Introduction
Non-small cell lung cancers (NSCLCs) with activating
mutations in the tyrosine kinase domain (TKD) of EGFR have been
reported to exhibit ‘oncogene addiction’ to reflect their
dependence on EGFR-mediated malignant biological behavior (1,2).
Several clinical trials have shown that epidermal growth factor
receptor-tyrosine kinase inhibitors (EGFR-TKIs) (e.g., gefitinib
and erlotinib) are the best frontline options for patients with
sensitive EGFR mutations (EGFR-mts), resulting in a 2–3-fold
prolongation of survival time compared with standard chemotherapy
(3–5). For patients with wild-type EGFR
(EGFR-wt) status, data from randomized trials suggested that some
of these patients will derive a modest benefit from these
agents.
Currently, first-line use of these agents should be
restricted to EGFR-mt-positive patients as a clinical practice
guideline in the treatment of NSCLC (6). In practice, however, some EGFR
status-unknown patients might also benefit from empirical use of
initial treatment with EGFR-TKIs.
Most patients with NSCLC are diagnosed at stages III
and IV (7). For those with
advanced lung cancer that cannot be removed surgically,
chemotherapy or molecular-targeting treatments are typically
recommended.
It has been reported that EGFR-mts creating
sensitivity to EGFR-TKIs are more common in Asian populations,
particularly in patients with lung adenocarcinoma (8). Fine-needle aspirates for diagnosis,
which are now commonly used, are often insufficient for molecular
analysis. Accordingly, a number of technical issues may confound
the analysis of EGFR-mts. In addition, >60% of NSCLCs show
overexpression of EGFR (9), and
numerous investigations have shown that EGFR-TKIs can inhibit TKD
activation of EGFR-wt in vitro. Moreover, many NSCLC
patients are more inclined to undergo EGFR-TKI treatment because
they fear chemotherapy toxicity, especially patients with poor
Eastern Cooperative Oncology Group (ECOG) performance status. In
view of this, some oncologists usually offer EGFR-TKIs as a
tentative treatment lasting for ~1 month in these patients.
Although details of subsequent treatments and
response rates for chemotherapy (as the second-line treatment)
following first-line EGFR-TKI treatment in patients with EGFR-wt
NSCLC are not available from the IPASS and First-SIGNAL trials, the
overall survival (OS) advantage of patients with standard
first-line chemotherapy indirectly suggests that prior treatment
with EGFR-TKIs might result in unwanted effects (8). The TORCH study, a phase III trial
performed in unselected NSCLC patients, most of whom were EGFR-wt,
addressed a sequence question by using a crossover design that
compared first-line erlotinib followed by cisplatin
(DDP)-gemcitabine at progression and comparing this with the
reverse strategy (10). This study
found that starting with erlotinib not only decreased the objective
response rate (ORR), but also led to worse survival in EGFR-wt
NSCLC patients (mOS: 6.5 vs. 9.6 months). Moreover, a retrospective
study to investigate the prognosis of patients with NSCLC receiving
second-line antitumor treatment after gefitinib therapy showed that
no survival benefits from platinum-based combination regimens
existed in patients with EGFR-wt NSCLC (11). These findings led us to investigate
whether initial EGFR-TKI treatment has an adverse effect on the
sensitivity to subsequent chemotherapy of EGFR-wt NSCLC, and to
explore the underlying mechanisms.
The tentative treatment may increase the risk of
patients with EGFR-wt having an unfavorable prognosis, including a
significantly reduced total progression-free survival (PFS) and OS.
Here, we describe the first study focusing on the effectiveness of
chemotherapy following continuous exposure of EGFR-wt NSCLC to
EGFR-TKIs in vitro.
Materials and methods
Reagents
RPMI-1640 medium, fetal bovine serum, trypsin,
penicillin and streptomycin were obtained from Gibco/Life
Technologies (Shanghai, China). Gefitinib (Iressa) was provided by
AstraZeneca (London, UK), erlotinib (Tarceva) was a gift from Roche
Pharmaceuticals (Basel, Switzerland), pemetrexed (Alimta) and
gemcitabine (Gemzar) were a gift from Eli Lilly and Company
(Indianapolis, IN, USA). DDP and paclitaxel (Taxol) were purchased
from Sigma-Aldrich (St. Louis, MO, USA). The drugs were dissolved
in dimethyl sulfoxide (DMSO) or sterile water and diluted in
culture medium before use. NSC 74859, an inhibitor of signal
transducer and activator of transcription 3 (STAT3) was purchased
from Selleck Chemicals (Houston, TX, USA). LY294002, AS605240 and
leptomycin B an inhibitor of nuclear export were purchased from
Sigma-Aldrich. Recombinant human EGF was purchased from BioLegend
(San Diego, CA, USA).
Cell culture and long-term exposure to
TKI
The NSCLC cell lines, HCC827 [lung adenocarcinoma
with an acquired mutation in the EGFR TKD (E746–A750 deletion)],
NCI-H1299 (established from a lymph node metastasis of the lung
from a patient who had received prior radiation therapy and with
EGFR-wt) and NCI-H1975 (primary adenocarcinoma harboring EGFR L858R
and T790M mutations) were purchased from the American Type Culture
Collection (ATCC) (Manassas, VA, USA) and maintained in RPMI-1640
medium supplemented with 10% FBS and antibiotics. We also
established a model of EGFR-TKIs exposure of lung cancer by
culturing H1299 in 10 μM gefitinib and 5 μM erlotinib respectively
for 4 weeks as well as H1975; HCC827 was incubated in 2 μM
gefitinib for 6 months.
Growth inhibition assay
The number of viable cells was estimated using the
Cell Counting kit-8 (Dojindo, Kumamoto, Japan) assay that provided
effective and reproducible determination of the proliferative
activity of cells. Human cells were seeded into flat-bottomed
96-well microplates at a density of 104 cells/well in
100 μl of culture medium and allowed to attach to the wells
overnight before 100 μl medium containing 2x indicated
concentration of EGFR-TKIs, with or without a STAT3 inhibitor, was
added to each well. After 24 h, the media were separately replaced
with fresh medium containing each cytotoxic drug (pemetrexed,
gemcitabine, DDP, paclitaxel) which dissolved at variously gradient
concentrations. Cells were treated with chemotherapeutic drugs for
48 h. Controls without cytotoxic drug exposure were included in
each experiment. Five replicate wells were used for each drug
concentration and each experiment was carried out independently
three times. To measure the proliferative activity of cells in
96-well microplates, CCK-8 reagent was added (20 μl/well) and
incubation continued for 2 h. Absorbance of the reduced formazan
was measured at 450 nm using a microplate reader (Multiskan MK3;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) with a reference
wavelength of 650 nm.
Caspase-3 activity assay
Caspase-3 activity was determined after treatment of
cells with TKI and cytotoxic drugs as described for the growth
inhibition assay. Cell lysates were prepared by the PathScan
Sandwich ELISA Lysis buffer and the activity of caspase-3 was
determined using a Caspase-3 Activity Assay kit (both from Cell
Signaling Technology, Inc., Danvers, MA, USA) that assesses
cleavage of the fluorogenic peptide. After treatment with cytotoxic
drugs, cells (including those loosely attached to the plate) were
collected and rinsed with ice-cold PBS. The fluorescence of cleaved
AMC was assessed after 1 h at 37°C incubation in the dark.
Caspase-3 activity values were calculated from a standard curve
generated by using varying concentrations of AMC (Microsoft Excel;
Microsoft, Redmond, WA, USA).
FACS analysis and apoptosis assay
Cells were fixed in ice-cold 70% ethanol and stained
with propidium iodide (50 mg/ml in PBS; Sigma-Aldrich) in the
presence of RNase A (100 mg/ml) for DNA content analysis by flow
cytometry using a FACSCalibur system (BD Biosciences, San Diego,
CA, USA). For each data point, 8,000 cells were analyzed. The
percentage of cells in various phases of the cell cycle was
calculated using FlowJo software version 7.6.1 (Tree Star, Inc.,
Ashland, OR, USA). Apoptosis was quantified using an Annexin V-FITC
Apoptosis kit (BD Biosciences) in accordance with the
manufacturer’s instructions. In brief, cells were trypsinized,
pelleted by centrifugation (1,500 rpm for 5 min, Heraeus Multifuge
X3; Thermo Fisher Scientific, Inc.) and resuspended in Annexin
V-binding buffer. FITC-conjugated Annexin V and propidium iodide
were added to cells and incubated for 30 min at room temperature in
the dark. Analyses were done on a FACSCalibur system (BD
Biosciences) and FlowJo software version 7.6.1 (Tree Star,
Inc.)
Preparation of nuclear and cytoplasmic
protein extracts
Nuclear extracts from cells were isolated using a
Nuclear and Cytoplasmic Protein Extraction kit in accordance with
the manufacturer’s instructions (Beyotime Institute of
Biotechnology, Jiangsu, China). In brief, cells were washed in
ice-cold PBS then collected and resuspended by pipet-ting up and
down 10 times in 200 μl of ice-cold cell lysis buffer. After
resting on ice for 15 min, nuclei were pelleted in a
microcentrifuge (Sorvall Legend Micro; Thermo Fisher Scientific,
Inc.) at 12,000 rpm for 5 min at 4°C and the cytoplasmic
supernatants were aliquoted and stored at -80°C for western blot
analysis when needed. Pelleted nuclei were then resuspended in 50
μl of nuclear extraction buffer. After intermittently vortexing
(vortex 30 sec, rest 30 sec) the mixing for 30 min and
centrifugation at 12,000 rpm for 10 min at 4°C, nuclear extracts
were aliquoted and stored at −80°C until use. The concentration of
proteins in the cytoplasmic and nuclear extracts were measured
using a BCA Protein Assay kit (Beyotime Institute of
Biotechnology).
Western blot analyses
Cells were lysed using the PhosphoSafe Extraction
Reagent (Novagen; EMD Biosciences, San Diego, CA, USA) supplemented
with a cocktail of protease inhibitors and 1 mM
phenylmethylsulfonyl fluoride (Sigma-Aldrich). Protein extracts
were heated in protein loading buffer (Beyotime Institute of
Biotechnology) at 95°C for 5 min and separated by SDS-PAGE. After
electrophoresis, separated proteins were electrotransblotted onto a
PVDF membrane and then blocked using 1% BSA in TBS-Tween-20 for 2 h
at room temperature. The membrane was incubated overnight at 4°C
with a primary antibody prior to use of a horseradish peroxidase
(HRP)-labeled secondary antibody, and visualization of bands by
chemiluminescence, recorded with X-OMAT BT film [Kodak (China) Co.,
Ltd., Fujian, China]. Details of the primary antibodies used are
given in Table I.
 | Table IDetails of the primary
antibodies. |
Table I
Details of the primary
antibodies.
| Primary
antibody | Clone | Dilution (WB) | Catalog | Supplier |
|---|
| FGFR2 | Polyclonal
rabbit | 1:2,000 | ab10648 | Abcam |
| IGF-1R | Monoclonal
rabbit | 1:2,000 | ab172965 | Abcam |
| IGFBP3 | Polyclonal
rabbit | 1:2,000 | ab76001 | Abcam |
| IRS-1 | Polyclonal
rabbit | 1:2,000 | ab52167 | Abcam |
| mTOR | Polyclonal
rabbit | 1:2,000 | ab2732 | Abcam |
| mTOR (phospho
S2448) | Polyclonal
rabbit | 1:2,000 | ab84400 | Abcam |
| P-STAT3
(Ser727) | Monoclonal
rabbit | 1:2,000 | ab32143 | Abcam |
| Bcl-2 | Monoclonal
rabbit | 1:1,000 | no. 2870 | CST |
| Bcl-xL | Monoclonal
rabbit | 1:1,000 | no. 2764 | CST |
| Cleaved PARP | Monoclonal
rabbit | 1:1,000 | no. 5625 | CST |
| c-MET | Polyclonal
rabbit | 1:1,000 | no. 4560 | CST |
| c-Myc | Monoclonal
rabbit | 1:1,000 | no. 5605 | CST |
| Cyclin D1 | Monoclonal
rabbit | 1:1,000 | no. 2978 | CST |
| E-cadherin | Monoclonal
rabbit | 1:1,000 | no. 3195 | CST |
| EGFR | Monoclonal
rabbit | 1:1,000 | no. 4267 | CST |
| GAPDH | Monoclonal
rabbit | 1:1,000 | no. 5174 | CST |
| HER-2 | Monoclonal
rabbit | 1:1,000 | no. 4290 | CST |
| Histone H3 | Monoclonal
rabbit | 1:1,000 | no. 4499 | CST |
| Mcl-1 | Monoclonal
rabbit | 1:1,000 | no. 5453 | CST |
| NF-κB p65 | Monoclonal
rabbit | 1:1,000 | no. 4764 | CST |
| P27Kip1 | Monoclonal
rabbit | 1:1,000 | no. 3686 | CST |
| P-AKT (Ser473) | Monoclonal
rabbit | 1:1,000 | no. 4060 | CST |
| P-Erk
(Thr202/Tyr204) | Monoclonal
rabbit | 1:1,000 | no. 4370 | CST |
| P-IGF-1Rβ
(Tyr1131) | Polyclonal
rabbit | 1:1,000 | no. 3021 | CST |
| P-IGF-1Rβ
(Tyr1316) | Polyclonal
rabbit | 1:1,000 | no. 6113 | CST |
| P-JNK
(Thr183/Tyr185) | Polyclonal
rabbit | 1:1,000 | no. 4668 | CST |
| P-MET
(Tyr1234/1235) | Monoclonal
rabbit | 1:1,000 | no. 3077 | CST |
| P-NF-κB p65
(Ser536) | Monoclonal
rabbit | 1:1,000 | no. 3033 | CST |
| P-p38
(Thr180/Tyr182) | Polyclonal
rabbit | 1:1,000 | no. 9211 | CST |
| P-SFK (Tyr416) | Monoclonal
rabbit | 1:1,000 | no. 6943 | CST |
| P-STAT1
(Tyr701) | Monoclonal
rabbit | 1:1,000 | no. 6943 | CST |
| P-STAT1
(Tyr727) | Polyclonal
rabbit | 1:1,000 | no. 9177 | CST |
| P-STAT3
(Tyr705) | Monoclonal
rabbit | 1:1,000 | no. 9145 | CST |
| | IHC 1:200 | | |
| P-β-catenin
(Ser552) | Polyclonal
rabbit | 1:1,000 | no. 9566 | CST |
| Snail | Monoclonal
rabbit | 1:1,000 | no. 3879 | CST |
| Total AKT | Polyclonal
rabbit | 1:1,000 | no. 9272 | CST |
| Total Erk | Monoclonal
rabbit | 1:1,000 | no. 4695 | CST |
| Total JNK | Monoclonal
rabbit | 1:1,000 | no. 9258 | CST |
| Total p38 | Polyclonal
rabbit | 1:1,000 | no. 9212 | CST |
| Total STAT1 | Polyclonal
rabbit | 1:1,000 | no. 9172 | CST |
| Total STAT3 | Monoclonal
rabbit | 1:1,000 | no. 4904 | CST |
| Vimentin | Monoclonal
rabbit | 1:1,000 | no. 5741 | CST |
| β-catenin | Monoclonal
rabbit | 1:1,000 | no. 9582 | CST |
| β-tubulin | Monoclonal
rabbit | 1:1,000 | no. 2128 | CST |
| Survivin | Monoclonal
mouse | 1:250 | sc-17779 | SCB |
Xenograft model
Female nude mice with a BALB/c genetic background
were purchased from HuaFukang Biological Technology Co., Ltd.
(Beijing, China). Mice aged 4–6 weeks, 18–22 g in weight, were
maintained under specific pathogen-free conditions with 12-h
light/12-h dark cycles at 26–28°C and 50–65% humidity in the
Experimental Animal Centre of the Sichuan University State Key
Laboratory of Biotherapy (Sichuan, China) for these experiments.
Each five animals were housed in plastic containers with lids. All
animals were checked daily; containers were changed once a week
during the entire length of the experiment. Animal feed and
underpad, which were purchased from the HuaFukang Biological
Technology Co., Ltd., were autoclaved and vacuum packed. The water
was sterilized and then adjusted to room temperature before use.
H1299 cells were used for the xenograft experiment. In brief, H1299
cells (1×107 cells/each mouse) were implanted
subcutaneously in the right axilla of nude mice. Drug treatments
were started on day 28. Gefitinib (100 mg/kg) or erlotinib (100
mg/kg) was given by oral gavage 5 times/week. In total three
treatment cycles were conducted. Each treatment group contained 10
mice. Mice were euthanized by cervical dislocation, and tumor
tissues were rapidly dissected; part of them flash-frozen in liquid
nitrogen, for later protein extraction, the others formalin-fixed
24 h and then paraffin-embedded. All procedures were approved by
the Animal Care and Use Committee of Sichuan University.
Immunohistochemical staining
The formalin-fixed paraffin-embedded tissue samples
of the tumor were cut into sections of 4 μm, which were mounted on
silanized slides. The sections were deparaffinized in xylene then
rehydrated through a graded series of ethanol/water. Antigen
retrieval was accomplished using pH 6.0 sodium citrate buffer (0.01
M) and microwave heating for 10 min at 95°C. After cooling, the
sections were incubated with a primary antibody at 4°C overnight
(Table I). The PowerVision 6000
immunohistochemistry detection reagent (ZSJQ Biotechnology,
Beijing, China) was used as a second antibody by incubating for 1 h
at 37°C and 3,3′-diaminobenzidine (DAB) was used as a chromogen.
Hematoxylin was used as a nuclear counterstaining agent.
Cytokine assays
EGFR-TKI-exposed or parental cells were plated in
their respective growth media at 1×105 cells/well and
incubated overnight for attachment. The media were replaced with
fresh serum-free medium for serum-starved and EGFR-TKI-exposed
(parental cells without EGFR-TKIs).
After 48 h of EGFR-TKI exposure, conditioned medium
was then harvested and stored at −80°C. Culture medium incubated
without cells served as the control. The conditioned medium was
thawed and centrifuged briefly before assay.
Quantification of IL-6 and -22 in cell culture
supernatants was carried out using an ELISA Development kit
(Quantikine Colorimetric Sandwich ELISAs; R&D Systems,
Minneapolis, MN, USA) in microplate format, measuring absorbance at
450 nm and with wavelength correction at 570 nm for correct optical
imperfections in the plates.
Immunoprecipitation
The physical interaction between STAT3 and EGFR was
detected by immunoprecipitation. Cells were lysed in non-denaturing
lysis buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton
X-100, supplemented with a protease and phosphatase inhibitor
cocktail (nos. P8340 and P0044; Sigma-Aldrich). Samples were
precleared with rabbit IgG (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA). Each sample supernatant was then incubated with
antibodies at a dilution ratio indicated in the instructions at 4°C
overnight with gentle agitation. The samples were further incubated
with 40 μl of Protein A/G PLUS Agarose beads (Santa Cruz
Biotechnology, Inc.) for 4 h at 4°C and the resulting immune
complexes were washed three times with lysis buffer by
centrifugation (800 rpm, 3 min). Samples were heated in SDS loading
buffer at 95°C for 5 min and analyzed by western blot analysis.
Results
Continuous exposure to EGFR-TKIs induces
chemoresistance to cytotoxic agents in EGFR-wt NSCLC cell
lines
To investigate the effects of EGFR-TKI exposure on
chemotherapy, EGFR-wt NSCLC H1299 cells were continuously treated
with gefitinib or erlotinib for 4 weeks. Chemosensitivity to DDP,
paclitaxel, gemcitabine or pemetrexed in these NSCLC cells, both
parental and TKI-exposed was evaluated. For each cytotoxic drug,
the IC50 values obtained from TKI-exposed cells were
significantly higher than from the parental cells (Fig. 1A, Table II). The difference was especially
marked for gemcitabine after continuous exposure to TKI for 4
weeks.
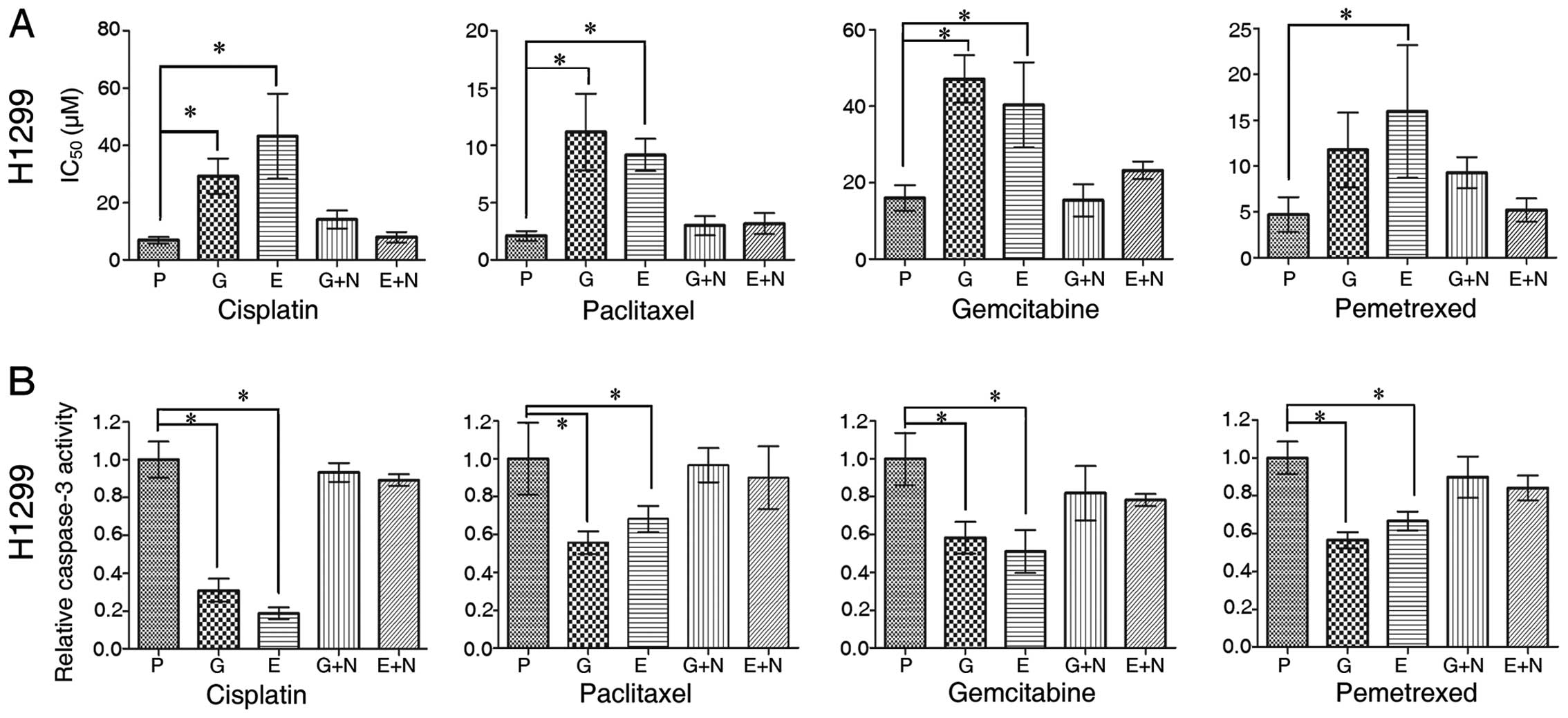 | Figure 1TKI-exposed-negative effect on
cytotoxic drugs in wild-type EGFR (EGFR-wt) non-small cell lung
cancer (NSCLC) cells. (A) The assessment of IC50
[cisplatin (DDP), paclitaxel, gemcitabine and pemetrexed] using the
CCK-8 assay on parental and TKI (gefitinib and erlotinib)-exposed
H1299 cells in the absence or presence of 20 μM NSC 74859 for 24 h.
(B) H1299 NSCLC cells (parental, TKI-exposure and addition of NSC
74859 20 μM for 24 h) were treated with DDP (10 μM), paclitaxel (1
μM), gemcitabine (40 μM) and pemetrexed (20 μM) for 24 h and
subjected to caspase-3 assay. P, parental; G, gefitinib-exposed; E,
erlotinib-exposed; G+N, Gefitinib-exposed + 24 h NSC 74859; E+N,
Erlotinib-exposed + 24 h NSC 74859. Data are shown as mean ± SD;
*P<0.05; statistical difference: one-way
ANOVA/Dunnett’s test, compared with the parental group. Experiments
were repeated three times. |
| Table IIIC50 of H1299 for four
cytotoxic drugs (μM). |
Table II
IC50 of H1299 for four
cytotoxic drugs (μM).
| P | G | E | G+N | E+N |
|---|
| DDP | 6.92±1.15 | 29.25±6.1 | 43.25±14.87 | 14.12±3.13 | 7.95±1.85 |
| Taxol | 2.09±0.44 | 11.16±3.36 | 9.16±1.41 | 2.99±0.84 | 3.16±0.91 |
| Gemzar | 16.00±3.38 | 47.18±6.2 | 40.36±11.1 | 15.37±4.2 | 23.23±2.3 |
| Alimta | 4.72±1.9 | 11.78±4.07 | 15.97±7.23 | 9.3±1.7 | 5.22±1.28 |
To further assess the chemoresistance induced by TKI
exposure, we assessed apoptosis induced by cytotoxic drugs. The
proportion of apoptotic cells (including early and late phase)
labeled with Annexin V(+) was decreased in all three TKI-exposed
cell lines compared with their parental lines (Fig. 2A).
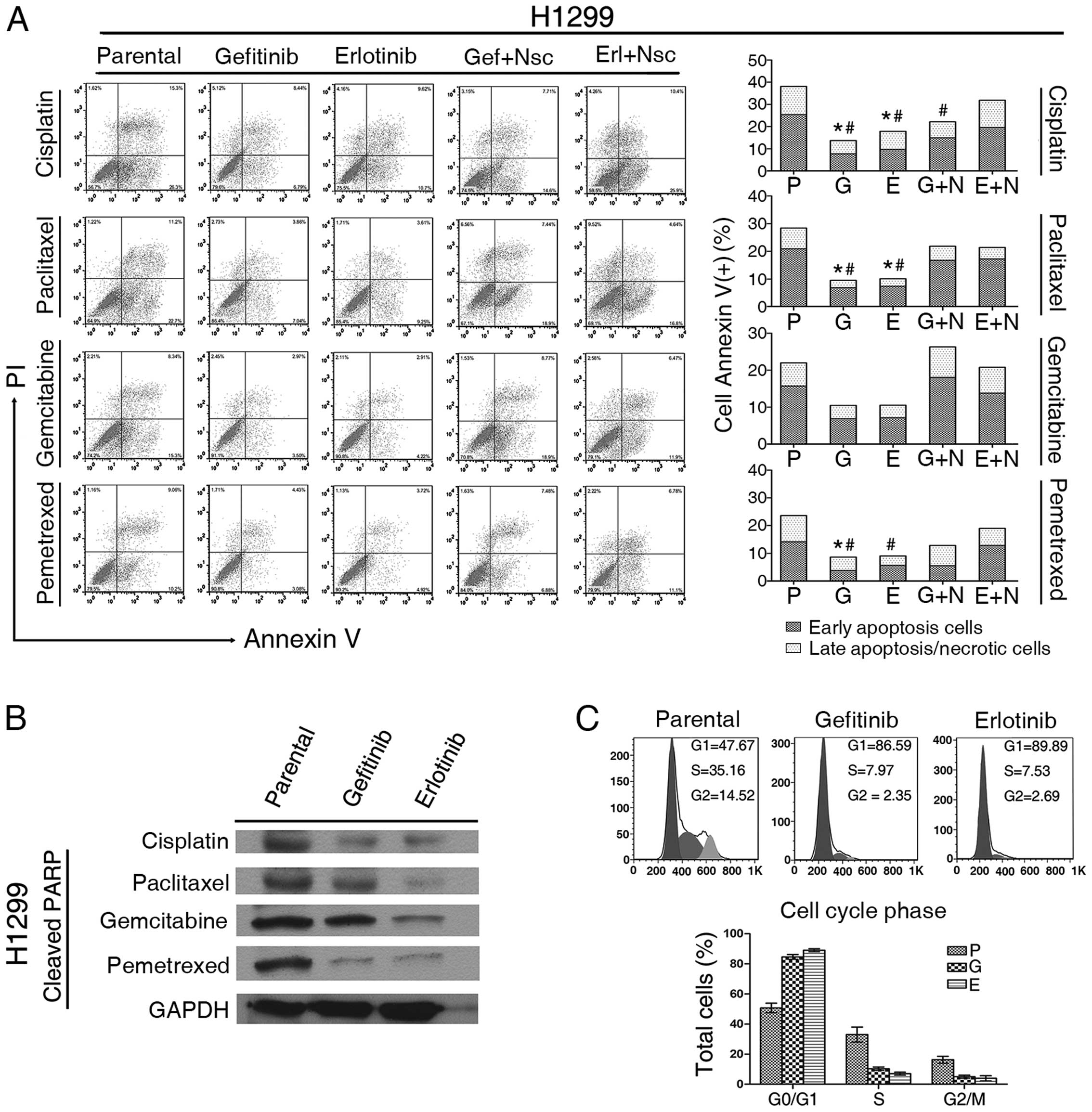 | Figure 2Analysis of apoptosis and cell cycle
phase in cultured non-small cell lung cancer (NSCLC). (A) The
apoptotic incidence for four chemotherapeutic agents [10 μM
cisplatin (DDP), 1 μM paclitaxel, 10 μM gemcitabine or 5 μM
pemetrexed] after 48 h exposure of H1299 cells in five
pre-treatment groups as indicated (P, parental; G,
gefitinib-exposed; E, erlotinib-exposed; G+N, gefitinib-exposed +
24 h NSC 74859; E+N, erlotinib-exposed + 24 h NSC 74859). Data are
shown as the mean from three independent
experiments.*Early apoptotic cells, P<0.05;
#total cells positive for Annexin V, P<0.05.
Statistical difference: One-way ANOVA/Dunnett’s test, compared with
the parental group. Representative graphs obtained by flow
cytometry analysis after double staining with Annexin V-FITC and
propidium iodide. (B) Effect of chemotherapy on poly(ADP-ribose)
polymerase (PARP) cleavage. H1299 parental and TKI-exposed cells
were incubated with four chemotherapeutic agents for 48 h (10 μM
DDP, 1 μM paclitaxel, 40 μM gemcitabine or 20 μM pemetrexed),
separation of cell proteins (30 μg) by SDS/PAGE and reaction with
antibodies against cleaved PARP. GAPDH was used as a loading
control. (C) The cell cycle phases (G0/G1, G2/M and S phase) of
both parental and TKI-exposed NSCLC cells were analyzed by flow
cytometry using propidium iodide staining of DNA content. |
We also measured the caspase-3 activity of H1299
cells, and detected the expression of cleaved poly(ADP-ribose)
polymerase (PARP) by western blot analysis. High levels of active
caspase-3 and proteolytic cleavage of PARP are two characteristic
biochemical markers of apoptosis. The level of active caspase-3
induced by each of the four cytotoxic drugs was attenuated after
TKI exposure compared with the parental group (Fig. 1B). The cells pre-exposed to TKI for
4 weeks showed a reduced level of cleaved PARP when treated with
cytotoxic drugs compared with their parental cells (Fig. 2B).
TKI exposure induces high-level
activation of STAT3 in EGFR-wt NSCLC cells
To gain insights into the mechanisms underlying the
resistance of cytotoxic agents after TKI exposure of EGFR-wt NSCLC
cells, proteins of the EGFR signaling pathway were detected by
western blot analysis. Given that EGFR signaling activation
stimulates intracellular cascades, including the MAPK, PI3K/AKT,
and STAT signaling pathways (12,13),
we analyzed the activity of several major EGFR downstream
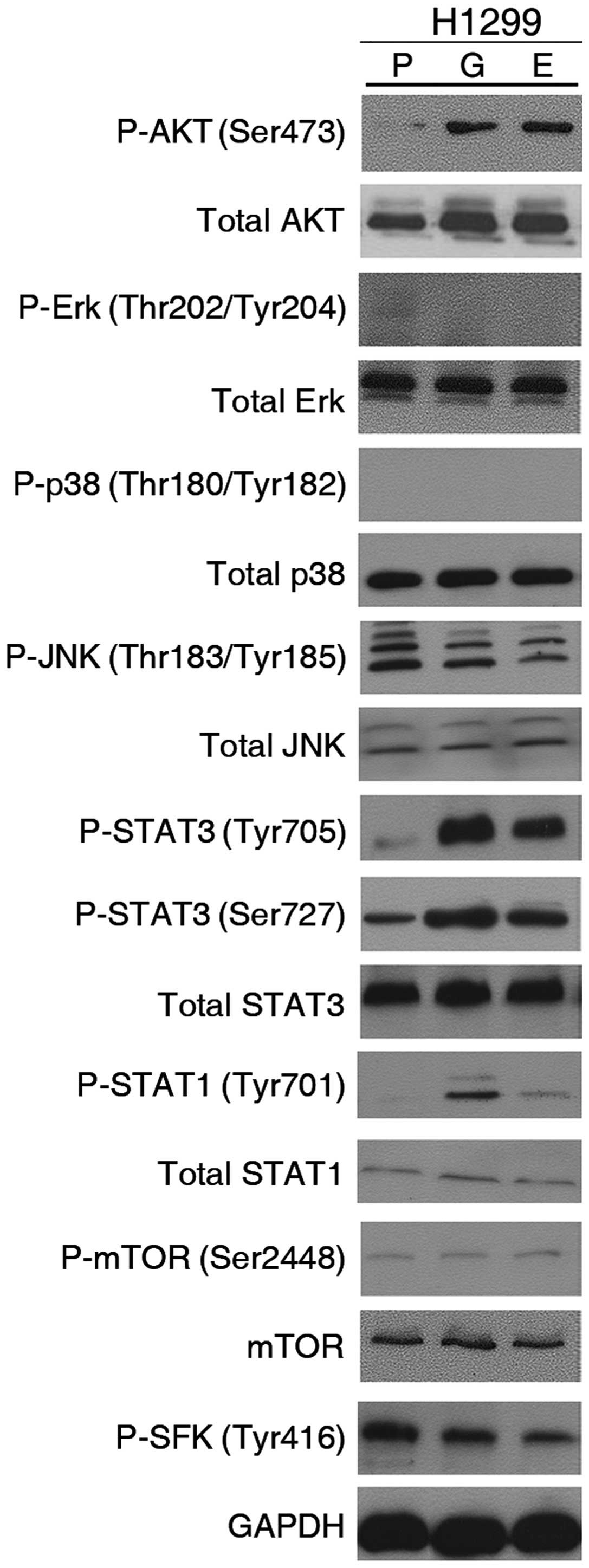
molecules: AKT, MAPK family (Erk, p38, JNK), STAT3, etc. (Fig. 3). Interestingly, we observed that
phosphorylated AKT and STAT3 (at both Ser727 and Tyr705 sites)
levels were substantially increased after exposure to TKI for 4
weeks, compared with parental H1299 cells. However, there was no
significant increase in the level of the other phosphorylated
molecules including Erk, p38, JNK and mTOR.
To investigate the relationship between AKT and
STAT3 in the signal pathway, we used two PI3K inhibitors together
(LY294002, 2 μM and AS605240, 10 nM) and STAT3 inhibitor (NSC
74859, 20 μM) to treat H1299 parental cells with or without EGF (50
ng/ml). PI3K inhibition was associated with a significant reduction
in P-AKT regardless of adding EGF or not, whereas level of P-STAT3
(Tyr705) showed obvious upregulation. Conversely, after incubation
of STAT3 inhibitor, P-STAT3 (Tyr705) was considerably decreased,
whereas P-AKT upregulated (Fig.
4A). Our data indicated that these two molecules were
compensatory to each other in H1299 parental cells. To further
investigate the relationship between these two proteins, along with
exposure of TKI, we analyzed P-STAT3 and P-AKT expression in
TKI-exposed cells. Our study revealed interesting data on these two
molecules interaction (Fig. 4B).
Similar to parental cells, PI3K inhibitors result in a
downregulation of P-AKT and increase of P-STAT3, while NSC 74859
treatment resulted in downregulation of both P-STAT3 and P-AKT.
These data suggest that EGFR-TKIs exposure results in role changes
of STAT3 and AKT by which STAT3 becomes a regulator of the AKT
signal. This also indicates that STAT3 plays a more important role
in response to EGFR inhibition in EGFR-wt NSCLC cells.
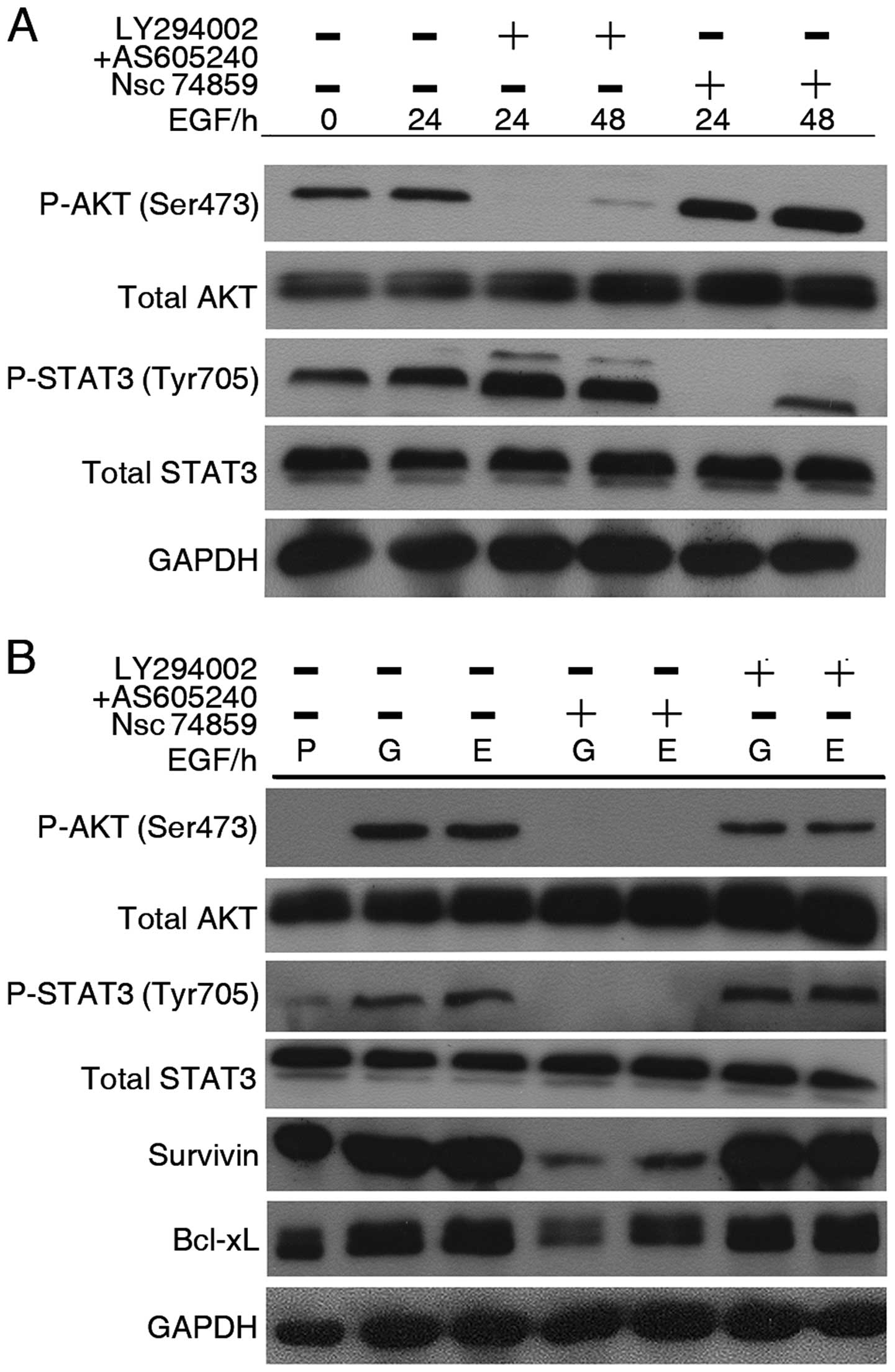 | Figure 4Chemical inhibitor of signal
transducer and activator of transcription 3 (STAT3) suppress the
activation of AKT in TKI-exposed non-small cell lung cancer (NSCLC)
cells, but not in parental cells. (A) Time-course analysis of AKT
and STAT3 molecule activation in EGF (50 ng/ml)-treated H1299
parental cells. H1299 cells were treated with combination of two
PI3K inhibitors (LY294002, 2 μM and AS605240, 10 nM) or STAT3
inhibitor (NSC 74859, 20 μM). (B) H1299 TKI-exposed cells were
treated with combination of two PI3K inhibitors (LY294002, 2 μM and
AS605240, 10 nM) or STAT3 inhibitor (NSC 74859, 20 μM) in the
presence of gefitinib or erlotinib. After 48 h, cell lysed analyzed
by western blot analysis. Equal loading and transfer were shown by
repeat probing with GAPDH. P, parental; G, gefitinib-exposed; E,
erlotinib-exposed. |
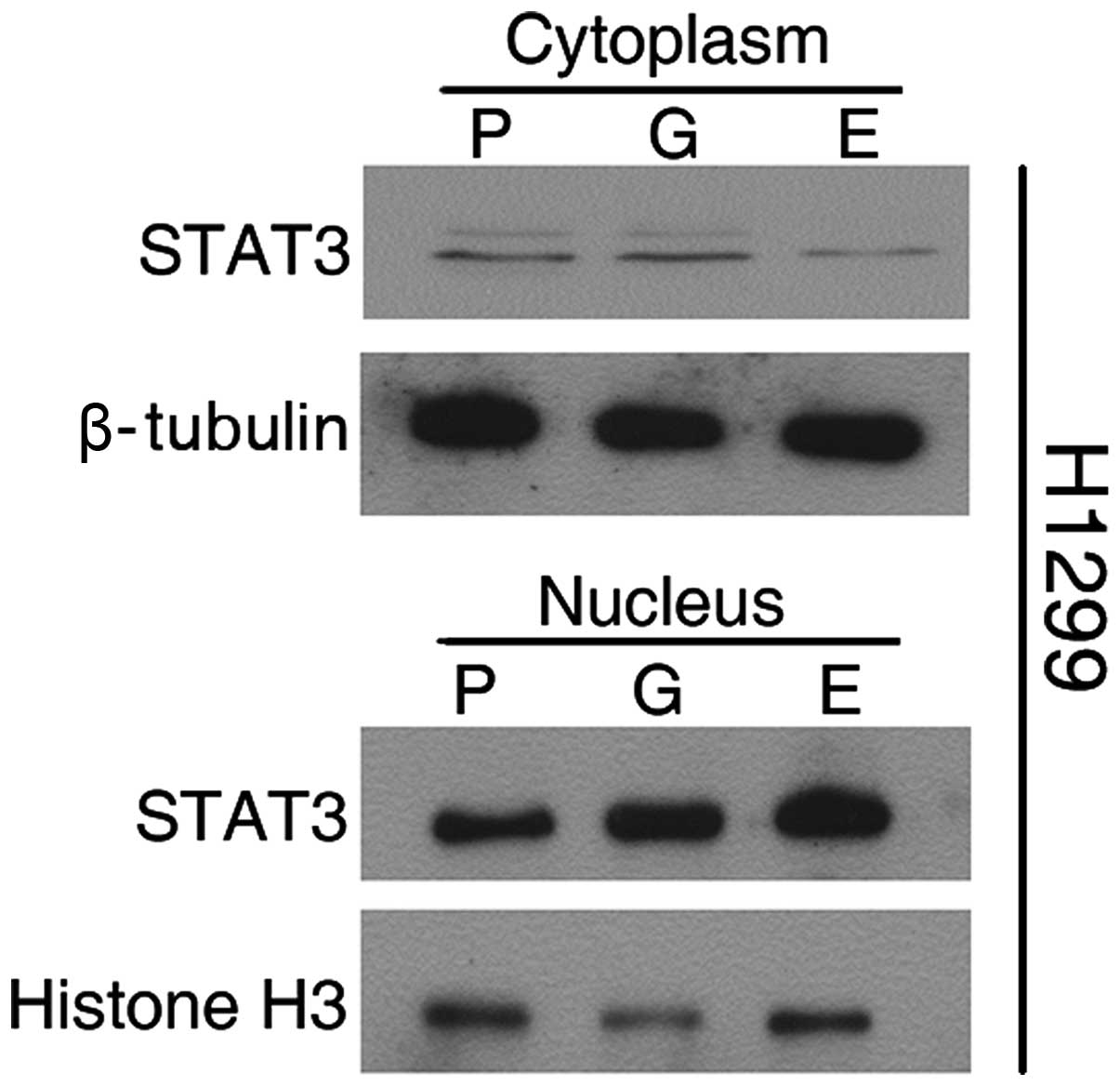
To further assess the involvement of STAT3, we
isolated nuclear and cytosolic fractions for immunoblotting assays.
As shown in Fig. 5, a basal level
of STAT3 was detectable in the nuclei of H1299 parental cells, as
well as in the cytosol. Extracts from cells exposed to TKI for 4
weeks showed decreased cytosolic STAT3 and increased nuclear
translocation.
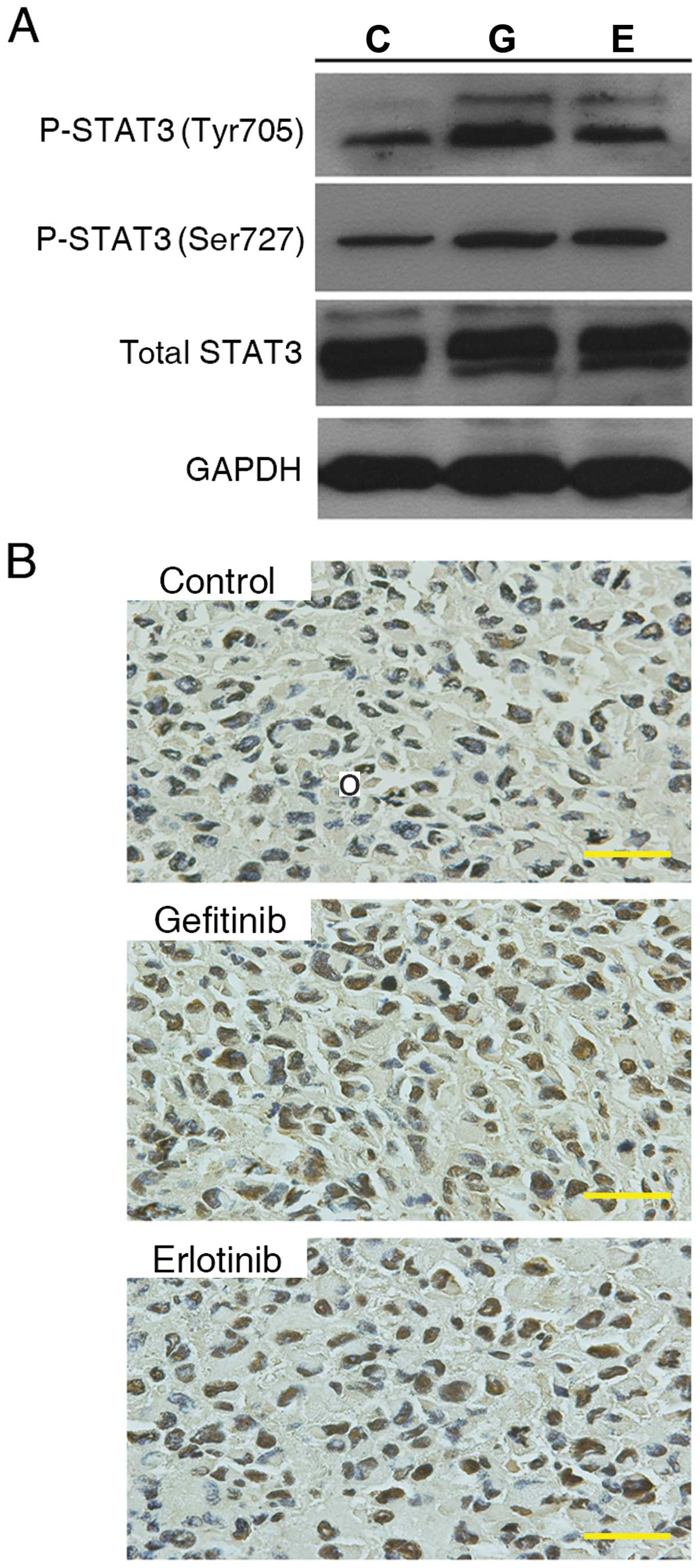
To validate whether P-STAT3 increased in EGFR-wt
NSCLC in vivo, we established a model using tumor xenografts
with subcutaneously implanted H1299 cells. The tumor-bearing mice
were gavaged once daily with gefitinib (100 mg/kg) or erlotinib
(100 mg/kg) for 4 weeks. The levels of P-STAT3 (Tyr705) and P-STAT3
(Ser727) as well as total STAT3 in tumors were analyzed by western
blot analysis (Fig. 6A) and
immunohistochemistry (Fig. 6B).
Consistent with the results obtained in vitro, we observed
increased levels of P-STAT3 (Tyr705) and P-STAT3 (Ser727) in
xenografts exposed to EGFR-TKIs in comparison to the group gavaged
daily with normal saline.
STAT3 activation results in
chemoresistance by increasing anti-apoptotic signals, cell cycle
arrest and epithelial-mesen-chymal transition (EMT) in EGFR-wt
NSCLC cells
To determine the role of STAT3 in chemoresistance
caused by TKI exposure, we first examined the effect of STAT3
inhibition with the pharmacological inhibitor NSC 74859 on
sensitivity to the four cytotoxic agents. STAT3 inhibition induced
by 24 h incubation with 20 μM NSC 74859 greatly recovered the
cytotoxic effect of the different cytotoxic agents as indicated in
Fig. 1A. We also found that
caspase-3 activity induced by cytotoxic agents had a significant
recovery after STAT3 inhibition by NSC 74859 (Fig. 1B). Moreover, flow cytometric
analysis of Annexin V-stained cells demonstrated that STAT3
inhibition increased apoptosis induced by cytotoxic agents
(Fig. 2A). Therefore, targeting
STAT3 with a specific inhibitor actually reversed chemoresistance,
and this indicates that STAT3 activation may play a vital role in
altering the signal pathways operating after TKI exposure in
EGFR-wt NSCLC cell lines.
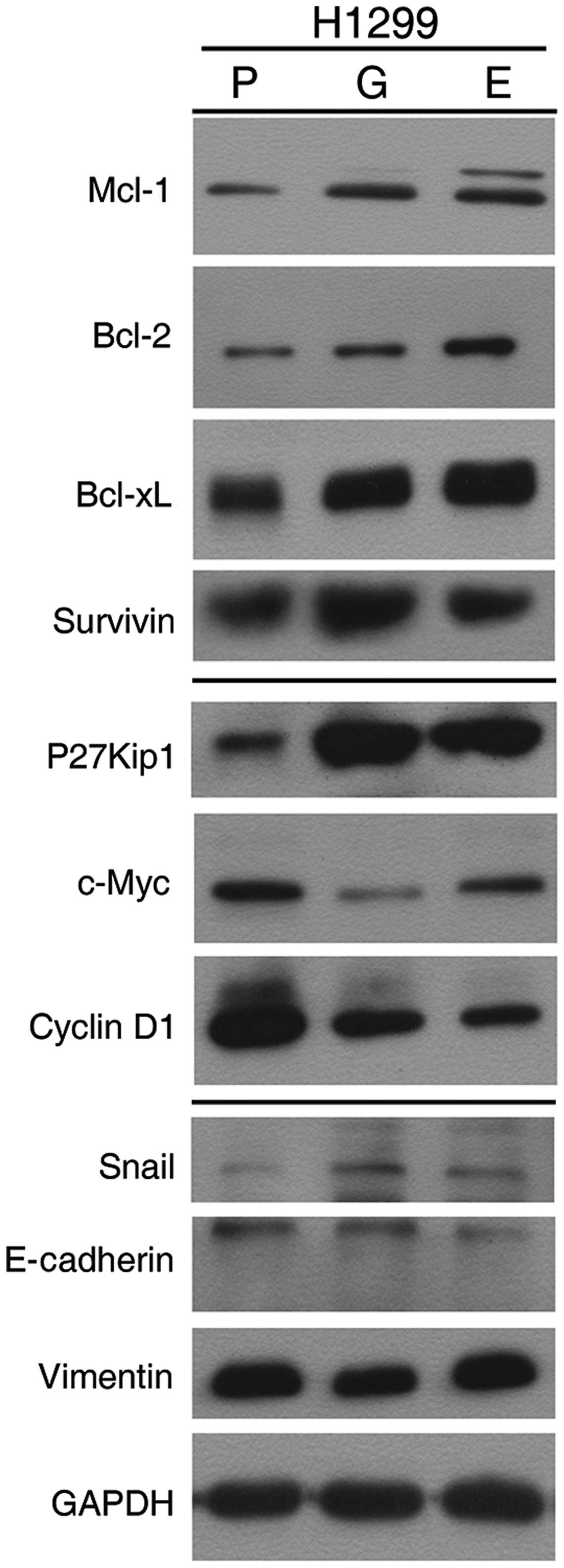
To explore the potential mechanisms underlying STAT3
activation-induced drug resistance, we assessed the abundance of
proteins of several STAT3-targeted genes (Fig. 7). The expression levels of four
anti-apoptotic proteins (Mcl-1, Bcl-2, Bcl-xL and survivin) were
greater in H1299 cells following prolonged TKI exposure. This
suggests that activating STAT3 by prolonged TKI exposure impairs
the ability of cytotoxic agents through the effects of these
anti-apoptotic proteins. The levels of, P27Kip1, c-Myc and cyclin
D1 also were measured; both cyclin D1 and c-Myc became less
abundant, whereas increased level of P27KIP1 was detected in cells
exposed to TKI. These findings may explain the G1-S phase arrest by
TKIs (Fig. 2C). In addition, we
also observed that P-STAT1 (Tyr701) levels in all three TKI-exposed
cell lines were markedly higher than parental cells. In this study,
we observed that Snail, a key regulator of EMT, expression in
TKI-exposed cells was slightly higher in the exposed compared to
the parental cells. We also detected decreased levels of E-cadherin
and increased levels of vimentin in TKI-exposed cells.
STAT3 activation does not depend on
EGFR
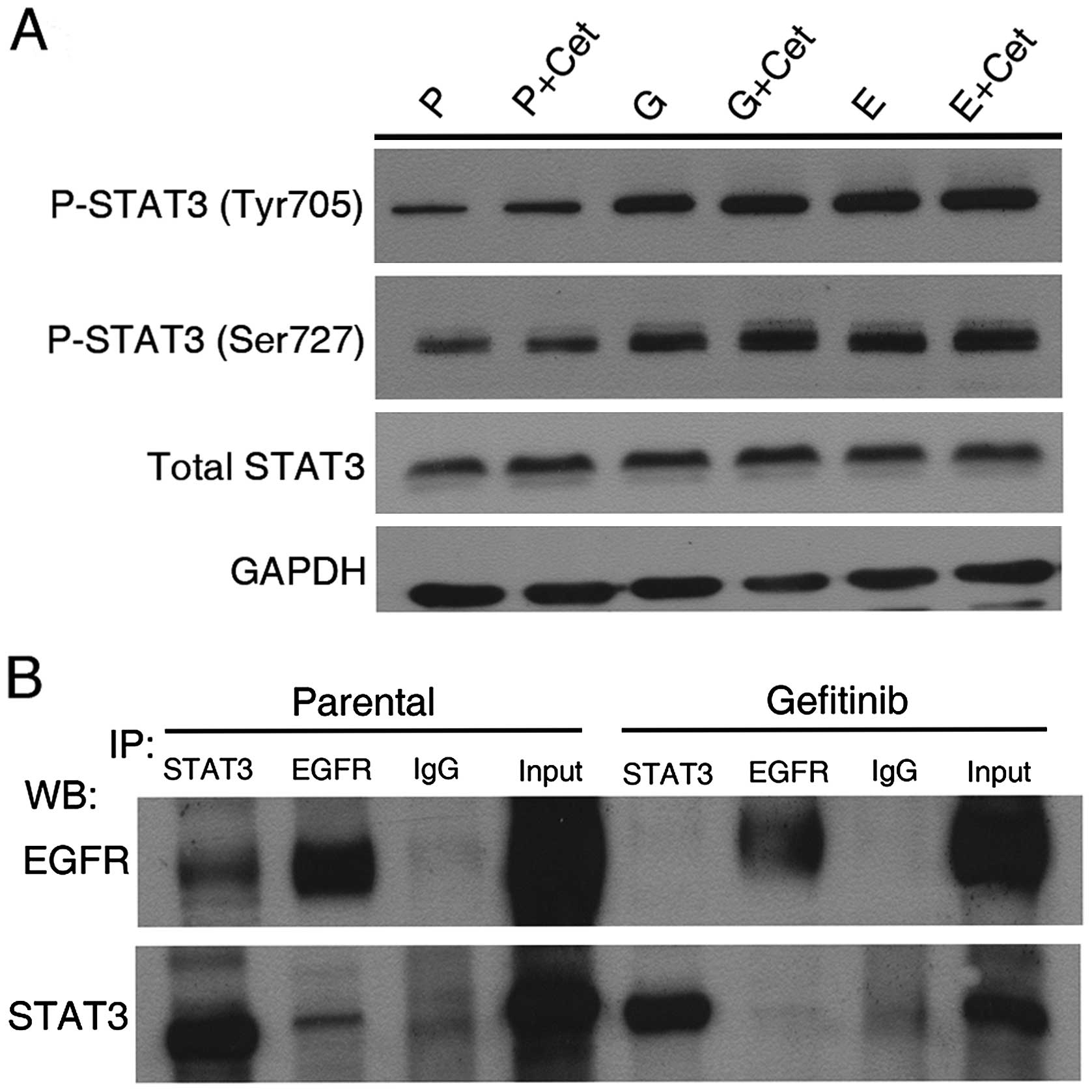
To examine the possibility that STAT3 is activated
through a direct physical interaction between STAT3 and EGFR, an
immunoprecipitation assay was performed. As shown in Fig. 8B, parental H1299 cells exhibit
slight binding between STAT3 and EGFR in the normal physiological
state, however, after long-term exposure to EGFR-TKI the binding of
STAT3 to EGFR was inhibited when identical amounts of total
proteins were used for pulldown by an anti-STAT3 or -EGFR antibody.
To further explore whether the mechanism of STAT3 activation was
independent of EGFR, we used cetuximab as a treatment to block EGFR
dimerization in EGFR-TKI-exposed cells. As shown in Fig. 8A, cetuximab did not affect the
abundance of P-STAT3.
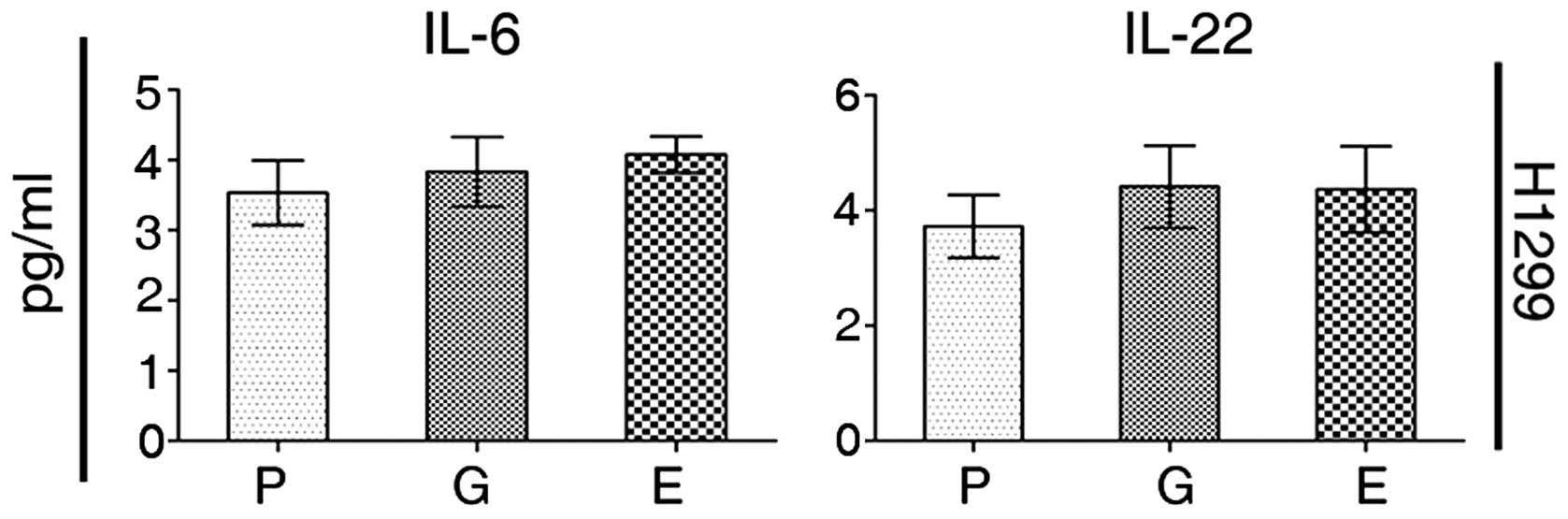
STAT3 activation is independent of IL-6
and -22
In order to explain the mechanisms of activation of
STAT3, we measured the level of IL-6 and -22 in the supernatant of
culture media harvested from our cell experiments. For H1299 cells
there was no significant difference between the levels of these
cytokines released from TKI-exposed and parental cells (Fig. 9).
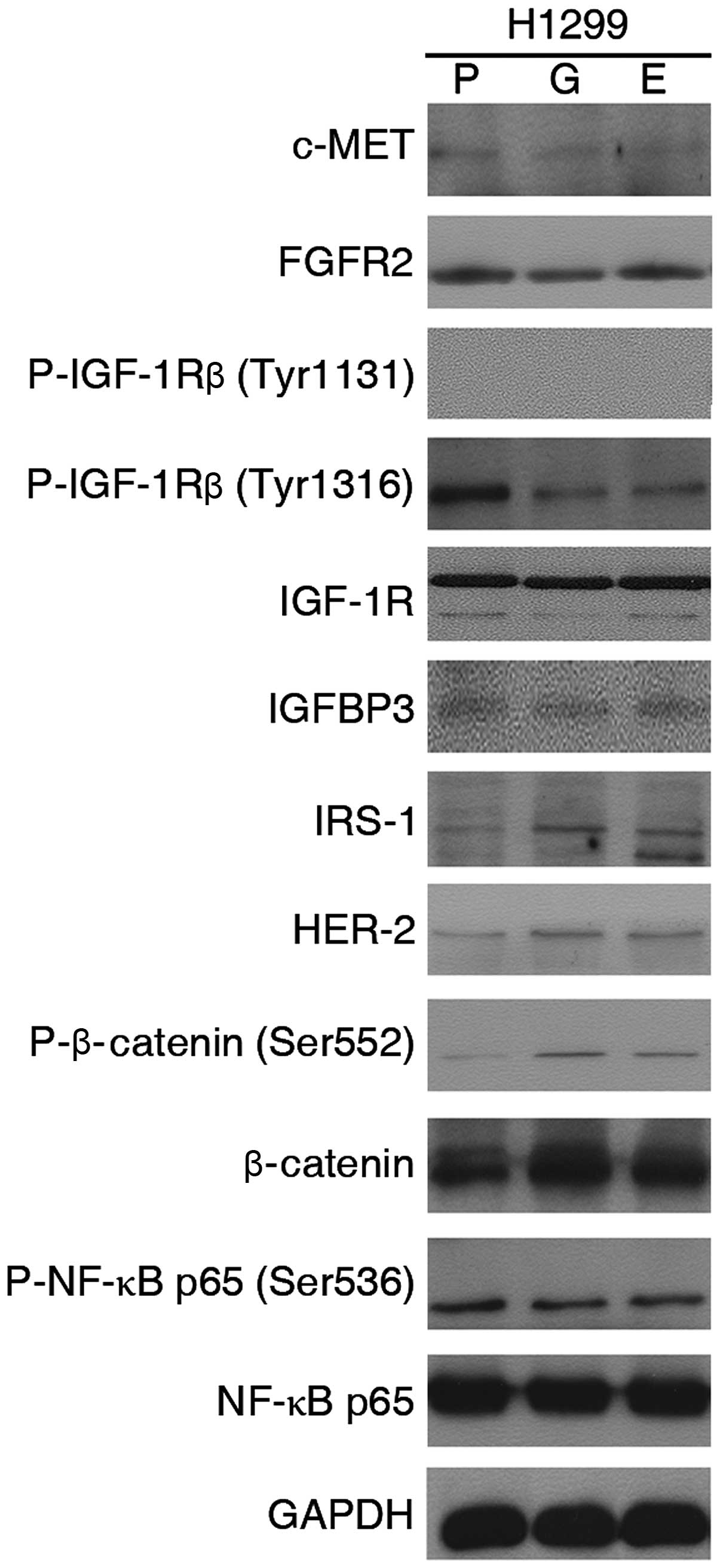
IGF-1R and c-MET are not involved in
chemoresistance
We examined whether there were other mechanisms,
which had been reported to potentially contributed to
chemoresistance, including several major proteins of IGF-1R
signaling, c-MET, phosphorylated NF-κB p65 and so on (Fig. 10). However, our studies revealed
there were no significant difference between parental and
TKI-exposed cells.
Targeting STAT3 augments the efficacy of
cytotoxic drugs against cells possessing EGFR with both L858R and
T790M mutations
Given that chemotherapy is a primary treatment
choice following EGFR-TKI treatment failure, we investigated
whether EGFR-mt NSCLC cells with resistance to EGFR-TKI generate
chemoresistance by similar mechanisms. H1975 cells (harboring two
mutations of EGFR) were treated with gefitinib for 4 weeks and
HCC827 cells (in which resistance to EGFR-TKI is due to c-MET
amplification) were treated with gefitinib for 6 months to simulate
clinical acquired TKI resistance. When, we assessed STAT3 after
gefitinib treatment, we found that its phosphorylation was
increased in H1975 cells (a representative of acquired EGFR-TKI
resistance with the EGFR T790M mutation) but not in HCC827 cells
(in which resistance to EGFR-TKI via c-MET amplification) (Fig. 11A and B). Subsequently, H1975
cells, gefitinib-exposed for 4 weeks were treated in three groups:
i) chemotherapeutic drug alone; ii) chemotherapeutics drugs in
combination with gefitinib; and iii) NSC 74859 and cytotoxic drugs
with gefitinib together (Fig.
11C). Compared with cytotoxic drugs alone, all cells treated
with gefitinib and cytotoxic drugs concurrently exhibited increased
cytotoxicity. The IC50 values were decreased, suggesting
a synergistic or addictive interaction between gefitinib and the
heterogeneous group of the four cytotoxic drugs (DDP, paclitaxel,
gemcitabine and pemetrexed). The addition of NSC 74859 to the
combination of gefitinib and cytotoxic drugs resulted in distinctly
enhanced cytotoxic effects (Fig.
11D). These data suggest that failure of EGFR-TKI treatment may
also result in activation of STAT3, and thus targeting the STAT3
pathway maybe helpful for subsequent chemotherapy.
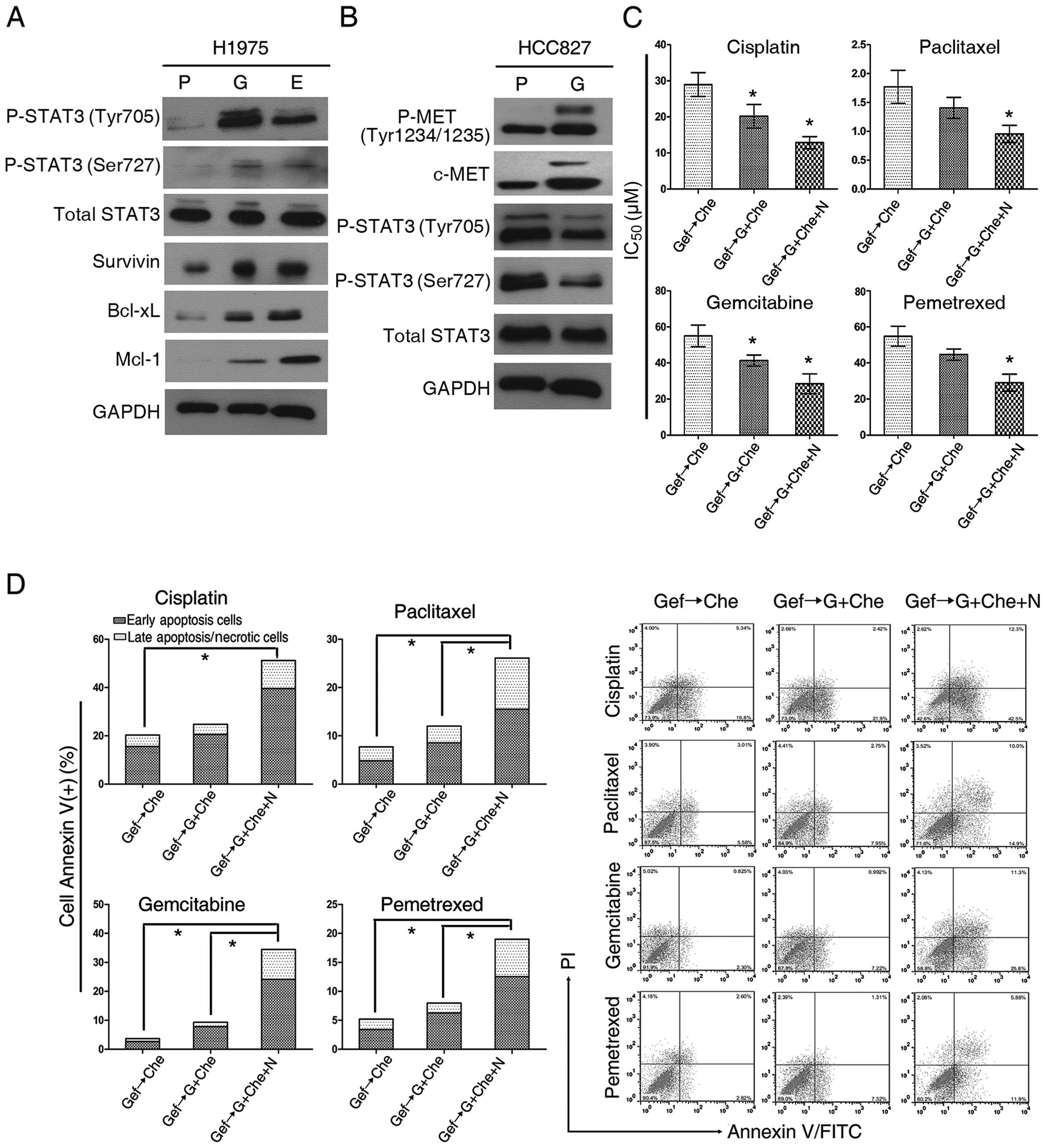 | Figure 11Signal transducer and activator of
transcription 3 (STAT3) inhibition enhances the antitumor effect of
combining gefitinib with cytotoxic drugs in non-small cell lung
cancer (NSCLC) cells bearing the EGFR T790M mutation. (A and B)
STAT3 activation was observed in H1975 cells exposed to TKI, but
not in HCC827 cells long-term exposed to gefitinib. P, parental; G,
gefitinib-exposed; E, erlotinib-exposed. (C) Determination of
IC50 using the CCK-8 cell number assay in H1975 cells
long-term exposed to gefitinib. Gef→Che, gefitinib removed and
replaced by chemotherapeutic agents; Gef→Gef+Che, gefitinib
continued and supplemented with chemotherapeutic agents;
Gef→Gef+Che+N, gefitinib continued and supplemented with
chemotherapeutic agents and NSC 74859. (D) Analysis of H1975 cells
by flow cytometry to assess apoptosis. Groups as in (C).
Concentrations of chemotherapeutic agents used: cisplatin (DDP) 10
μM, paclitaxel 1 μM, gemcitabine 20 μM or pemetrexed 20 μM. Data
shown as mean ± SD; *P<0.05; statistical difference:
one-way ANOVA/Dunnett’s test, compared with the control group.
Experiments were repeated three times. |
Discussion
Increased expression of EGFR has been found in
40–80% of NSCLC cases (14–16).
Therefore, multiple approaches have been developed in order to
inhibit EGFR, such as competition for the extracellular domain by
monoclonal antibodies (cetuximab) or the inhibition of EGFR
tyrosine kinase activity by small molecules interacting with the
intracellular domain (erlotinib, gefitinib and afatinib).
The characterization of EGFR-mts was a crucial
discovery associated with high efficacy of biomarker-driven
treatment (17). As a result,
EGFR-TKIs are now the treatment of choice for patients with
EGFR-mutated tumors (18,19).
For chemotherapy-naive advanced NSCLC patients,
several clinical trials with biomarker-driven selection (EURTAC,
OPTIMAL, WJTOG3405, and NEJ002) have proven that a statistically
significant and clinically relevant increase in PF was obtained
using TKIs compared to chemotherapy (4,20–22).
Nevertheless, subgroup analysis based on molecular analyses (IPASS
and First-SIGNAL) revealed that chemotherapy was significantly
better than EGFR-TKIs in EGFR-wt patients (3,8). It
has been proposed that some EGFR-wt or status-unknown NSCLC
patients who undergo first-line EGFR-TKI treatment have a worse
prognosis and lower response rate to chemotherapy, according to the
results of Gridelli et al in the TORCH study (Tarceva or
chemotherapy) (10).
Despite the fact that EGFR-TKIs are not generally
more efficacious than chemotherapy for unselected patients, and is
not recommended to treat patients whose EGFR status is unknown, in
practice it is reasonable for gefitinib or erlotinib to be used as
an exploratory treatment for patients whom have never smoked or
have been light smokers. For instance, EGFR-mts occur in ~50% of
Asian patients with NSCLC (23).
Standard EGFR mutation analysis requires a minimum amount of tumor
tissue, however, for a large proportion of advanced NSCLC patients
this may not be available. In addition, methods such as ‘liquid
biopsy’ that study circulating lung cancer cells or that analyze
‘free tumor DNA’ in the plasma still have a lot of problems to
conquer, including a low concordance rate between plasma and in
situ biopsy (24). Usually,
after undergoing a 4-week exploratory treatment, the tumor response
will be reassessed using response evaluation criteria in solid
tumors (RECIST) compared with base-line data. Following a tumor
response of partial response (PR) or stable disease (SD), TKI
treatment will continue, otherwise, TKI will be replaced with
chemotherapy.
Due to the likelihood that chemotherapy-naive
patients with EGFR-wt could possibly be treated with EGFR-TKIs, we
decided to evaluate the influence of EGFR-TKIs on subsequent
chemotherapy in a culture system model. The outcomes showed that
EGFR-TKIs had an adverse effect on the subsequent chemotherapy for
any of the four agents we tested: DDP, paclitaxel, gemcitabine and
pemetrexed. Our findings strongly support that continuous exposure
to EGFR-TKIs before chemotherapy results in chemoresistance in
EGFR-wt NSCLC cells.
Our study found that continuous EGFR-TKI exposure
actually induces high-level activation of STAT3 signaling and
rescue of AKT/mTOR. Interestingly, the inhibition of STAT3
completely deleted the phosphorylation of AKT, but not vice versa.
Moreover, inhibition of PI3K did not affect the level of
phospho-AKT. These results showed that sustaining EGFR-TKI exposure
deprived function of PI3K, an upstream regulator of AKT, while
overactivation of STAT3 replaced the role of PI3K to re-foster the
AKT/mTOR pathway. Our data support previous findings that STAT3
activation regulates AKT activation upstream of AKT pathway in
EGFR-wt NSCLC when exposed to EGFR-TKI (25), and indicates that overactivation of
STAT3 plays a critical role in response to long-term EGFR-TKI
exposure.
During consecutive, long-term exposure to EGFR-TKIs,
STAT3 is activated, as shown by increased levels of P-STAT3, DNA
binding, and transcriptional activity (26,27).
The finding in our present study is that the activation of STAT3 is
tightly correlated with the signals for survival and growth arrest.
H1299 cells responded with an upregulation of Bcl-2, Bcl-xL, Mcl-1
and survivin which represent anti-apoptotic signals. Additionally,
downregulation of c-Myc, cyclin D1 and an increase of P27KIP1
indicated cell growth arrest. These findings contradict recent
reports of a direct correlation among cyclin D1, c-Myc and STAT3
(28), but the downregulation of
c-Myc by activation of STAT3 in tumor tissues has also been
reported by other researchers (29), and cyclin D1 potentially creates a
negative feedback loop onto STAT3 (30). In addition, STAT1 has been
demonstrated to suppress c-Myc and cyclin D1 expression as a
negative transcriptional regulator which relates to cell cycle
arrest and an increase of P-STAT1 was observed in TKI-exposed cells
in our experiments (31–33). Previous investigations have
reported that STAT3 could lead to EMT, which may be helpful for
chemoresistance (34–36), EMT was observed in TKI-exposed
cells. Therefore, our study showed that STAT3 activation in
response to continuous EGFR-TKI exposure further resulted in
chemoresistance via multiple mechanisms.
To support the hypothesis that STAT3 was the major
effector molecule, we used an inhibitor of STAT3 (NSC 74859) to
treat cells long-term exposed to TKIs, and examined the sensitivity
of these cells to cytotoxic agents in vitro. We found that
the TKI preconditioned cells regained sensitivity to cytotoxic
agents, to a large degree. Considering our results obtained both
in vitro and in vivo, we believe that we can provide
a plausible explanation for these discordant results; that STAT3
play a major role in the adverse effects.
It is well known that IGF-1R and NF-κB signaling, as
well as MET amplification involving in EGFR-TKI resistance both
de novo and acquired (37,38).
As we have shown here that STAT3 has negative effects on cytotoxic
agents, IGF-1R and MET were not essential partners for this in
H1299 cells.
Activation of STAT3 has been reported to occur
through binding of the IL-6 family of cytokines to the gp130
receptor (35). High levels of
IL-6, which was secreted by EGFR-TKI, was induced in several cell
lines (39,40). Inconsistent with the known effects
of IL-6 on STAT3 signaling (40–42),
we found IL-6 as well as IL-22 was not essential for activating
STAT3 in long-term EGFR-TKI-exposed NSCLC. Our study also showed a
reduction in the level of EGFR/STAT3 complex in continuously
TKI-exposed cells, differently from in short-exposed (25). As cetuximab has no effect on the
activation of STAT3, we incline to believe that a negative
correlation exists between activation of STAT3 and EGFR in this
study. It will be important to further examine how STAT3 be
activated in our follow-up studies.
The EGFR TKI-resistant cell line H1975 harbors a
double mutation (L858R and T790M) in the EGFR gene. T790M is
sometimes present as a minor allele before EGFR-TKI therapy and
accounts for about half of the acquired resistance cases.
Several clinical trials have suggested that
second-line erlotinib treatment was effective in those who had
prior disease control with first-line gefitinib. Other research
shows that continuation of an EGFR-TKI with chemotherapy compared
to chemotherapy alone significantly increases the ORR but not PFS
and OS in patients with advanced NSCLC and acquired TKI resistance
(43). Indeed, in our in
vitro results, EGFR TKI with chemotherapy was more effective
than chemotherapy alone against H1975 TKI-exposed cells.
Considering that the activation of the STAT3 signaling pathway has
also been demonstrated both in H1975 TKI-exposed and parental
cells, we combined NSC 74859 with gefitinib and chemotherapy agents
in H1975 TKI-exposed cells. There was a significant synergistic
killing effect from combination treatment with NSC 74859, which is
in accordance with results from several other researchers.
In our study, we focused on EGFR TKIs as frontline
agents prior to chemotherapy. Our results raise the possibility
that exposure to EGFR-TKIs possibly activates STAT3. Similarly,
Haura et al (44) found
that patients with early-stage NSCLC who received 4 weeks of
treatment with gefitinib (250 mg daily) before surgical resection
had abundant expression of P-STAT3 in their surgically resected
tumors. Thus, the use of EGFR-TKI as exploratory treatment on
patients with unknown EGFR-mt status must be considered with
caution and prudence.
In conclusion, whether there is de novo or
acquired resistance to chemotherapy by persistent activation of
STAT3, a combination strategy of chemotherapeutic with STAT3
inhibitor may be beneficial for NSCLC patients. We believe that our
in vitro and in vivo xenograft models sufficiently
support that targeting STAT3 is a strategy worth considering for
circumventing EGFR-TKI resistance in patients.
Acknowledgements
This study was supported by the National Major
Project of China (2011ZX09302-001-01) and the National Natural
Science Foundation of China (Beijing, China) (81472197).
References
|
1
|
Riely GJ, Politi KA, Miller VA and Pao W:
Update on epidermal growth factor receptor mutations in non-small
cell lung cancer. Clin Cancer Res. 12:7232–7241. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ono M and Kuwano M: Molecular mechanisms
of epidermal growth factor receptor (EGFR) activation and response
to gefitinib and other EGFR-targeting drugs. Clin Cancer Res.
12:7242–7251. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mok TS, Wu YL, Thongprasert S, et al:
Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N
Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou C, Wu YL, Chen G, et al: Erlotinib
versus chemotherapy as first-line treatment for patients with
advanced EGFR mutation-positive non-small-cell lung cancer
(OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase
3 study. Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maemondo M, Inoue A, Kobayashi K, et al:
Gefitinib or chemotherapy for non-small-cell lung cancer with
mutated EGFR. N Engl J Med. 362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Laurie SA and Goss GD: Role of epidermal
growth factor receptor inhibitors in epidermal growth factor
receptor wild-type non-small-cell lung cancer. J Clin Oncol.
31:1061–1069. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gatzemeier U, Pluzanska A, Szczesna A, et
al: Results of a phase III trial of erlotinib (OSI-774) combined
with cisplatin and gemcitabine (GC) chemotherapy in advanced
non-small cell lung cancer (NSCLC). J Clin Oncol ASCO. 22:abs.
7010. 2004.
|
|
8
|
Han JY, Park K, Kim SW, et al:
First-SIGNAL: first-line single-agent Iressa versus gemcitabine and
cisplatin trial in never-smokers with adenocarcinoma of the lung. J
Clin Oncol. 30:1122–1128. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pirker R, Pereira JR, von Pawel J, et al:
EGFR expression as a predictor of survival for first-line
chemotherapy plus cetuximab in patients with advanced
non-small-cell lung cancer: analysis of data from the phase 3 FLEX
study. Lancet Oncol. 13:33–42. 2012. View Article : Google Scholar
|
|
10
|
Gridelli C, Ciardiello F, Gallo C, et al:
First-line erlotinib followed by second-line cisplatin-gemcitabine
chemotherapy in advanced non-small-cell lung cancer: the TORCH
randomized trial. J Clin Oncol. 30:3002–3011. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu JY, Shih JY, Yang CH, et al:
Second-line treatments after first-line gefitinib therapy in
advanced nonsmall cell lung cancer. Int J Cancer. 126:247–255.
2010. View Article : Google Scholar
|
|
12
|
Kumar A, Petri ET, Halmos B and Boggon TJ:
Structure and clinical relevance of the epidermal growth factor
receptor in human cancer. J Clin Oncol. 26:1742–1751. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kolch W and Pitt A: Functional proteomics
to dissect tyrosine kinase signalling pathways in cancer. Nat Rev
Cancer. 10:618–629. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sebastian S, Settleman J, Reshkin SJ,
Azzariti A, Bellizzi A and Paradiso A: The complexity of targeting
EGFR signalling in cancer: from expression to turnover. Biochim
Biophys Acta. 1766:120–139. 2006.PubMed/NCBI
|
|
15
|
Normanno N, De Luca A, Bianco C, et al:
Epidermal growth factor receptor (EGFR) signaling in cancer. Gene.
366:2–16. 2006. View Article : Google Scholar
|
|
16
|
Irmer D, Funk JO and Blaukat A: EGFR
kinase domain mutations - functional impact and relevance for lung
cancer therapy. Oncogene. 26:5693–5701. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kobayashi S, Boggon TJ, Dayaram T, et al:
EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Garassino MC, Martelli O, Broggini M, et
al: Erlotinib versus docetaxel as second-line treatment of patients
with advanced non-small-cell lung cancer and wild-type EGFR tumours
(TAILOR): a randomised controlled trial. Lancet Oncol. 14:981–988.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang J, Cheng Y, Zhao M, Zhou K, Yan H and
Zhang L: A phase II trial comparing pemetrexed with gefitinib as
the second-line treatment of nonsquamous NSCLC patients with
wild-type EGFR (CTONG0806). J Clin Oncol ASCO. 31:abs. 8042.
2013.
|
|
20
|
Rosell R, Carcereny E, Gervais R, et al:
Erlotinib versus standard chemotherapy as first-line treatment for
European patients with advanced EGFR mutation-positive
non-small-cell lung cancer (EURTAC): a multicentre, open-label,
randomised phase 3 trial. Lancet Oncol. 13:239–246. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mitsudomi T, Morita S, Yatabe Y, et al:
Gefitinib versus cisplatin plus docetaxel in patients with
non-small-cell lung cancer harbouring mutations of the epidermal
growth factor receptor (WJTOG3405): an open label, randomised phase
3 trial. Lancet Oncol. 11:121–128. 2010. View Article : Google Scholar
|
|
22
|
Kobayashi K, Inoue A, Maemondo M, et al:
First-line gefitinib versus first-line chemotherapy by carboplatin
(CBDCA) plus paclitaxel (TXL) in non-small cell lung cancer (NSCLC)
patients (pts) with EGFR mutations: a phase III study (002) by
North East Japan Gefitinib Study Group. J Clin Oncol ASCO. 27:abs.
8016. 2009.
|
|
23
|
Sequist LV, Bell DW, Lynch TJ and Haber
DA: Molecular predictors of response to epidermal growth factor
receptor antagonists in non-small-cell lung cancer. J Clin Oncol.
25:587–595. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Maheswaran S, Sequist LV, Nagrath S, et
al: Detection of mutations in EGFR in circulating lung-cancer
cells. N Engl J Med. 359:366–377. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu K, Chang Q, Lu Y, et al: Gefitinib
resistance resulted from STAT3-mediated Akt activation in lung
cancer cells. Oncotarget. 4:2430–2438. 2013.PubMed/NCBI
|
|
26
|
Barré B, Vigneron A, Perkins N, Roninson
IB, Gamelin E and Coqueret O: The STAT3 oncogene as a predictive
marker of drug resistance. Trends Mol Med. 13:4–11. 2007.
View Article : Google Scholar
|
|
27
|
Dauer DJ, Ferraro B, Song L, et al: Stat3
regulates genes common to both wound healing and cancer. Oncogene.
24:3397–3408. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ai T, Wang Z, Zhang M, et al: Expression
and prognostic relevance of STAT3 and cyclin D1 in non-small cell
lung cancer. Int J Biol Markers. 27:e132–e138. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yamanaka Y, Nakajima K, Fukada T, Hibi M
and Hirano T: Differentiation and growth arrest signals are
generated through the cytoplasmic region of gp130 that is essential
for Stat3 activation. EMBO J. 15:1557–1565. 1996.PubMed/NCBI
|
|
30
|
Germain D and Frank DA: Targeting the
cytoplasmic and nuclear functions of signal transducers and
activators of transcription 3 for cancer therapy. Clin Cancer Res.
13:5665–5669. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dimberg A, Karlberg I, Nilsson K and Oberg
F: Ser727/Tyr701- phosphorylated Stat1 is required for the
regulation of c-Myc, cyclins, and p27Kip1 associated with
ATRA-induced G0/G1 arrest of U-937 cells. Blood. 102:254–261. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ramana CV, Chatterjee-Kishore M, Nguyen H
and Stark GR: Complex roles of Stat1 in regulating gene expression.
Oncogene. 19:2619–2627. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dimco G, Knight RA, Latchman DS and
Stephanou A: STAT1 interacts directly with cyclin D1/Cdk4 and
mediates cell cycle arrest. Cell cycle. 9:4638–4649. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yamashita S, Miyagi C, Fukada T, Kagara N,
Che YS and Hirano T: Zinc transporter LIVI controls
epithelial-mesenchymal transition in zebrafish gastrula organizer.
Nature. 429:298–302. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yadav A, Kumar B, Datta J, Teknos TN and
Kumar P: IL-6 promotes head and neck tumor metastasis by inducing
epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling
pathway. Mol Cancer Res. 9:1658–1667. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang H, Zhang G, Zhang H, et al:
Acquisition of epithelial-mesenchymal transition phenotype and
cancer stem cell-like properties in cisplatin-resistant lung cancer
cells through AKT/β-catenin/Snail signaling pathway. Eur J
Pharmacol. 723:156–166. 2014. View Article : Google Scholar
|
|
37
|
Lin L and Bivona TG: Mechanisms of
resistance to epidermal growth factor receptor inhibitors and novel
therapeutic strategies to overcome resistance in NSCLC patients.
Chemother Res Pract. 2012:8172972012.PubMed/NCBI
|
|
38
|
Chen Y-F and Fu L-W: Mechanisms of
acquired resistance to tyrosine kinase inhibitors. Acta Pharm Sin
B. 1:197–207. 2011. View Article : Google Scholar
|
|
39
|
Yeh HH, Lai WW, Chen HH, Liu HS and Su WC:
Autocrine IL-6-induced Stat3 activation contributes to the
pathogenesis of lung adenocarcinoma and malignant pleural effusion.
Oncogene. 25:4300–4309. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ and
Settleman J: Drug resistance via feedback activation of Stat3 in
oncogene-addicted cancer cells. Cancer Cell. 26:207–221. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim SM, Kwon OJ, Hong YK, et al:
Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance
to irreversible EGFR inhibitors in non-small cell lung cancer with
T790M resistance mutation. Mol Cancer Ther. 11:2254–2264. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Song L, Rawal B, Nemeth JA and Haura EB:
JAK1 activates STAT3 activity in non-small-cell lung cancer cells
and IL-6 neutralizing antibodies can suppress JAK1-STAT3 signaling.
Mol Cancer Ther. 10:481–494. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Goldberg SB, Oxnard GR, Digumarthy S, et
al: Chemotherapy with Erlotinib or chemotherapy alone in advanced
non-small cell lung cancer with acquired resistance to EGFR
tyrosine kinase inhibitors. Oncologist. 18:1214–1220. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Haura EB, Sommers E, Song L, Chiappori A
and Becker A: A pilot study of preoperative gefitinib for
early-stage lung cancer to assess intratumor drug concentration and
pathways mediating primary resistance. J Thorac Oncol. 5:1806–1814.
2010. View Article : Google Scholar : PubMed/NCBI
|