Introduction
The epidermal growth factor receptor (EGFR) is a
member of the ErbB family of tyrosine kinases, and plays an
important role in the pathogenesis of different tumors. Therefore,
therapies that target EGFR function may prove effective as
anticancer treatments (1,2). Upon ligand binding, the EGFR
dimerizes, and the intracellular tyrosine kinase region is
activated; this causes receptor tyrosine autophosphorylation and
transphosphorylation of another receptor monomer (3,4).
These events lead to the recruitment and phosphorylation of several
intracellular substrates and the subsequent transmission of
extracellular signals to the nucleus via an intracellular signaling
network (4–6). Specifically, the EGFR pathway
involves the downstream mediators RAS/MAPK, phosphatidylinositol
3-kinase (PI3K)/serine/threonine kinase AKT, and STAT signaling
pathways; this leads to increased cellular survival, proliferation,
invasion, and inhibition of apoptosis (4,7).
After EGFR activation, the epidermal growth factor
(EGF)-EGFR complexes are internalized via clathrin-coated vesicles
and delivered to the early endosomal tubulovesicular compartment
commonly referred to as the ‘sorting endosomes’. In this
compartment, ubiquitinated receptors are recognized by the
endosomal sorting complex required for transport (ESCRT) machinery;
the ESCRT complexes generate multivesicular bodies (MVBs) by
packaging cargo into small vesicles that bud off from the limiting
membrane into the lumen of the endosomes (8). This sorting pathway terminates
EGF/EGFR signaling by delivering receptors to the lysosomes for
degradation, a process known as down-regulation (4). Alternatively, EGFR is recycled to the
plasma membrane by endosomal recycling pathways.
Selective EGFR-tyrosine kinase inhibitors
(EGFR-TKIs) such as gefitinib and erlotinib can block the signal
transduction pathways implicated in the proliferation and survival
of cancer cells (9–12). These EGFR-TKIs are effective in
treating non-small cell lung cancers (NSCLCs) that have activating
mutations in the EGFR gene (13,14).
Although most EGFR mutant NSCLCs initially respond to EGFR
inhibitors, the majority of these tumors eventually develop
resistance. Amplification of MET induces gefitinib
resistance by driving ERBB3 (HER3)-dependent activation of PI3K, a
pathway thought to be specific to EGFR/ERBB family receptors in
lung cancer (15). At present, the
mechanisms that contribute to gefitinib resistance in the remaining
tumors are unknown.
We reported previously that aberrations in certain
steps of EGF/phosphorylated epidermal growth factor receptor
(pEGFR) endocytic trafficking from the early endosomes to the late
endosomes/lysosomes occurs in gefitinib-resistant EGFR
wild-type NSCLC cells, whereas endocytosis of EGFR/pEGFR is normal
in gefitinib-sensitive EGFR mutant NSCLC cells (16,17).
We observed that large amounts of sorting nexin 1 (SNX1) colocalize
with internalized pEGFR in aggregated early endosomes (18).
Furthermore, we demonstrated that silencing of
endogenous SNX1 by siRNA stimulates endocytosis and the
ligand-induced downregulation of EGFR/pEGFR or MET/pMET, and
concomitantly increases pEGFR and phosphorylated MET (pMET) protein
expression in gefitinib-resistant cells (19,20).
These data indicate that SNX1 negatively regulates EGF-dependent
downregulation of EGFR and its phosphorylation via the early/late
endocytic pathway in human lung cancer cells. SNX1 was originally
identified as a protein that interacts with EGFR (21) and is localized to early endosomes
through its phospholipid-binding motif, which is termed the phox
homology (PX) domain (22).
Together with the observation that SNX1 enhances degradation of the
receptor upon EGF stimulation, these data indicate that SNX1 plays
a role in endosome-lysosome trafficking (21,23).
However, other reports do not support an intracellular
colocalization of SNX1 with EGFR or its direct role in EGFR
degradation (24,25); therefore, alternative mechanisms
must link SNX1 function with EGFR recycling. However, the specifics
of such mechanisms remain to be elucidated.
On the basis of these observations, we designed the
current study to investigate the role of SNX1 on EGF-stimulated
pEGFR endocytosis. We examined the effect of SNX1 knockdown and
treatment with monensin (an inhibitor of membrane recycling), on
EGF-stimulated EGFR endocytosis, and on phosphorylation of AKT. In
a gefitinib-resistant NSCLC cell line, we observed a rapid
phosphorylation of EGFR along with a substantial increase in
phosphorylated AKT (pAKT) after EGF stimulation. On the other hand,
monensin significantly suppressed AKT phosphorylation in the
EGF-treated cells. We therefore suggest that EGF-stimulated
recycling of EGFR to the plasma membrane is an important step that
activates the PI3K-AKT pathway, and that SNX1 is a negative
regulator of EGFR recycling.
Materials and methods
Materials
Texas Red-labeled human transferrin, and SlowFade
antifade reagent were purchased from Molecular Probes (Eugene, OR,
USA). Recombinant human EGF and hepatocyte growth factor (HGF) were
purchased from PeproTech, Inc. (London, UK). Monensin, bafilomycin
A1, cycloheximide (CHX), and DAPI were obtained from Sigma (St.
Louis, MO, USA). Other chemicals were of reagent grade and were
obtained from commercial sources.
Cell culture
Cell line A549 (National Kyushu Cancer Center,
Fukuoka, Japan) was cultured in RPMI supplemented with 10% fetal
bovine serum (FBS). Cells were maintained under standard cell
culture conditions at 37°C and 5% CO2 in a humid
environment. A549 cells transfected with siRNA-control, -SNX1, or
-Rab11a were starved for 12 h with RPMI-1640 without FBS at 37°C.
The serum-starved cells were preincubated with 10 μM monensin for 2
h or 0.17 μM bafilomycin A1 for 30 min and then stimulated with EGF
(100 ng/ml) at 37°C for the indicated times, after which further
analysis was performed.
Small interfering RNA
siRNAs corresponding to the nucleotide sequences of
the SNX1 (5′-AAGAACAAGACCAAGAGC CAC-3′) and Rab11a
(5′-AATGTCAGACAGACGCGAAA AT-3′) were purchased from Dharmacon
Research, Inc. (Boulder, CO, USA). Scramble sequence was used as a
control. A549 cells were transfected with Lipofectamine 2000 (Life
Technologies, Inc., Gaithersburg, MD, USA) in the presence of 40 nM
siRNAs according to the manufacturer’s instructions. Knock-down
efficiency was determined by RT-qPCR, and western blot
analysis.
RT-qPCR analysis
A549 cells transfected with siRNA- control, -SNX1,
or -Rab11a were incubated at 37°C for 48 h, and total RNA was
extracted from each transfectant using an RNeasy RNA isolation kit
(Qiagen, Hilden, Germany) according to the manufacturer’s
instructions. Transcription into cDNA was done in a 20-μl volume
using ThermoScript RT-PCR System with random hexamer (Invitrogen
Life Technologies, Carlsbad, CA, USA) according to the
manufacturer’s instructions. All PCR reactions were carried out in
a final volume of 25 μl and were performed in the ABI PRISM 7000
Sequence Detection System (Applied Biosystems, Inc., Foster City,
CA, USA) according to the manufacturer’s instructions.
Sequence-specific primers were quoted from an official website
‘PrimerBank’ (http://pga.mgh.harvard.edu/primerbank/) for the
indicated genes (Table I). The
reaction mixture consisted of SYBR Premix Ex Taq (2X) 12.5 μl, ROX
Reference Dye (50X) (both from Takara Bio, Inc., Shiga, Japan) 0.5
μl, 0.2 μM of each specific forward and reverse primer, and 9 μl of
diluted cDNA (equivalent to 0.03–2.85 ng of total RNA).
Amplifications were done under standard conditions (10 sec at 95°C
followed by 40 cycles of 5 sec at 95°C and 31 sec at 60°C). The
number of PCR cycles needed to reach the fluorescence threshold was
determined in triplicate for each cDNA, averaged and then
normalized to a reference gene (β-actin). A standard curve
was generated with serial 3-fold dilutions of a representative
cDNA. For all assays tested, the PCR reaction was linear over the
range studied (20–40 cycles of amplification). All RT-PCR reactions
gave a single band when analyzed by gel electrophoresis.
 | Table IPrimers for RT-qPCR for SNX1, Rab11a
and β-actin. |
Table I
Primers for RT-qPCR for SNX1, Rab11a
and β-actin.
| PCR primer | Nucleotide
sequence |
|---|
| SNX1 | F:
5′-AGCCCCAGCCAACCTATGA-3′
R: 5′-TCAGGATCAGTTATACCGACTGT-3′ |
| Rab11a | F:
5′-CAACAAGAAGCATCCAGGTTGA-3′
R: 5′-GCACCTACAGCTCCACGATAAT-3′ |
| β-actin | F:
5′-CATGTACGTTGCTATCCAGGC-3′
R: 5′-CTCCTTAATGTCACGCACGAT-3′ |
Antibodies
Alexa Fluor 488-labeled goat anti-mouse and goat
anti-rabbit secondary antibodies were obtained from Molecular
Probes. Normal rabbit IgG and normal mouse monoclonal IgG1 were
purchased from Imgenex Corp. (San Diego, CA, USA) and
Angio-Proteomie (Boston, MA, USA), respectively. Normal goat serum
was purchased from Sigma. Mouse monoclonal antibody to SNX1 was
purchased from BD Biosciences (San Jose, CA, USA). Anti-EGFR,
pEGFR, MET, pMET, AKT, pAKT (Ser473), pAKT (Thr308), p44/42 MAPK
(MAPK) and anti-phospho-p44/42 MAPK (p-MAPK) antibodies were
obtained from Cell Signaling Technology, Inc. (Beverly, MA, USA),
and anti-β-actin antibody was obtained from Sigma. Anti-cathepsin D
was affinity-purified by protein A Sepharose CL-4B (Sigma),
followed by immunoaffinity chromatography using antigen-conjugated
Sepharose 4B as described previously (26,27).
Immunofluorescence microscopy
Immunofluorescence microscopy was described
previously (16–20). A549 cells were grown for 2 days on
glass coverslips in 6-well plates in RPMI with 10% FBS. At 48 h
after the transfection of A549 cells with siRNA-control or -SNX1,
cells were starved for 12 h with RPMI without FBS at 37°C and the
cells were incubated in the presence of Texas Red-transferrin in
prewarmed medium, and the serum-starved cells were then incubated
with EGF (100 ng/ml) at 37°C for 30 min or 1 h. The cells were
fixed with 3.7% formaldehyde in phosphate-buffered saline (PBS), pH
7.4, permeabilized in PBS containing 0.1% saponin. After washing
with PBS, cells were blocked with PBS-10% normal goat serum. All
subsequent antibody and wash solutions contained 0.1% saponin. The
fixed cells were double-stained for pEGFR with anti-pEGFR
monoclonal antibody and for cathepsin D with anti-cathepsin D
antibody or for LIMPII with anti-LIMPII antibody followed by washes
with PBS containing 0.1% saponin and incubation for 1 h with the
secondary antibodies at 20 μg/ml. Each cell line was stained with
DAPI to reveal nuclei. Controls for antibody specificity were
non-immune normal mouse IgG1 or non-immune normal rabbit IgG. Late
endosomes/lysosomes were stained with anti-LIMPII antibody, since
the LIMPII protein is distributed within endocytic organelles and
is at its highest concentration in the late endosomes/lysosomes, as
observed for other lysosomal glycoproteins, namely,
lysosomal-associated membrane protein 1 (LAMP1) and 2 (28,29).
The distribution of the labeled proteins was then analyzed by
confocal immunofluorescence microscopy of the fixed cells. Slides
were mounted with SlowFade antifade reagent and observed on a Zeiss
LSM 510 META confocal laser scanning microscope (Carl Zeiss,
Oberkochen, Germany), equipped with krypton/argon laser sources.
Colocalization of pEGFR and Texas Red-labeled transferrin,
cathepsin D or LIMPII was quantified using ImageJ software and
MacSCOPE X software (Mitani Corp., Osaka, Japan).
Western blot analysis
Protein samples were separated by sodium dodecyl
sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and then
transferred to polyvinylidene difluoride membranes (Millipore,
Billerica, MA, USA). Following blocking, the membrane was blotted
with the appropriate antibody, and subsequently, horseradish
peroxidase-conjugated anti-mouse or anti-rabbit IgG (GE Healthcare
Bio-Sciences Corp., Tokyo, Japan) was applied. The final signal was
revealed by ECL chemiluminescence (Pierce Biotechnology, Inc.,
Rockford, IL, USA). Digital images were analyzed with NIH Image
software to measure the density of each band without a saturated
signal. For EGF-stimulated pEGFR degradation, A549 cells
transfected with siRNA-control or -SNX1 were starved for 12 h with
RPMI without FBS at 37°C. The serum-starved cells were then
pre-incubated for 30 min in the presence of CHX (20 μg/ml ) before
incubation with EGF (100 ng/ml) at 37°C for the indicated times.
The cells were then washed with ice-cold PBS and lysed, followed by
SDS-PAGE and western blot analysis. Monensin (10 μM) or bafilomycin
A1 (0.17 μM) was added when the cells were incubated with RPMI.
Statistical analysis
Data are expressed as mean ± SD unless otherwise
noted. Significance (p<0.05) was determined by using Student’s
t-test, since all data met all the assumptions for parametric
statistical analysis.
Results
Knockdown of SNX1 stimulates pEGFR
transport after EGF stimulation in gefitinib-resistant NSCLC cell
line
To examine the role of SNX1 in regulating
EGF-stimulated EGFR signaling and endocytosis, we knocked down
endogenous of SNX1 in the gefitinib-resistant NSCLC cell line,
A549. Fig. 1A (left) shows that
endogenous SNX1 mRNA was successfully depleted using
specific siRNA in these cells; the level of SNX1 transcripts
was 14.7±3.0% of that observed in control cells (p<0.001).
Consistent with this, western blot analysis (Fig. 3B) revealed that endogenous SNX1
protein was almost completely depleted following siRNA
transfection.
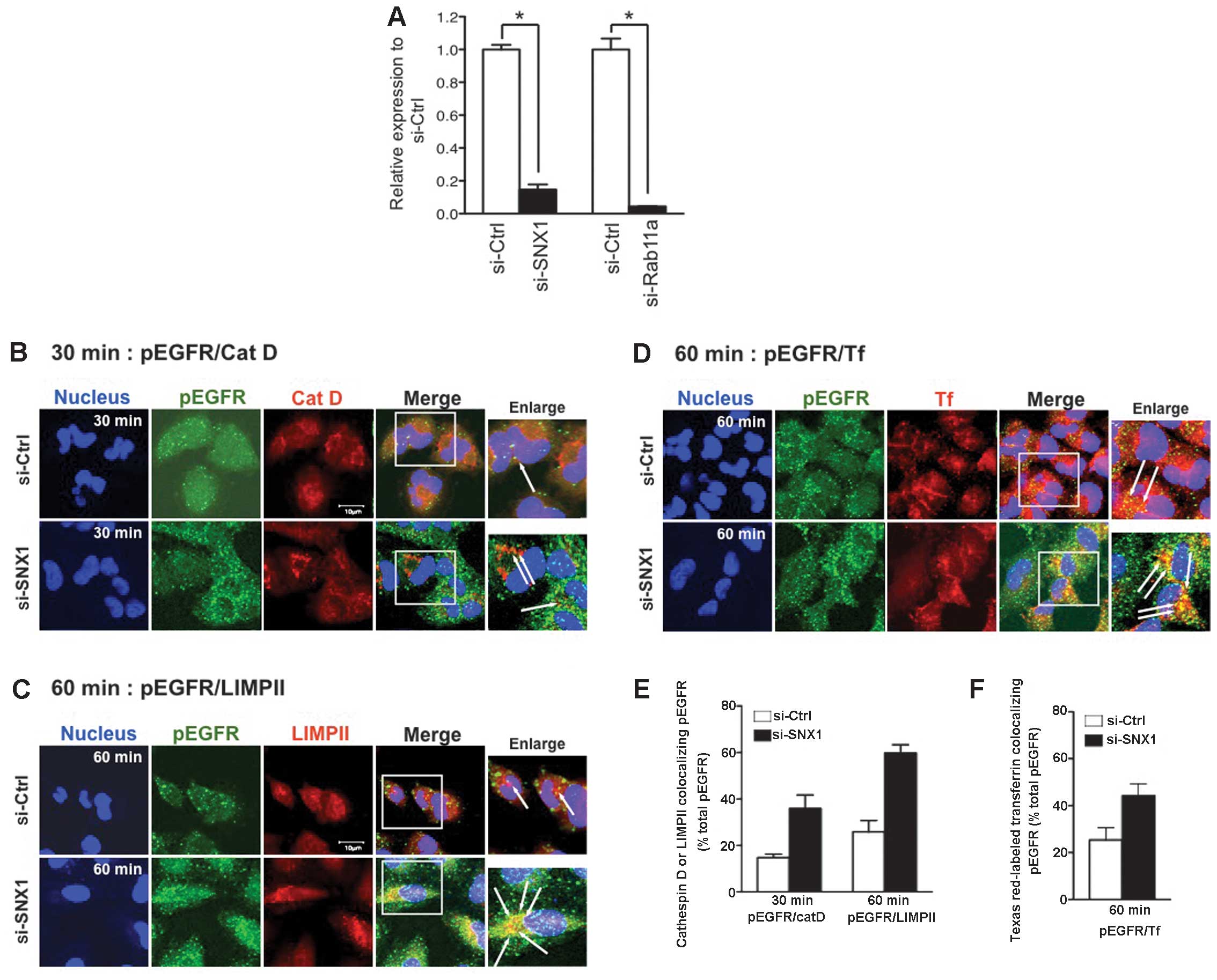 | Figure 1Epidermal growth factor receptor
(EGFR) phosphorylation and endocytosis are upregulated in sorting
nexin 1 (SNX1)-defient gefitinib-resistant A549 cells. (A) RT-qPCR
analysis of SNX1 and Rab11a mRNAs in A549 cells transfected with
siRNA-control (si-Ctrl), siRNA-SNX1 (si-SNX1), or siRNA-Rab11a
(si-Rab11a). Expression of SNX1 and Rab11a mRNAs was normalized
with that of β-actin. The error bar denotes SD from three separate
experiments, and significance (*p<0.05) was
determined using Student’s t-test. (B–F) A549 cells transfected
with si-Ctrl or -SNX1 were pre-treated at 37°C for 30 min in the
presence of Texas Red-transferrin in prewarmed medium, and the
cells were then incubated with epidermal growth factor (EGF) (100
ng/ml) at 37°C for 30 or 60 min. Internalized phosphorylated
epidermal growth factor receptor (pEGFR), lysosomes, and late
endosomes/lysosomes were then detected as described in Materials
and methods. Superimposed images of (B) pEGFR (green) with
cathepsin D (red) at 30 min, (C) pEGFR (green) with LIMPII (red) at
60 min, or (D) pEGFR (green) with the endocytosed Texas Red-labeled
transferrin (red) at 60 min, are also shown. Cells were stained
with DAPI (blue) to reveal nuclei. Scale bar, 10 μm. Right column
shows the merged images for double staining of pEGFR (green) and
each organelle marker (red), and white squares indicate enlarged
regions. The long white arrows indicate the merged confocal images
of pEGFR and each organelle marker. (E) Quantitative analysis of
colocalization between pEGFR and cathepsin D (at 30 min) or pEGFR
and LIMPII (at 60 min) in the si-Ctrl-transfected cells (white
column) or the si-SNX1-transfected cells (black column). Values are
the percentage of the integrated density of cathepsin D- or
LIMPII-colocalizing pEGFR compared to that of total pEGFR. (F)
Colocalization of pEGFR and Texas Red-labeled transferrin in the
si-Ctrl-transfected cells (white column) or the si-SNX1-transfected
cells (black column) are shown. Values are the percentage of the
integrated density of Texas Red-labeled transferrin-colocalized
pEGFR compared to that of total pEGFR. |
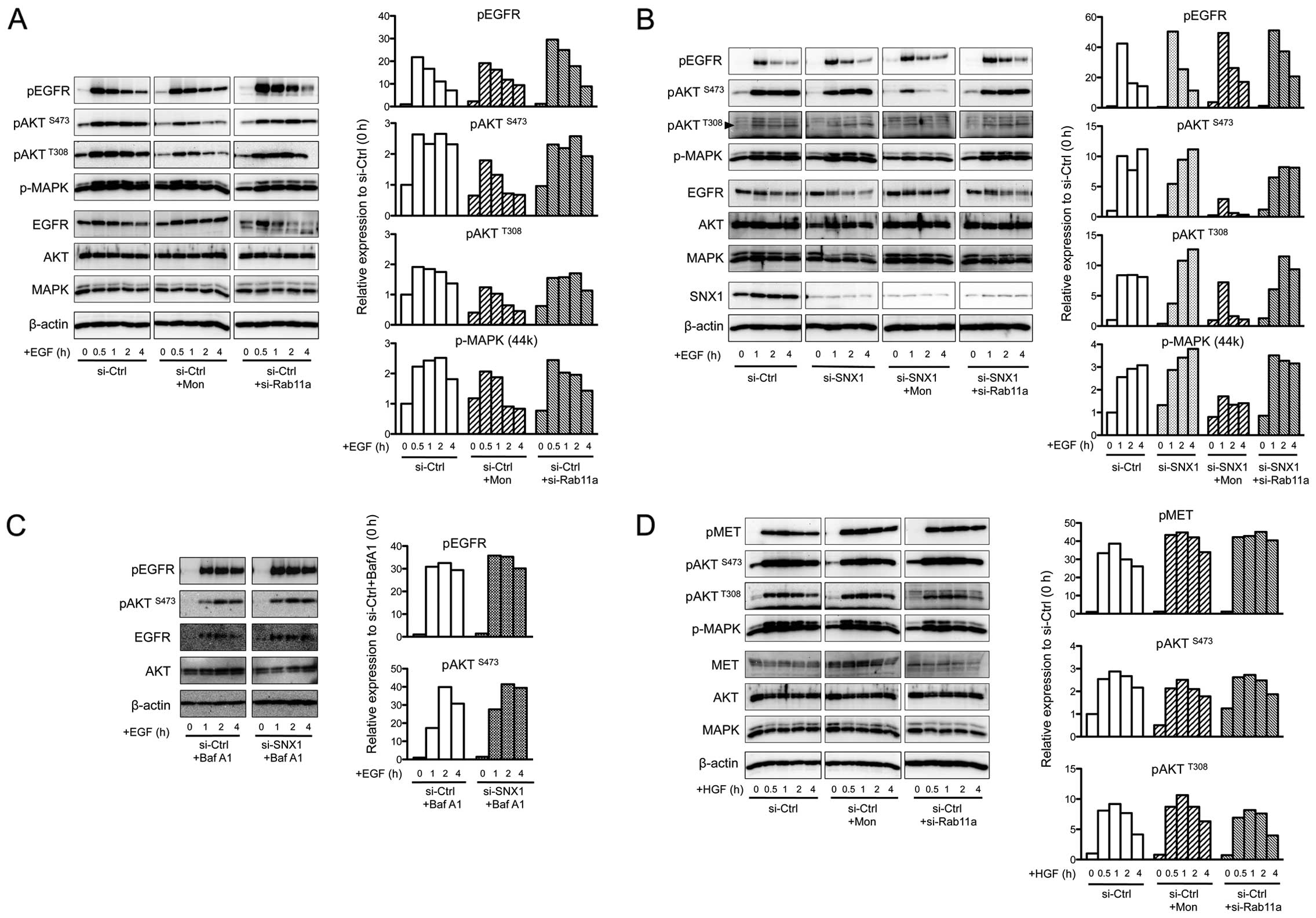 | Figure 3Effect of membrane recycling
inhibition on the activation of tyrosine kinase receptors and
downstream signaling in sorting nexin 1 (SNX1)-deficient cells. (A)
Left: expression of phosphorylated epidermal growth factor receptor
(pEGFR), phosphorylated AKT (pAKT) (Ser473), pAKT (Thr308),
phospho-p44/42 MAPK (p-MAPK), total epidermal growth factor
receptor (EGFR), total AKT, and total p44/42 MAPK (MAPK) before and
after 0.5, 1, 2, and 4 h of epidermal growth factor (EGF)
stimulation in the absence or presence of monensin (Mon) (10 μM) in
A549 cells transfected with siRNA-control (si-Ctrl). Effect of
Rab11a knockdown on the activation of growth factor receptors and
the downstream molecules in the si-Ctrl-transfected A549 cells was
also determined by western blot analysis. β-actin was used as a
loading control. Right: the quantitative western blot analysis is
shown. Relative expression levels of pEGFR, pAKT (Ser473), pAKT
(Thr308), and p-MAPK are shown and the values are normalized to
each phosphorylated protein before EGF stimulation in the
si-Ctrl-transfected cells. In the case of p-MAPK, the relative
expression levels of phosphorylated form of MAPK (upper band) was
determined. All results shown are representative of three
independent experiments. pEGFR, phosphorylated human EGFR; pAKT,
phosphorylated AKT (protein kinase B). (B) Left: expression of
pEGFR, pAKT (Ser473), pAKT (Thr308), p-MAPK, total EGFR, total AKT,
and total MAPK before and after 1, 2, and 4 h of EGF stimulation in
the absence or presence of Mon (10 μM) in cells transfected with
si-Ctrl or -SNX1 was determined by western blot analysis of protein
lysates. Effect of Rab11a knockdown on the activation of growth
factor receptors and the downstream molecules in the
si-SNX1-transfected A549 cells was also determined. pAKT (Thr308)
is indicated by arrowhead on the left. Knock-down efficiency of
SNX1 was examined by western blot analysis. β-actin was used as a
loading control. Right: the quantitative western blot analysis is
shown. Relative expression levels of pEGFR, pAKT (Ser473), pAKT
(Thr308), and p-MAPK are shown and the values are normalized to
each phosphorylated protein before EGF stimulation in the
si-Ctrl-transfected cells. All results shown are representative of
three independent experiments. (C) Effect of bafilomycin A1 (Baf
A1) on the activation of EGFR and AKT upon EGF stimulation in
si-Ctrl- or si-SNX1-transfected cells. Left: the expression of
pEGFR, pAKT (Ser473), total EGFR, and total AKT before and after 1,
2, and 4 h of EGF stimulation in the presence of Baf A1 in A549
cells transfected with si-Ctrl or -SNX1. The activation of growth
factor receptors and the downstream targets was determined by
western blot analysis. β-actin was used as a loading control.
Right: the quantitative western blot analysis is shown. Relative
expression levels of pEGFR and pAKT (Ser473) are shown and the
values are normalized to each phosphorylated protein before EGF
stimulation (in the absence of EGF stimulation) in the
si-Ctrl-transfected A549 cells. All results shown are
representative of three independent experiments. (D) Left:
expression of phosphorylated MET (pMET), pAKT (Ser473), pAKT
(Thr308), p-MAPK, total MET, total AKT, and total MAPK before and
after 0.5, 1, 2, and 4 h of hepatocyte growth factor (HGF)
stimulation in the absence or presence of monensin in cells
transfected with si-Ctrl was determined by western blot analysis of
protein lysates. Effect of Rab11a knockdown on the activation of
MET and the downstream molecules in the si-Ctrl-transfected A549
cells was also determined by western blot analysis. β-actin was
used as a loading control. Right: quantitative western blot
analysis is shown. Relative expression levels of pMET, pAKT
(Ser473), and pAKT (Thr308) are shown and the values are normalized
to each phosphorylated protein before HGF stimulation in the
si-Ctrl-transfected A549 cells. All results shown are
representative of three independent experiments. |
We recently reported that the gefitinib-resistant
cells accumulate internalized EGFR in the aggregated early
endosomes, whereas the gefitinib-sensitive cells show efficient
endocytosis of EGFR, and this is associated with SNX1 (17–19).
Accordingly, we inferred that deregulated SNX1 function might
underlie the alterations to EGFR endocytosis, leading to
gefitinib-resistance in NSCLC cell lines. In order to test this
hypothesis, we investigated the fate of internalized pEGFR in early
endosomes or late endosomes/lysosomes in A549 cells. Cells
transfected with siRNA-control or -SNX1 were incubated with EGF for
30 min and 1 h, and the colocalization of pEGFR with cathepsin D,
LIMPII, or Texas Red-labeled transferrin was then assessed by
confocal immunofluorescence microscopy as described in Materials
and methods. As shown in Fig. 1B and
C, no colocalization of pEGFR with cathepsin D- or
LIMPII-positive vesicles was seen in cells transfected with
siRNA-control until 1 h post-EGF treatment. In cells transfected
with siRNA-SNX1, however, the amount of pEGFR was markedly induced
and a significant increase in the colocalization of pEGFR with
cathepsin D and LIMPII was also observed. Robust accumulation of
pEGFR was also observed in the endocytosed transferrin-positive
early endosomes of the siRNA-SNX1-transfected cells (Fig. 1D). These results confirm not only
that SNX1 depletion increases the amounts of pEGFR, but also that
it stimulates ligand-induced pEGFR endocytosis via the early/late
endocytic pathway in a gefitinib-resistant NSCLC cell line. These
results are consistent with our previous observations (19). Quantitative analysis of the
colocalization confirmed that the amount of cathepsin D- or
LIMPII-colocalized pEGFR was greater in the cells transfected with
siRNA-SNX1 than in control cells after 30 min (36.0±5.8% vs.
14.8±1.5% total pEGFR) and 1 h (59.7±3.6% vs. 25.8±4.9% total
pEGFR) of EGF stimulation, respectively (Fig. 1E). In addition, the amount of pEGFR
that colocalized with transferrin was greater in cells transfected
with siRNA-SNX1 compared to controls after 1 h (44.2±5.1% vs.
25.4±5.2% total pEGFR) of EGF stimulation (Fig. 1F). These data demonstrate that
silencing of SNX1 stimulates rapid endocytosis and endocytic
trafficking of EGFR/pEGFR via the early/late endocytic pathway in
the gefitinib-resistant A549 cells.
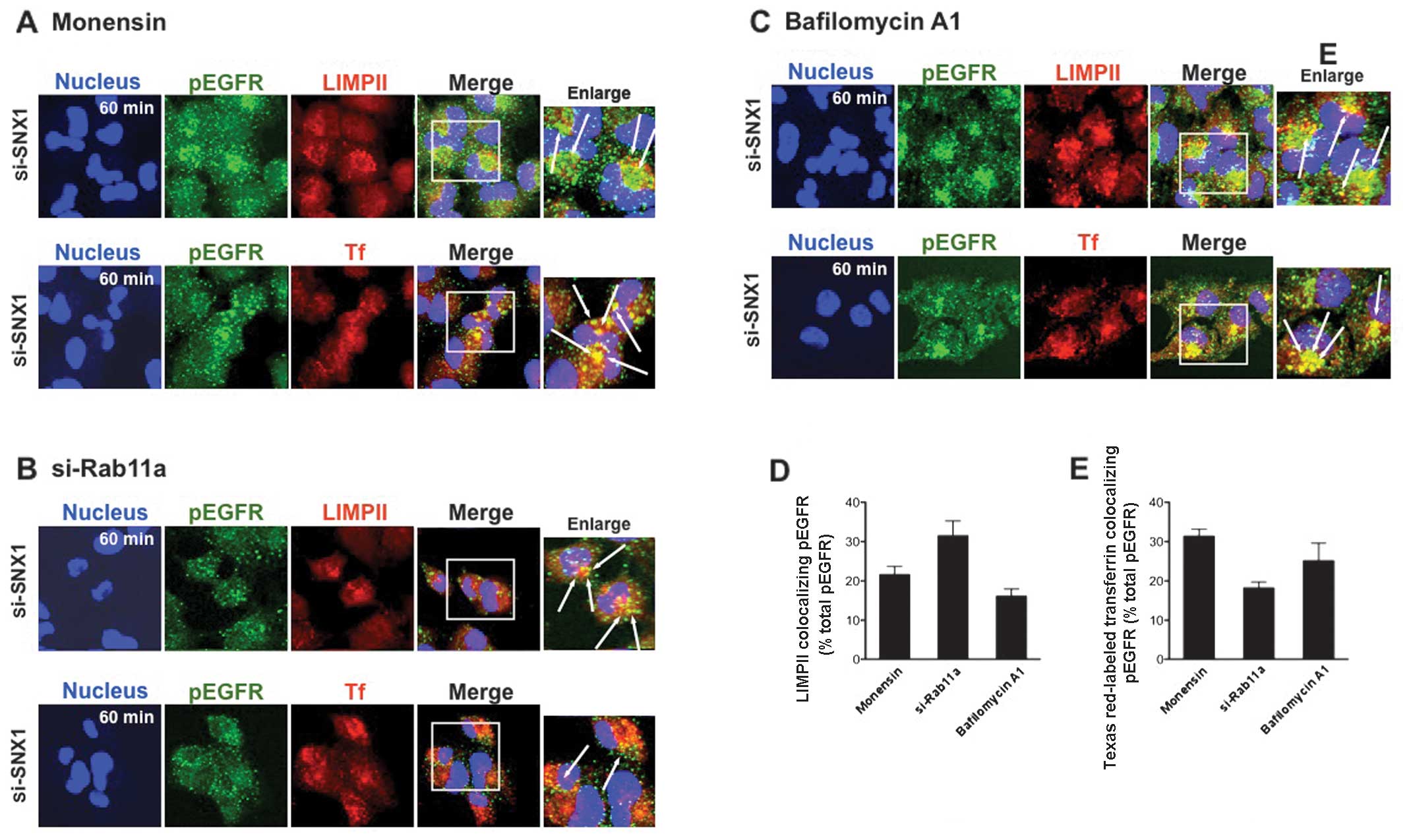
Inhibition of membrane recycling leads to
accumulation of pEGFR in endosomes in the absence of SNX1
Upon EGF stimulation, a fraction of endocytosed EGFR
is recycled back to the plasma membrane instead of being sorted to
the late endosomes/lysosomes (30). Therefore, we assumed that
non-degraded/non-pEGFR might undergo extensive recycling to the
cell membranes from early endosomes via the early endocytic
pathway. To further explore the stage at which SNX1 modulates EGFR
recycling, we treated SNX1 knock-down cells with monensin, an
inhibitor of EGFR recycling between early endosomes and cell
membranes (31).
Cell transfected with siRNA-SNX1 were pre-treated
with 10 μM monensin for 2 h and followed by stimulation for 1 h
with EGF. Cells were then fixed and double-labeled with antibodies
to pEGFR and LIMPII or to pEGFR and the endocytosed Texas
Red-labeled transferrin. The change in the intracellular
distribution of pEGFR was examined by confocal immunofluorescence
microscopy. Monensin increased pEGFR accumulation in the swollen
vacuoles that overlapped with the LIMPII-positive late endosomes
after 1 h of EGF stimulation (Fig.
2A). Robust accumulation of pEGFR was also observed in the
endocytosed transferrin-positive early endosomes of the
monensin-treated cells (Fig. 2A).
These effects were also reproduced by treatment with bafilomycin
A1, a lysosomal inhibitor (Fig.
2C). Despite the similarities between monensin and bafilomycin
A1, we inferred that pEGFR accumulation induced by monensin was
mostly due to the suppression of membrane recycling from early
endosomes to plasma membranes. We further investigated the role of
Rab11a, a small GTPase of the Rab family that regulates the
trafficking of intracellular vesicles from the early endosomes
through the trans-Golgi network compartments during the recycling
process (32). In addition, Rab11a
reportedly modulate the recycling of cell surface proteins
including the transferrin receptor.
To examine the role of Rab11a on the EGFR recycling
in A549 cells, the siRNA-SNX1-transfected cells were depleted of
Rab11a using specific siRNA and the intracellular distribution of
pEGFR along with early/late endosomes at 1 h after EGF stimulation
was then examined. The depletion of endogenous Rab11a mRNA was
verified by RT-qPCR analysis as shown in Fig. 1A; the Rab11a mRNA level was reduced
to 4.38±0.15% of that observed in controls (p<0.001, Fig. 1A, right). As shown in Fig, 2B, the
stimulation of endocytic delivery of pEGFR from early endosomes to
late endosomes triggered by SNX1 knockdown was not affected by the
loss of Rab11a. This indicates that Rab11a does not block EGFR
recycling, and that pEGFR is mostly recycled back to the plasma
membranes from early endosomes, but not from recycling endosomes.
Quantitative analysis of the colocalization confirmed that
LIMPII/pEGFR colocalization is greater in cells transfected with
siRNA-Rab11a than in the cells treated with monensin (31.4±3.9% vs.
21.5±2.2% total pEGFR) or bafilomycin A1 (31.4±3.9% vs. 16.1±1.9%
total pEGFR). Further, the amount of colocalized transferrin and
pEGFR was lower in cells transfected with siRNA-Rab11a than in the
cells treated with monensin (18.2±1.6% vs. 31.3±1.8% total pEGFR)
or bafilomycin A1 (18.2±1.6% vs. 25.1±4.6% total pEGFR) (Fig. 2D and E). These results indicate
that endocytosed pEGFR accumulates in early endosomes before being
sorted to late endosomes/lysosomes in the siRNA-SNX1-transfected
cells treated with monensin or bafilomycin A1. In contrast, pEGFR
is sorted to late endosomes/lysosomes efficiently in cells
transfected with siRNA-Rab11a.
AKT phosphorylation is inhibited by
monensin following EGF stimulation of gefitinib-resistant human
NSCLC cells
We described above that the pEGFR was not
efficiently trafficked to late endosomes/lysosomes in A549 cells
after EGF stimulation, but that SNX1 knockdown increased
phosphorylation of EGFR and stimulated its endocytosis via the
early endocytic pathway. Since a fraction of internalized
non-degraded EGFR is recycled to the plasma membrane from endosomes
via the endocytic pathway (30,31),
we hypothesized that EGF stimulated EGFR activation and consequent
PI3K-AKT pathway might be mediated by recycling of EGFR from early
endosomes to the plasma membranes.
To investigate this, we examined whether treatment
of SNX1 knock-down cells with monensin affected AKT
phosphorylation. Cells transfected with siRNA-control or -SNX1 were
pre-incubated with 10 μM monensin for 2 h and then stimulated with
EGF at 37°C in the absence or presence of monensin for the
indicated times. The lysates were analyzed with anti-pEGFR,
anti-pAKTS473, anti-pAKTT308, or
anti-p-p44/42 MAPK antibody by western blot analysis.
AKT is activated by phospholipid binding, which
triggers phosphorylation in the activation loop at Thr308 residue
by PDK1, which is essential for AKT catalytic activity (33). On the other hand, PDK2
phosphorylates a Ser473 residue in the carboxy terminus of AKT,
which also contributes to activation, which has been identified as
mammalian target of rapamycin (mTOR) in a rapamycin-insensitive
complex with Rictor and Sin1 (34,35).
As shown in Fig.
3B, the expression of pEGFR in SNX1 knock-down cells was
1.2-fold higher within 1-h incubation than in control cells after
EGF stimulation. pEGFR levels then gradually fell during 4-h
incubation period (Fig. 3A and B),
and the drop was suppressed by bafilomycin A1 (Fig. 3C). This indicates that
intracellular degradation of EGF-induced pEGFR proceeds via an
endosomal/lysosomal pathway, and is consistent with our previous
data (19). These results
accordingly imply that SNX1 plays a suppressive role in the
phosphorylation and downregulation of pEGFR via the endocytic
pathway in the gefitinib-resistant NSCLC cell line A549.
A rapid increase of pAKTS473 was seen in
the siRNA- control-transfected cells, with the level reaching
10-fold that of basal at 1 h; this was sustained during the
following 4-h incubation (Fig.
3B). In SNX1 knock-down cells, the increase in
pAKTS473 was ~5-fold at 1 h, but reached 10-fold by 4 h
post-treatment (Fig. 3B).
Similarly, the increased levels of pAKTT308 were
observed in both control and SNX1 knock-down cells, with increments
of 8- and 11-fold, respectively (Fig.
3A and B).
Importantly, monensin treatment markedly suppressed
AKT phosphorylation; pAKTS473 decreased by ~55 and 85%
after 1 h of EGF stimulation in the control and SNX1 knock-down
cells, respectively (Fig. 3A and
B), Similarly, pAKTT308 levels were significantly
suppressed by monensin, falling to 63 and 85% of vehicle control
following 2 h of EGF stimulation in control and SNX1 knock-down
cells, respectively (Fig. 3A and
B). These results indicate that monensin has a stronger
inhibitory effect on AKT phosphorylation in the absence of
SNX1.
Depletion of Rab11a does not inhibit
EGF-induced phosphorylation of AKT in gefitinib-resistant human
NSCLC cells
We next used siRNA to examine the effect of
depletion of Rab11a, which regulates recycling pathway from the
recycling compartment to plasma membranes (32), on AKT activation. As shown in
Fig. 3A and B, the degree of
pAKTS473 phosphorylation was unaffected by Rab11a
depletion, as it reached 2.5- and 6.5-fold at 1 h in control and
SNX1 knock-down cells, respectively. Also the increased levels of
pAKTS473 and pAKTS308 proteins remained in
cell during 4-h incubation. Hence, the Rab11a-dependent recycling
pathway is not involved in EGFR-mediated AKT activation.
Furthermore, AKT phosphorylation was not affected by treatment with
bafilomycin A1, as pAKTS473 reached 39- and 42-fold at 1
h following EGF stimulation in control and SNX1 knock-down cells,
respectively (Fig. 3C).
HGF-stimulated AKT phosphorylation is not
suppressed by monensin in gefitinib-resistant human NSCLC
cells
We next used monensin to determine whether endosomal
recycling was involved in signaling from the receptor tyrosine
kinase, MET. Cells transfected with siRNA-control were stimulated
with HGF in the absence or presence of monensin at 37°C for the
indicated times, and then the lysates were analyzed with anti-pMET,
anti-pAKTS473, or anti-pAKTT308 antibody by
western blot analysis. As shown in Fig. 3D, MET phosphorylation following HGF
stimulation increased 40- and 44-fold in control cells in the
absence and presence of monensin, respectively. Furthermore,
monensin treatment did not affect phosphorylation of AKT-following
HGF treatment, as the increase in pAKTT308 level was 9-
and 12-fold at 1 h in the absence and presence of drug,
respectively. An increase in pAKTS473 was also noted in
the monensin-treated cells, and depletion of Rab11a did not inhibit
phosphorylation of AKT and MET-following HGF treatment. Together,
these data indicate that Rab11a-dependent recycling is not required
for EGFR-mediated AKT activation.
Discussion
Here, we investigated the mechanisms associated with
EGFR endosomal recycling, and AKT activation in the
gefitinib-resistant NSCLC cell line A549. We verified that
knock-down of SNX1 markedly stimulates ligand-induced pEGFR
endocytosis via the early/late endocytic pathway, which is
accompanied by increasing amounts of pEGFR in cells. This confirms
that SNX1 suppresses EGF-stimulated endocytosis of pEGFR via the
early/late endocytic pathway. We also found that monensin led to an
accumulation of pEGFR in swollen vacuoles that colocalize with
LIMPII-positive late endosomes and with the transferrin-positive
early endosomes after EGF stimulation. Although these observations
are analogous to those of the cells treated with bafilomycin A1, a
lysosomal inhibitor, which blocks the efficient EGF-induced
degradation of pEGFR, we assumed that pEGFR accumulation in the
endosomes of monensin-treated cells might be mostly due to the
suppression of membrane recycling from the early endosomes to
plasma membranes. Furthermore, knockdown of Rab11a, which regulates
intracellular vesicle trafficking from recycling endosomes to
plasma membranes, did not affect pEGFR endocytosis or accumulation
of pEGFR in the early/late endosomes. Monensin suppresses membrane
recycling from early endosomes or recycling endosomes to the plasma
membranes (31). Our data
accordingly suggest that the majority of endocytosed EGFR/pEGFR in
EGF-stimulated cells is recycled back to the plasma membranes from
early endosomes, but not from the recycling endosomes.
Furthermore, depletion of SNX1 led to increased
expression of pEGFR-following EGF stimulation, and the level of
pEGFR then decreased. These observations are consistent with our
earlier report (19), and suggest
that the early/late endocytic trafficking of EGF-induced pEGFR and
subsequent downregulation is accelerated in SNX1 knock-down cells.
We conclude that SNX1, either directly or indirectly, is a negative
regulator of EGFR/pEGFR endocytosis, and that its downregulation
via an ESCRT-dependent pathway is required for activation of the
degradation or recycling pathways (8).
We also examined the effect of depletion of SNX1
expression by siRNA or of pharmacological effect such as monensin
on the EGF-stimulated AKT phosphorylation by using western blot
analysis, and then assessed whether EGF-stimulated AKT
phosphorylation might be affected by enhanced recycling. To
distinguish the phosphorylation status between the activation loop
at Thr308 residue by PDK1 and the carboxy-terminus of AKT at Ser473
residue phosphorylated by the elusive PDK2, two different
antibodies, anti-pAKTT308 and anti-pAKTS473
antibodies were used for western blot analysis. We found that AKT
phosphorylation was rapidly induced in both control and SNX1
knock-down cells after EGF treatment. Importantly, monensin
inhibited AKT phosphorylation in both cases, although the effect
was much stronger in the absence of SNX1. Thus, SNX1-dependent
inhibition of pEGFR recycling from the early endosomes to the
plasma membranes is the most probable cause of AKT activation in
these cells. Interestingly, depletion of endogenous Rab11a did not
alter phosphorylation of either AKT or EGFR. We thus infer that the
Rab11a-dependent recycling pathway does not participate in
EGF-induced EGFR recycling.
Notably, MET-dependent activation of AKT was not
affected by monensin. We infer that MET is not recycled back to the
plasma membranes following ligand binding. Rather, we propose that
pMET directly activates the PI3K-AKT pathway at the plasma
membranes, independently of membrane recycling.
Taken together, our data suggest that upregulation
of SNX1 may contribute to the survival of gefitinib-resistant cells
by inhibiting endosomal recycling of EGFR to the plasma membranes.
This leads to constitutive AKT activation and mTOR signaling;
ultimately these signaling cascades positively regulate survival,
growth, and proliferation. Thus, targeting the EGFR recycling
pathway, in combination with current TKIs, should be considered as
a therapeutic strategy, particularly in lung cancer. Further
studies are required to delineate the precise molecular mechanisms
by which SNX and other proteins regulate the recycling
pathways.
Abbreviations:
|
EGF
|
epidermal growth factor
|
|
pEGFR
|
phosphorylated epidermal growth factor
receptor
|
|
pAKT
|
phosphorylated AKT
|
|
HGF
|
hepatocyte growth factor
|
|
pMET
|
phosphorylated MET
|
|
SNX1
|
sorting nexin 1
|
|
NSCLC
|
non-small cell lung cancer
|
References
|
1
|
de Bono JS and Rowinsky EK: The ErbB
receptor family: a therapeutic target for cancer. Trends Mol Med.
8(Suppl 4): S19–S26. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mendelsohn J and Baselga J: The EGF
receptor family as targets for cancer therapy. Oncogene.
19:6550–6565. 2000. View Article : Google Scholar
|
|
3
|
Carpenter G and Cohen S: Epidermal growth
factor. J Biol Chem. 265:7709–7712. 1990.PubMed/NCBI
|
|
4
|
Yarden Y: The EGFR family and its ligands
in human cancer. Signaling mechanisms and therapeutic
opportunities. Eur J Cancer. 37(Suppl 4): S3–S8. 2001. View Article : Google Scholar
|
|
5
|
Ullrich A and Schlessinger J: Signal
transduction by receptors with tyrosine kinase activity. Cell.
61:203–212. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schlessinger J: Common and distinct
elements in cellular signaling via EGF and FGF receptors. Science.
306:1506–1507. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Engelman JA and Cantley LC: The role of
the ErbB family members in non-small cell lung cancers sensitive to
epidermal growth factor receptor kinase inhibitors. Clin Cancer
Res. 12:4372s–4376s. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Henne WM, Buchkovich NJ and Emr SD: The
ESCRT pathway. Dev Cell. 21:77–91. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Arteaga CL and Johnson DH: Tyrosine kinase
inhibitors-ZD1839 (Iressa). Curr Opin Oncol. 13:491–498. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barker AJ, Gibson KH, Grundy W, Godfrey
AA, Barlow JJ, Healy MP, Woodburn JR, Ashton SE, Curry BJ, Scarlett
L, Henthorn L and Richards L: Studies leading to the identification
of ZD1839 (IRESSA): an orally active, selective epidermal growth
factor receptor tyrosine kinase inhibitor targeted to the treatment
of cancer. Bioorg Med Chem Lett. 11:1911–1914. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Baselga J and Averbuch SD: ZD1839
(‘Iressa’) as an anticancer agent. Drugs. 60(Suppl 1): S33–S42.
2000. View Article : Google Scholar
|
|
12
|
Woodburn JR: The epidermal growth factor
receptor and its inhibition in cancer therapy. Pharmacol Ther.
82:241–250. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J and
Haber DA: Activating mutations in the epidermal growth factor
receptor underlying responsiveness of non-small-cell lung cancer to
gefitinib. N Engl J Med. 350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ,
Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE and
Meyerson M: EGFR mutations in lung cancer: correlation with
clinical response to gefitinib therapy. Science. 304:1497–1500.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C,
Johnson BE, Cantley LC and Jänne PA: MET amplification leads to
gefitinib resistance in lung cancer by activating ERBB3 signaling.
Science. 316:1039–1043. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nishimura Y, Bereczky B and Ono M: The
EGFR inhibitor gefitinib suppresses ligand-stimulated endocytosis
of EGFR via the early/late endocytic pathway in non-small cell lung
cancer cell lines. Histochem Cell Biol. 127:541–553. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nishimura Y, Yoshioka K, Bereczky B and
Itoh K: Evidence for efficient phosphorylation of EGFR and rapid
endocytosis of phosphorylated EGFR via the early/late endocytic
pathway in a gefitinib-sensitive non-small cell lung cancer cell
line. Mol Cancer. 7:422008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nishimura Y, Yoshioka K, Takiguchi S,
Bereczky B, Nakabeppu Y and Itoh K: A role for SNX1 in the
regulation of EGF-dependent phosphorylated EGFR endocytosis via the
early/late endocytic pathway in a gefitinib-sensitive human lung
cancer cells. Curr Signal Transduct Ther. 6:383–395. 2011.
View Article : Google Scholar
|
|
19
|
Nishimura Y, Takiguchi S, Yoshioka K,
Nakabeppu Y and Itoh K: Silencing of SNX1 by siRNA stimulates the
ligand-induced endocytosis of EGFR and increases EGFR
phosphorylation in gefitinib-resistant human lung cancer cell
lines. Int J Oncol. 41:1520–1530. 2012.PubMed/NCBI
|
|
20
|
Nishimura Y, Takiguchi S, Ito S and Itoh
K: Evidence that depletion of the sorting nexin 1 by siRNA promotes
HGF-induced MET endocytosis and MET phosphorylation in a
gefitinib-resistant human lung cancer cell line. Int J Oncol.
44:412–426. 2014.
|
|
21
|
Kurten RC, Cadena DL and Gill GN: Enhanced
degradation of EGF receptors by a sorting nexin, SNX1. Science.
272:1008–1010. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Worby CA and Dixon JE: Sorting out the
cellular function of sorting nexins. Nat Rev Mol Cell Biol.
3:919–931. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhong Q, Lasar CS, Tronchère H, Sato T,
Meerloo T, Yeo M, Songyang Z, Emr SD and Gill GN: Endosomal
localization and function of sorting nexin 1. Proc Natl Acad Sci
USA. 99:6767–6772. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Carlton J, Bujny M, Peter BJ, Oorschot VM,
Rutherford A, Mellor H, Klumperman J, McMahon HT and Cullen PJ:
Sorting nexin-1 mediates tubular endosome-to-TGN transport through
coincidence sensing of high-curvature membranes and
3-phosphoinositides. Curr Biol. 14:1791–1800. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gullapalli A, Garrett TA, Paing MM,
Griffin CT, Yang Y and Trejo J: A role for sorting nexin 2 in
epidermal growth factor receptor down-regulation: evidence for
distinct functions of sorting nexin 1 and 2 in protein trafficking.
Mol Biol Cell. 15:2143–2155. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nishimura Y, Higaki M and Kato K:
Identification of a precursor form of cathepsin D in microsomal
lumen: characterization of enzymatic activation and proteolytic
processing in vitro. Biochem Biophys Res Commun. 148:335–343. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nishimura Y, Kawabata T and Kato K:
Identification of latent procathepsins B and L in microsomal lumen:
characterization of enzymatic activation and proteolytic processing
in vitro. Arch Biochem Biophys. 261:64–71. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kornfeld S and Mellman I: The biogenesis
of lysosomes. Ann Rev Cell Biol. 5:483–525. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sandoval IV, Arredondo JJ, Alcalde J,
Gonzalez-Noriega A, Vandekerckhove J, Jimenez MA and Rico M: The
residues Leu(Ile)475-Ile(Leu, Val, Ala)476, contained in the
extended carboxyl cytoplasmic tail, are critical for targeting of
the resident lysosomal membrane protein LIMP II to lysosomes. J
Biol Chem. 269:6622–6631. 1994.PubMed/NCBI
|
|
30
|
Jones MC, Caswell PT and Norman JC:
Endocytic recycling pathways: emerging regulators of cell
migration. Curr Opin Cell Biol. 18:549–557. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Felder S, Miller K, Moehren G, Ullrich A,
Schlessinger J and Hopkins CR: Kinase activity controls the sorting
of the epidermal growth factor receptor within the multivesicular
body. Cell. 61:623–634. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ullrich O, Reinsch S, Urbé S, Zerial M and
Parton RG: Rab11 regulates recycling through the pericentriolar
recycling endosome. J Cell Biol. 135:913–924. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Alessi DR, James SR, Downes CP, Holmes AB,
Gaffney PR, Reese CB and Cohen P: Characterization of a
3-phosphoinositide-dependent protein kinase which phosphorylates
and activates protein kinase Bα. Curr Biol. 7:261–269. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sarbassov DD, Guertin DA, Ali SM and
Sabatini DM: Phosphorylation and regulation of Akt/PKB by the
rictor-mTOR complex. Science. 307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jacinto E, Facchinetti V, Liu D, Soto N,
Wei S, Jung SY, Huang Q, Qin J and Su B: SIN1/MIP1 maintains
rictor-mTOR complex integrity and regulates Akt phosphorylation and
substrate specificity. Cell. 127:125–137. 2006. View Article : Google Scholar : PubMed/NCBI
|