Introduction
Osteosarcoma (OS) is the most common primary
malignant tumor of the bone, occurring mainly in children and young
adolescents. With combination therapy, the 5-year survival of
affected patients is 60–70%. However, the survival rates of
patients with metastasis or recurrence are far worse, at <30%
and <20%, respectively (1). The
lungs are the predominant site of OS metastasis, and pulmonary
involvement is the most common cause of death. Unfortunately, the
survival rates have not improved over the last 20 years, despite
the significant increase in clinical trials.
Epigenetic modifications have been shown to have
critical roles in regulating gene expression. DNA methylation and
histone modifications are also important mediators of epigenetic
gene silencing and essential steps in diverse biological processes
(2). Increasing evidence
implicates histone deacetylase (HDAC) inhibitors as some of the
most promising epigenetic anticancer agents (3,4).
Valproic acid (VPA), an HDAC inhibitor, is a well-established
long-term treatment for epilepsy and bipolar disorder (5,6).
However, in recent years, VPA and trichostatin A (TSA) have also
been shown to have the potential ability to modulate the biology of
several tumor cell types by inducing differentiation, apoptosis and
immunogenicity and subsequently decreasing the rates of metastasis
and angiogenesis (3). Angiogenesis
is a key biological process that occurs during embryonic
development and tissue repair and is required for tumor progression
and metastasis. Vascular endothelial growth factor (VEGF), a
well-known pro-angiogenic factor, plays an important role in
tumor-induced angiogenesis in various human tumors. The tumor
angiogenesis mediated by hypoxia inducible factor-1α (HIF-1α) and
its transcriptional target, VEGF, is increased, and the
anti-angiogenic function of HDAC inhibitors is to suppress the VEGF
gene expression via HIF-1α inhibition (7).
Vascular endothelial growth inhibitor (VEGI), also
known as TL1A or TNFSF15, is a novel member of the tumor necrosis
factor (TNF) superfamily (8). VEGI
was originally thought to be exclusively expressed in endothelial
cells and abundantly expressed in the kidneys, lungs, prostate,
placenta and liver (8,9). However, it was subsequently reported
that VEGI is also expressed in a wide variety of human cancer cell
lines, including breast, prostate, bladder, colorectal and liver
cancers (10). VEGI contains 251
amino acids in the full-length product of TNF ligand-related
molecule 1 (TL1), and three isoforms of VEGI have been reported,
all sharing a common 151 C-terminal amino acid sequence with
different N-terminal regions (11,12).
The biological activity of VEGI is dependent on the solubilized
extracellular domain of these isoforms, and the secreted or
recombinant soluble forms of VEGI are potent inhibitors of
endothelial cell proliferation and angiogenesis as well as tumor
growth and neovascularization (9,11,13–15).
This inhibition is mediated by death receptor 3 (DR3) (TNFRSF25),
known to be a functional receptor for VEGI (11,16),
which contains a death domain in its cytoplasmic tail capable of
inducing apoptosis that can be blocked by decoy receptor 3 (DcR3)
(12). DcR3 is a soluble receptor
lacking transmembrane domains that belongs to the TNFR superfamily.
The overexpression of DcR3 noted in the some malignant cells blocks
the suppression of apoptosis by interfering with the binding of
VEGI, FasL and LIGHT to receptors (12,17).
In addition, DcR3 enhances angiogenesis by blocking the
anti-angiostatic function of VEGI (17).
In this study, we investigated the effects of HDAC
inhibitors on the endogenous anti-angiogenic factor VEGI and its
receptors and found that treatment with VPA and TSA increased the
expression of VEGI and its receptor, DR3, without inducing
increased DcR3 in OS cells. Moreover, VPA-treated OS cell culture
medium containing soluble VEGI inhibited vascular tube formation,
while the overexpression of VEGI in the OS cells induced cell
death. Therefore, HDAC inhibitors result in enhanced tumor and
vascular endothelial cell death via the soluble VEGI/DR3 autocrine-
and paracrine pathways.
Materials and methods
Reagents and antibodies
Sodium valproate (VPA) and trichostatin A (TSA) were
purchased from Wako (Osaka, Japan), anti-acetyl-histone H3 rabbit
polyclonal antibodies were purchased from Upstate (Temecula, CA,
USA), anti-human VEGI mouse monoclonal antibody (IgG1),
anti-human DR3 mouse monoclonal antibody (IgG2b) and
horseradish peroxidase-conjugated goat anti-rabbit IgG1
antibody were purchased from Santa Cruz (Santa Cruz, CA, USA),
biotin-conjugated anti-human VEGI rabbit polyclonal antibodies
(IgG) and anti-human VEGI rabbit polyclonal antibodies (IgG) were
purchased from Abcam (Tokyo, Japan) and anti-actin rabbit
polyclonal antibodies and horseradish peroxidase-conjugated rabbit
anti-mouse IgG antibodies were purchased from Sigma (St. Louis, MO,
USA).
Cells
U-2 OS and SaOS-2 human osteosarcoma cells were
purchased from the American Type Culture Collection (ATCC,
Manassas, VA, USA), and Riken BRC Cell Bank (Tsukuba, Ibaragi,
Japan), respectively. The U-2 OS and SaOS-2 cells were cultured in
McCoy’s 5A modified medium (Invitrogen, Carlsbad, CA, USA). Primary
normal human dermal microvascular endothelial (HMVE) cells were
purchased from the Cell Systems Corp. (CSC, Kirkland, WA, USA) and
cultured using the CS-C medium kit (DS Pharma Biomedical, Osaka,
Japan). All media contained 10% fetal bovine serum (FBS) (MP
Biomedical; Morgan Irvine, CA, USA), penicillin (100 U/ml) and
streptomycin (100 μg/ml), and all cells were cultured in a
humidified atmosphere of 5% CO2 in air at 37°C.
Reverse transcription polymerase chain
reaction (RT-PCR)
Total RNA was extracted from the U-2 OS, SaOS-2 and
HMVE cells using TRIzol reagent (Invitrogen) according to the
manufacturer’s instructions. Reverse transcription of 2 μg of total
RNA was performed at 42°C for 1 h using random primer (Roche
Diagnostics, Tokyo, Japan) and reverse transcriptase (Roche
Diagnostics), and the cDNAs thus produced were sequentially
amplified by PCR with Takara Ex Taq™ DNA polymerase (Takara Bio,
Ohtsu, Shiga, Japan) using the following specific primer sets:
sense, 5′-CTCTGCACTG GGAACATGAA-3′ and antisense, 5′-TTGGCTCAGGGTA
GCTGTCT-3′ for VEGI; sense, 5′-CGCAGA GATACTGACTG TGG-3′ and
antisense, 5′-AGGAGGTGCTAGAAGGGTGT-3′ for DR3; sense,
5′-GCTACTGCAACGTCCTCTG-3′ and antisense, 5′-ACACTCCTCAGCTCCTGGTA-3′
for DcR3; sense, 5′-GTCATCAATGGAAATCCCATCACC-3′ and antisense,
5′-GCTCAGGGATGACCTTGCCC-3′ for GAPDH. The amplification conditions
for PCR of VEGI, DR3 and DcR3 included 35 cycles at 95°C for 30
sec, 57°C for 1 min and 72°C for 1 min, followed by heating at 72°C
for 7 min, while that for GAPDH included 30 cycles at 95°C for 30
sec, 57°C for 30 sec and 72°C for 1 min, followed by heating at
72°C for 7 min. The amplified fragments were resolved by
electrophoresis in 1.5% agarose gel and detected using ethidium
bromide staining.
Quantitative real-time polymerase chain
reaction (real-time PCR)
U-2 OS, SaOS-2 cells and HMVE cells were cultured in
medium with or without 1.0 mM VPA and 30 nM TSA with a change of
the medium on day 3. Total RNA was extracted from the cells in each
culture dish on days 3 and 7 using TRIzol reagent (Invitrogen). An
aliquot of RNA was reverse transcribed using the High Capacity RNA
to cDNA kit (Applied Biosystems, Foster City, CA, USA), according
to the manufacturer’s instructions, and real-time PCR for VEGI, DR3
and DcR3 mRNAs was performed using TaqMan Gene Expression assays
(Applied Biosystems). The primer set was Hs00270802_ml for VEGI
mRNA, Hs00600930_ml for DR3 mRNA and Hs00187070_ml for DcR3 mRNA,
respectively (Applied Biosystems). The amount of GAPDH mRNA was
estimated as an internal reference using human GAPDH endogenous
control (Applied Biosystems), and the amount of VEGI, DR3 and DcR3
mRNA in each sample was corrected for the amount of GAPDH mRNA in
corresponding samples. The expression levels of each gene in the
treated cultures are expressed as the ratio of the average value in
the control cultures.
Enzyme-linked immunosorbent assay
(ELISA)
U-2 OS, SaOS-2 and HMVE cells were seeded at
2×105 cells/dish in 10-cm tissue culture dishes
containing 5 ml of medium per dish. After 24 h (day 0), VPA (1.0
mM) or TSA (30 nM) was added to the medium, and the cells were
cultured for 7 days with a change of the medium on day 3. After the
culture, medium was collected for assays of the soluble VEGI and
DcR3. For the detection of soluble VEGI, briefly, 96-well plates
were coated with capture antibody, anti-human VEGI mouse monoclonal
antibody (2.0 μg/ml), overnight at room temperature and then washed
three times with washing buffer (PBS containing 0.05% Tween-20)
after each incubation period. A standard protein, the recombinant
human TL1A/TNFSF15 (R&D Systems, Minneapolis, MN, USA), and the
samples were incubated in the wells for 2 h at room temperature.
After washing, biotin-conjugated anti-human VEGI rabbit polyclonal
antibodies (0.5 μg/ml) were incubated in the wells for 2 h at room
temperature. Following horseradish peroxidase-conjugated
streptavidin (R&D Systems) incubation, the substrate solution
(R&D Systems) was subsequently incubated in the wells for 20
min at room temperature. The detection of soluble DcR3 was
performed using the ELISA development system (R&D Systems),
according to the manufacturer’s instructions. In order to determine
the optical density in each well, the absorbance at 450 nm against
a reference wavelength of 570 nm was measured using a microplate
reader (Bio-Rad, Tokyo, Japan). The effects of VPA and TSA were
evaluated by determining the amount of soluble forms per
104 viable cells in the treated cultures and expressed
as the ratio of the average value in the untreated control
cultures.
Western blot analysis
U-2 OS cells cultured with or without VPA or TSA and
pVEGI-transfected cells were washed twice with PBS and lysed in
radioimmunoprecipitation assay (RIPA) buffer [50 mM Tris-HCl (pH
7.4), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS]
supplemented with a complete protease inhibitor cocktail (Roche
Diagnostics). Briefly, the cell lysates were first incubated on ice
for 30 min and then sonicated several times prior to centrifugation
for 10 min at 10,000 rpm. The supernatant was obtained and analyzed
for the protein concentrations according to the Bradford method,
with bovine serum albumin as the standard. An aliquot of the
supernatant (equivalent to 30 μg of proteins) was mixed with a
3-fold volume of the SDS sample buffer (BioLab, Beverly, MA, USA)
containing 10% β-mercaptoethanol and subsequently heated at 95°C
for 10 min, then electrophoresed on 4–12% Bis-Tris gel with the MES
SDS running buffer (Invitrogen) and transferred onto nitrocellulose
membranes. The membranes were blocked for 30 min at room
temperature in blocking buffer containing 5% skim milk in
Tris-buffered saline with Tween-20 (TBST) [10 mM Tris, 150 mM NaCl
(pH 7.4), 1% Tween-20] and then incubated for 90 min with a 1:500
dilution of anti-acetyl-histone H3 rabbit polyclonal antibodies, a
1:100 dilution of anti-human VEGI rabbit polyclonal antibodies or a
1:200 dilution of anti-human DR3 mouse monoclonal antibody in TBST
buffer, after which the membranes were washed with TBST three times
(10 min each). The membranes were then incubated for 90 min at room
temperature with a 1:10,000 dilution of horseradish
peroxidase-conjugated goat anti-rabbit IgG antibodies for
acetyl-histone H3 or VEGI and horseradish peroxidase-conjugated
rabbit anti-mouse IgG2b antibody for DR3 and
subsequently detected and visualized using the SuperSignal West
Pico chemiluminescence detection system (Pierce, Rockford, IL,
USA). The same membranes were stripped in stripping buffer (Pierce)
and reprobed using the action on the membrane detected with
anti-actin rabbit polyclonal antibodies at 1:200 dilution in TBST
buffer.
Plasmid construction and
transfection
In order to construct a VEGI cDNA plasmid (pVEGI),
the RT-PCR fragment amplified from the U-2 OS total RNA using the
primer sets sense, 5′-caccATGGCCGATCTGGGA-3′ and antisense,
5′-CTATAG TAAGAAGGCTCCAAAGAAGGT-3′ was cloned. Following gel
purification (Promega, Madison, WI, USA), the amplified DNA
fragment was inserted into an expression vector,
pcDNA3.1/V5-His-TOPO (Invitrogen). The plasmid inserts were
subsequently verified by sequencing. The siRNA designed for the
VEGI mRNAs was 5′-ACCUGACAGUUG UGAGACAtt-3′ (sense strand),
synthesized by Applied Biosystems. For transfection, U-2 OS cells
were seeded at 1×105 cells/well in 6-well tissue culture
plates and cultured in 2 ml of medium for 24 h. The culture medium
was changed to Opti-MEM medium (Invitrogen), and the cells were
transfected with 1 μg of plasmid DNA or 20 nM of siRNA using
Lipofectamine™ 2000 (Invitrogen) and RNAiMAX (Invitrogen),
respectively, according to the manufacturer’s instructions. Total
RNA, protein and cell culture media were prepared 48 h after
transfection. The RNA was examined by RT-PCR and real-time PCR, and
the proteins were evaluated using western blot analysis. The cell
culture medium was assessed according to an ELISA assay in order to
determine the soluble VEGI and DcR3 levels, as described above.
Images of the transfected OS cells were examined using an inverted
microscope (Nikon, Tokyo, Japan).
Human microvascular endothelial (HMVE)
cell proliferation assay
HMVE cells (1×103 cells per well) were
seeded in 96-well tissue culture plates containing 100 μl of CS-C
complete medium in each well. After 24 h (day 0), VPA (1.0 mM) or
TSA (30 nM) was added to the medium, and the cells were cultured
for another 7 days with a change of the medium on day 3. The number
of viable cells in each well was estimated using a CellTiter
96® AQueous One solution cell proliferation assay kit
(Promega), according to the manufacturer’s instructions and
presented as the ratio to the average level of optical density in
the control cultures.
In vitro tube formation assay
HMVE cells were subjected to an in vitro
endothelial tube formation assay (Cell Biolabs Inc., San Diego, CA,
USA), according to the manufacturer’s instructions. Briefly, HMVE
cells (3.0×103 cells) were seeded in ECM gel pre-coated
96-well plates and cultured with HMVE cells in the culture media
following the description provided above. After 16 h, the medium
was changed to with or without 1.0 mM VPA and VPA-treated or
untreated OS cell culture medium, and the culture was extended for
additional 16 h. After washing with 1× staining buffer, Calcein AM
staining was performed, the endothelial cells and tubes were
examined using a fluorescence microscope (Nikon).
Statistical analysis
The data are presented as the mean ± SE. Data for
three groups or more were analyzed using the two-tailed Dunnett’s
t-test for multiple comparisons. A P-value of <0.05 was
considered to be significant.
Results
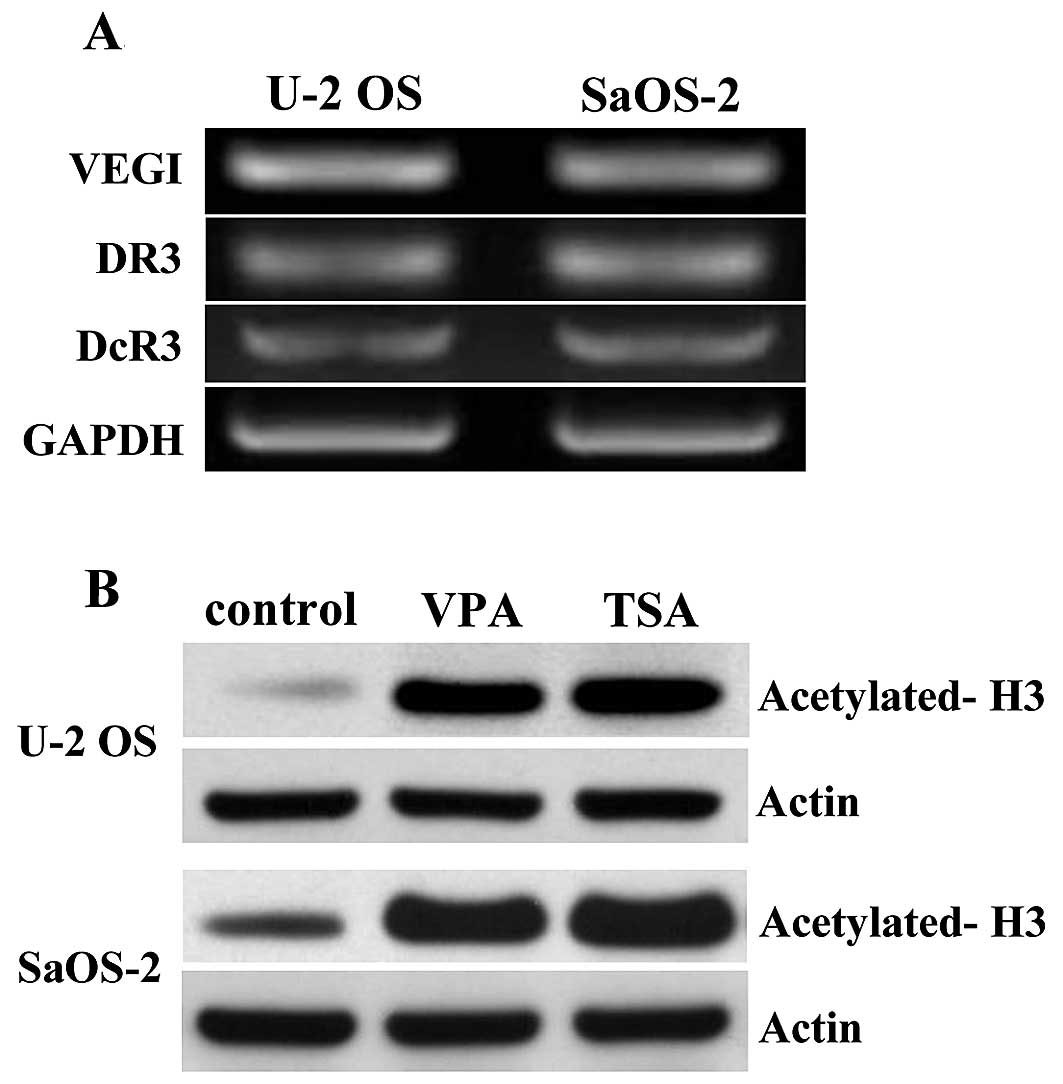
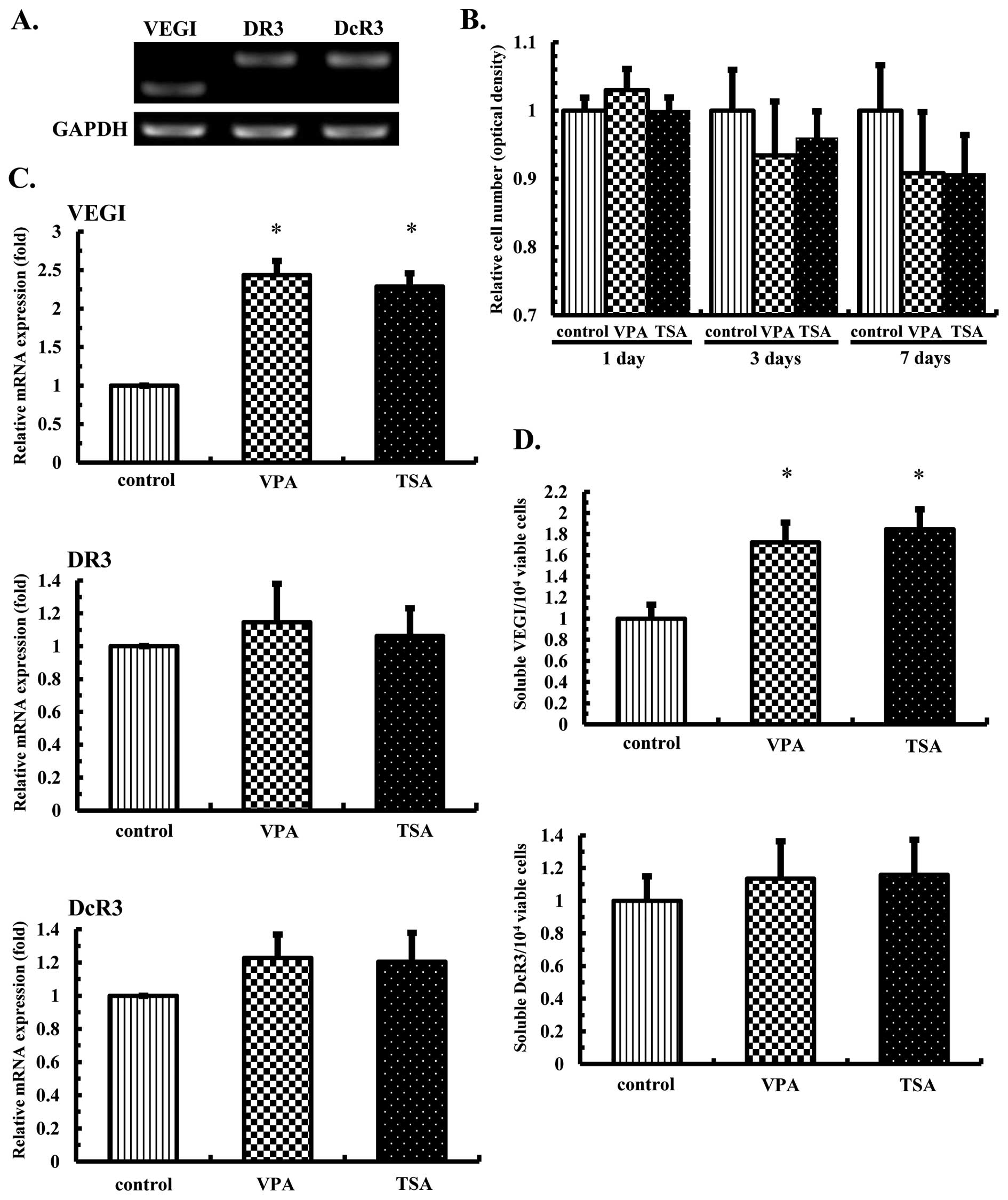
Detection of the transcription of VEGI
and its related receptors and effects of the HDAC inhibitors on
histone acetylation in the OS cells
The expression levels of VEGI, DR3 and DcR3 in OS
cells (U-2 OS and SaOS-2 cells) were examined using RT-PCR. All
three gene transcripts were detected in the OS cell lines (Fig. 1A). In order to confirm the actions
of VPA and TSA as histone deacetylase (HDAC) inhibitors in the OS
cells, U-2 OS and SaOS-2 cells were cultured in the presence or
absence of VPA at 1.0 mM or TSA at 30 nM for 7 days, after which
the histone H3 expression was examined using western blot analysis.
The results showed significantly increased protein levels of
acetylated histone H3 in both the VPA and TSA-treated OS cell lines
(Fig. 1B).
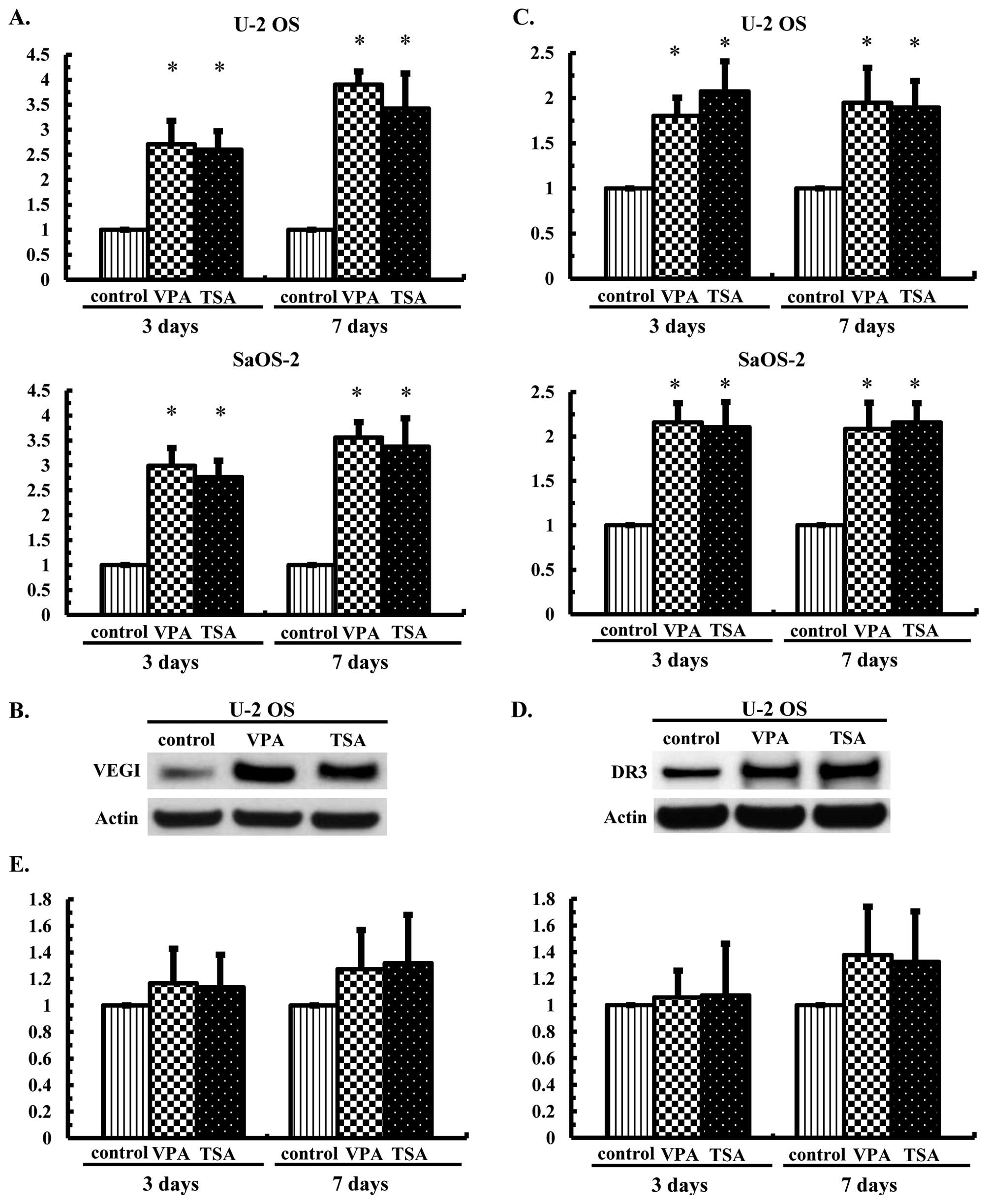
Effects of the HDAC inhibitors on the
mRNA and protein expression levels of VEGI and its related
receptors in the OS cells
U-2 OS and SaOS-2 cells were cultured in the
presence or absence of VPA at 1.0 mM or TSA at 30 nM for 3 or 7
days, after which the mRNA expression levels of VEGI, DR3 and DcR3
were examined using real-time PCR and the protein expression levels
of VEGI and DR3 were examined using western blot analysis. The mRNA
transcription of VEGI was increased 2.6- to 3.7-fold (Fig. 2A), while that of DR3 was increased
1.7- to 2.4-fold (Fig. 2C) in the
U-2 OS and SaOS-2 cells, respectively. Increased protein expression
levels were also observed (Fig. 2B and
D), whereas treatment with VPA or TSA did not significantly
increase DcR3 mRNA transcription (Fig.
2E).
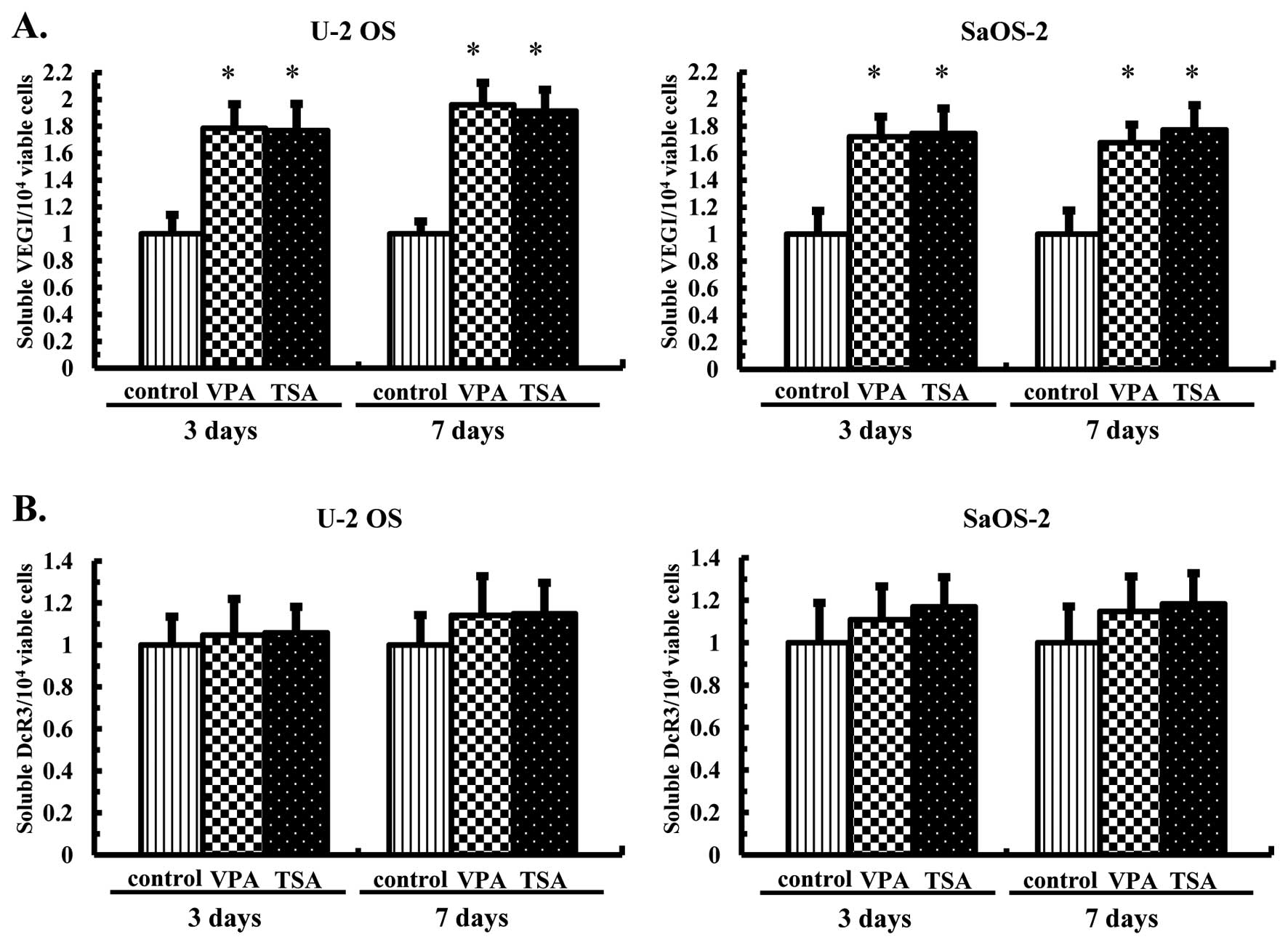
Effects of the HDAC inhibitors on the
production of soluble VEGI and secretion of DcR3 by the OS
cells
In order to determine effects of the HDAC inhibitors
on the production of soluble VEGI and secretion of DcR3, U-2 OS and
SaOS-2 cells were cultured in the presence of VPA at 1.0 mM or TSA
at 30 nM for 3 or 7 days, and the levels of soluble VEGI and DcR3
accumulated in the medium during the first 3 days and the next 4
days were analyzed using ELISA assays. Consequently, the soluble
VEGI levels were significantly increased in the OS cells on 3 and 7
days (Fig. 3A). However, the
levels of DcR3 remained essentially unchanged in the U-2 OS and
SaOS-2 cells at 3 and 7 days following treatment with both HDAC
inhibitors (Fig. 3B).
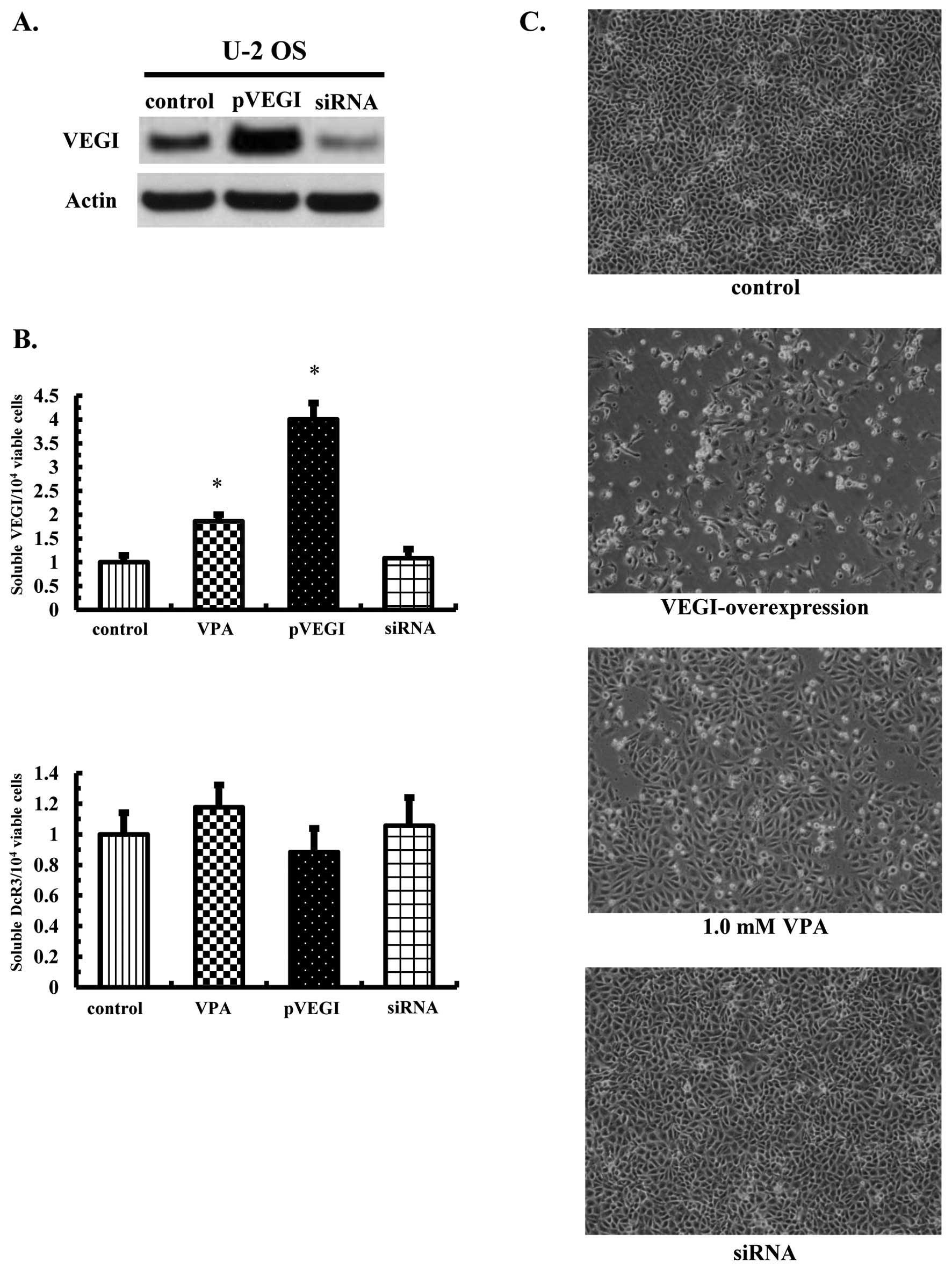
VEGI overexpression directly induces
osteosarcoma cell death
Because treatment with VEGI induced apoptosis in the
vascular endothelial cells (13),
we attempted to confirm whether VEGI caused apoptosis in tumor
cells. Either VEGI full-length gene expression or control vectors
or siRNA for VEGI were transfected into U-2 OS cells, and the
overexpression of VEGI was confirmed based on the protein
expression (Fig. 4A). An ~4-fold
increase in soluble VEGI secretions, without DcR3 productions, was
also observed (Fig. 4B). Moreover,
the transfection of VEGI siRNA did not change either the soluble
VEGI or DcR3 levels (Fig. 4B),
whereas the overexpression of VEGI significantly induced OS cell
death compared with that seen in the control and siRNA-transfected
cells (Fig. 4C).
Transcription of VEGI and its related
receptors and effects of the HDAC inhibitors on cell proliferation
in the HMVE cells
The expression levels of VEGI, DR3 and DcR3 were
examined in the HMVE cells using RT-PCR. All three genes were
transcribed in the HMVE cells (Fig.
5A), and an analysis of HMVE cell growth showed that neither
treatment with VPA at 1.0 mM or TSA at 30 nM had an effect
different from that observed in the control cells (Fig. 5B). However, a real-time PCR
analysis revealed that VEGI mRNA transcription was increased
approximately 2.5-fold, although no significant difference was
noted in the DR3 or DcR3 levels (Fig.
5C).
Effects of the HDAC inhibitors on the
production of soluble VEGI and secretion of DcR3 in the HMVE
cells
Although the HDAC inhibitors increased the soluble
VEGI levels in the OS cells, we further examined the HMVE cells on
day 7. Notably, increased soluble VEGI production was detected
after VPA or TSA treatment (Fig.
5D). However, the DcR3 levels remained essentially unchanged by
both HDAC inhibitors (Fig.
5D).
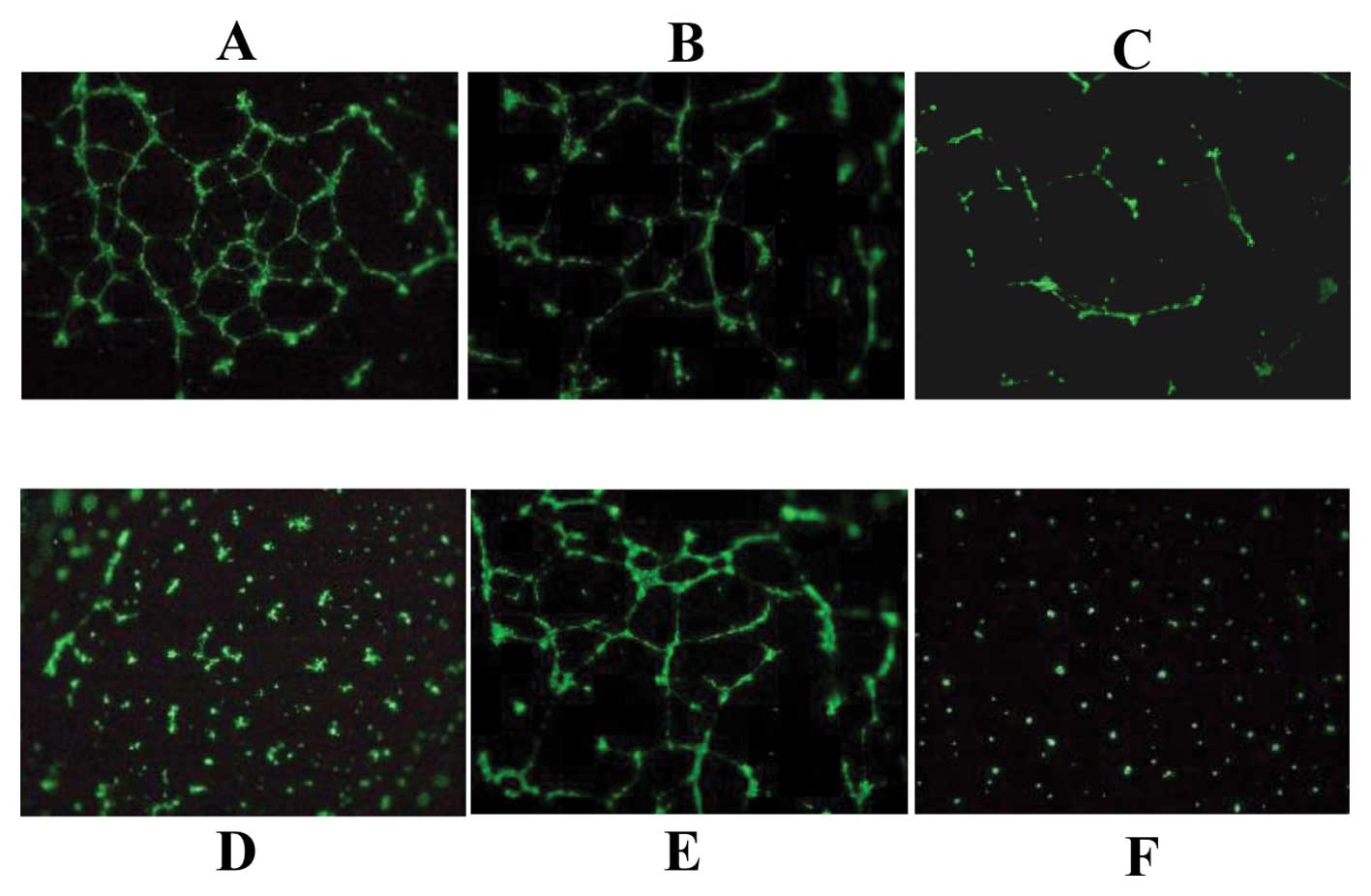
The VPA-induced soluble VEGI in the OS
cells inhibits vascular tube formation
In order to confirm whether the production of
soluble VEGI from the tumor cells inhibited neovascularization, we
assessed the effects of OS cell culture medium treated with VPA as
well as pVEGI-transfected cells. The results showed some tube
formation in the HMVE cells treated with 1.0 mM VPA, although it
was less than that seen in the control cells (Fig. 6B). Surprisingly, tube formation was
markedly decreased by the VPA-treated OS cell culture medium
(Fig. 6C), and the
pVEGI-transfected medium completely suppressed tube formation
(Fig. 6D).
Discussion
The present findings showed that treatment with VPA
and TSA, histone deacetylase (HDAC) inhibitors, increased the
expression of endogenous vascular endothelial growth inhibitor
(VEGI) and its receptor, death receptor 3 (DR3), without
stimulating decoy receptor 3 (DcR3) secretion, via the effects of
histone acetylation. Moreover, VEGI overexpression resulted in
increased OS cell death, and the VPA-treated soluble VEGI in the OS
cell culture medium were associated with the inhibition of vascular
tube formation in the HMVE cells. Recent studies suggest that the
main role of VEGI in human cancer cells is the anticancer effect
via the direct inhibition of cancer cell proliferation as well as
anti-angiogenic effects on endothelial cells (11,18).
DR3, a member of tumor necrosis factor receptor family (TNFRSF25),
has been shown to be capable of stimulating caspase-dependent cell
apoptosis, and the activation of caspases contributes to
VEGI-induced cell death (8,9,11,19).
Therefore, VEGI may serve as a potential target for the development
of angiogenesis-based cancer therapy.
Although the VEGI and DR3 mRNA expression was
detected in the OS cells, the corresponding proteins were expressed
at low levels. This reduction in the tumor VEGI and DR3 expression
suggests a shift in balance to pro-angiogenic conditions. Such loss
of balance may be conducive to tumor growth and survival (10,14).
The VEGI expression is regulated by the central transcription
factor NF-κB (20), and HDAC
inhibition increases the NF-κB transcriptional activity, which
regulates the expression levels of a variety of genes (21). In order to recover an abnormal
angiogenic condition in the tumor cells, we applied VPA and TSA as
HDAC inhibitors and found that both VPA and TSA increased VEGI and
DR3 mRNA transcription and also their protein expression levels.
This observation indicates that HDAC inhibitors promote the
recovery of angiogenic imbalance in tumor cells. The enhanced
expression of DcR3 has been indicated to protect against induced
apoptosis in various tumors and is positively correlated with tumor
progression (22–25). Our data suggest that treatment with
VPA and TSA is not accompanied by increased DcR3 secretion in OS
cells. These results prompted us to explore whether VPA-induced
soluble VEGI inhibits vascular tube formation in HMVE cells.
Consequently, the growth of HMVE cells was not changed by VPA and
TSA treatment. Although both VPA and TSA were confirmed to enhance
VEGI mRNA transcription accompanied by soluble VEGI production,
neither DR3 or DcR3 mRNA transcription nor soluble DcR3 production
was increased. Furthermore, the effect of VPA treatment on vascular
tube formation was slight compared with that observed in the
control cells. Surprisingly, in vitro vascular tube
formation was inhibited by the VPA-treated or VEGI-overexpressing
OS cell culture conditioned media. The results showed that the
soluble VEGI concentration increased ~2.0-fold by the VPA-treated
OS cell medium versus that seen in the control cells and the
overexpression of VEGI in the culture medium was significant.
Hence, the VPA-induced soluble VEGI in OS cells induced HMVE cell
death by binding of VEGI to DR3 in a paracrine manner. HDAC
inhibitors have been reported to inhibit tumor angiogenesis
(7). This process may be in part
mediated by HIF-1α downregulation in both tumor and vascular
endothelial cells and the consequent downregulation of VEGF and
other HIF-1α regulated angiogenesis-related genes (26–29).
We evaluated the effects of VPA and TSA on the VEGFA gene
expression using real-time PCR. Although treatment with VPA and TSA
reduced the VEGFA gene expression in both OS and HMVE cells (data
not shown), the in vitro vascular tube formation assay
showed that the effect of VPA on the HMVE cells was slight or
limited with respect to vascular tube formation. However, the
VPA-treated OS cell culture medium markedly reduced such formation,
suggesting that the anti-angiogenic effect of VPA involves VEGF as
well as modulation via the soluble VEGI/DR3 pathway. In addition,
evidence indicates that the antitumor activity of VEGI is more
likely the result of interference with the development of the
tumor-associated vasculature than the direct effects on tumor cells
(18–20). Our VEGI-overexpression model was
found to exhibit significant OS cell death. Xiao et al,
reported that the oncolytic adenovirus ZD55-VEGI-251 inhibits
angiogenesis via the paracrine actions of VEGI-251 and directly
induces the apoptosis of cancer cells via the autocrine actions of
VEGI-251 (30). Two fragments of
soluble VEGI are produced by ADAM17/10-mediated alternative
cleavage. Soluble TL1AL72-L251 acts mainly in a paracrine manner to
influence cells in the immune system, whereas the TL1AV84-L251
fragment acts via the autocrine pathway to induce the inhibition of
the proliferation and apoptosis of endothelial cells (31). Our previous report indicated that
VPA-treated OS cells do not exhibit increased ADAM17 gene
transcription (32). Hence, the
increase in the expression of soluble VEGI induced by HDAC
inhibitors may involve a different mechanism of action than that
mediating the shedding of membrane-bound VEGI.
In conclusion, VPA has anti-angiogenesis and
simultaneous antitumor activity as a result of enhancing soluble
VEGI/DR3-mediated apoptosis via both autocrine and paracrine
pathways. Targeted inhibition of the HDAC activity is a rational
treatment that is currently in clinical development. Collectively,
modulating the VEGI expression with HDAC inhibitors may eventually
be exploited as a therapeutic strategy in osteosarcoma
patients.
Acknowledgements
This study was supported in part by a Grant-in-Aid
for Young Scientists (B) (23792159) and Grant-in-Aid for Scientific
Research (C) (26462279) from the Ministry of Education, Culture,
Sports, Science, and Technology of Japan, Grantin-Aid for
Researchers, Hyogo College of Medicine, 2012.
References
|
1
|
Messerschmitt PJ, Garcia RM, Abdul-Karim
FW, Greenfield EM and Getty PJ: Osteosarcoma. J Am Acad Orthop
Surg. 17:515–527. 2009.PubMed/NCBI
|
|
2
|
Shen H and Laird PW: Interplay between the
cancer genome and epigenome. Cell. 153:38–55. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kristensen LS, Nielsen HM and Hansen LL:
Epigenetics and cancer treatment. Eur J Pharmacol. 625:131–142.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ferguson LR, Tatham AL, Lin Z and Denny
WA: Epigenetic regulation of gene expression as an anticancer drug
target. Curr Cancer Drug Targets. 11:199–212. 2011. View Article : Google Scholar
|
|
5
|
Rogawski MA and Löscher W: The
neurobiology of antiepileptic drugs. Nat Rev Neurosci. 5:553–564.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bialer M: Why are antiepileptic drugs used
for nonepileptic conditions? Epilepsia. 53(Suppl 7): S26–S33. 2012.
View Article : Google Scholar
|
|
7
|
Dickinson M, Johnstone RW and Prince HM:
Histone deacetylase inhibitors: Potential targets responsible for
their anticancer effect. Invest New Drugs. 28(Suppl 1): S3–S20.
2010. View Article : Google Scholar
|
|
8
|
Tan KB, Harrop J, Reddy M, Young P,
Terrett J, Emery J, Moore G and Truneh A: Characterization of a
novel TNF-like ligand and recently described TNF ligand and TNF
receptor superfamily genes and their constitutive and inducible
expression in hematopoietic and non-hematopoietic cells. Gene.
204:35–46. 1997. View Article : Google Scholar
|
|
9
|
Zhai Y, Ni J, Jiang GW, et al: VEGI, a
novel cytokine of the tumor necrosis factor family, is an
angiogenesis inhibitor that suppresses the growth of colon
carcinomas in vivo. FASEB J. 13:181–189. 1999.PubMed/NCBI
|
|
10
|
Parr C, Gan CH, Watkins G and Jiang WG:
Reduced vascular endothelial growth inhibitor (VEGI) expression is
associated with poor prognosis in breast cancer patients.
Angiogenesis. 9:73–81. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chew LJ, Pan H, Yu J, Tian S, Huang WQ,
Zhang JY, Pang S and Li LY: A novel secreted splice variant of
vascular endothelial cell growth inhibitor. FASEB J. 16:742–744.
2002.PubMed/NCBI
|
|
12
|
Migone TS, Zhang J, Luo X, Zhuang L, Chen
C, Hu B, Hong JS, Perry JW, Chen S-F and Zhou JXH: TL1A is a
TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell
costimulator. Immunity. 16:479–492. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Grimaldo S, Tian F and Li LY:
Sensitization of endothelial cells to VEGI-induced apoptosis by
inhibiting the NF-kappaB pathway. Apoptosis. 14:788–795. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ge Z, Sanders AJ, Ye L and Jiang WG:
Aberrant expression and function of death receptor-3 and death
decoy receptor-3 in human cancer. Exp Ther Med. 2:167–172.
2011.PubMed/NCBI
|
|
15
|
Liang PH, Tian F, Lu Y, Duan B, Stolz DB
and Li LY: Vascular endothelial growth inhibitor (VEGI; TNFSF15)
inhibits bone marrow-derived endothelial progenitor cell
incorporation into Lewis lung carcinoma tumors. Angiogenesis.
14:61–68. 2011. View Article : Google Scholar :
|
|
16
|
Chinnaiyan AM, O’Rourke K, Yu GL, Lyons
RH, Garg M, Duan DR, Xing L, Gentz R, Ni J and Dixit VM: Signal
transduction by DR3, a death domain-containing receptor related to
TNFR-1 and CD95. Science. 274:990–992. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang CR, Hsieh SL, Teng CM, Ho FM, Su WL
and Lin WW: Soluble decoy receptor 3 induces angiogenesis by
neutralization of TL1A, a cytokine belonging to tumor necrosis
factor superfamily and exhibiting angiostatic action. Cancer Res.
64:1122–1129. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hou W, Medynski D, Wu S, Lin X and Li LY:
VEGI-192, a new isoform of TNFSF15, specifically eliminates tumor
vascular endothelial cells and suppresses tumor growth. Clin Cancer
Res. 11:5595–5602. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Z and Li LY: TNFSF15 modulates
neovascularization and inflammation. Cancer Microenviron.
5:237–247. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xiao Q, Hsu CY, Chen H, Ma X, Xu J and Lee
JM: Characterization of cis-regulatory elements of the vascular
endothelial growth inhibitor gene promoter. Biochem J. 388:913–920.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Marks PA and Xu WS: Histone deacetylase
inhibitors: Potential in cancer therapy. J Cell Biochem.
107:600–608. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bai C, Connolly B, Metzker ML, et al:
Overexpression of M68/DcR3 in human gastrointestinal tract tumors
independent of gene amplification and its location in a four-gene
cluster. Proc Natl Acad Sci USA. 97:1230–1235. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takahama Y, Yamada Y, Emoto K, Fujimoto H,
Takayama T, Ueno M, Uchida H, Hirao S, Mizuno T and Nakajima Y: The
prognostic significance of overexpression of the decoy receptor for
Fas ligand (DcR3) in patients with gastric carcinomas. Gastric
Cancer. 5:61–68. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ge Z, Sanders AJ, Ye L, Wang Y and Jiang
WG: Expression of death decoy receptor-3 (DcR3) in human breast
cancer and its functional effects on breast cancer cells in vitro.
J Exp Ther Oncol. 9:109–118. 2011.PubMed/NCBI
|
|
25
|
Zong L, Chen P and Wang DX: Death decoy
receptor overexpression and increased malignancy risk in colorectal
cancer. World J Gastroenterol. 20:4440–4445. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Qian DZ, Kachhap SK, Collis SJ, Verheul
HM, Carducci MA, Atadja P and Pili R: Class II histone deacetylases
are associated with VHL-independent regulation of hypoxia-inducible
factor 1 alpha. Cancer Res. 66:8814–8821. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kong X, Lin Z, Liang D, Fath D, Sang N and
Caro J: Histone deacetylase inhibitors induce VHL and
ubiquitin-independent proteasomal degradation of hypoxia-inducible
factor 1alpha. Mol Cell Biol. 26:2019–2028. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kang FW, Que L, Wu M, Wang ZL and Sun J:
Effects of trichostatin A on HIF-1α and VEGF expression in human
tongue squamous cell carcinoma cells in vitro. Oncol Rep.
28:193–199. 2012.PubMed/NCBI
|
|
29
|
Zhao Y, Yu D, Wu H, Liu H, Zhou H, Gu R,
Zhang R, Zhang S and Wu G: Anticancer activity of SAHA, a potent
histone deacetylase inhibitor, in NCI-H460 human large-cell lung
carcinoma cells in vitro and in vivo. Int J Oncol. 44:451–458.
2014.
|
|
30
|
Xiao T, Fan JK, Huang HL, Gu JF, Li LY and
Liu XY: VEGI-armed oncolytic adenovirus inhibits tumor
neovascularization and directly induces mitochondria-mediated
cancer cell apoptosis. Cell Res. 20:367–378. 2010. View Article : Google Scholar
|
|
31
|
Mück C, Herndler-Brandstetter D, Micutkova
L, Grubeck-Loebenstein B and Jansen-Dürr P: Two functionally
distinct isoforms of TL1A (TNFSF15) generated by differential
ectodomain shedding. J Gerontol A Biol Sci Med Sci. 65:1165–1180.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yamanegi K, Yamane J, Kobayashi K, et al:
Downregulation of matrix metalloproteinase-9 mRNA by valproic acid
plays a role in inhibiting the shedding of MHC class I-related
molecules A and B on the surface of human osteosarcoma cells. Oncol
Rep. 28:1585–1590. 2012.PubMed/NCBI
|