Introduction
The 14-3-3 proteins are a family of highly conserved
acidic polypeptides that are expressed in all eukaryotic cells
(1–3). Seven isoforms, encoded by seven
distinct genes have been identified in mammals (named β, γ, ɛ, η,
σ, τ and ζ). The 14-3-3 family proteins are thought to act as
scaffolding proteins and interact with a wide variety of protein
partners which participate in many biological processes, such as
cell cycle progression (4–7), apoptosis (8–10),
cell adhesion and migration (11,12)
and signal transduction regulation (13–15).
Importantly, high expression of 14-3-3 family proteins have been
observed in a number of human cancers, including lung (16,17),
prostate (18,19), pancreatic (20,21),
gastric (22) and colon cancer
(23) which suggests that they may
have a critical role as tumor oncogenes (24). However, few studies concerning
regulation of 14-3-3 protein expression on transcriptional or
post-transcriptional levels have been published.
The p53 tumor suppression protein has been defined
as a critical component of the DNA damage checkpoint machinery
(25). p53 can transcriptionally
activate or silence a number of target genes (26,27).
One of the seven human 14-3-3 isoforms 14-3-3σ is directly
transactivated by p53 after DNA damage (28,29).
14-3-3σ induces cell cycle arrest in G2 by sequestering proteins in
the cytoplasm which are required for entry into mitosis (29,30).
Noyably, 14-3-3σ can bind to p53 and leads to p53 stabilization and
enhances transcriptional activity of p53 (31). Therefore, as a target gene of p53,
14-3-3σ appears to have a positive feedback effect on p53 activity.
In addition to 14-3-3σ, several 14-3-3 isoforms, including 14-3-3γ
have been demonstrated to interact with p53 both in vitro
and in vivo and in turn, this interaction increases the DNA
binding activity of p53 (32,33).
In a previous study, we found that the 14-3-3γ
protein is elevated in human lung cancerous tissues (16) and overexpression of this protein in
lung cancer cells affects cell cycle progression and results in DNA
polyploidization (34). In
addition, wild-type p53 can bind the 14-3-3γ promoter and reduce
its mRNA (35). In the present
study we investigated the expression of p53 and 14-3-3γ in human
lung cancer tissues and discovered that 14-3-3γ expression is
significantly correlated with p53 mutation. Further investigation
using cultured colon and lung cancer cells showed wild-type of p53
but not mutant p53 (R175H) could suppress 14-3-3γ expression.
Importantly, inhibition of 26S proteasome by MG132 blocked the
reduction of 14-3-3γ by p53. These results indicate that p53
negatively regulates 14-3-3γ by stimulating proteasome-mediated
14-3-3γ protein degradation.
Materials and methods
Antibodies and reagents
Anti-14-3-3γ (specific) antibody was obtained from
Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies to flag
the β-actin were purchased from Sigma (St. Louis, MO, USA).
Anti-p53 (DO-1) and GFP antibodies were obtained from Oncogene (La
Jolla, CA, USA) and BD Pharmingen (San Diego, CA, USA),
respectively. Cycloheximide and MG132 were purchased from
Calbiochem (La Jolla, CA, USA).
Plasmid construction and cell
transfection
cDNA encoding human 14-3-3γ was cloned behind the
CMV promoter in pCMV-Tag2 (with flag tag at N-terminal) expression
vector (Stratagene, La Jolla, CA, USA). Human non-small cell lung
carcinoma cell line H358 (p53 null) from the American Type Culture
Collection (ATCC, Rockville, MD, USA) was cultured in Dulbecco’s
modified Eagle’s medium (DMEM; Gibco-BRL) supplemented with 10%
fetal bovine serum. Transfection was conducted using LipoTAXI
mammalian transfection kit (Stratagene, La Jolla, CA, USA)
according to the manufacturer’s instructions. In brief,
3×105 H358 cells were seeded in 60-mm dish and the cells
cultured until they reached a confluence of 50–70%. The cells were
washed with serum/antibiotic-free DMEM medium prior to
transfection. Plasmid pCMV-flag-14-3-3γ (5 μg) purified with
Perfectprep Plasmid midi (Eppendorf, Hamburg, Germany) and 50 μl of
LipoTAXI reagent diluted in 900 μl of serum/antibiotic-free DMEM
medium were mixed gently and incubated at room temperature for 30
min. After adding 1.5 ml of serum/antibiotic-free DMEM medium the
mixture was placed onto the cells. Transfection was performed at
37°C in an incubator under 5% CO2 for 5 h and then
further 2.5 ml DMEM containing 20% serum was added. After
incubation overnight the DNA mixture was replaced by fresh,
complete medium and the cells were incubated for 48 h and split
into three 10-cm plates. When the cells grew to 95% confluence, 0.5
μg/ml of G418 (Invitrogen, Carlsbad, CA, USA) was added to medium
to select the clones. After three weeks, 24 clones were collected
and expression of 14-3-3γ protein was assessed by immunoblotting
using anti-Flag antibody (Sigma).
Adenovirus construction and
infections
Ad-GFP, Ad-p53 (wild-type) and Ad-p53 (mutant,
R175H) expressed GFP, wild-type and mutant p53 were kindly provided
by Dr Cyrus Vaziri (Boston University School of Medicine) and Dr
Bernard Futscher (Arizona Cancer Center, The University of
Arizona), respectively. HCT116 cells with p53 knocked out were
plated in 60-mm culture dishes (in DMEM containing 10% fetal bovine
serum) and infected by adding virus directly to the medium when the
cultures had attained a confluence of ~60%. After 24-, 32- and 48-h
infection, the cells were harvested and endogenous 14-3-3γ protein
level was analyzed by immunoblotting. H358 cells stably expressing
flag-14-3-3γ were also infected by adenovirus-GFP, wild-type and
mutant p53 for 24 h and then treated with cycloheximide alone or
together with proteasome inhibitor MG132. Eight and 16 h later,
cells were lysed and exogenous flag-14-3-3γ was analyzed by
immunoblotting.
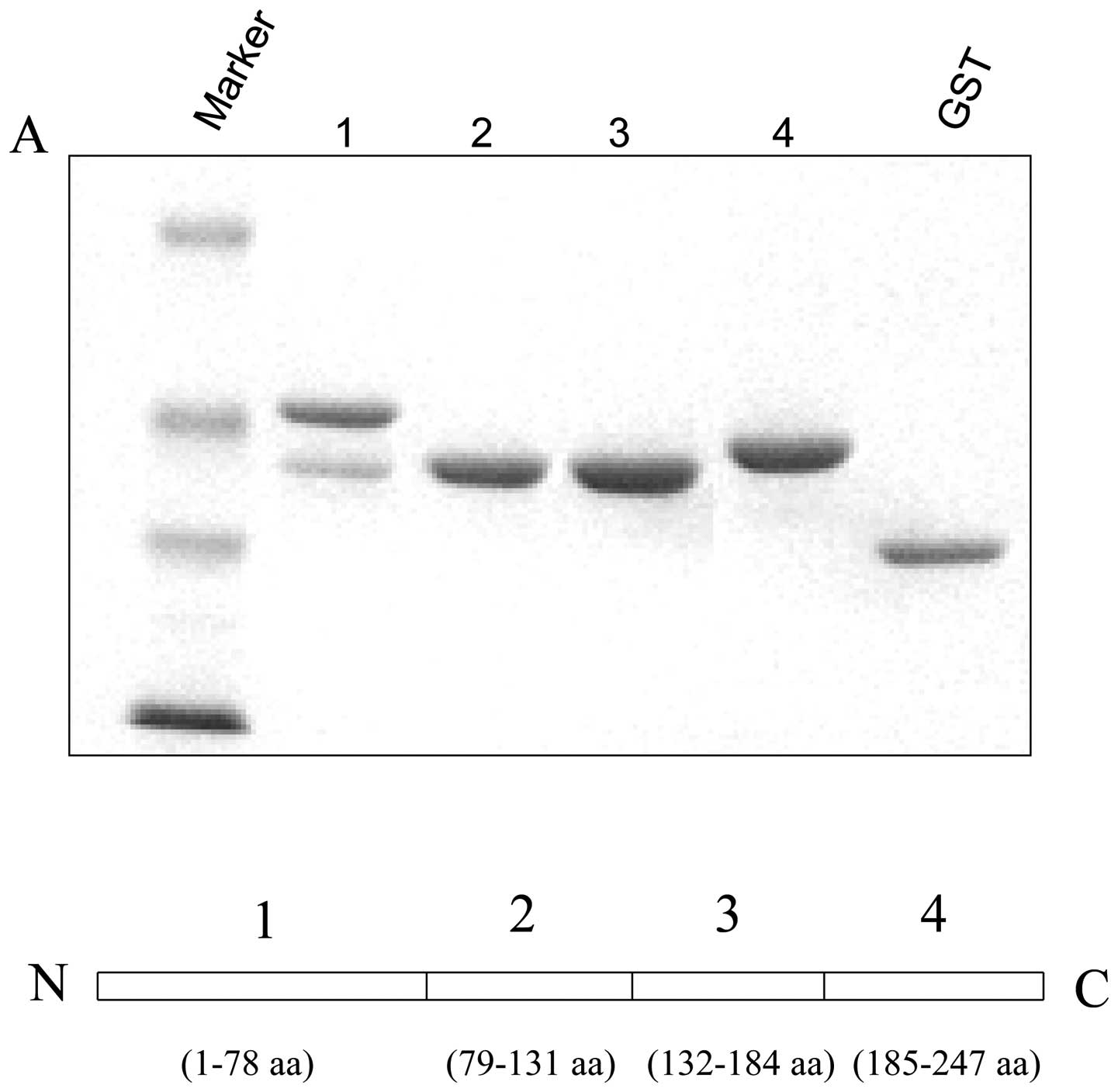
The 14-3-3γ truncated constructs and
protein expression
Human 14-3-3γ fragments (1:1-78 amino acid,
N-terminal; 2:79-131 amino acid, central part; 3:132-184 amino
acid, central part; and 4:185-247 amino acid, C-terminal) were
amplified by PCR and inserted in-frame into pGEX2T vector (Amersham
Pharmacia Biotech) for expression of GST fusion proteins. All
constructs were sequenced to make sure the sequences were correct.
The recombinant glutathione-S-transferase (GST) tagged proteins
were expressed in E. coli strain DH5α with 0.5 mM
isopropyl-β-L-thiogalactopyranoside (IPTG) at 37°C for 3 h. Cells
were collected and lysed in B-PER bacterial protein extraction
reagent (Pierce). After incubation on ice 30 min, the samples were
centrifuged and the supernatants were purified using glutathione
sepharose 4B beads (Amersham Pharmacia Biotech).
Glutathione-S-transferase pull-down assay
and western blotting
Confluent cells (70%) were harvested and lysed in
0.5% Nonidet P-40 lysis buffer (50 mM Tris-Cl, pH 7.4, 0.25 M NaCl,
0.5% NP-40, 50 mM NaF and ptotease inhibitors). The pull-down assay
was conducted by incubating cell extracts in lysis buffer with
GST-14-3-3s or GST bound to 40 μl glutathione sepharose beads for 2
h at 4°C. After five washes with lysis buffer, the bound proteins
were subjected to SDS-PAGE, transferred to nitrocellulose membrane,
and immunoblotted with primary antibodies.
Protein analysis
Frozen human lung cancerous (squamous cell carcinoma
and adenocarcinoma) and normal tissues were obtained from the
Department of Pathogenic Biology, Affiliated Hospital of Jiangsu
University (Zhenjiang, China). Frozen tissues and cultured cells
were lysed in NP-40 lysis buffer containing 50 mM Tris-Cl (pH 7.4),
0.15 M NaCl, 0.5% NP-40, 1 mM DTT, 50 mM sodium fluoride, and 2
μl/ml protease inhibitor cocktail (Sigma). Protein concentrations
were determined using the Bio-Rad protein assay kit (Bio-Rad
Laboratories, Hercules, CA, USA) and 50 μg protein was resolved by
electrophoresis on a 10% SDS-PAGE gel. The proteins were then
transferred onto a nitrocellulose membrane and non-specific binds
were blocked by incubating with 5% non-fat milk in TBST buffer
(0.01 M Tris-Cl, pH 8.0, 0.15 M NaCl, 0.5% Tween-20) at room
temperature for 1 h. The membrane was subjected to the indicated
antibodies and the proteins were detected by the SuperSignal West
Pico detection system (Pierce, Rockford, IL, USA).
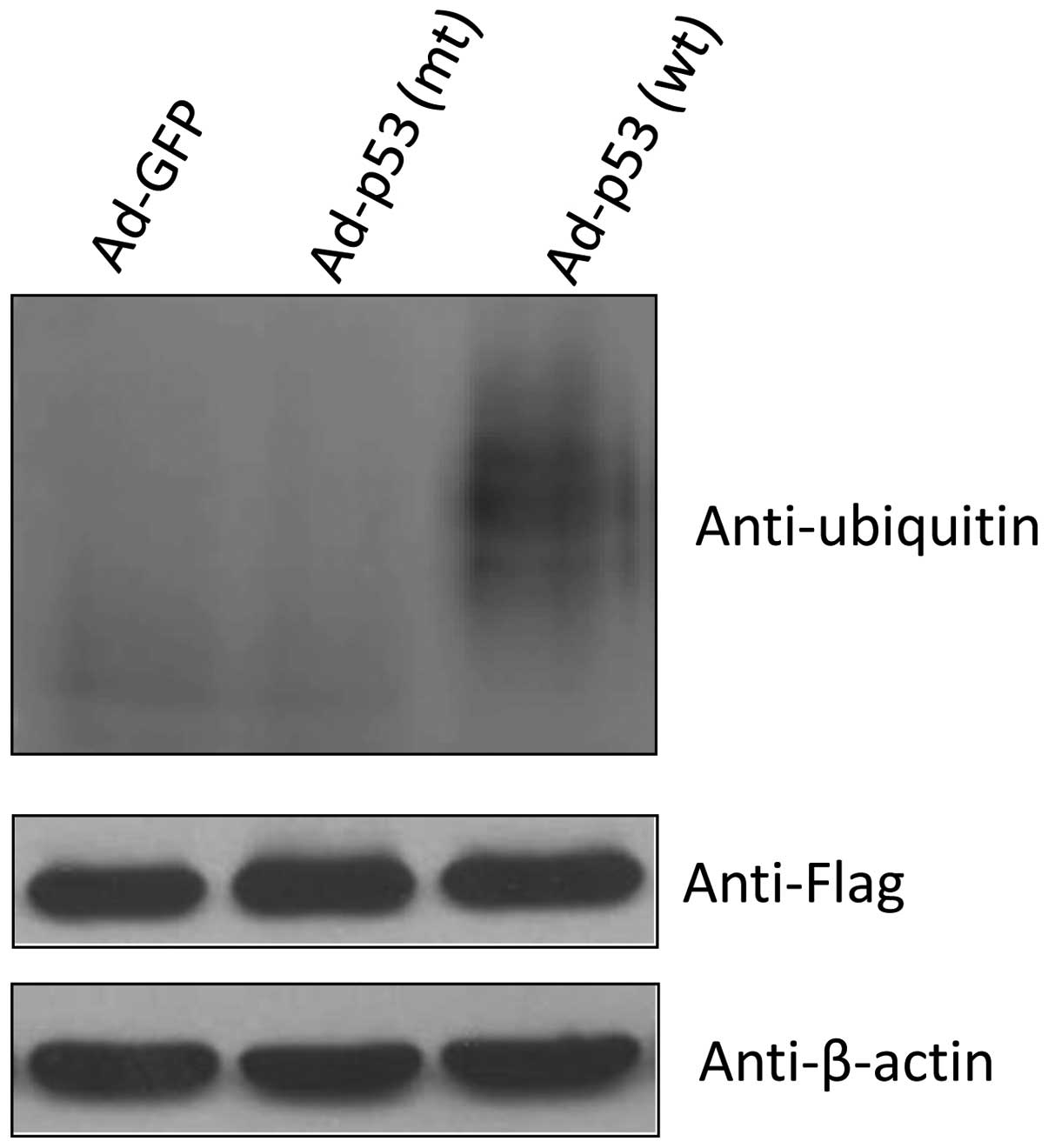
Ubiquitination assay
H358 cells were co-transfected with Flag-tagged
14-3-3γ expression vector and His-tagged ubiquitin expression
vector. At 16 h after transfection, cells were infected with
Ad-GFP, Ad-p53 (wt) and Ad-p53 (mt) for 32 h and then harvested.
Cells were lysed in NP-40 lysis buffer (50 mM Tris-Cl, pH 7.4, 0.25
M NaCl, 0.5% NP-40, 50 mM NaF and ptotease inhibitors).
Co-immunoprecipitation was conducted using Anti-Flag M2 affinity
gel (Sigma). Briefly, 200 μg of total protein in 500 μl lysis
buffer was incubated with 20 μl Anti-Flag M2 affinity gel at 4°C
overnight with rotation and the beads were then washed four times
with lysis buffer. The proteins were eluted by boiling in SDS-PAGE
sample buffer and applied to SDS-PAGE gels. 14-3-3γ ubiquitin was
detected by immunoblotting using the anti-ubiquitin antibody
(Upstate).
Statistical analysis
The Chi-squared test was performed to determine the
correlation between mutant p53 and 14-3-3γ protein expression and
P<0.05 is considered significant.
Results
14-3-3γ protein expression correlates
with overexpression of p53 in human lung cancerous tissues
Our previous study indicates that high levels of
14-3-3γ protein exist in human lung cancer tissues as compared with
the normal lung (16).
Importantly, overexpression of 14-3-3γ protein can cause genomic
instability by inducing polyploidization in human lung cancer cells
(34). p53, a tumor suppressor
protein, is reported to interact with several 14-3-3 isoforms
including 14-3-3γ and this interaction appears to be functionally
significant (32,33). In an attempt to understand the
functional relationship between p53 and 14-3-3γ in lung cancers we
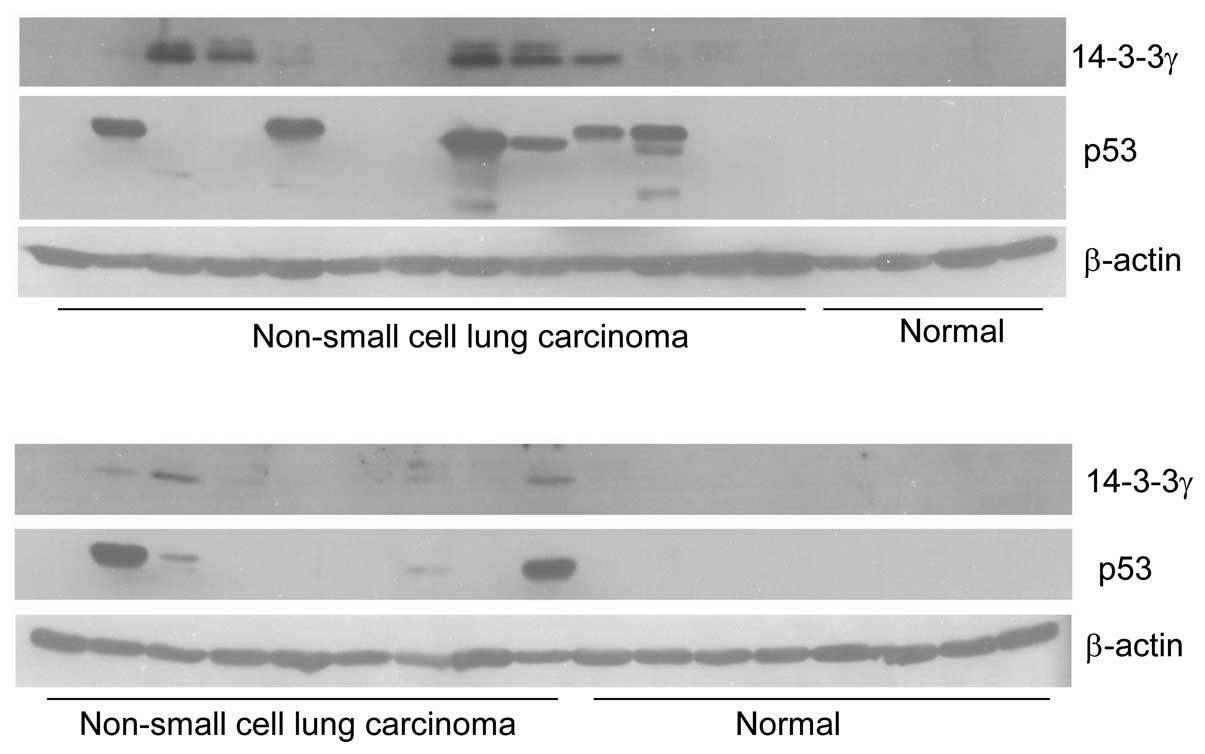
determined the expression of p53 and 14-3-3γ by western blotting.
In total 80 lung cancer tissues and 35 lung normal tissues were
used in this experiment. A typical representative expression
profile was shown in Fig. 1. As
expected, we could not detect any signals of p53 in the 35 normal
tissues. Consistent with our previous study, only one of 35 normal
tissues had a band of 14-3-3γ. This normal tissue was probably
contaminated with cancer cells. However, out of 80 cancer tissues
there were 35 (43.8%) with p53 overexpression of (Fig. 1). In these 35 specimens, 33
expressed 14-3-3γ protein. Statistical analysis between 14-3-3γ and
p53 proteins expression showed that 14-3-3γ expression
significantly correlated with overexpression of p53 protein
(P=0.0001; Table I). This result
indicated that 14-3-3γ expression in lung cancers was dependent on
overexpression of p53.
 | Table IExpression of 14-3-3γ correlates with
overexpression of p53 in human lung cancer tissues. |
Table I
Expression of 14-3-3γ correlates with
overexpression of p53 in human lung cancer tissues.
| No. of cancer
tissues | % of 14-3-3γ
expression in p53 overexpressed tissues | P-value |
|---|
| p53 positive | 35 | | |
| 14-3-3γ
positive | 33 | 94.3% | 0.0001 |
| 14-3-3γ
negative | 2 | | |
Wild-type p53 protein suppresses the
4-3-3γ protein level
p53 acts as a transcription factor to activate or
silence a number of downstream target genes (27,28).
To investigate the effect of p53 on 14-3-3γ protein level we
introduced both wild-type p53 and mutant p53 (R175H) into human
colon cancer cell line HCT116 in which endogenous wild-type p53
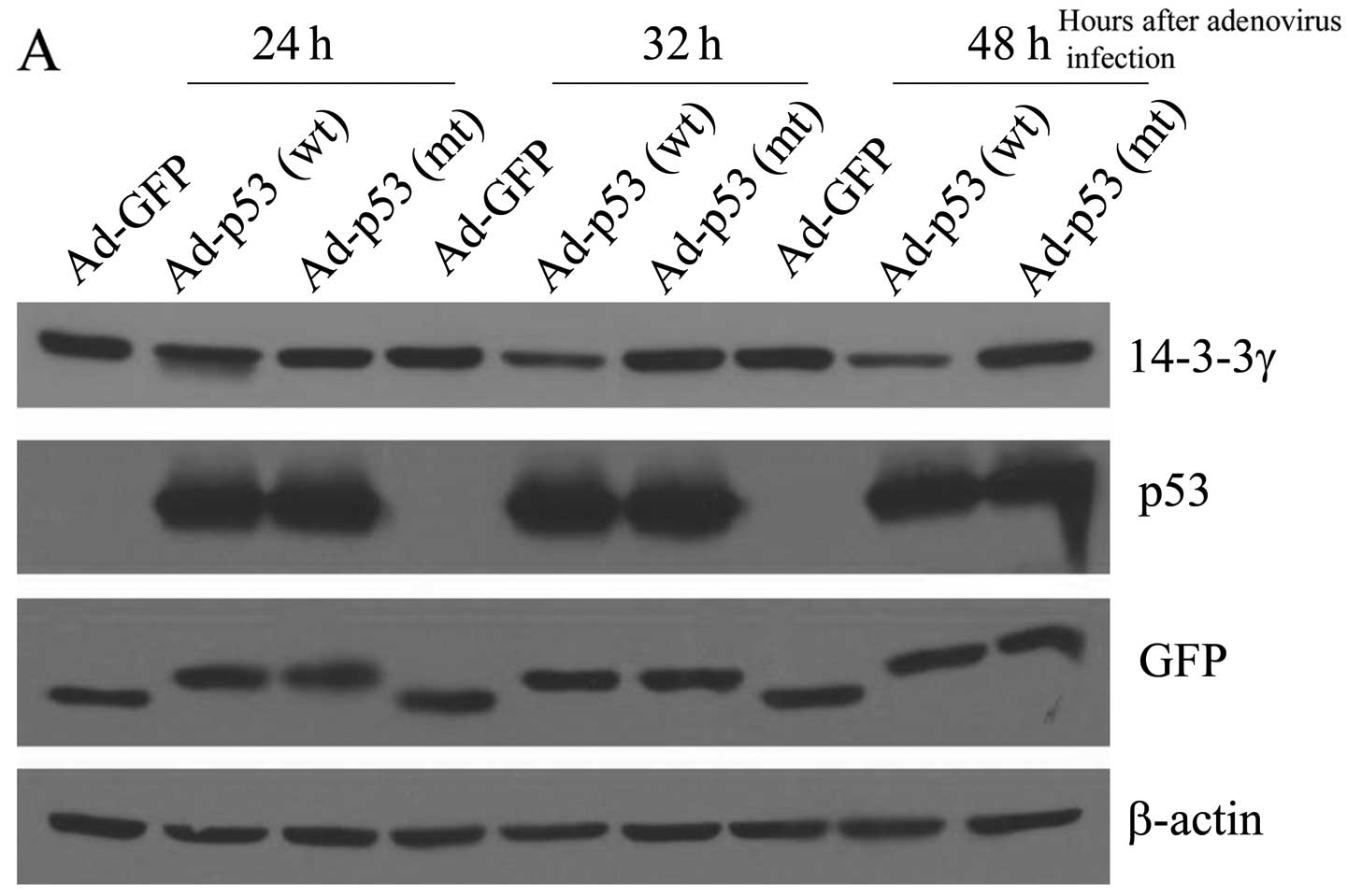
gene had been knocked out. We infected the cells with adenovirus
containing GFP, wild-type p53 (wt) and mutant p53 (mt) cDNA for 24,
32 and 48 h, and then the endogenous 14-3-3γ protein level was
examined by immunoblot analysis using a specific antibody against
14-3-3γ. The same blot was also probed with antibodies against GFP
and p53 to detect the efficiency of adenovirus infection.
Anti-human β-actin antibody was also used on the same blot to
control for variations in loading. The results showed that after 32
h the wild-type p53 caused a considerable decrease (~50%) in
14-3-3γ protein level (Fig. 2).
However, neither GFP nor mutant p53 (R175H) had any effect on
levels of 14-3-3γ in HCT116 cells (p53−/−). Hence,
wild-type p53, but not mutant p53 (R175H) suppressed endogenous
14-3-3γ protein levels in colon cancer cells.
p53 suppresses 14-3-3γ through protein
degradation by proteasome
Like other genes, 14-3-3γ regulation may occur at
multiple levels, including transcription, post-transcriptional
regulation of mRNA, translation and post-translational modification
(36). Our previous study
demonstrated that one of mechanism of p53 suppression of 14-3-3γ is
through binding its promoter and inhibiting 14-3-3γ transcription
(35). In order to clarify whether
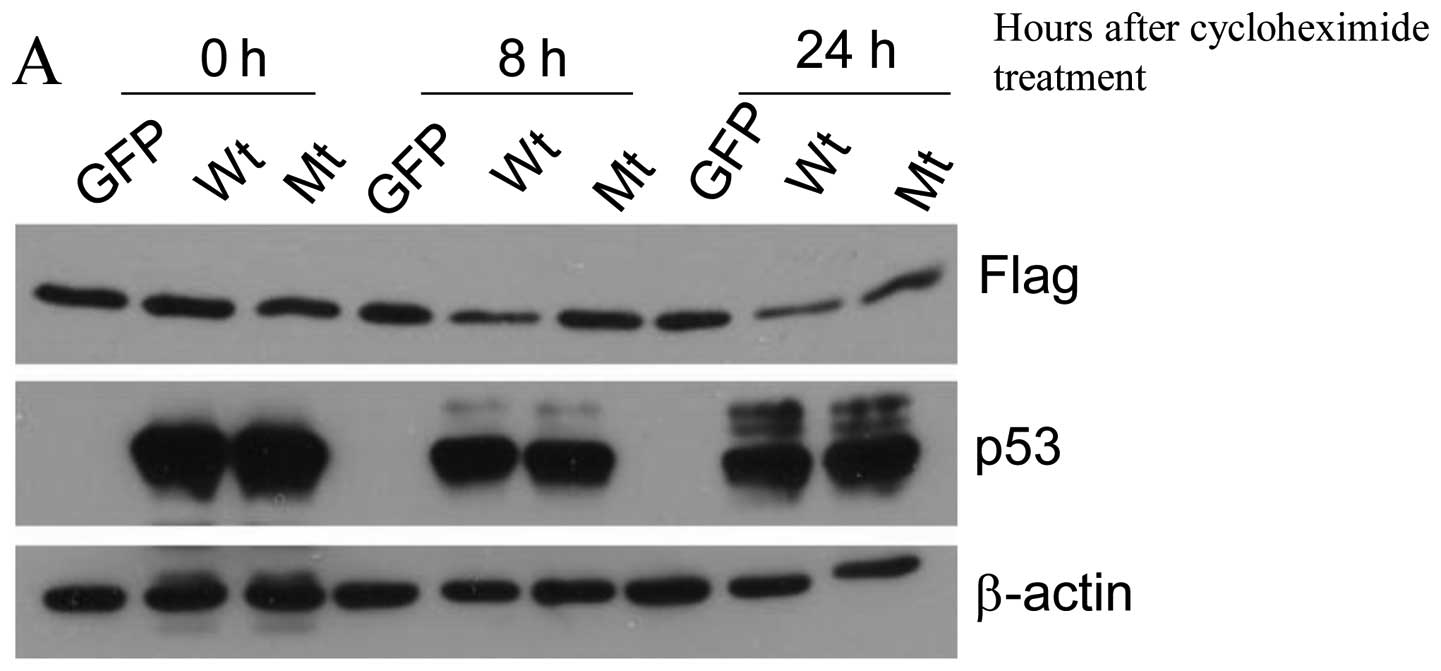
p53 suppresses 14-3-3γ post-transcription we stably transfected
14-3-3γ with Flag tag into human lung cancer cell line H358 which
is p53 null and then examine whether p53 suppresses this exogenous
14-3-3γ protein. H358 cells which constitutively express
Flag-14-3-3γ protein were infected by adenovirus-GFP, wild-type p53
and mutant p53 (R175H) for 24 h and cycloheximide was added to
inhibit protein synthesis. At 0, 8 and 24 h after cycloheximide
treatment, the Flag-14-3-3γ protein levels were determined by
immunoblot analysis using an antibody against Flag. Similarly to
the effect of p53 on the endogenous 14-3-3γ in colon cancer cell
line HCT116, wild-type p53 decreased 50% of Flag-14-3-3γ protein
(Fig. 3). This result clearly
indicates that p53 can negatively regulate 14-3-3γ by
post-translational modification.
p53 suppression of 14-3-3γ could be
through stimulating proeasome-mediated protein degradation
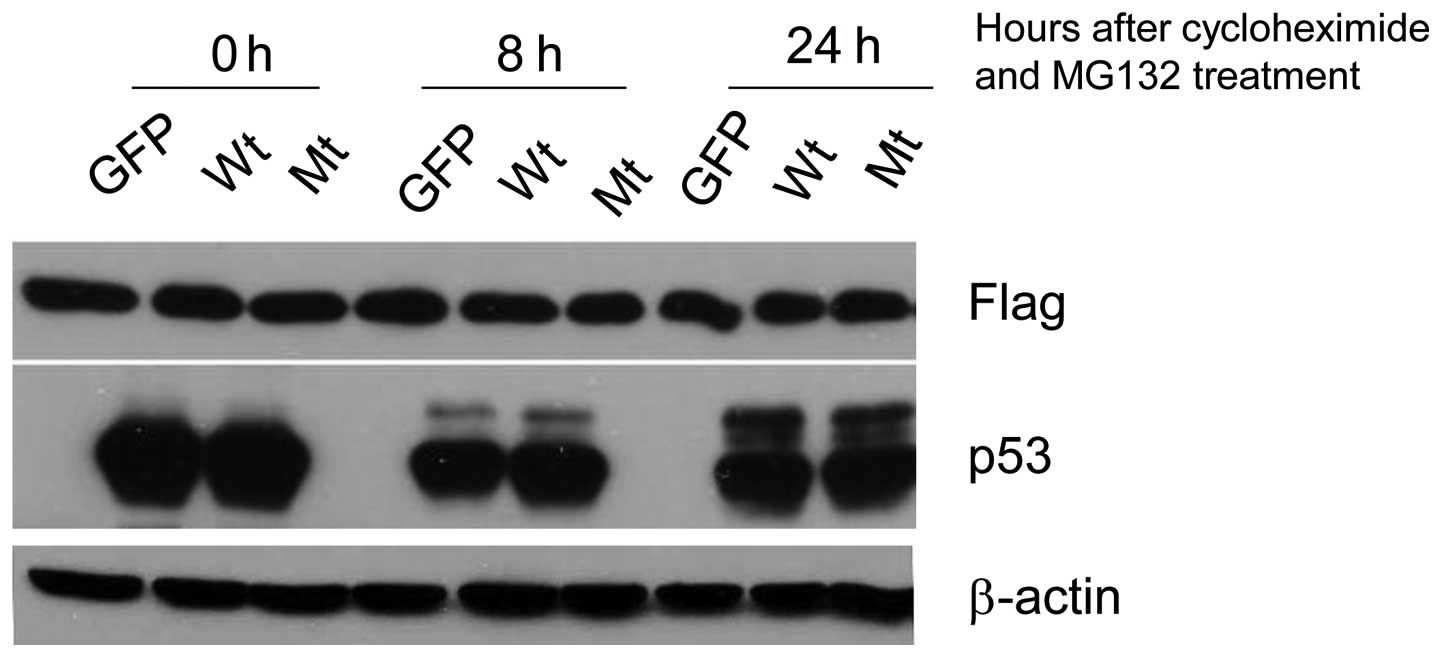
To determine whether p53 mediated 14-3-3γ reduction
by stimulating proteasomal degradation of 14-3-3γ a specific
inhibitor of proteasomal activity, MG132, was used and its effect
on p53-mediated 14-3-3γ reduction was examined. The results showed
that MG132 treatment completely abolished the p53-mediated
reduction of 14-3-3γ protein (Fig.
4) which suggests that p53-mediated suppression of 14-3-3γ is
accomplished by stimulating the process of proteasomal degradation
of the protein.
C-terminal of 14-3-3γ binds to p53
It has been reported that wild-type of p53 can bind
to 14-3-3γ (32,33). This interaction may lead to
proteasomal degradation of 14-3-3 protein. However, the detailed
binding domain of 14-3-3γ is still unclear. To further explore the
possible region of 14-3-3γ that is involved in binding to p53, we
made GST-14-3-3γ truncated fusion proteins including N-terminal,
GST-14-3-3γ1 (1-78 amino acids); central regions, GST-14-3-3γ2 and
3 (79-131 and 132-184 amino acids); and C-terminal GST-14-3-3γ4
(185-247 amino acids) (Fig. 5A).
HCT116 cells were lysed and pull-down assay experiment was
performed to determine the interaction. Fig. 5B showed that the C-terminal region
of 14-3-3γ bound to p53 very efficiently compared with other
domains, demonstrating that p53 could interact with C-terminal of
14-3-3γ.
p53 enhances 14-3-3γ ubiquitination
Since ubiquitination is required in the process of
protein degradation by 26S protesome which can be blocked by MG132,
we next determined whether p53 increases 14-3-3γ protein ubiquitin.
H358 cells were co-transfected with Flag-tagged 14-3-3γ expression
vector and His-tagged ubiquitin expression vector and then infected
with Ad-GFP, Ad-p53 (wt) and Ad-p53 (mt) viruses. After
immunoprecipitation by anti-Flag antibody, 14-3-3γ ubiquitination
was evaluated, and Fig. 6 clearly
shows that wild-type p53 stimulated 14-3-3γ ubiquitination in
vivo. Taken together, these data indicated that p53 could bind
to 14-3-3γ, and enhanced its ubiquitin and proteasome-mediated
degradation.
Discussion
14-3-3γ protein is overexpressed in human lung
cancer tissues (16) and plays a
role in the development of cancer (37). Notably, 14-3-3γ also has been
demonstrated to interact with tumor suppression protein p53 and
enhance transcriptional activity of p53 (33). Therefore, it is important to
characterize the expression regulation of 14-3-3γ. In the present
study, we investigated the expression profile of both 14-3-3γ and
p53 in human lung cancer tissues and found that 14-3-3γ protein
expression correlated with overexpression of p53. Since wild-type
p53 protein level is usually too low to detect we supposed the p53
see in the bands on the blot to be mutated. Therefore, it is
possible that 14-3-3γ expression correlated with p53 mutation in
lung cancer patients. Ecotopic expression experiments showed
wild-type p53, but not mutant p53 (R175H) could reduce both
endogenous and exogenous 14-3-3γ protein levels in colon and lung
cancer cells. In a previous study we showed that wild-type p53
inhibited 14-3-3γ on mRNA level (35). In the present study, we further
demonstrated that p53 also can suppress 14-3-3γ by stimulating
proteasome-mediated 14-3-3γ protein degradation. Together, these
data indicate that p53 negatively regulate 14-3-3γ on both of
transcriptional and post-translational level.
p53 has been studied intensely for its function as a
genome stability guardian by regulating cell cycle arrest,
apoptosis and DNA repair after DNA damage (26,38).
It is also well known that p53 is the most commonly mutated gene
with up to 50% mutations in different types of cancers. Consistent
with previous studies, we found here p53 was overexpressed in 35
out of 80 human lung cancerous tissues. Importantly, high
expression of 14-3-3γ was observed in 94% of these tissues with
overexpression of p53 (33 of 35). Because overexpression of 14-3-3γ
protein can cause DNA polyploidization in human lung cancer
(34) and induced cell oncogenic
transformation (37), it is
possible that p53 guards genome stability and prevents
tumorigenesis through inhibiting 14-3-3γ protein in the lung.
Therefore, 14-3-3γ could be a novel target of p53 for maintaining
genomic stability.
As a tumor suppressor gene, 14-3-3σ expression
levels are significantly reduced or totally lost in a number of
cancers, such as oral squamous cell carcinomas (39), primary bladder tumors (40), most types of breast cancer
(41), gastric cancer (42) and hepatocellular carcinoma
(43). One of the mechanisms
resulting in 14-3-3σ reduction in cancers is due to p53 mutation
because 14-3-3σ is transactivated directly by p53 (29). On the contrary, p53 negatively
regulates 14-3-3γ by promoting its protein degradation. Taken
together, these data suggest that p53 has diverse effects on 14-3-3
proteins in an isoform specific manner. p53 has been demonstrated
to be mutated in almost half of all human cancers. In response to
this, tumor suppression protein 14-3-3σ will be lost and the
potential oncoprotein 14-3-3γ will progress. Therefore, p53 may act
as the center of a complex network in tumorigenesis by regulating
differently the 14-3-3 family proteins.
Recently, 14-3-3 proteins were identified to bind to
an E3 ubiquitin-protein ligase, tripartite motif-containing protein
32 (TRIM32) and to prevent TRIM32 auto-ubiquitylation (44). Notably, Sato et al (45) also discovered the ubiquitin ligase
ATL31 associated with and ubiquitinated 14-3-3 proteins for
degradation via the ubiquitin-proteasome system during the response
to cellular carbon (C)/nitrogen (N) status in Arabidopsis.
Therefore, it is possible that p53 can regulate the interaction
between 14-3-3γ and ubiquitin ligase. However, the mechanism of
14-3-3γ ubiquitination, especially enhanced by p53 is still unclear
and further studies are needed.
Acknowledgements
The present study is supported by the fund from the
Health Administration of Jiangsu Province.
References
|
1
|
Aitken A: 14-3-3 proteins: a historic
overview. Semin Cancer Biol. 16:162–172. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dougherty MK and Morrison DK: Unlocking
the code of 14-3-3. J Cell Sci. 117:1875–1884. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hermeking H: The 14-3-3 cancer connection.
Nat Rev Cancer. 3:931–943. 2003. View
Article : Google Scholar
|
|
4
|
Hermeking H and Benzinger A: 14-3-3
proteins in cell cycle regulation. Semin Cancer Biol. 16:183–192.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang Y, Karas M, Zhao H, Yakar S and
LeRoith D: 14-3-3-sigma mediation of cell cycle progression is
p53-independent in response to insulin-like growth factor-I
receptor activation. J Biol Chem. 279:34353–34360. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kasahara K, Goto H, Enomoto M, Tomono Y,
Kiyono T and Inagaki M: 14-3-3gamma mediates Cdc25A proteolysis to
block premature mitotic entry after DNA damage. EMBO J.
29:2802–2812. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Du J, Chen L, Luo X, Shen Y, Dou Z, Shen
J, Cheng L, Chen Y, Li C, Wang H and Yao X: 14-3-3zeta cooperates
with phosphorylated Plk1 and is required for correct cytokinesis.
Front Biosci (Schol Ed). 4:639–650. 2012. View Article : Google Scholar
|
|
8
|
Masters SC and Fu H: 14-3-3 proteins
mediate an essential antiapoptotic signal. J Biol Chem.
276:45193–45200. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mhawech P: 14-3-3 proteins - an update.
Cell Res. 15:228–236. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yan Y, Xu Y, Gao YY, Zong ZH, Zhang Q, Li
C and Wang HQ: Implication of 14-3-3epsilon and 14-3-3theta/tau in
proteasome inhibition-induced apoptosis of glioma cells. Cancer
Sci. 104:55–61. 2005. View Article : Google Scholar
|
|
11
|
Han DC, Rodriguez LG and Guan JL:
Identification of a novel interaction between integrin beta1 and
14-3-3beta. Oncogene. 20:346–357. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rodriguez LG and Guan JL: 14-3-3
regulation of cell spreading and migration requires a functional
amphipathic groove. J Cell Physiol. 202:285–294. 2005. View Article : Google Scholar
|
|
13
|
Luk SC, Ngai SM, Tsui SK, Fung KP, Lee CY
and Waye MM: In vivo and in vitro association of 14-3-3 epsilon
isoform with calmodulin: implication for signal transduction and
cell proliferation. J Cell Biochem. 73:31–35. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Matitau AE, Gabor TV, Gill RM and Scheid
MP: MEKK2 association with 14-3-3 regulates activation of c-Jun
N-terminal kinase. J Biol Chem. 288:28293–28302. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Radhakrishnan VM, Putnam CW and Martinez
JD: Activation of phosphatidylinositol 3-kinase (PI3K) and
mitogen-activated protein kinase (MAPK) signaling and the
consequent induction of transformation by overexpressed 14-3-3gamma
protein require specific amino acids within 14-3-3gamma N-terminal
variable region II. J Biol Chem. 287:43300–43311. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qi W, Liu X, Qiao D and Martinez JD:
Isoform-specific expression of 14-3-3 proteins in human lung cancer
tissues. Int J Cancer. 113:359–363. 2005. View Article : Google Scholar
|
|
17
|
Fan T, Li R, Todd NW, Qiu Q, Fang HB, Wang
H, Shen J, Zhao RY, Caraway NP, Katz RL, Stass SA and Jiang F:
Up-regulation of 14-3-3zeta in lung cancer and its implication as
prognostic and therapeutic target. Cancer Res. 67:7901–7906. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Titus MA, Tan JA, Gregory CW, Ford OH,
Subramanian RR, Fu H, Wilson EM, Mohler JL and French FS: 14-3-3η
amplifies androgen receptor actions in prostate cancer. Clin Cancer
Res. 15:7571–7581. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Han B, Xie H, Chen Q and Zhang JT:
Sensitizing hormone-refractory prostate cancer cells to drug
treatment by targeting 14-3-3sigma. Mol Cancer Ther. 5:903–912.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li Z, Dong Z, Myer D, Yip-Schneider M, Liu
J, Cui P, Schmidt CM and Zhang JT: Role of 14-3-3sigma in poor
prognosis and in radiation and drug resistance of human pancreatic
cancers. BMC Cancer. 10:5982010. View Article : Google Scholar
|
|
21
|
Naidoo K, Jones R, Dmitrovic B, Wijesuriya
N, Kocher H, Hart IR and Crnogorac-Jurcevic T: Proteome of
formalin-fixed paraffin-embedded pancreatic ductal adenocarcinoma
and lymph node metastases. J Pathol. 226:756–763. 2012. View Article : Google Scholar
|
|
22
|
Nishimura Y, Komatsu S, Ichikawa D, Nagata
H, Hirajima S, Takeshita H, Kawaguchi T, Arita T, Konishi H,
Kashimoto K, Shiozaki A, Fujiwara H, Okamoto K, Tsuda H and Otsuji
E: Overexpression of YWHAZ relates to tumor cell proliferation and
malignant outcome of gastric carcinoma. Br J Cancer. 108:1324–1331.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
O’Dwyer D, Ralton LD, O’Shea A and Murray
GI: The proteomics of colorectal cancer: identification of a
protein signature associated with prognosis. PLoS One.
6:e277182011. View Article : Google Scholar
|
|
24
|
Yang X, Cao W, Zhang L, Zhang W, Zhang X
and Lin H: Targeting 14-3-3zeta in cancer therapy. Cancer Gene
Ther. 19:153–159. 2012. View Article : Google Scholar
|
|
25
|
Kaufmann WK and Paules RS: DNA damage and
cell cycle checkpoints. FASEB J. 10:238–247. 1996.PubMed/NCBI
|
|
26
|
Mendoza-Rodriguez CA and Cerbon MA: Tumor
suppressor gene p53: mechanisms of action in cell proliferation and
death. Rev Invest Clin. 53:266–273. 2001.(In Spanish).
|
|
27
|
Yang X, Taylor L and Polgar P: p53
down-regulates human brady-kinin B1 receptor gene expression. J
Cell Biochem. 82:38–45. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
el-Deiry WS: Regulation of p53 downstream
genes. Semin Cancer Biol. 8:345–357. 1998. View Article : Google Scholar
|
|
29
|
Hermeking H, Lengauer C, Polyak K, He TC,
Zhang L, Thiagalingam S, Kinzler KW and Vogelstein B: 14-3-3 sigma
is a p53-regulated inhibitor of G2/M progression. Mol Cell. 1:3–11.
1997. View Article : Google Scholar
|
|
30
|
Laronga C, Yang HY, Neal C and Lee MH:
Association of the cyclin-dependent kinases and 14-3-3 sigma
negatively regulates cell cycle progression. J Biol Chem.
275:23106–23112. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang HY, Wen YY, Chen CH, Lozano G and Lee
MH: 14-3-3 sigma positively regulates p53 and suppresses tumor
growth. Mol Cell Biol. 23:7096–7107. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rajagopalan S, Sade RS, Townsley FM and
Fersht AR: Mechanistic differences in the transcriptional
activation of p53 by 14-3-3 isoforms. Nucleic Acids Res.
38:893–906. 2010. View Article : Google Scholar :
|
|
33
|
Stavridi ES, Chehab NH, Malikzay A and
Halazonetis TD: Substitutions that compromise the ionizing
radiation-induced association of p53 with 14-3-3 proteins also
compromise the ability of p53 to induce cell cycle arrest. Cancer
Res. 61:7030–7033. 2001.PubMed/NCBI
|
|
34
|
Qi W, Liu X, Chen W, Li Q and Martinez JD:
Overexpression of 14-3-3gamma causes polyploidization in H322 lung
cancer cells. Mol Carcinog. 46:847–856. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Radhakrishnan VM, Putnam CW, Qi W and
Martinez JD: P53 suppresses expression of the 14-3-3 gamma
oncogene. BMC Cancer. 11:3782011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Qiao D, Gaitonde SV, Qi W and Martinez JD:
Deoxycholic acid suppresses p53 by stimulating proteasome-mediated
p53 protein degradation. Carcinogenesis. 22:957–964. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Radhakrishnan VM and Martinez JD:
14-3-3gamma induces oncogenic transformation by stimulating MAP
kinase and PI3K signaling. PLoS One. 5:e114332010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Golubovskaya VM and Cance WG: Targeting
the p53 pathway. Surg Oncol Clin N Am. 22:747–764. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bhawal UK, Sugiyama M, Nomura Y, Kuniyasu
H and Tsukinoki K: Loss of 14-3-3 sigma protein expression and
presence of human papillomavirus type 16 E6 in oral squamous cell
carcinoma. Arch Otolaryngol Head Neck Surg. 134:1055–1059. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ostergaard M, Rasmussen HH, Nielsen HV,
Vorum H, Orntoft TF, Wolf H and Celis JE: Proteome profiling of
bladder squamous cell carcinomas: identification of markers that
define their degree of differentiation. Cancer Res. 57:4111–4117.
1997.PubMed/NCBI
|
|
41
|
Ferguson AT, Evron E, Umbricht CB, Pandita
TK, Chan TA, Hermeking H, Marks JR, Lambers AR, Futreal PA,
Stampfer MR and Sukumar S: High frequency of hypermethylation at
the 14-3-3 sigma locus leads to gene silencing in breast cancer.
Proc Natl Acad Sci USA. 97:6049–6054. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Suzuki H, Itoh F, Toyota M, Kikuchi T,
Kakiuchi H and Imai K: Inactivation of the 14-3-3 sigma gene is
associated with 5′ CpG island hypermethylation in human cancers.
Cancer Res. 60:4353–4357. 2000.PubMed/NCBI
|
|
43
|
Iwata N, Yamamoto H, Sasaki S, Itoh F,
Suzuki H, Kikuchi T, Kaneto H, Iku S, Ozeki I, Karino Y, Satoh T,
Toyota J, Satoh M, Endo T and Imai K: Frequent hypermethylation of
CpG islands and loss of expression of the 14-3-3 sigma gene in
human hepatocellular carcinoma. Oncogene. 19:5298–5302. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ichimura T, Taoka M, Shoji I, Kato H, Sato
T, Hatakeyama S, Isobe T and Hachiya N: 14-3-3 proteins sequester a
pool of soluble TRIM32 ubiquitin ligase to repress
autoubiquitylation and cytoplasmic body formation. J Cell Sci.
126:2014–2026. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sato T, Maekawa S, Yasuda S, Domeki Y,
Sueyoshi K, Fujiwara M, Fukao Y, Goto DB and Yamaguchi J:
Identification of 14-3-3 proteins as a target of ATL31 ubiquitin
ligase, a regulator of the C/N response in Arabidopsis. Plant J.
68:137–146. 2011. View Article : Google Scholar : PubMed/NCBI
|