Introduction
Gastric cancer (GC) is a significant health problem
worldwide, and is the second most common cause of cancer-associated
mortality (1). It is well known
that GC results from combination of a complex series of genetic and
epigenetic events. It is reported that epigenetic alteration
contributes to the progression of GC via regulating the expression
of tumor suppressor genes (TSGs). The silencing of TSGs by these
epigenetic regulators is recognized as a vital mechanism in GC
formation (2–4). Therefore, a better understanding of
the abnormal epigenetic modification of GC is key for determining
how to inhibit GC progression effectively.
DNA methylation and histone modifications are key
players in epigenetic modification, and appear to be linked to each
other (5,6). Early studies have shown that DNA
methyl-transferases (DNMTs), together with the methyl-CpG-binding
protein MECP2, localize to DNA-methylated promoters and recruit a
protein complex that contains histone deacetylases (HDACs) and
histone methyltransferases (7–9).
Extinction of DNA methylation affecting H3 methylation and other
histone modifications have also been found in Arabidopsis
and human cells (10,11). These studies suggest DNA
methylation induces chromatin structural changes through alteration
of histone modifications. DNA methylation and histone modifications
likely have a mutually reinforcing relationship and both are
required for stable and long-term epigenetic silencing of TSGs.
Aberrant DNA methylation and histone modifications lead to
downregulation or silencing of TSGs and are often found in CpG-rich
sites, known as CpG islands, located in the promoter region of many
TSGs (12–14). The 5′-end of the PSD gene contains
five CpG islands, suggesting that its expression may be controlled
by epigenetic modification.
In our previous studies (15,16),
genome-wide analysis of histone modifications, using a chromatin
immunoprecipitation microarray (ChIP-chip) investigated inactive
genes in GC, with the aim of identifying targets of genes
controlled by histone modifications. We identified the PSD gene as
a possible TSG in GC. PSD is a guanine nucleotide exchange factor
for ADP-ribosylation factor 6 (15), which regulates the membrane
trafficking of small G proteins (16). It is located on human chromosome
10q24 and encodes a 71-kDa protein (17). Okada et al (18) reported that DNA methylation at the
PSD promoter is more frequently methylated in ulcerative colitis
(UC)-associated colorectal cancer tissues compared to
non-neoplastic UC epithelia. In addition, PSD mRNA is positively
correlated with the methylation status of PSD, however, little is
known about the association between epigenetic alteration and PSD
expression in GC.
In the present study, we investigated PSD expression
in GC cells and tissues, and aimed to verify whether decreased PSD
expression in GC is related to DNA methylation and repressive
histone modification at the PSD promoter. Four GC cell lines and
one normal gastric cell line were used to examine PSD mRNA
expression and epigenetic alteration. In addition, 40 GC tissue
specimens and 40 corresponding non-malignant gastric tissues were
used to observe the mRNA and protein expression of PSD, and its
clinical significance. Suppression of PSD in SGC7901 cells by siRNA
transfection was utilized to detect the function of PSD in GC
progression.
Materials and methods
Cell culture and treatment with
epigenetic agents
GES1, an immortalized normal gastric cell line, was
obtained from the Oncology Institute of China Medical University
(Shenyang, China). Four human GC cell lines, HGC27, AGS, BGC823 and
SGC7901 were obtained from the Institute of Biochemistry and Cell
Biology, Chinese Academy of Sciences (Shanghai, China). These cells
were cultured in RPMI-1640 medium (Gibco BRL, Grand Island, NY,
USA) supplemented with 10% fetal bovine serum (Gibco) and incubated
at 37°C in a humidified 5% CO2 atmosphere. Four GC lines
were incubated in culture medium with 5 μM DNMT inhibitor
5-aza-2′-de-oxycytidine (DAC; Sigma, St. Louis, MO, USA) for 3
days, and 0.3 μM of the histone deacetylase inhibitor trichostatin
A (TSA; Sigma) for 1 day. The time, dose and sequence of DAC and/or
TSA were based on previous studies (19,20).
Tissue samples
Human GC samples were collected from 40 patients who
underwent gastrectomy at Cancer Institute of China Medical
University (Shenyang, China) between January 2009 and June 2011.
All GC cases were pathologically confirmed. Non-malignant gastric
tissues that were ≥5 cm away from the tumor were obtained from the
patients. This study was approved by the Institutional Review Board
of China Medical University.
Chromatin immunoprecipitation (ChIP)
assay
Five million cells were crosslinked with 1%
formaldehyde for 10 min at 37°C, and then 0.125 M glycine was added
to stop the cross-linking. After washing with ice-cold PBS, the
cell pellets were resuspended in lysis buffer, and sonicated to
generate 200–1,000-bp DNA fragments. The lysate was then divided
into three fractions. The first lysate was precipitated using
antibodies against Lys-9 trimethylated histone H3 (05-1242;
Millipore, Billerica, MA, USA), Lys-9 acetylated histone H3
(07-352; Millipore) or Lys-4 trimethylated histone H3 (07-472;
Millipore) at 4°C overnight. The second lysate was incubated with
normal rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA)
as a negative control. The third lysate was used as an input
control. We added protein G-Sepharose beads to collect the
immunoprecipitated complexes and left them to incubate for 1 h at
4°C. After washing, the beads were treated with RNase (50 mg/ml)
for 30 min at 37°C and then proteinase K overnight. The crosslinks
were then reversed by heating the sample at 65°C for 6 h. DNA was
extracted by the phenol/chloroform method, ethanol precipitated,
and resuspended in 20 μl water.
PCR analysis of immunoprecipitated
DNA
A total of 2 μl of immunoprecipitated DNA, DNA input
control and negative control were used for PCR. The following
primer set for PCR were designed to amplify the overlapping
fragments of 190 bp along the PSD promoter: sense: 5′-gactggctt
ctgtc-gtcctc-3′ and antisense: 5′-ggcagacagtaagagcctgg-3′. PCR
products were subjected to 2.5% agarose gel electrophoresis at 120
V for 40 min and quantified using the Fluor Chen 2.0 system
(Bio-Rad, Hercules, CA, USA). For quantitation, PCR amplification
was performed on an ABI 7700 real-time PCR (Applied Biosystems,
Foster City, CA, USA). PCR conditions included an initial
denaturation step of 4 min at 95°C, followed by 35 cycles of 5 sec
at 95°C, 30 sec at 60°C and 20 sec at 72°C. Quantitative ChIP-PCR
values were normalized against values from a standard curve
constructed using input DNA that was extracted for the ChIP
experiment. The ChIP real-time PCR experiments were repeated three
times.
RNA extraction and real-time reverse
transcriptase polymerase chain reaction (RT-PCR)
Total RNA was extracted from cells and tissues with
TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer’s protocol. The quality and concentration of RNA were
measured by ultraviolet absorbance at 260 and 280 nm
(A260/A280 ratio) and checked by agarose gel
electrophoresis individually. Total RNA was reverse transcribed
into cDNA using an Expand Reverse Transcriptase kit (Takara,
Dalian, China). Expression of PSD mRNA was detected using real-time
PCR with the following program: 95°C for 30 sec and 35 cycles of
95°C for 5 sec and 60°C for 30 sec. The reaction mixture contained
12.5 μl SYBR Green (Takara), 1 μl each primer, 2 μl cDNA, and 8.5
μl diethylpyrocarbonate (DEPC)-treated water. Primers used were
5′-CTGGGCAAGAACAATGACTTC-3′ (sense) and 5′-GAGGACAGGGCTTCAGGATT-3′
(antisense) for PSD; and 5′-CATGAGAAGTATGACAACAGCCT-3′ (sense) and
5′-AGTCCTTCCACGATACCAAAGT-3′ (antisense) for
glyceraldehyde-3-phosphate dehydrogenase (GADPH). The negative
control used DEPC-treated water to replace cDNA templates for every
PCR. The PSD level was expressed as Ct after normalization to the
levels of GAPDH mRNA. The experiment was done in triplicate.
PSD gene knockdown by siRNA
Three pairs of the PSD siRNA sequence were designed
and synthesized by GenePharma (Shanghai, China). As a result of
relative effectiveness and stability, the following siRNA sequence
was selected in our experiments: 5′-CCAAGCUCAGGGUGUUUTT-3′, 5′-AAA
CACCCUGAGAGCUUGGTT-3′; it was transfected into SGC7901 cells. A
non-silencing siRNA sequence was used as a negative control
(5′-UUCUCCGAACGUGUCACGUTT-3′, 5′-ACGUGACACGUUCGGAGAATT-3′). siRNA
transfections were performed according to the manufacturer’s
instructions. SGC7901 cells were seeded in 6-well plates in medium
containing 10% serum 24 h before the experiment. PSD or negative
control siRNA of 100 pmol was diluted in 250 μl Opti-MEM I medium.
Diluted siRNA was mixed with diluted Lipofectamine 2000 for 20 min.
The oligomer-Lipofectamine complexes were applied to the SGC7901
cells. Following transfection, the PSD mRNA levels were assessed 48
h later.
Methylation-specific PCR (MSP)
Genomic DNA was extracted from cells and tissues
with phenol-chloroform-isoamyl alcohol and collected by ethanol
precipitation. The bisulfite treatment was performed using the EZ
DNA Methylation-Gold kit (Zymo Research, Los Angeles, CA, USA)
according to the manufacturer’s protocol. The CpG map of the PSD
promoter and the location of primers used in this study were
analyzed by http://www.urogene.org/methprimer/index1.html
(21). We found five CpG islands
in the PSD promoter region. The primers for the methylated PSD CpG
island were 5′-GTTG TAGGGAAGCGGTTC-3′ (sense) and 5′-CGACCACGAAA
AAAAACC-3′ (antisense). The primers for the unmethylated PSD CpG
islands were 5′-AGGGTTGTAGGGAAGTGG TTT-3′ (sense) and
5′-CAACCACAAAAAAAAACCTA-3′ (antisense). Peripheral blood cell DNA
from healthy adults treated with SssI methyltransferase (New
England Biolabs, Ipswich, MA, USA) and untreated DNA were used as
positive and negative controls, respectively. PCR products were
separated by electrophoresis on 2% agarose gels.
Western blotting
Total protein was extracted from cells and tissues
in a lysate buffer: 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.5%
Nonidet P40, 0.5% sodium deoxycholate and phenylmethylsulfonyl
fluoride (all from Beyotime Institute of Biotechnology, Shanghai,
China). Each sample (60 μg) was electrophoresed in 10%
polyacrylamide gel and transferred to a polyvinylidene difluoride
membrane (Millipore, Bedford, MA, USA). After blocking with 5% BSA
in Tris-buffered saline-Tween-20 (20 mM Tris-HCl, 500 mM NaCl,
0.05% Tween-20) for 2 h at room temperature and then incubated with
primary antibodies for PSD (1:500 dilution; Novus International,
Littleton, MA, USA), caspase-3 (1:500 dilution; Millipore),
caspase-7 (1:500 dilution; Millipore) or β-actin (1:2,000 dilution;
ZSGB-Bio, Beijing, China) overnight at 4°C. The next day, after
incubation with a 1:2,000 dilution of secondary antibodies at 37°C
for 2 h, and after washing, the immunoreactive protein bands were
visualized using an electrochemiluminescence (ECL) detection kit
(Thermol Biotech, Rockford, IL, USA). The ratio between the optical
density of the protein of interest and β-actin was calculated as
the relative content of the protein detected. Each experiment was
repeated three times.
Wound healing assay
Cells were plated in 6-well plates and maintained in
RPMI-1640 medium containing 10% fetal calf serum. A wound was
created in the center of the cell monolayer by a sterile plastic
pipette tip. The cells were allowed to migrate for 24 h. Images
were taken at 0, 12 and 24 h after wounding to assess the ability
of the cells to migrate into the wound area using an inverted
microscope (IX-71; Olympus, Tokyo, Japan).
Matrigel invasion assay
Approximately 5×104 cells cultured in 200
μl serum-free RPMI-1640 medium were seeded onto Matrigel-coated
8-μm pore size Transwell filters (Corning Life Sciences, Corning,
NY, USA) in the upper chambers. Five hundred microliters of
RPMI-1640 containing 10% fetal calf serum was added to the lower
chambers as a chemoattractant. Cells were incubated at 37°C in a
humidified 5% CO2 atmosphere for 24 h. Cells that had
successfully invaded through the inserts were fixed in 4%
paraformaldehyde for 30 min, and stained with methylrosanilinium
chloride. The invading cells were counted from five preselected
microscopic fields of view at ×200 magnification. The assay was
performed from three independent experiments.
Cell counting kit-8 (CCK-8) assay
Cell proliferation was evaluated by CCK-8 assay
(Beyotime Institute of Biotechnology). Cells were seeded in 96-well
plates (3×103 per well). CCK-8 solution (10/100 μl
medium) was added to each well, and cells were incubated for 1 h at
37°C. Absorbance was measured at 450 nm using Synergy2 Multi-Mode
Microplate Reader (BioTek, Winooski, VT, USA). The assay was
conducted in five replicate wells for each sample and three
parallel experiments were performed.
Statistical analysis
All statistical analyses were performed using SPSS
version 17.0 (SPSS, Chicago, IL, USA). We used the χ2
test and Fisher’s exact-test to analyze the correlations between
PSD expression and clinicopathological characteristics. Student’s
t-test or one-way ANOVA were used for continuous variables. Data
are expressed as mean ± SD. A value of p<0.05 was considered
statistically significant.
Results
PSD expression is downregulated in GC
cells and tissues
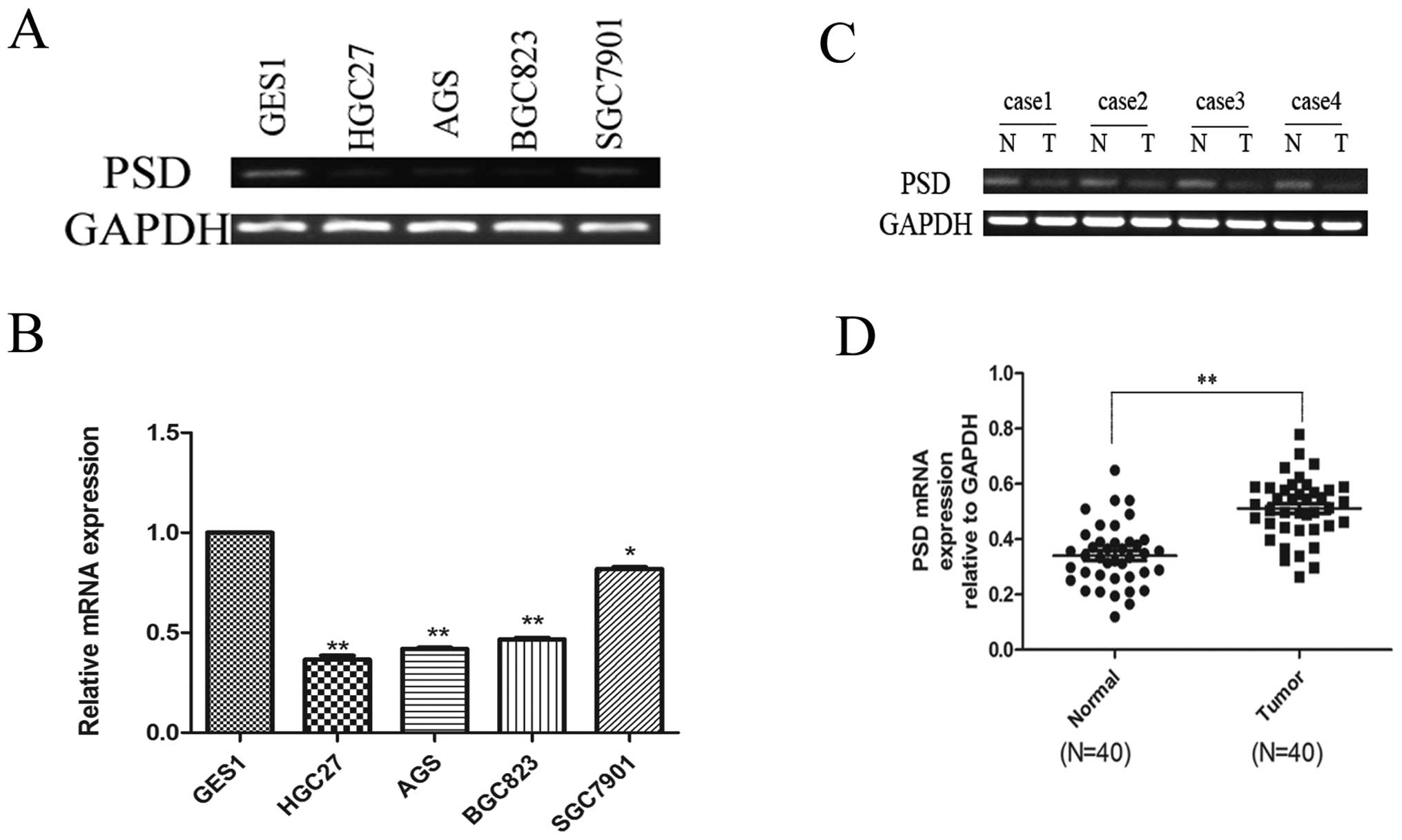
Using real-time RT-PCR to assess mRNA expression of
PSD, we found that PSD was downregulated in HGC27 (0.3651±0.020),
AGS (0.4198±0.053), BGC823 (0.467±0.077) and SGC7901 (0.8188±0.009)
cells compared to the normal mucosal line, GES1 (1-fold as the
control) (p<0.05; Fig. 1A and
B). We also analyzed PSD mRNA expression in 40 paired GC
specimens and corresponding normal tissues. PSD mRNA expression was
significantly lower in GC tissues than in their corresponding
normal tissues (0.3406±0.017 vs. 0.5115±0.018), (p<0.01,
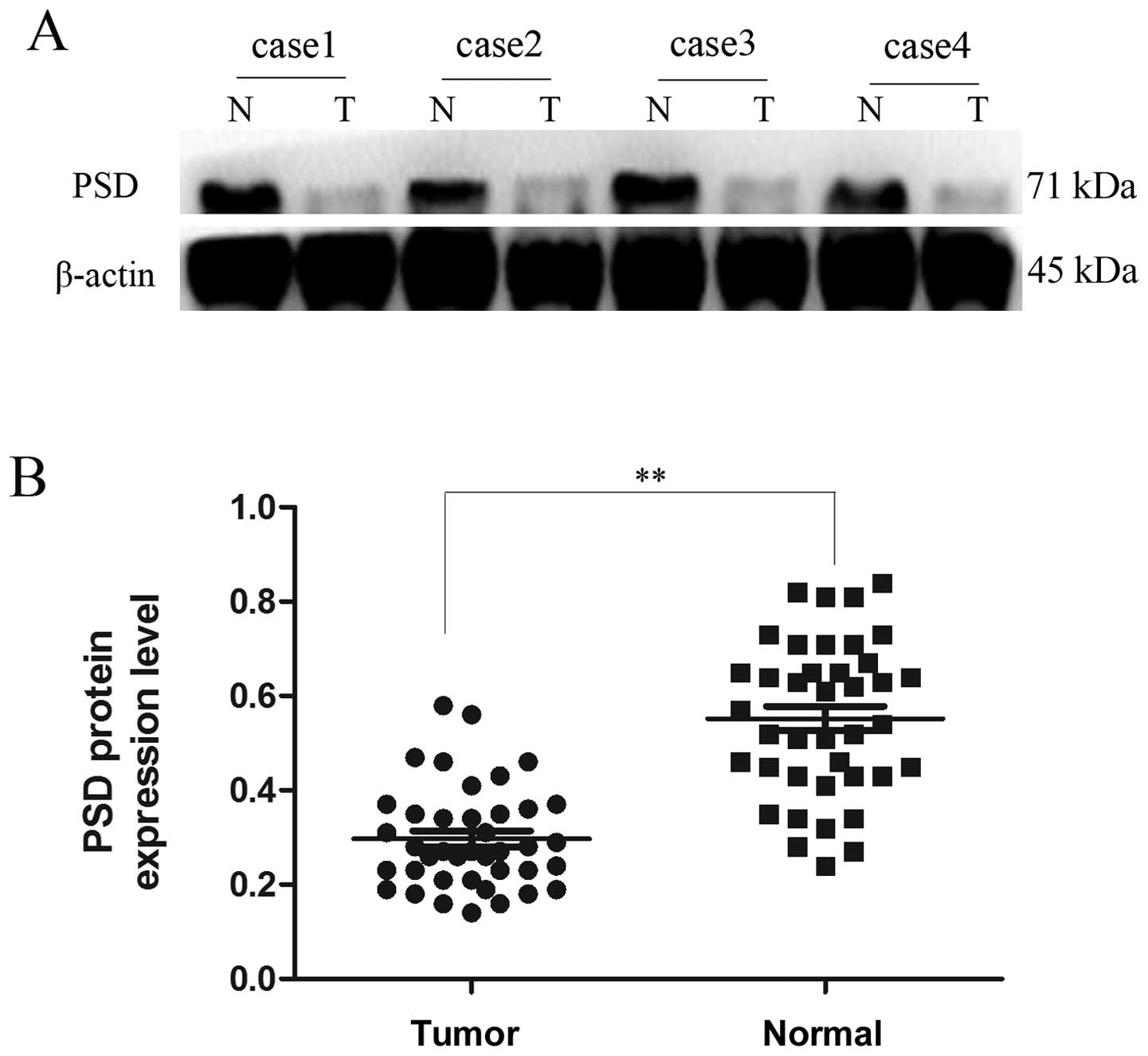
Fig. 1C and D). Western blotting
showed that protein expression of PSD in GC tissues was also lower
than in their corresponding non-tumor tissues (0.2970±0.01706 vs.
0.5523±0.02593), (p<0.01, Fig.
2).
Clinical significance of PSD protein
expression in GC tissues
We further investigated the relationship between PSD
expression and clinicopathological factors. PSD mRNA and PSD
protein levels were both related to tumor differentiation and lymph
node metastasis (Tables I and
II). There was no correlation
with any of the clinicopathological features, including age, sex,
tumor size, invasion depth and TNM stage.
 | Table ICorrelation of PSD mRNA expression and
clinicopathological parameters of gastric cancer samples. |
Table I
Correlation of PSD mRNA expression and
clinicopathological parameters of gastric cancer samples.
| | PSD mRNA
expression | |
|---|
| |
| |
|---|
| Variable | Patients (n),
N=40 | Low (%) | High (%) | P-value |
|---|
| Age (years) | | | | |
| <65 | 18 | 15 (83.3) | 3 (16.7) | 0.289 |
| ≥65 | 22 | 17 (77.3) | 5 (22.7) | |
| Gender | | | | |
| Male | 21 | 17 (81.0) | 4 (19.0) | 0.724 |
| Female | 19 | 15 (78.9) | 4 (21.1) | |
| Tumor location | | | | |
| Upper + middle | 15 | 11 (73.3) | 4 (26.7) | 0.058 |
| Lower | 25 | 21 (84.0) | 4 (16.0) | |
| Tumor size (cm) | | | | |
| <3 | 22 | 17 (77.3) | 5 (22.7) | 0.289 |
| ≥3 | 18 | 15 (83.3) | 3 (16.7) | |
| Depth of
invasion | | | | |
| T1+T2 | 17 | 14 (82.4) | 3 (17.6) | 0.480 |
| T3+T4 | 23 | 18 (78.3) | 5 (21.7) | |
| Differentiation | | | | |
| Well/moderate | 18 | 13 (72.2) | 5 (27.8) | 0.015a |
| Poor | 22 | 19 (86.4) | 3 (13.6) | |
| TNM stage | | | | |
| I+II | 15 | 12 (80.0) | 3 (20.0) | 1.000 |
| III+IV | 25 | 20 (80.0) | 5 (20.0) | |
| Lymph node
metastasis | | | | |
| No | 18 | 12 (66.7) | 6 (33.3) | 0.000a |
| Yes | 22 | 20 (90.9) | 2 (9.1) | |
| Table IICorrelation of PSD protein expression
and clinicopathological parameters of gastric cancer samples. |
Table II
Correlation of PSD protein expression
and clinicopathological parameters of gastric cancer samples.
| | PSD protein
expression | |
|---|
| |
| |
|---|
| Variable | Patients (n),
N=40 | Low (%) | High (%) | P-value |
|---|
| Age (years) | | | | |
| <65 | 18 | 14 (77.8) | 4 (22.2) | 0.665 |
| ≥65 | 22 | 14 (63.6) | 8 (36.4) | |
| Gender | | | | |
| Male | 21 | 15 (71.4) | 6 (28.6) | 0.645 |
| Female | 19 | 13 (68.4) | 6 (31.6) | |
| Tumor location | | | | |
| Upper +
middle | 15 | 11 (73.3) | 4 (26.7) | 0.880 |
| Lower | 25 | 17 (68.0) | 8 (32.0) | |
| Tumor size
(cm) | | | | |
| <3 | 22 | 16 (72.7) | 6 (27.3) | 0.874 |
| ≥3 | 18 | 12 (66.7) | 6 (33.3) | |
| Depth of
invasion | | | | |
| T1+T2 | 17 | 11 (64.7) | 6 (35.3) | 0.635 |
| T3+T4 | 23 | 17 (73.9) | 6 (26.1) | |
|
Differentiation | | | | |
| Well/moderate | 18 | 11 (61.1) | 7 (38.9) | 0.014a |
| Poor | 22 | 17 (77.3) | 5 (22.7) | |
| TNM stage | | | | |
| I+II | 15 | 10 (66.7) | 5 (33.3) | 0.443 |
| III+IV | 25 | 18 (72.0) | 7 (28.0) | |
| Lymph node
metastasis | | | | |
| No | 18 | 10 (55.6) | 8 (44.4) | 0.000a |
| Yes | 22 | 18 (81.8) | 4 (18.2) | |
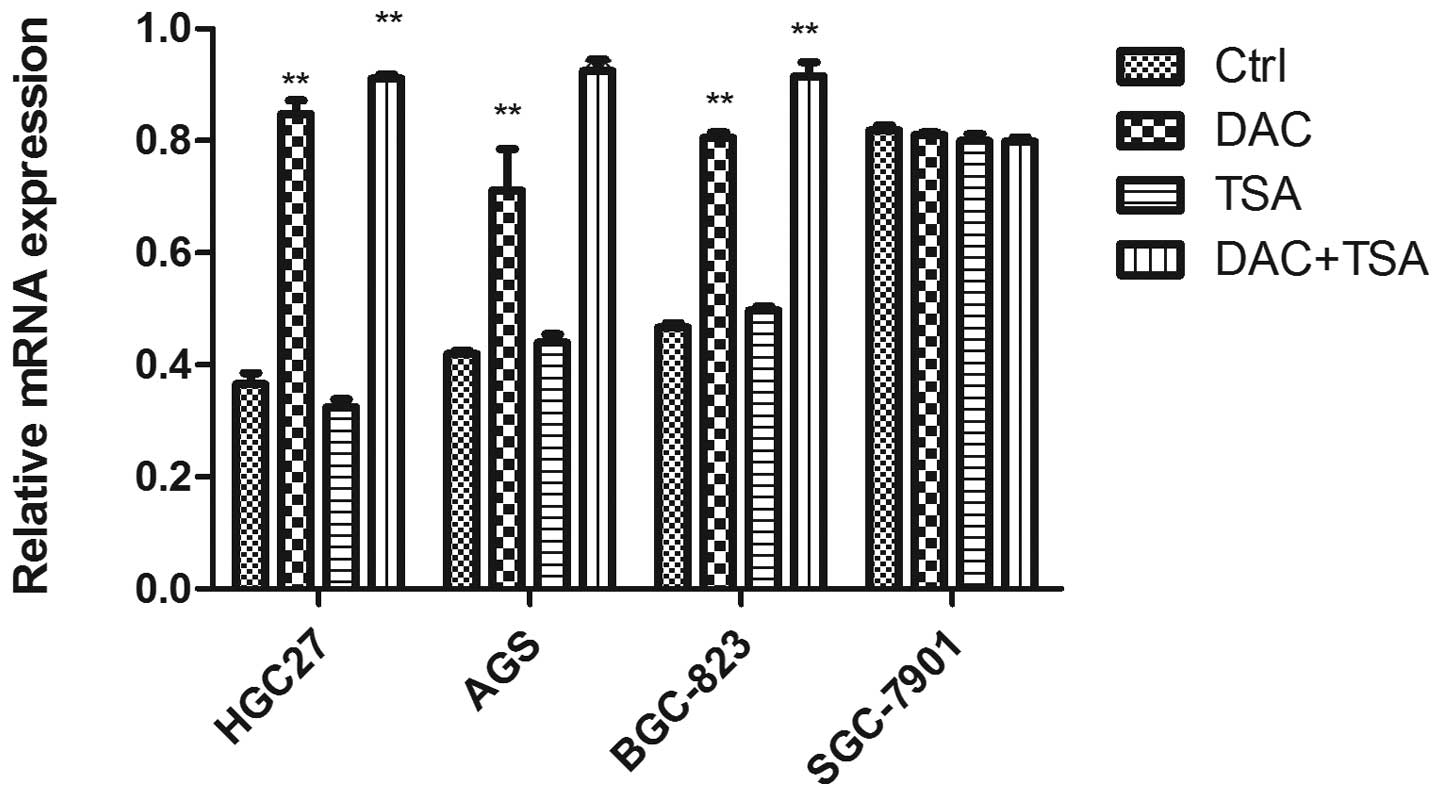
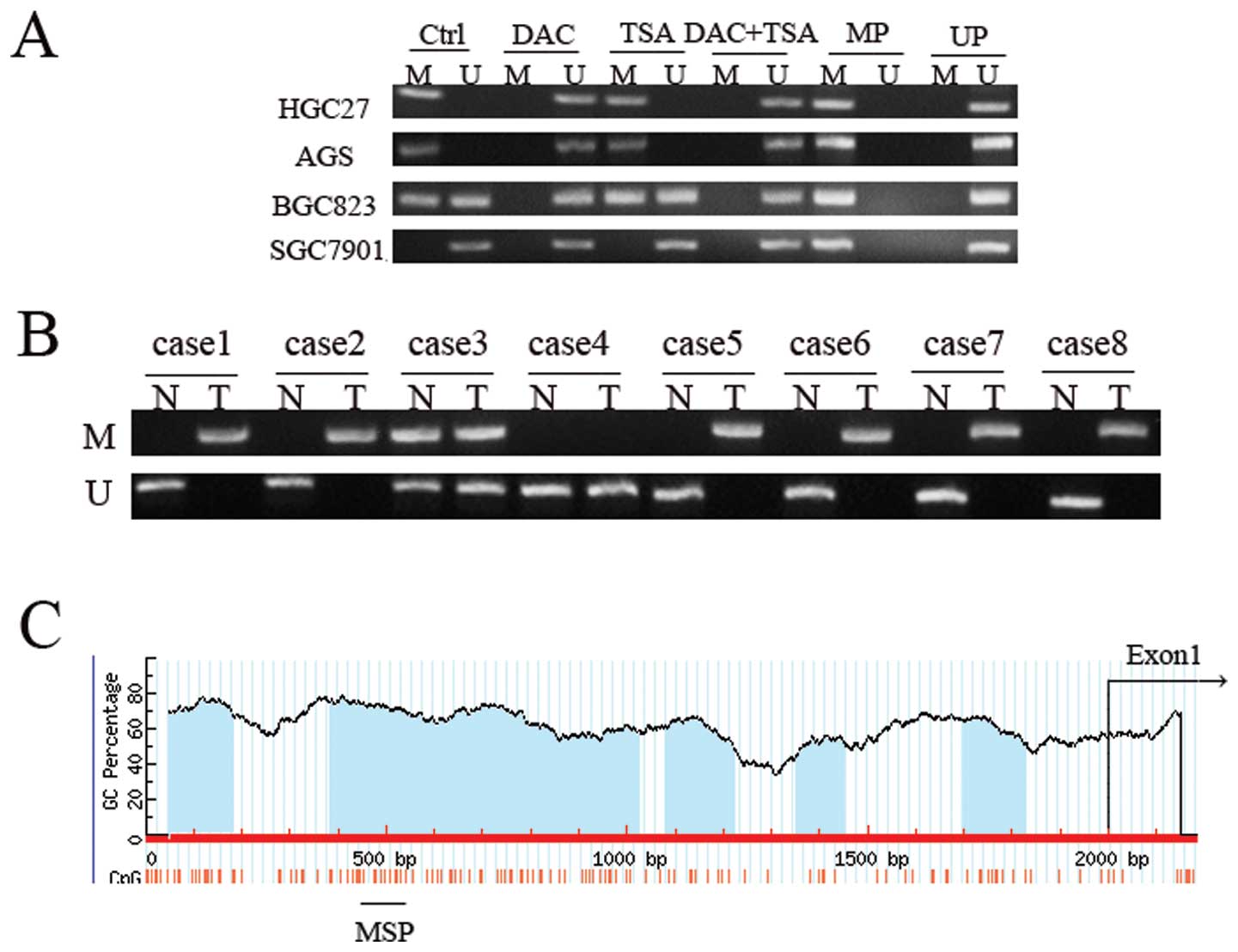
Abnormal histone modification is
associated with PSD gene silencing in GC cell lines
To elucidate whether the aberrant DNA methylation
and histone modification of PSD were associated with loss of PSD,
we treated GC cell lines HGC27, AGS, BCG823 and SGC7901 with
epigenetic agents (DAC and TSA). DAC and TSA had different effects
on PSD expression in the PSD-positive cell line (SGC7901) and
PSD-negative cell lines (HGC27, AGS and BCG823). After treatment
with DAC or TSA alone, the PSD mRNA was induced in HGC27, AGS and
BCG823 cells. Combined treatment with both agents restored PSD
expression to a significantly greater degree than did treatment
with either agent alone. In SGC7901 cells, treatment with DAC and
TSA, alone or in combination, had no significant effect on the
restoration of PSD expression (Fig.
3).
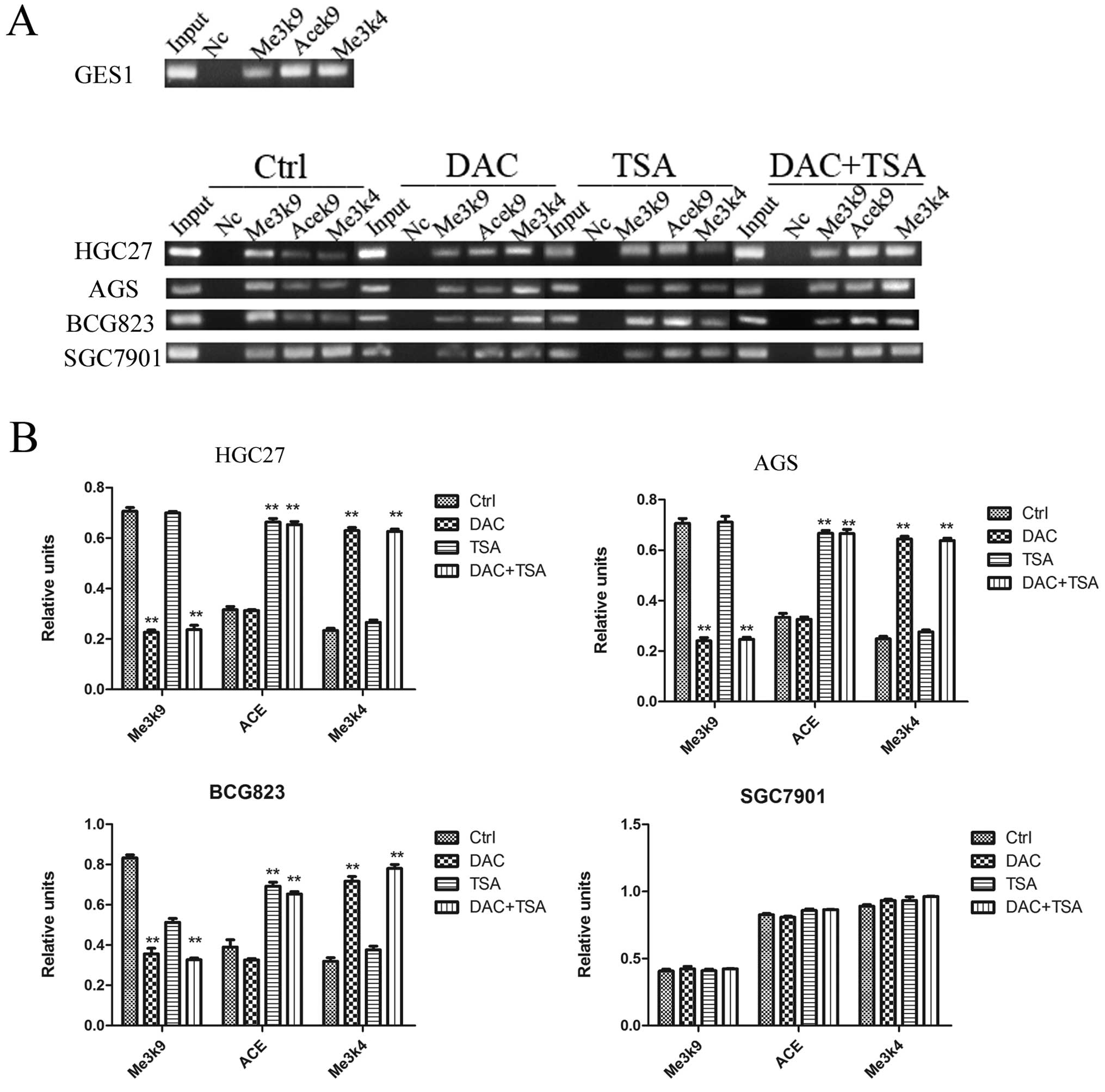
To probe the role of histone modifications in the
regulation of the PSD gene expression, we examined the histone
markers H3-K9 trimethylation, H3-K9 acetylation and H3-K4
trimeth-ylation with the PSD promoter region using ChIP. As shown
in Fig. 4, in SGC7901 cells, where
PSD was expressed, the level of H3-K9 trimethylation in the
promoter regions was minimal. In comparison, H3-K9 trimethylation
levels in the PSD gene promoter were high in HGC27, AGS and BCG823
cells. We found that the levels of both H3-K9 acetylation and H3-K4
trimethylation at PSD promoter regions were minimal in HGC27, AGS
and BCG823 cell lines. However, these levels were higher in SGC7901
cells. After treatment with DAC, H3-K9 trimethylation in the PSD
promoter was decreased significantly and H3-K4 trimethylation was
increased significantly in HGC27, AGS and BGC823 cells. We also
found that TSA increased H3-K9 acetylation in the PSD promoter, but
had no effect on H3-K9 trimethylation and H3-K4 trimethylation in
HGC27, AGS and BGC823 cells. Combined treatment significantly
decreased H3-K9 trimethylation, and increased H3-K9 acetylation and
H3-K4 trimethylation in the PSD promoter in PSD-negative cell
lines. The efficiency of combined treatment was similar to DAC or
TSA alone. In the PSD-positive cell line (SGC7901), treatment with
DAC, TSA or both had no significant effect on the histone markers
H3-K9 trimethylation, H3-K9 acetylation and H3-K4
trimethylation.
PSD gene silencing by DNA methylation in
GC cell lines and primary GC
The PSD promoters contain five CpG islands, so we
can suppose that low PSD expression is due to DNA methylation. In
order to test this hypothesis, we first examined the DNA
methylation of PSD in GC cell lines using MSP. We observed
hypermethylation of PSD in HGC27 and AGS cells, partial methylation
in BGC823, and no methylation in SGC7901 cells (Fig. 5). In PSD-negative cell lines
(HGC27, AGS and BGC823), treatment with DAC resulted in DNA
demethylation. TSA had no effect on DNA methylation of PSD, and
combined treatment had no additional effect on DNA demethylation
beyond that produced by treatment with DAC alone. In unmethylated
SGC7901 cells, treatment with DAC, TSA or both did not have a
significant effect on DNA methylation. MSP was performed to detect
methylation status of PSD gene in 40 paired tumor and corresponding
non-malignant gastric tissues. DNA methylation in gastric tissues,
which included methylated and partially methylated tissues,
occurred in 60% (24/40) of primary GC tissues and 27.5% (11/40) of
non-malignant gastric tissues. No methylation was observed in 16
(40%) primary GC tissues and 29 (72.5%) non-malignant gastric
tissues. The difference in methylation status of PSD between
primary GC and non-malignant gastric tissue specimens was
significant (p<0.01, Table
III).
| Table IIIMethylation status of PSD between
gastric tissues (T) and non-malignant gastric tissues (N). |
Table III
Methylation status of PSD between
gastric tissues (T) and non-malignant gastric tissues (N).
| Group | Case | Methylation
(%) | No methylation
(%) | P-value |
|---|
| T | 40 | 24 (60.00) | 16 (40.00) | <0.01a |
| N | 40 | 11 (27.50) | 29 (72.50) | |
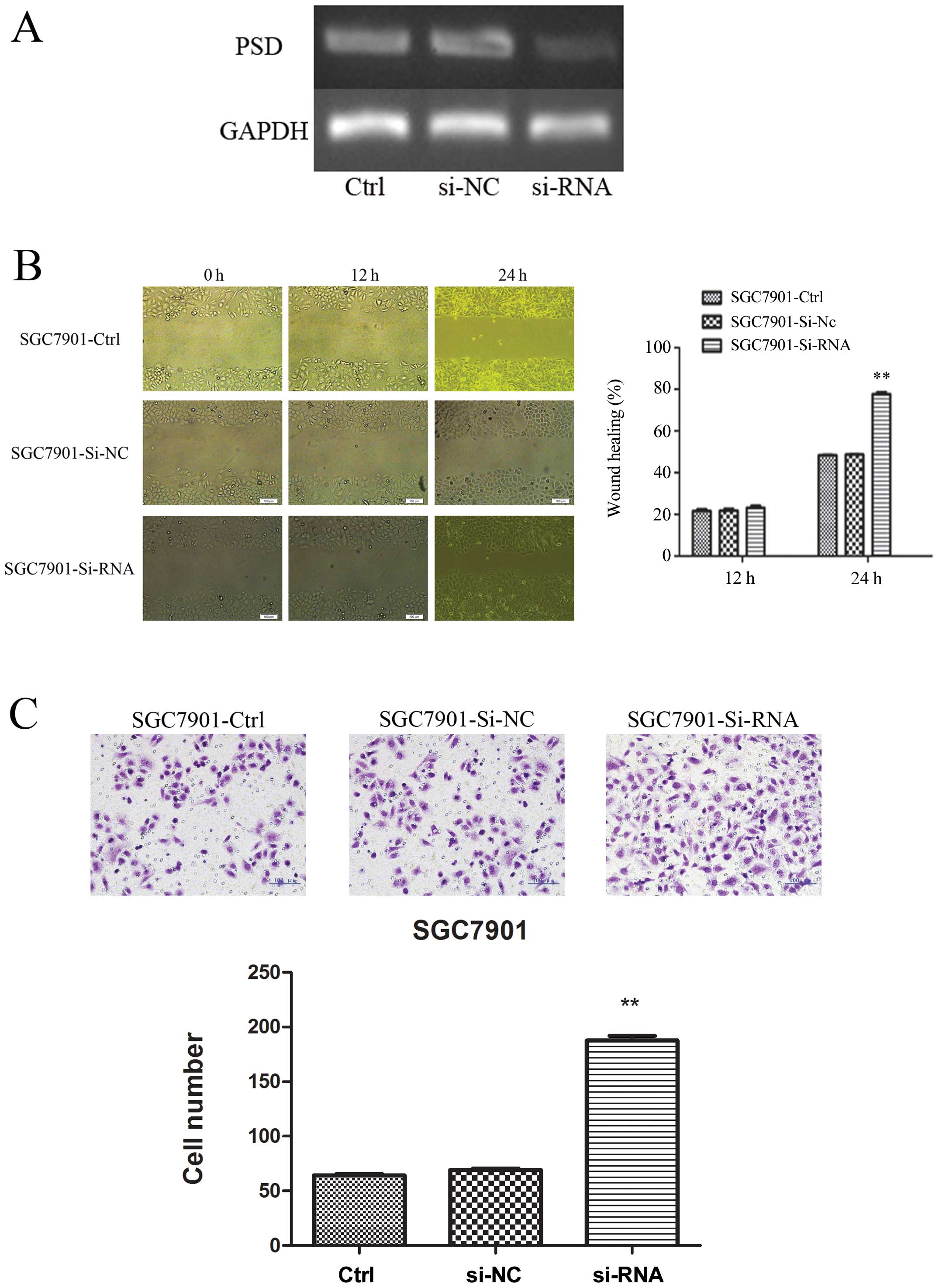
Suppression of PSD expression improved
migration and invasion of SGC7901 cells in vitro
To explore further the tumor-suppressive function of
PSD, we used PSD-specific siRNA to knock down PSD expression in the
SGC7901 cell line, in which the level of PSD was relatively high.
RT-PCR confirmed that PSD was downregulated after 48-h transfection
(Fig. 6A). Wound-healing assay
showed that in SGC7901 cells, knockdown of PSD led to cell
migration at 24 h after establishing the wound (Fig. 6B). We also examined whether cell
invasive capacity was altered in PSD-depleted cells. Matrigel
invasion assay showed that SGC7901 cells transfected with PSD siRNA
had more invasion (187.5±4.518) than cells transfected with control
siRNA (66.5±1.848) or untreated SGC7901 cells (64.2±1.250)
(p<0.01, Fig. 6C).
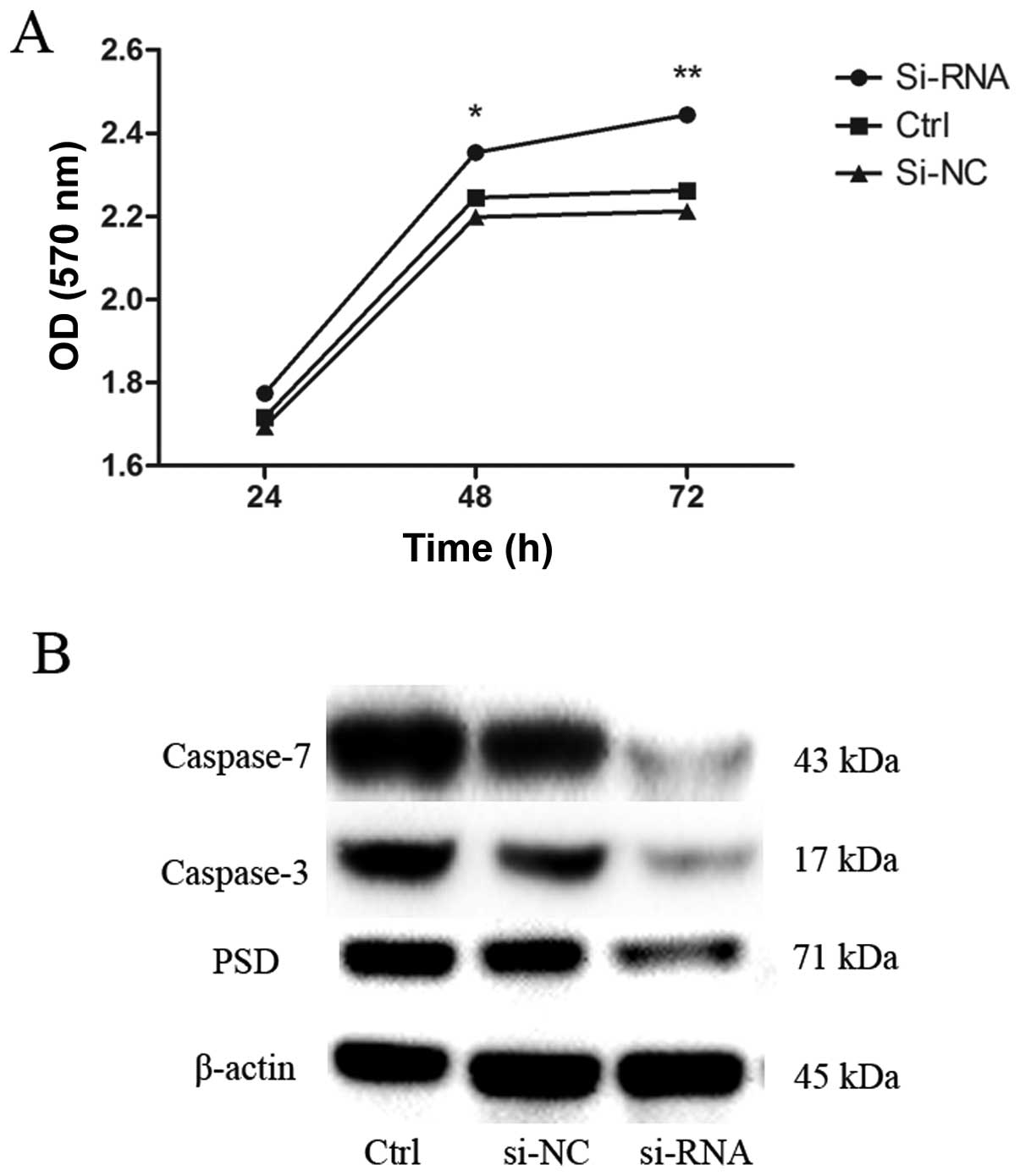
Depletion of PSD reduces apoptosis of
SGC7901 cells
To evaluate the potential effects of transfection
with PSD siRNA on growth of SGC7901 cells, we examined the growth
curve by CCK-8 assay. Depletion of PSD promoted growth of SGC7901
cells in a time-dependent manner (Fig.
7A). We then sought to determine the possible mechanisms that
underlie the depletion of PSD-promoted growth of SGC7901 cells.
Levels of apoptosis-related proteins (caspase-3 and -7) were
evaluated using western blot analysis. Caspase-3 and -7 protein
levels decreased after 48 h of transfection with PSD siRNA
(Fig. 7B).
Discussion
We found that PSD expression was significantly
reduced in GC cell lines and tissues compared to GES1 and normal
tissues. PSD mRNA expression in GC tissues was related to tumor
differentiation and lymph node metastasis. This is in agreement
with the result that differential PSD expression in the GC cell
lines may be related to cell differentiation. PSD mRNA expression
in human poorly differentiated gastric carcinoma cell line HGC27
was lower than in human moderately differentiated gastric carcinoma
cell line SGC7901. This suggests that the degree of malignancy of
GC may be higher when the expression of PSD is low.
We also identified three mechanisms underlying the
decreased expression of PSD: DNA hypermethylation of PSD promoter,
hypertrimethylation of histones H3-K9, and hypotrimethylation of
histones H3-K4 attached to the promoter. First, using ChIP
techniques in four GC cell lines and GES-1 cell line, we showed
that the level of H3-K9 trimethylation in the promoter regions of
PSD-negative cell lines (HGC27, AGS and BCG823) was higher than in
GES-1 and PSD-positive cell line SGC7901. In contrast, the level of
H3-K9 acetylation and H3-K4 trimethylation in the PSD promoter
region was lower than in GES-1 and PSD-positive cell line SGC7901.
Second, we examined the DNA methylation of PSD in GC cell lines and
tissues by MSP. The level of DNA methylation in the promoter
regions of PSD-negative cell lines (HGC27, AGS and BCG823) was
higher than in PSD-positive cell line SGC7901. Lastly, we treated
the GC cell lines with DAC and TSA and found that DAC or combined
treatment restored PSD expression to a significantly greater degree
by means of reversing the level of H3-K9 trimethylation, H3-K4
trimethylation and DNA hypermethylation. TSA significantly
increased H3-K9 acetylation but the effect on restored PSD
expression was limited. Thus, we have reason to believe that
silencing of PSD genes is mainly due to H3-K9 trimethylation, H3-K4
trimethylation and DNA hypermethylation but not H3-K9 acetylation.
Our results are consistent with a model in which H3-K9 methylation
and DNA methylation closely collaborate in maintaining the
repressive state of tumor TSGs. H3-K4 trimethylation is associated
with an open chromatin configuration and activation of TSG
transcription. H3-K4 trimethylation in promoter regions of PSD was
inversely correlated with DNA methylation status (22–24).
To investigate further the tumor-suppressive
functions of PSD, SGC7901 cells were treated with PSD-specific
siRNA to knock down PSD expression. Cell migration and invasion
were markedly enhanced when PSD was silenced. These results are in
agreement with our present study that the expression of PSD was
statistically significantly inverse correlated with lymph node
metastasis. This provides further evidence that PSD may function as
a tumor suppressorin gastric cancer.
We also appraised the effect of PSD-specific siRNA
on SGC7901 cell growth, which indicated that depletion of PSD
promoted growth of SGC7901 cells. Next, we assessed the levels of
apoptosis-related proteins caspase-3 and -7, and found that they
decreased after 48-h transfection with PSD siRNA. These findings
were also supported by a similar study in a human promyelocytic
leukemia cell line, HL-60 (25).
The mechanism underlying modifications of depletion of PSD reducing
apoptosis is not clear. One possibility is that Fas-induced
apoptosis is mediated by the activation of a Ras-related C3
botulinum toxin substrate 1 (Rac1), and PSD can regulate Rac1
(26,27). Rac1 plays a pivotal role in
inducing apoptosis in response to stimuli such as UV light
(28), tumor necrosis factor-α
(16) and Fas (15). These findings strongly support our
data showing that PSD silencing can inhibited Rac1-mediated
apoptosis in siPSD-treated SGC7901 cells.
This is believed to be the first report of the
tumor-suppressor functions of PSD, which are epigenetically
silenced in GC, while silencing of PSD after transfection with
siRNA promotes GC progression. These findings provide new targets
for prognosis and pharmacological intervention in human GC.
Therefore, further in vitro and in vivo studies are
needed to determine the precise mechanism of PSD in the progression
of GC.
Acknowledgements
This study was supported in part by a grant from the
National Natural Science Foundation of China (grant no. 30572162),
the Foundation of Liaoning Province Science and Technology Plan
Project (grant no. 2013225021) and the Higher Specialized Research
Fund for Doctoral Program of Ministry of Education of China (grant
no. 20102104110001).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Park YS, Jin MY, Kim YJ, Yook JH, Kim BS
and Jang SJ: The global histone modificaton pattern correlates with
cancer recurrence and overall survival in gastric adenocarcinoma.
Ann Surg Oncol. 15:1968–1976. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Meng CF, Zhu XJ, Peng G and Dai DQ: Role
of histone modifications and DNA methylation in the regulation of
O6-methylguanine-DNA methyltransferase gene expression
in human stomach cancer cells. Cancer Invest. 28:331–339. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kim M, Jang HR, Kim JH, et al: Epigenetic
inactivation of protein kinase D1 in gastric cancer andits role in
gastric cancer cell migration and invasion. Carcinogenesis.
29:629–637. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jaenisch R and Bird A: Epigenetic
regulation of gene expression: how the genome integrates intrinsic
and environmental signals. Nat Genet. 33:245–254. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sharma S, Kelly TK and Jones PA:
Epigenetics in cancer. Carcinogenesis. 31:27–36. 2010. View Article : Google Scholar
|
|
7
|
Nan X, Ng HH, Johnson CA, Laherty CD,
Turner BM, Eisenman RN and Bird A: Transcriptional repression by
the methyl-CpG-binding protein MeCP2 involves a histone deacetylase
complex. Nature. 393:386–389. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hendrich B and Bird A: Identification and
characterization of a family of mammalian methyl-CpG binding
proteins. Mol Cell Biol. 18:6538–6547. 1998.PubMed/NCBI
|
|
9
|
Bird A: DNA methylation patterns and
epigenetic memory. Genes Dev. 16:6–21. 2002. View Article : Google Scholar
|
|
10
|
Tariq M, Saze H, Probst AV, Lichota J,
Habu Y and Paszkowski J: Erasure of CpG methylation in arabidopsis
alters patterns of histone H3 methylation in heterochromatin. Proc
Natl Acad Sci USA. 100:8823–8827. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Espada J, Ballestar E, Fraga MF, et al:
Human DNA methyltransferase 1 is required for maintenance of the
histone H3 modification pattern. J Biol Chem. 279:37175–37184.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Meng CF, Zhu XJ, Peng G and Dai DQ:
Promoter histone H3 lysine 9 di-methylation is associated with DNA
methylation and aberrant expression of p16 in gastric cancer cells.
Oncol Rep. 22:1221–1227. 2009.PubMed/NCBI
|
|
13
|
Kondo Y, Shen L, Yan PS, Huang TH and Issa
JP: Chromatin immunoprecipitation microarrays for identification of
genes silenced by histone H3 lysine 9 methylation. Proc Natl Acad
Sci USA. 101:7398–7403. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rubinek T1, Shulman M, Israeli S, et al:
Epigenetic silencing of the tumor suppressor klotho in human breast
cancer. Breast Cancer Res Treat. 133:649–657. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gulbins E, Coggeshall KM, Brenner B,
Schlottmann K, Linderkamp O and Lang F: Fas-induced apoptosis is
mediated by activation of a Ras and Rac protein-regulated signaling
pathway. J Biol Chem. 271:26389–26394. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Esteve P, Embade N, Perona R, et al:
Rho-regulated signals induce apoptosis in vitro and in vivo by a
p53-independent, but Bcl2 dependent pathway. Oncogene.
17:1855–1869. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Perletti L, Talarico D, Trecca D,
Ronchetti D and Fracchiolla NS: Identification of a novel gene,
PSD, adjacent to NFKB2/lyt-10, which contains Sec7 and
pleckstrin-homologydomains. Genomics. 46:251–259. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Okada S, Suzuki K, Takaharu K, et al:
Aberrant methylation of the Pleckstrin and Sec7 domain-containing
gene is implicated in ulcerative colitis-associated carcinogenesis
throughits inhibitory effect on apoptosis. Int J Oncol. 40:686–694.
2012.
|
|
19
|
Fahrner JA, Eguchi S, Herman JG and Baylin
SB: Dependence of histone modifications and gene expression on DNA
hypermethylation in cancer. Cancer Res. 62:7213–7218.
2002.PubMed/NCBI
|
|
20
|
Cameron EE, Bachman KE, Myohane S, Herman
JG and Baylin SB: Synergy of demethylation and histone deacetylase
inhibition in the re-expression of genes silenced in cancer. Nat
Genet. 21:103–107. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li LC and Dahiya R: MethPrimer: designing
primers for methylation PCRs. Bioinformatics. 18:1427–1431. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fuks F: DNA methylation and histone
modifications: teaming up to silence genes. Curr Opin Genet Dev.
15:490–495. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu J, Wang SH, Potter D, et al: Diverse
histone modifications on histone 3 lysine 9 andtheir relation to
DNA methylation in specifying gene silencing. BMC Genomics.
8:1312007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu J, Zhu X, Xu X and Dai D: DNA promoter
and histone H3 methylation downregulate NGX6 in gastric cancer
cells. Med Oncol. 31:8172014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kato T, Suzuki K, Okada S, et al: Aberrant
methylation of PSD disturbs. Rac1-mediated immune responses
governing neutrophil chemotaxis and apoptosis in ulcerative
colitis-associated carcinogenesis. Int J Oncol. 40:942–950.
2012.
|
|
26
|
Saez R, Chan AM, Miki T and Aaronson SA:
Oncogenic activation of human R-ras by point mutations analogous to
those of prototype H-ras oncogenes. Oncogene. 9:2977–2982.
1994.PubMed/NCBI
|
|
27
|
Nishida K, Kaziro Y and Satoh T:
Anti-apoptotic function of Rac in hematopoietic cells. Oncogene.
18:407–415. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Eom YW, Yoo MH, Woo CH, et al: Implication
of the small GTPase Rac1 in the apoptosis induced by UV in Rat-2
fibroblasts. Biochem Biophys Res Commun. 285:825–829. 2001.
View Article : Google Scholar : PubMed/NCBI
|