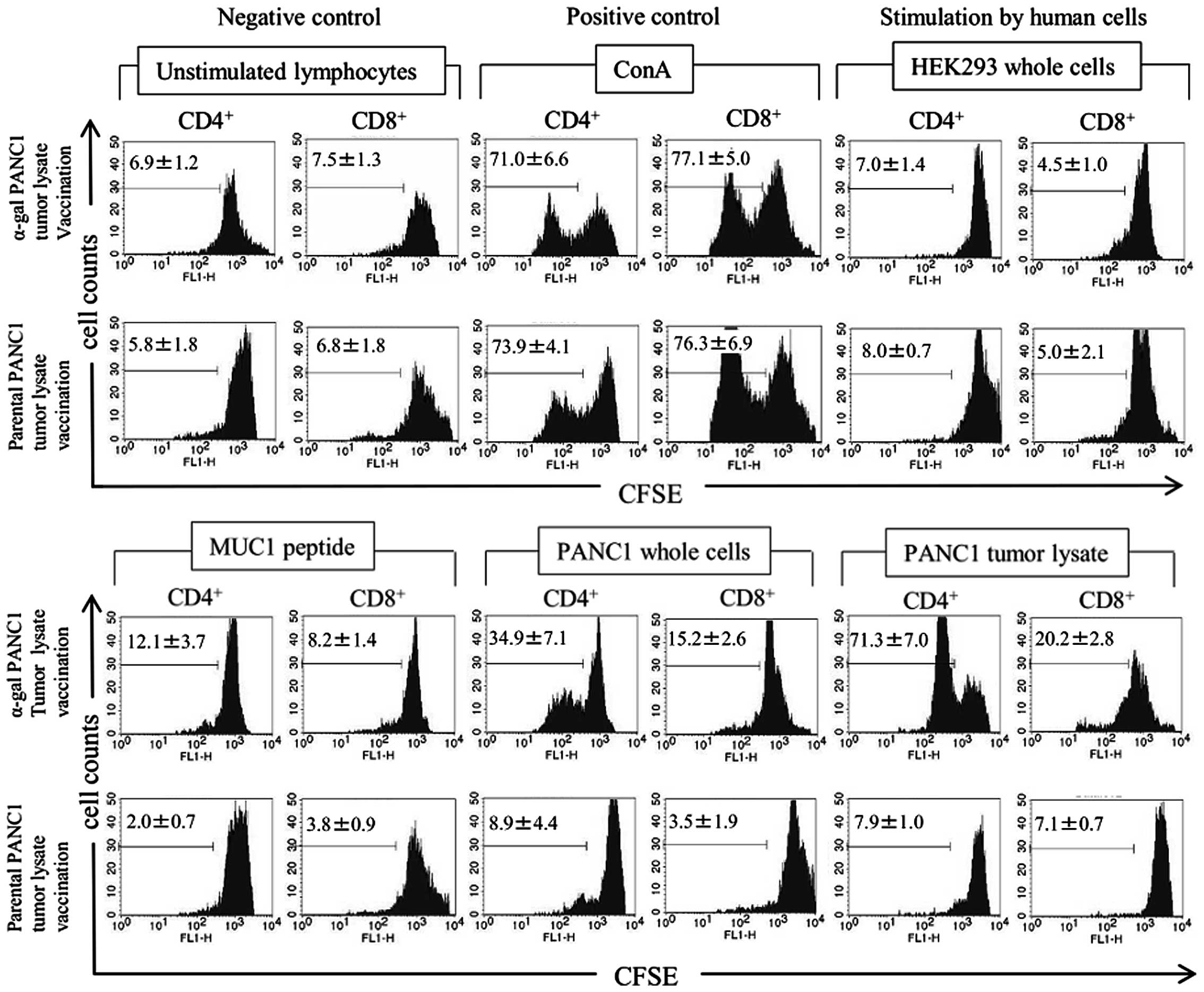
As shown in Fig. 6,
the negative control showed no significant difference in the
percentage of proliferated T cells, which appeared as CFSE-low
responder T cells (i.e., the CFSE intensity was <400), between
α-gal-t- and pt-lysate-vaccination. The positive control
(Concanavalin A stimulation) also showed no significant differences
in proliferated T cells. Proliferation of T cells was significantly
induced in the presence of PANC1 whole cells, PANC1 tumor lysate
and MUC1 peptide; whereas, no proliferation was elicited by HEK293
whole cell stimulation. Lymphocytes were also stimulated with other
kinds of irradiated cells, including monkey COS7 cells and mice
fibroblast NIH3T3 cells. However, these types of stimulatory cells
failed to induce significant proliferation (data not shown).
Moreover, the proliferation rate of T cells in
α-gal-t-lysate-vaccinated α1,3GT KO mice was significantly higher
than in pt-lysate-vaccinated α1,3GT KO mice (Fig. 6).
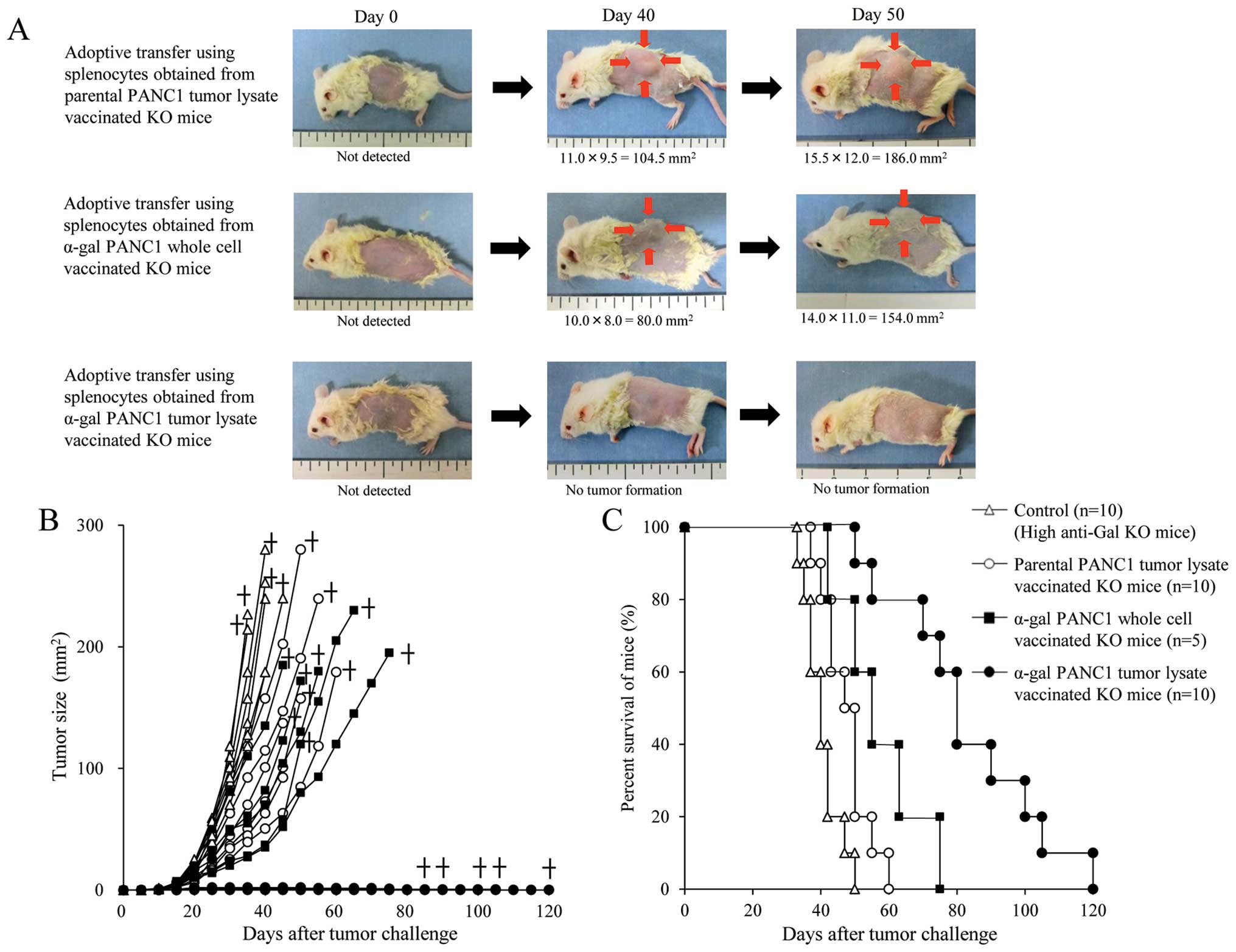
Adoptive transfer of splenocytes from
α-gal PANC1 tumor lysate-vaccinated α1,3GT KO mice induces an
effective anti-tumor response in NOD/SCID mice
The experimental design of in vivo studies is
shown in Fig. 1A. To confirm the
production of anti-PANC1 and anti-MUC1 IgG Abs in adoptively
transferred NOD/SCID mice, we performed an ELISA prior to tumor
challenge (Fig. 1B and C). Sera
from control NOD/SCID mice (without adoptive transfer of
splenocytes) showed no anti-PANC1 and anti-MUC1 IgG Ab production;
while, NOD/SCID mice who received pt-lysate-vaccinated splenocytes
showed small amounts of anti-PANC1 IgG Ab (Fig. 1B and C). In contrast, extremely
large amounts of both anti-PANC1 and anti-MUC1 IgG Abs were noted
in NOD/SCID mice who received α-gal-t-lysate-vaccinated-splenocytes
(Fig. 1B and C). Representative
pictures of mice treated with α-gal-whole-c, α-gal-t-lysate or
pt-lysate are shown in Fig. 7A.
Compared with untreated control mice (data not shown), pt-lysate-
and α-gal-whole-c-vaccinated mice developed large tumors; while, no
tumors were noted in the α-gal-t-lysate-vaccinated mice (Fig. 7A and B). The in vivo
results, including survival time, are summarized in Table I. As shown in Fig. 7B, we monitored tumor growth in
splenocyte-transferred mice. No significant differences in the time
to appearance of palpable tumor after tumor challenge were observed
in either the untreated control or pt-lysate group (untreated,
10.6±2.5 days; pt-lysate, 11.9±2.1 days). In contrast, the
development of tumors in the α-gal-whole-c-vaccination group was
significantly delayed compared with the untreated and pt-lysate
groups (α-gal-whole-c: 16.0±2.8 days, P=0.018 vs. control; P=0.004
vs. pt-lysate). In the untreated control group, the maximum tumor
size was 100 mm2 within 29 to 34 days (mean, 31.4±2.1
days). In comparison, tumor growth to a similar size was markedly
delayed in both the pt-lysate group (40.3±6.9 days, P=0.007 vs.
control) and α-gal-whole-c group (45.6±8.3 days, P=0.0013 vs.
control). The beneficial effects of vaccination with pt-lysate,
α-gal-t-lysate, or α-gal-whole-c were also noted in the
prolongation of survival after tumor challenge (Fig. 7C). As shown in Fig. 7C and Table I, the mean survival time of KO mice
vaccinated with α-gal-t-lysate was markedly prolonged (82.5±21.9
days) compared with non-vaccinated (41.0±5.7 days, P<0.001),
pt-lysate-vaccinated (48.0±6.7 days, P<0.001), and
α-gal-whole-c-vaccinated KO mice (57.0±12.6 days, P=0.01). The
final cause of death for adoptively transferred NOD/SCID mice from
non-vaccinated, pt-lysate-vaccinated and α-gal-whole-c-vaccinated
KO mice were indicated as cancer death, whereas mice transferred
from α-gal-t-lysate-vaccinated KO mice died a natural death without
the appearance of cancer. Notably, the mean survival time was
significantly improved in the pt-lysate-vaccinated group compared
with the non-vaccinated group (P=0.02), despite the lack of
synthesis of α-gal epitopes in the tumor lysate vaccine.
 | Table IThe in vivo antitumor response
against live parental PANC1 cells in adoptive transferred NOD/SCID
mice. |
Table I
The in vivo antitumor response
against live parental PANC1 cells in adoptive transferred NOD/SCID
mice.
| Type of vaccination
(n) | Control mice (no
vaccination) (n=10) | Parental PANC1
tumor lysate (n=10) | α-gal PANC1 whole
cell (n=5) | α-gal PANC1 tumor
lysate (n=10) |
|---|
| Time to appearance
of a palpable tumor (Mean ± SD, days) | 10.6±2.5a,b | 11.9±2.1c | 16.0±2.8 | No tumor
formation |
| Time to tumor size
reaching 100 mm2 (Mean ± SD, days) | 31.4±2.1d,e | 40.3±6.9f | 45.6±8.3 | No tumor
formation |
| Mean survival time
(Mean ± SD, days) | 41.0±5.7g,h,i | 48.0±6.7j,k | 57.0±12.6l | 82.5±21.9 |
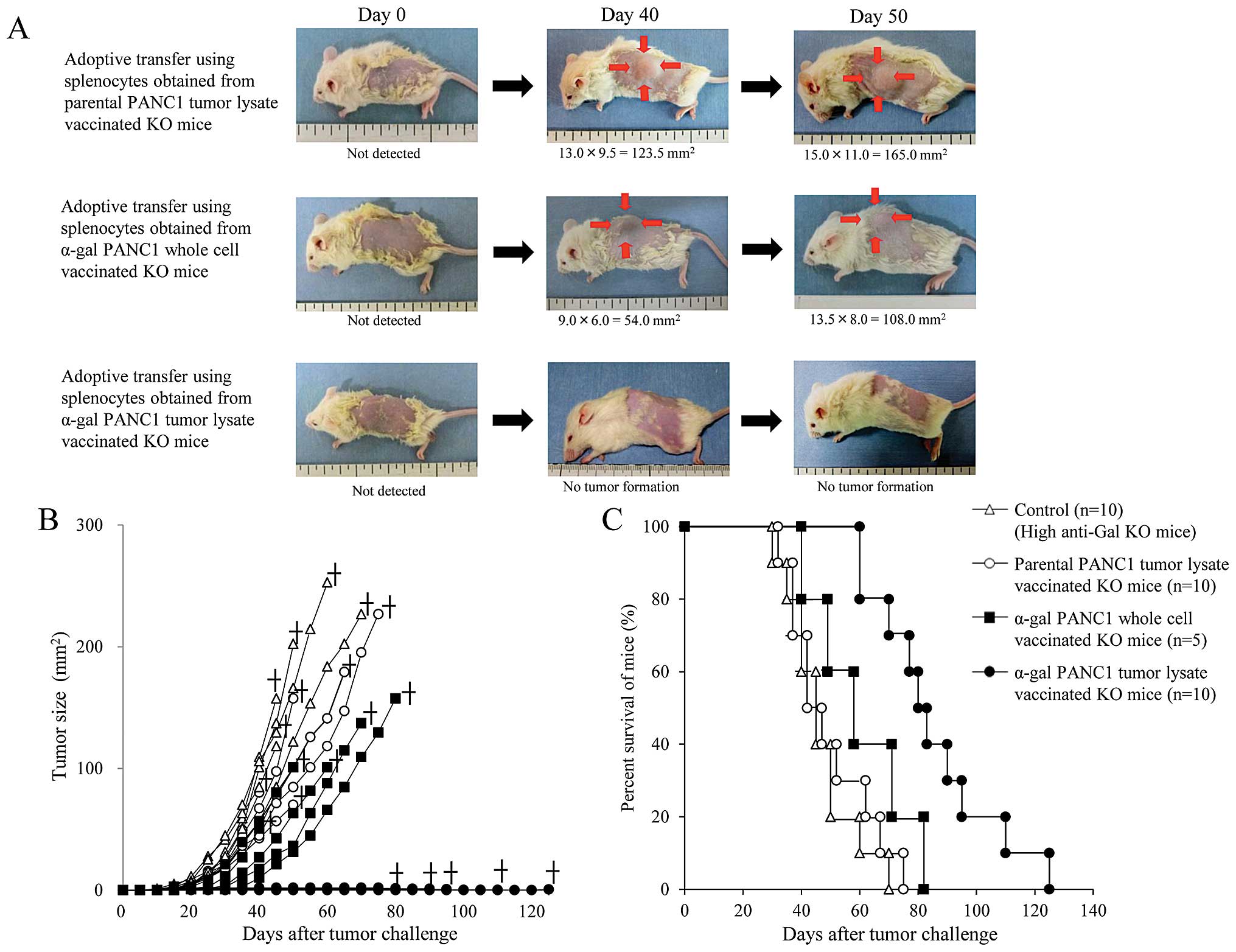
α-gal PANC1 tumor lysate vaccine protects
and prolongs survival of NOD/SCID mice harboring pancreatic cancer
stem cell tumors
Compared with untreated control mice (data not
shown), pt-lysate- or α-gal-whole-c-vaccinated mice developed large
tumors, but the tumorigenesis of pancreatic CSCs was completely
prevented in all α-gal-t-lysate-vaccinated mice (Fig. 8A and B). With the exception of the
α-gal-t-lysate group, there were no significant differences in the
time to appearance of palpable tumors after tumor challenge among
the groups (untreated, 13.1±3.3 days; pt-lysate, 14.4±3.4 days;
α-gal-whole-c, 17.0±3.8 days) (Table
II). The tumor size reached 100 mm2 in 40.6±1.8 and
48.0±4.4 days in the untreated and pt-lysate groups, respectively;
while, tumor growth to a similar size was significantly delayed in
the α-gal-whole-c group, (60.5±7.9 days; P<0.001, vs. control;
P=0.033, vs. pt-lysate) (Fig. 8B,
Table II). However, vaccination
with pt-lysate and α-gal-whole-c did not prolong the survival time
after tumor challenge (49.3±14.3 and 60.0±16.8 days, respectively),
compared with the non-vaccinated control mice (46.5±11.8 days)
(Fig. 8C, Table II). The final causes of death for
these mice were indicated as cancer death. In contrast, vaccination
using α-gal-t-lysate significantly improved survival after tumor
challenge and these treated mice died a natural death without the
appearance of cancer (85.0±20.8 days; P<0.001 vs. control;
P=0.002 vs. pt-lysate; P=0.018 vs. α-gal-whole-c) (Fig. 8C, Table II). The mean survival time was not
significantly different between mice vaccinated with pt-lysate and
the non-vaccinated control group (Fig.
8C, Table II), despite the
beneficial effects seen with live parental PANC1 cells.
| Table IIThe in vivo antitumor response
against pancreatic cancer stem cells in the adoptive transfer
NOD/SCID mice. |
Table II
The in vivo antitumor response
against pancreatic cancer stem cells in the adoptive transfer
NOD/SCID mice.
| Type of vaccination
(n) | Control mice (no
vaccination) (n=10) | Parental PANC1
tumor lysate (n=10) | α-gal PANC1 whole
cell (n=5) | α-gal PANC1 tumor
lysate (n=10) |
|---|
| Time to appearance
of a palpable tumor (Mean ± SD, days) | 13.1±3.3a,b | 14.4±3.4c | 17.0±3.8 | No tumor
formation |
| Time to tumor size
reaching 100 mm2 (Mean ± SD, days) | 40.6±1.8d,e | 48.0±4.4f | 60.5±7.9 | No tumor
formation |
| Mean survival time
(Mean ± SD, days) | 46.5±11.8g,h,i | 49.3±14.3j,k | 60.0±16.8l | 85.0±20.8 |
Vaccination with α-gal PANC1 tumor lysate
induces production of antibodies against parental PANC1 and
CD44+CD24+ isolated PANC1 cells
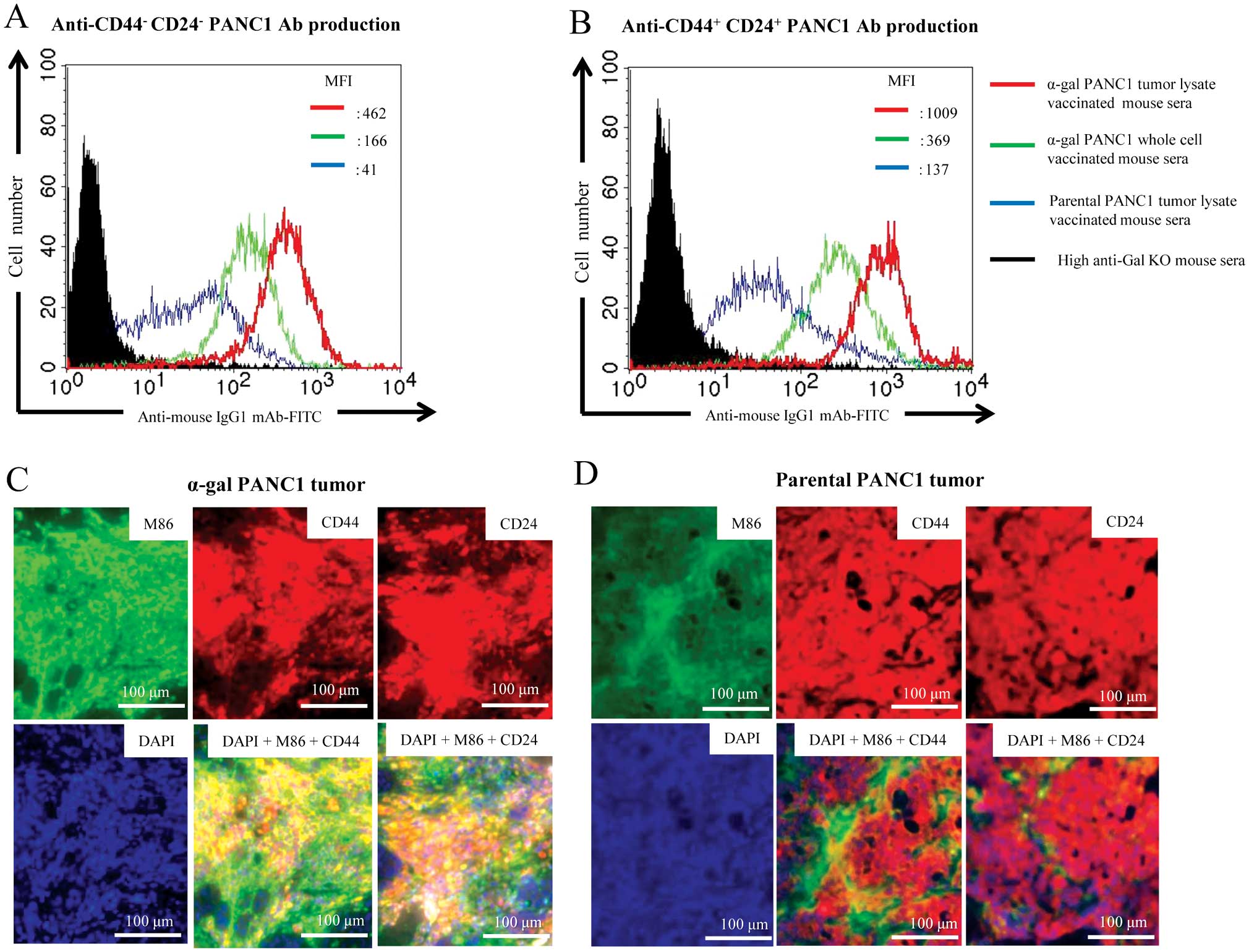
As shown in Fig.
9A, sera from both the α-gal-whole-c and α-gal-t-lysate groups
more strongly bound to CD44−CD24− PANC1 cells
than those from the pt-lysate group, as judged by the mean
fluorescence intensity. There was strong Ab production against
pancreatic CSCs (i.e., CD44+ CD24+ isolated
PANC1 cells) elicited by vaccination with α-gal-whole-c and
α-gal-t-lysate (Fig. 9B).
Importantly, vaccination with α-gal-t-lysate induced better Ab
production against both CD44−CD24− PANC1
cells and pancreatic CSCs than with α-gal-whole-c, as judged by the
mean fluorescence intensity (Fig. 9A
and B).
Evaluation of α-gal epitope expression (M86
staining) revealed there was abundant expression of α-gal epitopes
in α-gal PANC1 tumors; whereas, expression of α-gal epitopes was
scarcely observed in parental PANC1 tumors (Fig. 9C and D). Both CD44 and CD24
molecules were expressed in >90% of cancer cells in PANC1 tumors
on NOD/SCID mice. The expression levels of these CSC markers in
PANC1 tumor cells were markedly upregulated in comparison with the
levels in either α-gal or parental PANC1 cells (12,13).
Thus, the CSC components in PANC1 cells were enriched upon tumor
formation in NOD/SCID mice. In α-gal PANC1 tumor tissue, merged
microphotographs showed tissues stained positive for both M86 and
CD44 as well as for both M86 and CD24; thus, the tissues
simultaneously expressed the α-gal epitopes and CD44 or CD24 on the
cell surface (Fig. 9C, yellow
regions). However, no yellow regions were observed in parental
PANC1 tumor tissue (Fig. 9D).
These results suggest that the buildup of α-gal epitopes on the
carbohydrates of CSC-related molecules allows the internalization
and antigen-presentation of these molecules by APC. Furthermore,
the concentration of CSC-related molecules in the tumor lysate
vaccine seems greater than in the whole cell vaccine (i.e., α-gal
PANC1 whole cell vaccine).
Discussion
The three main findings of the present study were:
i) tumor lysate vaccines elicited strong antibody production
against pancreatic cancer cells, the MUC1 peptide, and
CD44+CD24+ PANC1 cells, and the latter were
isolated as a pancreatic CSC population; ii) tumor lysate
vaccination led to effective activation of T cells specific to both
the MUC1 peptide and endogenous TAA molecules derived from
pancreatic cancer cells; and iii) in vivo experiments on
challenge with either live pancreatic cancer cells or a
CD44+CD24+ pancreatic CSC population
demonstrated an immune response was induced that completely
prevented tumor development at local sites in the adoptive
transferred NOD/SCID mice. Moreover, the immune response against
live tumor cells, elicited by α-gal PANC1 tumor lysate vaccination,
was significantly stronger than that induced by the α-gal PANC1
whole cell vaccination.
For clinical application of this effective
immunotherapy, we need to assess the toxicity and safety of
injection of α-gal tumor lysate in humans. The major concern before
the start of the present study was that effective uptake of
anti-Gal opsonized tumor lysate by APC might induce an immune
response against both normal antigens of the tumor lysate and
normal cells, such as stromal cells in the tumor. Previous clinical
trials using lysate or whole cancer cells as a source of vaccine
showed no clinically relevant autoimmune responses (22–24).
This conclusion should be further examined in humans to verify
whether there is a lack of clinical evidence of auto-immunity
induced by α-gal tumor lysate vaccination. Although we plan to
primarily employ autologous tumor lysate, which is surgically
resected from patients with pancreatic cancer and enzymatically
processed in vitro to express α-gal epitopes, as the
vaccinating material (25), the
volume of tumor mass in the resected pancreas is small and limited.
Actually, the vast majority of patients are diagnosed as inoperable
because they present with incurable metastatic disease. To overcome
this critical situation, we propose to generate the tumors in mice
to create vaccinating material, as in the present study.
Pancreatic cancer-associated antigens that are
candidates for potential immune targeting include Her2/neu
(26), MUC1 (27), CEA (4), mesothelin (5,24),
telomerase (28) and survivin
(29). However, vaccinating
against a single antigen is disadvantageous because it is not known
what exact antigen can potentially induce a more effective
antitumor immune response. Furthermore, immunity against a single
antigen may be ineffective in tumors with heterogeneous cell
populations and carries the risk of inducing tumor antigen escape
variants (30,31). However, this strategy is sometimes
applicable to those patients with a specific HLA type. To overcome
the drawbacks of single antigen immunotherapy, several groups used
multiple-antigen vaccine platforms and reported successful
induction of antigen-specific immune responses (32,33).
However, these studies were conducted in animal models of tumors or
in in vitro (32,33). The use of unfractionated
tumor-derived antigens in the form of tumor lysates circumvents
these disadvantages because tumor lysates contain multiple known
and unknown antigens that can be presented to T cells by both MHC
class I- and class II-pathways (34–36).
Therefore, effective uptake of α-gal tumor lysate by APC is more
likely to induce a polyclonal expansion of T cells, including MHC
class II-restricted T-helper cells. These cells have been
recognized to play an important role in the activation of
CD8+ CTLs, probably the most important cells in any
antitumor immune response (22,23,30).
The generation of CTL clones with multiple specificities may be an
advantage in heterogeneous tumors and could also reduce the risk of
tumor escape variants.
The lethal nature of pancreatic cancer is due to the
ability of remnant cells, including differentiated cancer cells and
CSCs after surgery, chemotherapy and radiation therapy, to develop
into recurrent or metastatic tumors. However, these remnant
residual cancer cells might be destroyed by strong activation of
immunocytes, induced by vaccination with the α-gal tumor lysate
that can specifically attack and destroy TAA-expressing tumor
cells. The most encouraging results of immunotherapy in pancreatic
cancer have been in adjuvant settings, such as post-surgery
(37,38). Moreover, due to genome instability
and the heterogeneity in pancreatic cancer, the immunological
setting for the destruction of TAA-expressing tumor cells
frequently results in the appearance and expansion of tumor cell
subclones with no or low expression of the specific TAA (39–41).
Our previous study demonstrated the effect of using tumor cells as
a vaccine source to inhibit the development of transplanted
melanoma cell tumors in mice (12). However, the inhibition of tumor
formation was not complete. The weakness of such vaccine therapy
could be related to the use of melanoma cells rather than
pancreatic cancer cells, or the use of a less than optimal vaccine
therapy to overcome various types of CSCs due to the presence of
only a few TAAs in the whole cell vaccine. To achieve complete
destruction of CSCs, it may be necessary to target the tumor
microenvironment as well as tumor cells themselves. For this
purpose, tumor tissue lysate seems to offer a better option than
tumor cell lysate as a source of vaccine. A polyvalent tumor lysate
vaccine, engineered to express α-gal epitopes and prepared from
autologous tumors, is the most suitable material for immunotherapy.
Notably, recent studies demonstrated that the heterogeneity of
metastases reflects heterogeneity already existing within the
primary tumor, and that the primary carcinoma is a mixture of
numerous subclones, each of which independently expands to form a
large number of cells (42,43).
In summary, we plan to employ autologous tumor
lysate prepared from surgically resected pancreas cancer, which is
enzymatically processed in vitro to express α-gal epitopes,
as vaccinating material; although, the tumor mass in the resected
pancreas is often small and limited. The vast majority of patients
are diagnosed as inoperable because they present with incurable
metastatic disease. To overcome this problem, we propose using
tumors generated in mice as candidate vaccination material. We hope
that the use of a tumor lysate vaccine, engineered to express α-gal
epitopes, can elicit a strong immune response toward all pancreatic
cancer cells, including differentiated pancreatic cancer cells and
pancreatic CSCs, and may improve the prognosis for patients with
pancreatic cancer.
Acknowledgements
The authors thank Professor Uri Galili from the
University of Massachusetts Medical School (Worcester, MA) for
generously providing α1,3GT KO mice, and Dr Haruko Ogawa from
Obihiro University of Agriculture and Veterinary Medicine (Obihiro,
Hokkaido) for generously providing plasmids encoding the murine
α1,3GT gene. The authors thank Dr Paul Kretchmer, (San Francisco
Edit, CA, USA) for the careful reading and editing of the
manuscript. The present study was supported by a grant from the
Ministry of Education, Sports, and Culture of Japan to Masahiro
Tanemura (no. 22591520). The study was also supported in part by a
Takamatsuno-miya cancer grant to Eiji Miyoshi (no. 27324).
Abbreviations:
|
Ab
|
antibody
|
|
α1,3GT
|
α1,3-galactosyltransferase
|
|
α-gal epitopes
|
Galα1-3Galβ1-4 GlcNAc-R
|
|
anti-Gal
|
anti-Gal IgG antibody
|
|
APCs
|
antigen presenting cells
|
|
CSCs
|
cancer stem cells
|
|
CTLs
|
cytotoxic T lymphocytes
|
|
ELISA
|
enzyme-linked immunosorbent assay
|
|
ELISPOT
|
enzyme-linked immunospot
|
|
i.p.
|
intraperitoneal
|
|
KO
|
knockout
|
|
mAb
|
monoclonal antibody
|
|
MUC1
|
mucin-1
|
|
NOD/SCID mice
|
non-obese diabetic/severe combined
immunodeficiency mice
|
|
TAAs
|
tumor-associated antigens
|
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Lepisto AJ, Moser AJ, Zeh H, et al: A
phase I/II study of a MUC1 peptide pulsed autologous dendritic cell
vaccine as adjuvant therapy in patients with resected pancreatic
and biliary tumors. Cancer Ther. 6:955–964. 2008.PubMed/NCBI
|
|
3
|
Gjertsen MK, Bakka A, Breivik J, et al:
Vaccination with mutant ras peptides and induction of T-cell
responsiveness in pancreatic carcinoma patients carrying the
corresponding RAS mutation. Lancet. 346:1399–1400. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Laheru D and Jaffee EM: Immunotherapy for
pancreatic cancer - science driving clinical progress. Nat Rev
Cancer. 5:459–467. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Argani P, Iacobuzio-Donahue C, Ryu B, et
al: Mesothelin is overexpressed in the vast majority of ductal
adenocarcinomas of the pancreas: identification of a new pancreatic
cancer marker by serial analysis of gene expression (SAGE). Clin
Cancer Res. 7:3862–3868. 2001.PubMed/NCBI
|
|
6
|
Liyanage UK, Moore TT, Joo HG, et al:
Prevalence of regulatory T cells is increased in peripheral blood
and tumor microenvironment of patients with pancreas or breast
adenocarcinoma. J Immunol. 169:2756–2761. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Soares KC, Zheng L, Edil B and Jaffee EM:
Vaccines for pancreatic cancer. Cancer J. 18:642–652. 2012.
View Article : Google Scholar
|
|
8
|
Galili U, Clark MR, Shohet SB, Buehler J
and Macher BA: Evolutionary relationship between the natural
anti-Gal antibody and the Gal alpha 1→3Gal epitope in primates.
Proc Natl Acad Sci USA. 84:1369–1373. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tanemura M, Yin D, Chong AS and Galili U:
Differential immune responses to alpha-gal epitopes on xenografts
and allografts: implications for accommodation in
xenotransplantation. J Clin Invest. 105:301–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tanemura M, Saga A, Kawamoto K, et al: In
vitro and in vivo prevention of human CD8+ CTL-mediated
xenocytotoxicity by pig c-FLIP expression in porcine endothelial
cells. Am J Transplant. 8:288–297. 2008. View Article : Google Scholar
|
|
11
|
Galili U and LaTemple DC: Natural anti-Gal
antibody as a universal augmenter of autologous tumor vaccine
immunogenicity. Immunol Today. 18:281–285. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Deguchi T, Tanemura M, Miyoshi E, et al:
Increased immunogenicity of tumor-associated antigen, mucin 1,
engineered to express alpha-gal epitopes: a novel approach to
immunotherapy in pancreatic cancer. Cancer Res. 70:5259–5269. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Z, Li Y, Ahmad A, et al: Pancreatic
cancer: understanding and overcoming chemoresistance. Nat Rev
Gastroenterol Hepatol. 8:27–33. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li C, Heidt DG, Dalerba P, et al:
Identification of pancreatic cancer stem cells. Cancer Res.
67:1030–1037. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
LaTemple DC and Galili U: Adult and
neonatal anti-Gal response in knock-out mice for
alpha1,3-galactosyltransferase. Xenotransplantation. 5:191–196.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Thall AD, Maly P and Lowe JB: Oocyte Gal
alpha 1,3Gal epitopes implicated in sperm adhesion to the zona
pellucida glycoprotein ZP3 are not required for fertilization in
the mouse. J Biol Chem. 270:21437–21440. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sipos B, Moser S, Kalthoff H, Torok V,
Lohr M and Kloppel G: A comprehensive characterization of
pancreatic ductal carcinoma cell lines: towards the establishment
of an in vitro research platform. Virchows Arch. 442:444–452.
2003.PubMed/NCBI
|
|
18
|
LaTemple DC, Henion TR, Anaraki F and
Galili U: Synthesis of alpha-galactosyl epitopes by recombinant
alpha1,3galactosyl transferase for opsonization of human tumor cell
vaccines by anti-galactose. Cancer Res. 56:3069–3074.
1996.PubMed/NCBI
|
|
19
|
Tanemura M, Maruyama S and Galili U:
Differential expression of alpha-GAL epitopes
(Galalpha1–3Galbeta1–4GlcNAc-R) on pig and mouse organs.
Transplantation. 69:187–190. 2000. View Article : Google Scholar
|
|
20
|
Kawamoto K, Tanemura M, Nishida T,
Fukuzawa M, Ito T and Matsuda H: Significant inhibition of human
CD8(+) cytotoxic T lymphocyte-mediated xenocytotoxicity by
overexpression of the human decoy Fas antigen. Transplantation.
81:789–796. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
LaTemple DC, Abrams JT, Zhang SY and
Galili U: Increased immunogenicity of tumor vaccines complexed with
anti-Gal: studies in knockout mice for
alpha1,3-galactosyltransferase. Cancer Res. 59:3417–3423.
1999.PubMed/NCBI
|
|
22
|
Thomas AM, Santarsiero LM, Lutz ER, et al:
Mesothelin-specific CD8+ T cell responses provide
evidence of in vivo cross-priming by antigen-presenting cells in
vaccinated pancreatic cancer patients. J Exp Med. 200:297–306.
2004. View Article : Google Scholar
|
|
23
|
Jaffee EM, Hruban RH, Biedrzycki B, et al:
Novel allogeneic granulocyte-macrophage colony-stimulating
factor-secreting tumor vaccine for pancreatic cancer: a phase I
trial of safety and immune activation. J Clin Oncol. 19:145–156.
2001.
|
|
24
|
Hassan R and Ho M: Mesothelin targeted
cancer immunotherapy. Eur J Cancer. 44:46–53. 2008. View Article : Google Scholar
|
|
25
|
Galili U, Chen ZC and DeGeest K:
Expression of alpha-gal epitopes on ovarian carcinoma membranes to
be used as a novel autologous tumor vaccine. Gynecol Oncol.
90:100–108. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ladjemi MZ, Jacot W, Chardes T, Pelegrin A
and Navarro-Teulon I: Anti-HER2 vaccines: new prospects for breast
cancer therapy. Cancer Immunol Immunother. 59:1295–1312. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tang CK, Katsara M and Apostolopoulos V:
Strategies used for MUC1 immunotherapy: human clinical studies.
Expert Rev Vaccines. 7:963–975. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu JP, Chen W, Schwarer AP and Li H:
Telomerase in cancer immunotherapy. Biochim Biophys Acta.
1805:35–42. 2010.PubMed/NCBI
|
|
29
|
Ryan BM, O’Donovan N and Duffy MJ:
Survivin: a new target for anti-cancer therapy. Cancer Treat Rev.
35:553–562. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Saito H, Dubsky P, Dantin C, Finn OJ,
Banchereau J and Palucka AK: Cross-priming of cyclin B1, MUC-1 and
survivin-specific CD8+ T cells by dendritic cells loaded
with killed allogeneic breast cancer cells. Breast Cancer Res.
8:R652006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schnurr M, Scholz C, Rothenfusser S, et
al: Apoptotic pancreatic tumor cells are superior to cell lysates
in promoting cross-priming of cytotoxic T cells and activate NK and
gammadelta T cells. Cancer Res. 62:2347–2352. 2002.PubMed/NCBI
|
|
32
|
Bohnenkamp HR, Coleman J, Burchell JM,
Taylor-Papadimitriou J and Noll T: Breast carcinoma cell
lysate-pulsed dendritic cells cross-prime MUC1-specific
CD8+ T cells identified by peptide-MHC-class-I
tetramers. Cell Immunol. 231:112–125. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Palucka AK, Ueno H, Connolly J, et al:
Dendritic cells loaded with killed allogeneic melanoma cells can
induce objective clinical responses and MART-1 specific
CD8+ T-cell immunity. J Immunother. 29:545–557. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fields RC, Shimizu K and Mule JJ: Murine
dendritic cells pulsed with whole tumor lysates mediate potent
antitumor immune responses in vitro and in vivo. Proc Natl Acad Sci
USA. 95:9482–9487. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Brossart P and Bevan MJ: Presentation of
exogenous protein antigens on major histocompatibility complex
class I molecules by dendritic cells: pathway of presentation and
regulation by cytokines. Blood. 90:1594–1599. 1997.PubMed/NCBI
|
|
36
|
Shen Z, Reznikoff G, Dranoff G and Rock
KL: Cloned dendritic cells can present exogenous antigens on both
MHC class I and class II molecules. J Immunol. 158:2723–2730.
1997.PubMed/NCBI
|
|
37
|
Shakhar G and Ben-Eliyahu S: Potential
prophylactic measures against postoperative immunosuppression:
could they reduce recurrence rates in oncological patients? Ann
Surg Oncol. 10:972–992. 2003. View Article : Google Scholar
|
|
38
|
Weighardt H, Heidecke CD, Emmanuilidis K,
et al: Sepsis after major visceral surgery is associated with
sustained and interferon-gamma-resistant defects of monocyte
cytokine production. Surgery. 127:309–315. 2000. View Article : Google Scholar
|
|
39
|
Livingston P: The unfulfilled promise of
melanoma vaccines. Clin Cancer Res. 7:1837–1838. 2001.PubMed/NCBI
|
|
40
|
Khong HT and Restifo NP: Natural selection
of tumor variants in the generation of ‘tumor escape’ phenotypes.
Nat Immunol. 3:999–1005. 2002. View Article : Google Scholar
|
|
41
|
Dunn GP, Bruce AT, Ikeda H, Old LJ and
Schreiber RD: Cancer immunoediting: from immunosurveillance to
tumor escape. Nat Immunol. 3:991–998. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Campbell PJ, Yachida S, Mudie LJ, et al:
The patterns and dynamics of genomic instability in metastatic
pancreatic cancer. Nature. 467:1109–1113. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yachida S, Jones S, Bozic I, et al:
Distant metastasis occurs late during the genetic evolution of
pancreatic cancer. Nature. 467:1114–1117. 2010. View Article : Google Scholar : PubMed/NCBI
|