The phosphatidylinositol 3-kinase (PI3K)/Akt
signaling pathway plays a pivot role in the development and
survival of cancers. The upstream regulators and downstream
effectors of this pathway consists of a complex cellular signaling
network including crosstalk, feedback loops, and branch points that
controls a variety of cellular processes and functions. One of the
essential downstream signaling pathways of Akt is the c-Jun
N-terminal kinase (JNK) signaling pathway, which belongs to a
subgroup of mitogen-activated protein kinase (MAPK) signaling
pathways. The interaction between PI3K/Akt and JNK pathways is
complicated due to the dual roles of JNK signaling in apoptosis.
The interaction between these two pathways may determine the fate
of the cell: survival or apoptosis.
In this review, we first summarized the functional
characteristics, signal transduction and activation mechanisms of
the PI3K/Akt and JNK pathways, and then discussed the dual roles of
JNK pathway in apoptosis and tumor development. Upon this
background, the interaction between these two signaling pathways
was mapped to determine the potential targets and combination
therapeutic strategies for cancer treatment.
PI3Ks are lipid kinases involved in a variety of
biological processes, such as cell proliferation, differentiation,
motility, survival and angiogenesis (1,2).
PI3Ks are activated by receptor tyrosine kinases (RTKs) or
G-protein-coupled receptors (GPCRs). RTKs include a variety of cell
surface receptors that interact with growth factors, cytokines, or
hormones including insulin, epidermal growth factor (EGF),
platelet-derived growth factor (PDGF), and insulin-like growth
factor-1 (IGF-1). Under the circumstance of ligand stimulation,
RTKs undergo autophosphorylation, providing binding sites for PI3K
regulatory subunits which subsequently lead to PI3K activation.
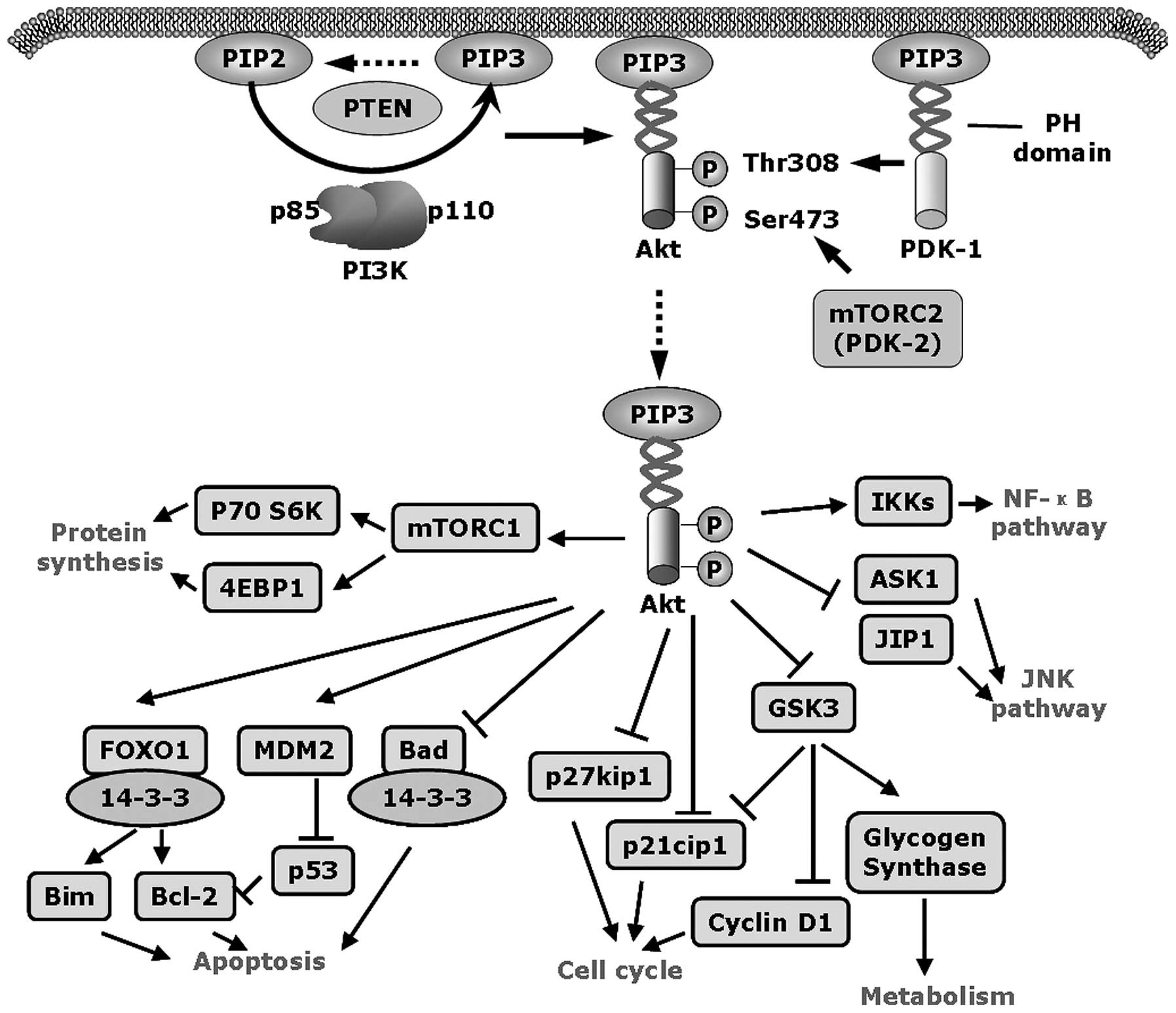
Activated PI3K phosphorylates the lipid phosphatidylinositol
4,5-bisphosphate [PtdIns(4,5)P2, PIP2] to generate
phosphatidylinositol 3,4,5-triphosphate [PtdIns(3,4,5)
P3, PIP3], which then binds to the Pleckstrin homology
(PH) domain of Akt and recruits it to the plasma membrane (Fig. 1). PIP3 also engages
phosphoinositide-dependent kinase-1 (PDK-1) to plasma membrane
through binding to its PH domain. PDK-1 then phosphorylates Akt at
residue threonine 308 (Thr308) in the activation loop to partially
activate Akt (3,4). Subsequently Akt is completely
activated through phosphorylation at serine 473 (Ser473) in the
hydrophobic motif by mTOR complex 2 (mTORC2) (5). Activated Akt and PDK-1 can
phosphorylate a number of downstream proteins such as mTOR,
glycogen synthase kinase 3 (GSK3), Bcl-2-associated death promoter
(Bad) and forkhead box protein O1 (FOXO1) to regulate cell growth,
motility, survival and metabolism (Fig. 1) (6).
Phosphatase and tensin homolog deleted on chromosome
10 (PTEN) is a tumor suppressor with lipid phosphatase and tyrosine
phosphatase activities. Wild-type PTEN is able to dephosphorylate
PIP3 back to PIP2, and interfere with the recruitment of Akt to the
plasma membrane, resulting in reduced Akt activation (7,8).
PTEN locates on chromosome 10q23, which is highly
susceptible to mutation in human cancers. Mutation or loss of
function of PTEN is frequent in a wide range of cancers
including glioblastoma multiforme (9), gastric cancer (10), endometrial cancer (11,12),
ovarian cancer (12) and lung
cancer (13). PTEN can decrease
the synthesis of IGF-I, -II and IGF-1R, which in turn generates an
autocrine loop to downregulate Akt activation (14,15).
Therefore, high Akt phosphorylation is frequently associated with
loss of PTEN, leading to chemorestistance and poor prognosis
in cancer patients (16,17). Convincing evidence shows that PTEN
inactivation confers higher EGFR phosphorylation in tumor cells and
their resistance to EGFR specific inhibitors (18). Wild-type PTEN promotes the
ubiquitination of activated EGFR through PIP3 and Akt-dependent
mechanism (18). These findings
indicate that PTEN is a key element of PI3K/Akt pathway and acts as
an important tumor suppressor through suppressing PI3K/Akt
signaling.
JNK, also known as stress-activated protein kinase
(SAPK), can be activated in response to a number of environmental
challenges including UV irradiation, heat shock, toxins, as well as
cytokines and growth factors (19). In mammals, JNK is encoded by three
genes (JNK1, JNK2 and JNK3), which are located on
three different chromosomes. Among these three genes, JNK1
and JNK2 are ubiquitously expressed, while JNK3 is
largely restricted to the brain, heart and testis (20). This family has at least ten
isoforms: JNK1 (four isoforms), JNK2 (four isoforms)
and JNK3 (two isoforms), which are generated by the
alternative splicing of the mRNA 3′-coding region. These JNK
isoforms are expressed as 54 kDa and 46 kDa proteins that recognize
and interact with different substrates. Although the function of
JNK splice variants is not clear so far, it is found that
JNK-interacting protein 1 (JIP1) can augment the expression of 46
kDa of JNK splice variants and increase the stability of JNK-JIP1
module (21). It indicates that
JNK variants have different affinity to the scaffold proteins,
which may result in the JNK variants recognizing different
substrates. JNK is a member of an evolutionarily conserved
sub-family of mitogen-activated protein kinases (MAPKs). In
mammals, several members of MAPK family have been identified,
including extracellular signal-regulated kinases (ERKs), SAPK/JNK,
the 38-kDa protein kinases (p38), and ERK5/ big mitogen activated
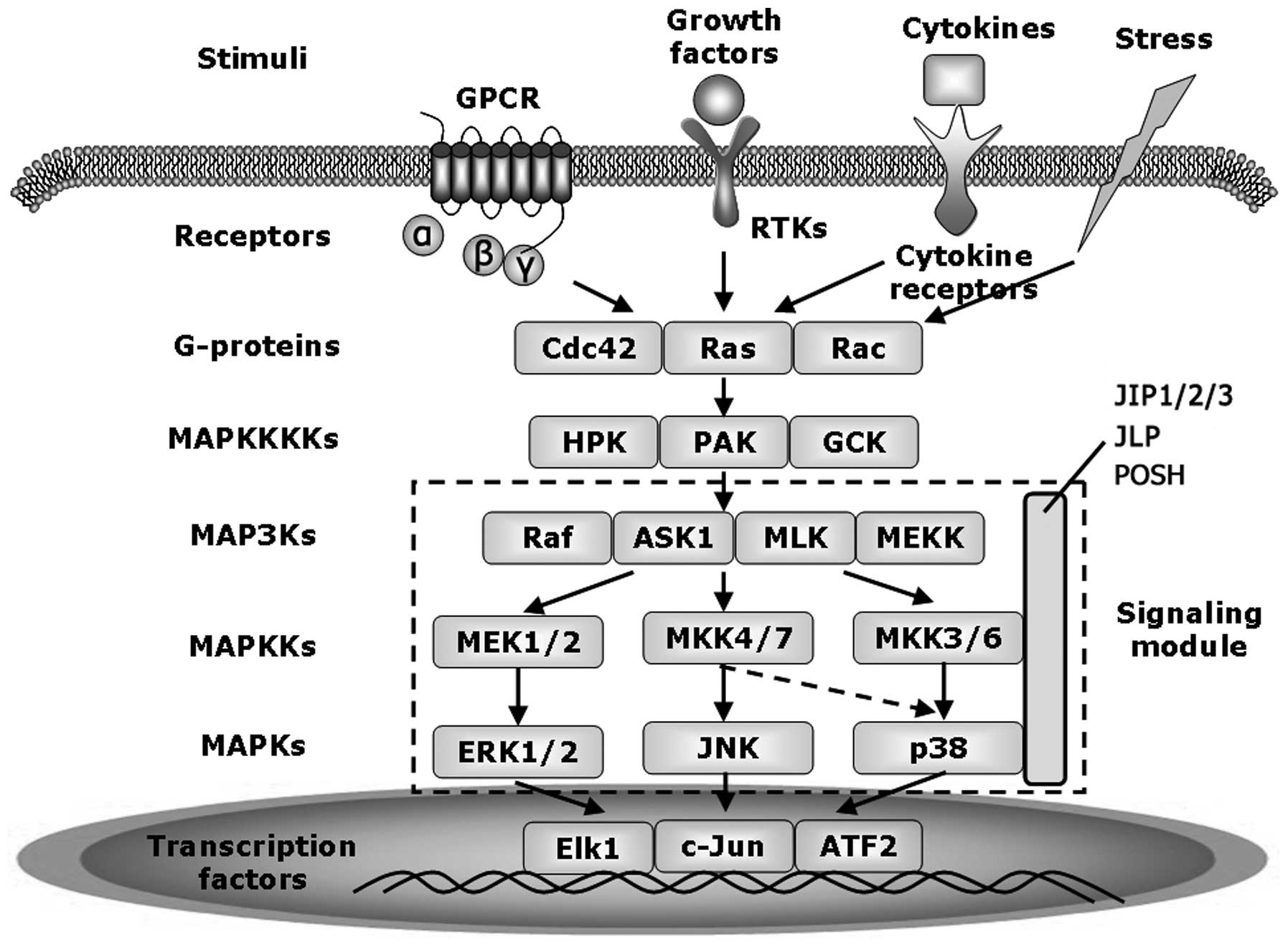
protein kinase 1 (BMK1). Generally, activation of MAPK signaling
pathways starts with receptor activation, leading to the
recruitment of adaptor proteins and activation of small GTP-binding
proteins (G-proteins) (22). The
protein kinase cascade that consists of up to four tiers of kinases
is subsequently activated. The MAPKKKKs phosphorylate and activate
the MAPKKKs (MAP3Ks or MEKKs) which, in turn, phosphorylate and
activate the MAPKKs (or MEKs). MAPKKs then phosphorylate and
activate the MAPKs, which phosphorylate and interact with their
specific substrates (Fig. 2)
(23).
JNK activation is mediated by two upstream MAPKKs:
mitogen-activated protein kinase kinase 4 (MKK4) (also known as
SAPK/ERK kinase 1, SEK1) and MKK7. MKK4 and MKK7 preferentially
phosphorylate JNK at tyrosine and threonine residues respectively,
leading to full activation of JNK (Fig. 2) (24,25).
MAP3Ks that activate MKK4 and MKK7 in JNK signaling pathway,
include the Raf kinase (26),
apoptosis signal-regulating kinase-1 (ASK1) (27), MAPK kinase kinase 1/4 (MEKK1/4)
(28) and mixed-lineage kinase
(MLK) (29). MAP3Ks are activated
by Rho family of GTPases, a family of small signaling G proteins
(~21 kDa) including the cell division control protein 42 (CDC42),
Rac, Ras and Ras homolog gene family member A (RhoA) (30). Activated JNK can translocate into
the nucleus and phosphorylate a variety of transcription factors
including c-Jun, JunB, JunD, ATF2, STAT3 and p53 (Fig. 2). The first identified substrate of
JNK is c-Jun, of which the activation and stability are mediated by
phosphorylation of Ser63 and Ser73 in the N-terminal region.
Further, phosphorylated c-Jun can interact with c-Fos to form
activator protein-1 (AP-1) complex, which can specifically bind to
the promoter or enhancer of numerous genes to mediate their
transcriptional activity (31).
Several proteins such as JNK interacting protein
(JIP), JNK-interacting leucine zipper protein (JLP), and plenty of
SH3 (POSH) have been identified as scaffold proteins that interact
with specific member of JNK signaling cascade and form a module to
facilitate signaling transduction (Fig. 2). The first identified scaffold
protein in JNK signaling pathway is JIP1, which only interacts
directly with SAPK/JNK, rather than other MAPKs, p38 and ERK
(32). JIP1 recruits JNK, MKK7,
MLK, and haematopoietic progenitor kinase-1 (HPK1) to form the
signaling module (33). The
activation of the JNK module requires the phosphorylation of JIP1
by JNK (34). However,
overexpression of JIP-1 could retain JNK in the cytoplasm and
prevent the activation of c-Jun and activating transcription factor
2 (ATF2), indicating that excess JIP1 may decrease JNK activity
through a negative feedback loop (32).
The JNK signaling pathway controls a variety of
cellular events including cell proliferation, development,
inflammation, and apoptosis. JNK can act as a pro-apoptotic or
anti-apoptotic molecule depending on the circumstance. Evidence
shows that apoptosis is mediated by JNK signaling in a
stimulus-dependent manner (35,36).
Numerous studies have demonstrated that the MLK/JNK/c-Jun axis
promotes apoptosis not only in sympathetic neurones (37,38),
but also in cancer cells (39–41).
Knockdown of dual leucine zipper-bearing kinase (DLK), a member of
the MLK family, is able to suppress JNK phosphorylation and
poly-ADP ribose polymerase (PARP) cleavage, leading to inhibition
of calphostin C-induced apoptosis in human breast cancer cells
(40). The JNK inhibitor SP600125
inhibits the phosphorylation of BCL2-associated X protein (Bax) and
prevents the translocation of JNK into mitochondria, resulting in
suppression of stress-induced apoptosis in human hepatoma cells
(42). In addition, JNK is able to
promote apoptosis by regulating p53 upregulated modulator of
apoptosis (PUMA) (43,44). PUMA is a pro-apoptotic BH3-only
protein that promotes stress- and growth factor-induced apoptosis
in either p53-dependent or -independent manner, through direct
interaction with B-cell lymphoma 2 (Bcl-2) family members and
antagonizing the anti-apoptotic effect of Bcl-2 family (45). JNK effectively enhances PUMA
activation and apoptosis induced by betulinic acid in
cisplatin-resistant ovarian cancer cells (44). Recent studies also found that p53
and its homologue p73 are the substrates of JNK and can be
phosphorylated by JNK (46,47).
Phosphorylated p53 or p73 subsequently binds to the promoter of
PUMA and regulates its expression. These findings indicate
that JNK may be a critical upstream regulator of PUMA and plays a
pro-apoptotic role in cancer cells in the context of a wide variety
of stimuli including genotoxic stress, cytokines and growth
factors.
Activation of JNK is also involved in cell survival
and anti-apoptosis in both Fas- and mitochondria-dependent manner
(48,49). Evidence shows that thymocytes and
peripheral T cells from MKK4−/− mice are
more susceptible to CD95 (Fas)- and CD3-mediated apoptosis,
indicating MKK4-induced JNK activation participates in the survival
of T-cell (49). Expression of a
constitutively active JNK mutant suppresses pro-B cell apoptosis
induced by IL-3 withdrawal. JNK phosphorylates BAD at Thr201, and
prohibits BAD from interacting with Bcl-xL, resulting in decreased
apoptosis (48). In addition,
downregulation of JNK2 expression induces significant apoptosis in
a variety of p53-deficient cancer cell lines (50).
The JNK pathway also plays different roles in tumor
development. A number of studies report that JNK is required for
tumor formation and development. Evidence shows that both JNK1 and
JNK2 are required for in vitro Ras-induced cellular
transformation and in vivo tumor formation of lung cancer,
which is correlated with increased c-Jun and AP-1 activities
(51–53). JNK2 can phosphorylate ATF-2 and
further protect c-Myc from proteasomal degradation during
Ras-induced transformation in mouse embryonic fibroblasts (MEFs)
(54). Furthermore, c-Jun is also
required for Ras-induced transformation. The
c-Jun−/− fibroblasts do not show
transformation induced by Ras, which can be reversed by
overexpression of wild-type c-Jun (55). These findings indicate that
JNK/c-Jun axis is essential in the Ras-induced transformation. The
evidence that JNK pathway promotes cellular transformation supports
that JNK signaling is also essential in tumor development.
Recently, constitutively active isoforms of JNKs have been found in
various cancers including gastric cancer (56), hepatocellular carcinoma (57), breast cancer (58) and glioma (59). Using an ATP-competitive JNK
inhibitor SP600125, the growth of pancreatic cancer in vitro
and in vivo were suppressed, and the survival of mice was
also prolonged (60). In addition,
decreased DNA damage and replicative stress response are found in
JNK2−/− mammary tumor mice, and
JNK2−/− mice exhibit shorter latency and
higher tumor multiplicity than wild-type JNK2, indicating
that JNK2 is required for tumor development and genetic stability
(61).
Supporting evidence shows that deficiency of both
JNK1 and JNK2 in hepatocytes promotes diethylnitrosamine
(DEN)-induced hepatocellular carcinoma (HCC) development in mice,
which may result from the anti-apoptotic role of JNK. Deficiency of
JNK causes increased apoptosis of hepatocytes, leading to increased
compensatory proliferation that contributes to HCC development. On
the contrary, deficiency of JNK in nonparenchymal cells suppresses
HCC development by increasing the expression of cytokines IL-6 and
TNF-α to provide an inflammatory environment (62). It indicates that the role of JNK in
tumor development is cell type-dependent.
Since the JNK pathway can also act a pro-apoptotic
role in cancer, JNK may be considered as a tumor suppressor. Loss
of JNK expression increases the number and growth of tumor nodules
in vivo and induces the transformed phenotype of fibroblasts
in vitro. Besides, JNK-null cells display less Ras-induced
apoptosis than cells with wild-type JNK (63). Other studies also show that
inactivation of JNK in the prostate epithelium results in rapid
development of invasive adenocarcinoma in vivo (64). The JNK3 is considered as the
tumor suppressor gene. Loss of JNK3 gene is found in a
variety of cancer cell lines including brain tumor (10 of 19),
non-Hodgkin’s lymphoma (15 of 16), Hodgkin’s lymphoma (3 of 6),
breast cancer (3 of 10), gastric cancer (6 of 10), and
hepatocellular carcinoma (8 of 12) (65,66).
Moreover, activation of JNK3, rather than JNK1 and JNK2, induces
mitochondrial dysfunction and promotes TNF-α-induced apoptosis in
human oligodendrocytes, suggesting a possible mechanism for the
tumor suppressor role of JNK3 (67).
Since the PI3K/Akt pathway plays essential roles in
tumor transformation, development and progression, the dual roles
of JNK signaling in apoptosis and tumor development may determine
different crosstalk between PI3K/Akt and JNK pathways. Increasing
evidence reveals that these two signaling pathways interact with
each other and consist of a regulatory network. The scaffold
protein JIP1, which is essential for formation and activation of
JNK module, can directly bind to the PH domain of Akt1, leading to
the formation of Akt1-JIP1 complex facilitating the activation of
Akt1 via PDK1 (68–70). Moreover, increasing interaction
between JIP1 and Akt1 leads to release of JNK from JIP-JNK module
and inhibition of JNK activity (70,71).
Overexpression of wild-type or constitutively active Akt1 also
inhibits JNK activity, especially JNK2, and decreases apoptosis in
a variety of cells such as normal neurons, 293T, PC12 and T cells
(70–73). On the contrary, since the
activation of Akt requires the activities of PI3K or RTKs,
withdrawal of RTK ligands such as insulin and IGF-1, or PI3K
inhibitor treatment promotes JNK activation by dephosphorylating
Akt and increasing the interaction between JIP1 and JNK (69,71–74).
Apart from interacting with JIP1, Akt can bind to
ASK1 and phosphorylate ASK1 at Ser83 (75,76).
ASK1 is a member of MAP3Ks and serves as an upstream activator of
JNK. Phosphorylation of Akt inhibits the oxidative stress-induced
activation of ASK1 in human embryonic kidney 293T cells, leading to
reduced apoptosis (76). In
addition, IGF-1 stimulation suppresses the activation of ASK1/JNK
induced by serum deprivation or cytokines through activation of
PI3K/Akt signaling, whereas PI3K inhibitors reverse the inhibitory
effect (76,77). Recent evidence indicates that
disabled-2 interacting protein (DAB2IP) is a scaffold protein
bridging both Akt and ASK1 via different domains, and its
overexpression suppresses Akt signaling but activates ASK1/JNK,
leading to enhanced TNF-α-induced apoptosis in prostate cancer
cells (78). Besides, MKK4 is also
a substrate of Akt in intact cells. Activated Akt induced by
insulin is able to phosphorylate MKK4 at Ser78 and inhibit the
activation of MKK4 and JNK in 293T cells, leading to prolonged cell
survival (79). In addition,
angiopoi-etin-1-induced Akt activation also phosphorylates MKK4 at
Ser80 and suppress apoptosis and oxidative stress-induced JNK
activation in vascular endothelial cells (80). Furthermore, Akt interacts with and
phosphorylate MLK3 on Ser674 to inhibit its kinase activity
(81,82). Insulin-induced phosphorylation of
Akt concomitant with reduced kinase activities of MLK3, MKK7 and
JNK is observed in human hepatoma HepG2 cells, indicating that
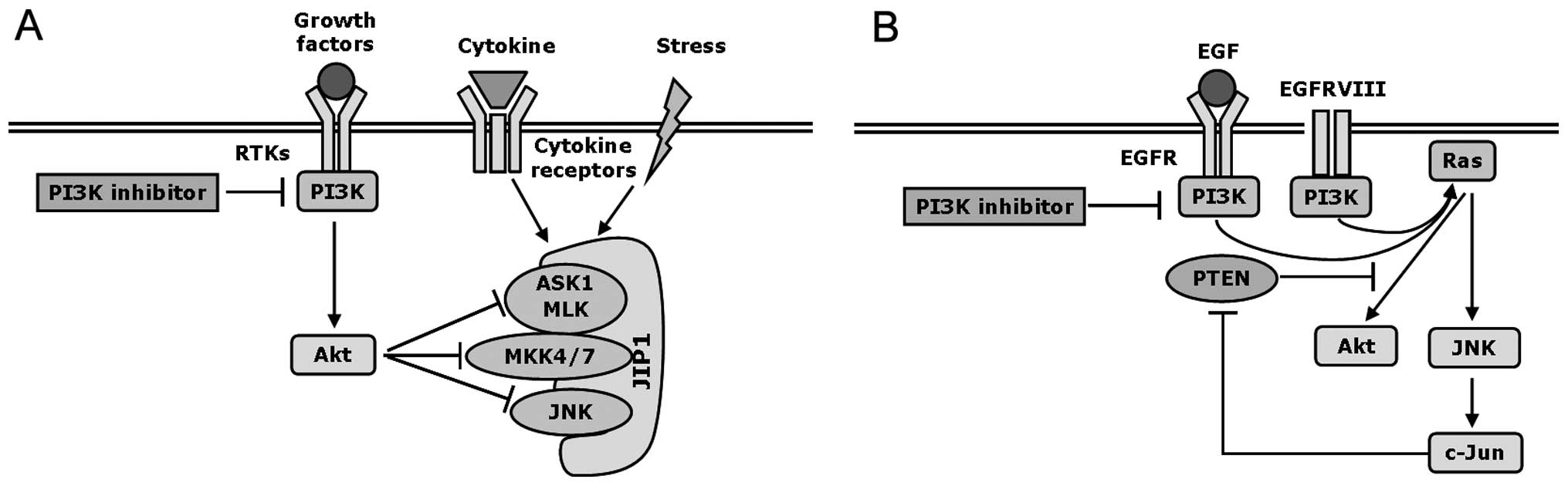
activation of Akt antagonizes MLK3-MKK7-JNK signaling (81). Thus, JNK signaling plays a
pro-apoptotic role, which is antagonized by Akt activation under
stimuli including stress, toxin and cytokines (Fig. 3A).
Interestingly, Song and Lee found that PI3K/Akt
signaling inhibits glucose deprivation induced-activation of JNK
through antagonizing JIP1-MKK4-JNK cascade in human prostate
carcinoma cells (83). However,
activated JNK2 phosphorylates JIP1 at Thr103, leading to increased
interaction between JIP1 and JNK2, and disassociation of Akt1 from
JIP1. Subsequently, the disassociated Akt1 binds to MKK4 and
inhibits its activity. The inhibition of MKK4 in turn suppresses
the activation of JNK, leading to the formation of a negative
regulatory feedback loop (83).
JNK preferentially takes on a pro-apoptosis role in
response to a variety of stimuli including stress, toxin and
cytokines. Activation of PI3K/Akt signaling inhibits the JNK
activity and leads to decreased apoptosis, which is consistent with
the finding that PI3K/ Akt signaling is positively correlated with
survival. However, a number of studies show that activation of Akt
or PI3K is frequently accompanied by JNK activation in cancers
including glioblastoma, cervical carcinoma and prostate cancer,
indicating that JNK may play an anti-apoptotic role under certain
circumstances (84–86). One possible explanation is that JNK
is constitutively active in some types of cancers. Reports show
that JNK2 isoforms are constitutively active in glioblastoma and
non-small cell lung carcinoma, and a positive correlation between
elevated JNK activity and higher histological grade is found
(86–89). If JNK plays a pro-apoptotic role in
apoptosis induced by extracellular stimuli, the constitutively
activated JNK may served as an anti-apoptotic protein contributing
to the survival of cancer cells. Another possible explanation is
that activation of both Akt and JNK signaling can be induced by
EGF/EGFR in cancers, which is correlated with the loss of
PTEN. Overexpression of wild-type EGFR or EGFRVIII (a
mutated variant of EGFR with constitutive activity), as well as EGF
stimulation activate both PI3K/Akt and JNK signaling pathways in
various cancer cell lines (90–92).
Aberrant expression of EGFR or EGFRVIII in
PTEN−/− glioblastoma cells induces
activation of JNK and transcription factor JunD/ AP-1, as well as
activation of Akt. Restoration of PTEN, which antagonizes PI3K
activity, diminishes activities of Akt and JNK (91). Using the PI3K inhibitor wortmannin
or a dominant-negative mutant of PI3K, Akt phosphorylation is
inhibited in HeLa cells, and the activation of JNK induced by EGF
is also suppressed, whereas no effect is observed on the JNK
activity induced by UV or osmotic stress (84). These findings suggest that EGF/EGFR
and constitutively active EGFRVIII are the upstream activators of
Akt and JNK signaling in cancers (Fig.
3B).
The molecular mechanism of PI3K/Akt and JNK pathway
activation is now well understood. Genetic alternations frequently
occur within PI3K/Akt signaling pathway in cancers, leading to the
constitutively active PI3K/Akt signaling, and then promoting cell
proliferation, survival, migration and invasion through interacting
with downstream effectors. JNK signaling pathway is one of the
downstream pathways of PI3K/Akt signaling, and plays dual roles in
apoptosis in a stimulus-dependent manner. The specific role that
JNK takes on, in apoptosis determines its involvement in tumor
development, as well as different interplays between these two
pathways. Activation of PI3K/Akt signaling can inhibit stress- and
cytokine-induced JNK activation through Akt antagonizing the
formation of the JIP1-JNK module, as well as the activities of
upstream kinases ASK1, MKK4/7 and MLK. On the other hand,
activation of JNK pathway is concomitant with activation of
PI3K/Akt pathway in cancer in the context of EGFR overexpression
stimulation or PTEN deficiency. Hence, the interaction between
PI3K/Akt and JNK pathways provides potential targets and
therapeutic strategies for cancer treatment. Inhibitors aimed at
PI3K/Akt and JNK pathways simultaneously may have synergistic
positive effects on improving the survival and prognosis of cancer
patients, which should be further investigated.
This study was supported by a Central Research Grant
from the Hong Kong Polytechnic University (RTH4).
|
1
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Katso R, Okkenhaug K, Ahmadi K, White S,
Timms J and Waterfield MD: Cellular function of phosphoinositide
3-kinases: Implications for development, homeostasis, and cancer.
Annu Rev Cell Dev Biol. 17:615–675. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vanhaesebroeck B and Alessi DR: The
PI3K-PDK1 connection: More than just a road to PKB. Biochem J.
346:561–576. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Toker A and Newton AC: Cellular signaling:
Pivoting around PDK-1. Cell. 103:185–188. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sarbassov DD, Ali SM and Sabatini DM:
Growing roles for the mTOR pathway. Curr Opin Cell Biol.
17:596–603. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sun H, Lesche R, Li DM, Liliental J, Zhang
H, Gao J, Gavrilova N, Mueller B, Liu X and Wu H: PTEN modulates
cell cycle progression and cell survival by regulating
phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B
signaling pathway. Proc Natl Acad Sci USA. 96:6199–6204. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Song MS, Salmena L and Pandolfi PP: The
functions and regulation of the PTEN tumour suppressor. Nat Rev Mol
Cell Biol. 13:283–296. 2012.PubMed/NCBI
|
|
9
|
Srividya MR, Thota B, Shailaja BC,
Arivazhagan A, Thennarasu K, Chandramouli BA, Hegde AS and Santosh
V: Homozygous 10q23/ PTEN deletion and its impact on outcome in
glioblastoma: A prospective translational study on a uniformly
treated cohort of adult patients. Neuropathology. 31:376–383. 2011.
View Article : Google Scholar
|
|
10
|
Chong ML, Loh M, Thakkar B, Pang B,
Iacopetta B and Soong R: Phosphatidylinositol-3-kinase pathway
aberrations in gastric and colorectal cancer: Meta-analysis,
co-occurrence and ethnic variation. Int J Cancer. 134:1232–1238.
2014. View Article : Google Scholar
|
|
11
|
Garcia-Dios DA, Lambrechts D, Coenegrachts
L, Vandenput I, Capoen A, Webb PM, Ferguson K, Akslen LA, Claes B,
Vergote I, et al; ANECS. High-throughput interrogation of PIK3CA,
PTEN, KRAS, FBXW7 and TP53 mutations in primary endometrial
carcinoma. Gynecol Oncol. 128:327–334. 2013. View Article : Google Scholar
|
|
12
|
McConechy MK, Ding J, Senz J, Yang W,
Melnyk N, Tone AA, Prentice LM, Wiegand KC, McAlpine JN, Shah SP,
et al: Ovarian and endometrial endometrioid carcinomas have
distinct CTNNB1 and PTEN mutation profiles. Mod Pathol. 27:128–134.
2014. View Article : Google Scholar :
|
|
13
|
Jin G, Kim MJ, Jeon HS, Choi JE, Kim DS,
Lee EB, Cha SI, Yoon GS, Kim CH and Jung TH: PTEN mutations and
relationship to EGFR, ERBB2, KRAS, and TP53 mutations in non-small
cell lung cancers. Lung Cancer. 69:279–283. 2010. View Article : Google Scholar
|
|
14
|
Kang-Park S and Lee YI and Lee YI: PTEN
modulates insulin-like growth factor II (IGF-II)-mediated
signaling; the protein phosphatase activity of PTEN downregulates
IGF-II expression in hepatoma cells. FEBS Lett. 545:203–208. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yi HK, Kim SY, Hwang PH, Kim CY, Yang DH,
Oh Y and Lee DY: Impact of PTEN on the expression of insulin-like
growth factors (IGFs) and IGF-binding proteins in human gastric
adenocarcinoma cells. Biochem Biophys Res Commun. 330:760–767.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oki E, Baba H, Tokunaga E, Nakamura T,
Ueda N, Futatsugi M, Mashino K, Yamamoto M, Ikebe M, Kakeji Y, et
al: Akt phosphorylation associates with LOH of PTEN and leads to
chemoresistance for gastric cancer. Int J Cancer. 117:376–380.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tang JM, He QY, Guo RX and Chang XJ:
Phosphorylated Akt overexpression and loss of PTEN expression in
non-small cell lung cancer confers poor prognosis. Lung Cancer.
51:181–191. 2006. View Article : Google Scholar
|
|
18
|
Vivanco I, Rohle D, Versele M, Iwanami A,
Kuga D, Oldrini B, Tanaka K, Dang J, Kubek S, Palaskas N, et al:
The phosphatase and tensin homolog regulates epidermal growth
factor receptor (EGFR) inhibitor response by targeting EGFR for
degradation. Proc Natl Acad Sci USA. 107:6459–6464. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weston CR and Davis RJ: The JNK signal
transduction pathway. Curr Opin Cell Biol. 19:142–149. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bode AM and Dong Z: The functional
contrariety of JNK. Mol Carcinog. 46:591–598. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang JY, Moulin N, van Bemmelen MX, Dubuis
G, Tawadros T, Haefliger JA, Waeber G and Widmann C: Splice
variant-specific stabilization of JNKs by IB1/JIP1. Cell Signal.
19:2201–2207. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barr RK and Bogoyevitch MA: The c-Jun
N-terminal protein kinase family of mitogen-activated protein
kinases (JNK MAPKs). Int J Biochem Cell Biol. 33:1047–1063. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tournier C, Dong C, Turner TK, Jones SN,
Flavell RA and Davis RJ: MKK7 is an essential component of the JNK
signal transduction pathway activated by proinflammatory cytokines.
Genes Dev. 15:1419–1426. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Haeusgen W, Herdegen T and Waetzig V: The
bottleneck of JNK signaling: Molecular and functional
characteristics of MKK4 and MKK7. Eur J Cell Biol. 90:536–544.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Leicht DT, Balan V, Kaplun A, Singh-Gupta
V, Kaplun L, Dobson M and Tzivion G: Raf kinases: Function,
regulation and role in human cancer. Biochim Biophys Acta.
1773:1196–1212. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ichijo H, Nishida E, Irie K, ten Dijke P,
Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K and Gotoh
Y: Induction of apoptosis by ASK1, a mammalian MAPKKK that
activates SAPK/JNK and p38 signaling pathways. Science. 275:90–94.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun BK, Kim JH, Nguyen HN, Oh S, Kim SY,
Choi S, Choi HJ, Lee YJ and Song JJ: MEKK1/MEKK4 are responsible
for TRAIL-induced JNK/p38 phosphorylation. Oncol Rep. 25:537–544.
2011.
|
|
29
|
Xu Z, Maroney AC, Dobrzanski P, Kukekov NV
and Greene LA: The MLK family mediates c-Jun N-terminal kinase
activation in neuronal apoptosis. Mol Cell Biol. 21:4713–4724.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lopez-Ilasaca M: Signaling from
G-protein-coupled receptors to mitogen-activated protein
(MAP)-kinase cascades. Biochem Pharmacol. 56:269–277. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shaulian E: AP-1 - The Jun proteins:
Oncogenes or tumor suppressors in disguise? Cell Signal.
22:894–899. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Whitmarsh AJ, Cavanagh J, Tournier C,
Yasuda J and Davis RJ: A mammalian scaffold complex that
selectively mediates MAP kinase activation. Science. 281:1671–1674.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Whitmarsh AJ, Kuan CY, Kennedy NJ, Kelkar
N, Haydar TF, Mordes JP, Appel M, Rossini AA, Jones SN, Flavell RA,
et al: Requirement of the JIP1 scaffold protein for stress-induced
JNK activation. Genes Dev. 15:2421–2432. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nihalani D, Meyer D, Pajni S and Holzman
LB: Mixed lineage kinase-dependent JNK activation is governed by
interactions of scaffold protein JIP with MAPK module components.
EMBO J. 20:3447–3458. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lin A: Activation of the JNK signaling
pathway: Breaking the brake on apoptosis. BioEssays. 25:17–24.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lin A and Dibling B: The true face of JNK
activation in apoptosis. Aging Cell. 1:112–116. 2002. View Article : Google Scholar
|
|
37
|
Besirli CG and Johnson EM Jr:
JNK-independent activation of c-Jun during neuronal apoptosis
induced by multiple DNA-damaging agents. J Biol Chem.
278:22357–22366. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huntwork-Rodriguez S, Wang B, Watkins T,
Ghosh AS, Pozniak CD, Bustos D, Newton K, Kirkpatrick DS and
Lewcock JW: JNK-mediated phosphorylation of DLK suppresses its
ubiquitination to promote neuronal apoptosis. J Cell Biol.
202:747–763. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Reno EM, Haughian JM, Jackson TA, Thorne
AM and Bradford AP: c-Jun N-terminal kinase regulates apoptosis in
endometrial cancer cells. Apoptosis. 14:809–820. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Robitaille K, Daviau A, Lachance G,
Couture JP and Blouin R: Calphostin C-induced apoptosis is mediated
by a tissue trans-glutaminase-dependent mechanism involving the
DLK/JNK signaling pathway. Cell Death Differ. 15:1522–1531. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Song J, Ko HS, Sohn EJ, Kim B, Kim JH, Kim
HJ, Kim C, Kim JE and Kim SH: Inhibition of protein kinase C α/βII
and activation of c-Jun NH2-terminal kinase mediate glycyrrhetinic
acid induced apoptosis in non-small cell lung cancer NCI-H460
cells. Bioorg Med Chem Lett. 24:1188–1191. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kim BJ, Ryu SW and Song BJ: JNK- and p38
kinase-mediated phosphorylation of Bax leads to its activation and
mitochondrial translocation and to apoptosis of human hepatoma
HepG2 cells. J Biol Chem. 281:21256–21265. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ambacher KK, Pitzul KB, Karajgikar M,
Hamilton A, Ferguson SS and Cregan SP: The JNK- and
AKT/GSK3β-signaling pathways converge to regulate Puma induction
and neuronal apoptosis induced by trophic factor deprivation. PLoS
One. 7:e468852012. View Article : Google Scholar
|
|
44
|
Zhao Z, Wang J, Tang J, Liu X, Zhong Q,
Wang F, Hu W, Yuan Z, Nie C and Wei Y: JNK- and Akt-mediated Puma
expression in the apoptosis of cisplatin-resistant ovarian cancer
cells. Biochem J. 444:291–301. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yu J and Zhang L: PUMA, a potent killer
with or without p53. Oncogene. 27(Suppl 1): S71–S83. 2008.
View Article : Google Scholar
|
|
46
|
Buschmann T, Potapova O, Bar-Shira A,
Ivanov VN, Fuchs SY, Henderson S, Fried VA, Minamoto T,
Alarcon-Vargas D, Pincus MR, et al: Jun NH2-terminal kinase
phosphorylation of p53 on Thr-81 is important for p53 stabilization
and transcriptional activities in response to stress. Mol Cell
Biol. 21:2743–2754. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jones EV, Dickman MJ and Whitmarsh AJ:
Regulation of p73-mediated apoptosis by c-Jun N-terminal kinase.
Biochem J. 405:617–623. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yu C, Minemoto Y, Zhang J, Liu J, Tang F,
Bui TN, Xiang J and Lin A: JNK suppresses apoptosis via
phosphorylation of the proapoptotic Bcl-2 family protein BAD. Mol
Cell. 13:329–340. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Nishina H, Fischer KD, Radvanyi L,
Shahinian A, Hakem R, Rubie EA, Bernstein A, Mak TW, Woodgett JR
and Penninger JM: Stress-signalling kinase Sek1 protects thymocytes
from apoptosis mediated by CD95 and CD3. Nature. 385:350–353. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Potapova O, Gorospe M, Dougherty RH, Dean
NM, Gaarde WA and Holbrook NJ: Inhibition of c-Jun N-terminal
kinase 2 expression suppresses growth and induces apoptosis of
human tumor cells in a p53-dependent manner. Mol Cell Biol.
20:1713–1722. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cellurale C, Sabio G, Kennedy NJ, Das M,
Barlow M, Sandy P, Jacks T and Davis RJ: Requirement of c-Jun
NH(2)-terminal kinase for Ras-initiated tumor formation. Mol Cell
Biol. 31:1565–1576. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xiao L and Lang W: A dominant role for the
c-Jun NH2-terminal kinase in oncogenic ras-induced morphologic
transformation of human lung carcinoma cells. Cancer Res.
60:400–408. 2000.PubMed/NCBI
|
|
53
|
Nielsen C, Thastrup J, Bøttzauw T,
Jäättelä M and Kallunki T: c-Jun NH2-terminal kinase 2 is required
for Ras transformation independently of activator protein 1. Cancer
Res. 67:178–185. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mathiasen DP, Egebjerg C, Andersen SH,
Rafn B, Puustinen P, Khanna A, Daugaard M, Valo E, Tuomela S,
Bøttzauw T, et al: Identification of a c-Jun N-terminal
kinase-2-dependent signal amplification cascade that regulates
c-Myc levels in ras transformation. Oncogene. 31:390–401. 2012.
View Article : Google Scholar
|
|
55
|
Johnson R, Spiegelman B, Hanahan D and
Wisdom R: Cellular transformation and malignancy induced by ras
require c-jun. Mol Cell Biol. 16:4504–4511. 1996.PubMed/NCBI
|
|
56
|
Shibata W, Maeda S, Hikiba Y, Yanai A,
Sakamoto K, Nakagawa H, Ogura K, Karin M and Omata M: c-Jun
NH2-terminal kinase 1 is a critical regulator for the development
of gastric cancer in mice. Cancer Res. 68:5031–5039. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chang Q, Chen J, Beezhold KJ, Castranova
V, Shi X and Chen F: JNK1 activation predicts the prognostic
outcome of the human hepatocellular carcinoma. Mol Cancer.
8:642009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang X, Chao L, Li X, Ma G, Chen L, Zang Y
and Zhou G: Elevated expression of phosphorylated c-Jun
NH2-terminal kinase in basal-like and ‘triple-negative’ breast
cancers. Hum Pathol. 41:401–406. 2010. View Article : Google Scholar
|
|
59
|
Li JY and Wang H, May S, Song X, Fueyo J,
Fuller GN and Wang H: Constitutive activation of c-Jun N-terminal
kinase correlates with histologic grade and EGFR expression in
diffuse gliomas. J Neurooncol. 88:11–17. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Takahashi R, Hirata Y, Sakitani K, Nakata
W, Kinoshita H, Hayakawa Y, Nakagawa H, Sakamoto K, Hikiba Y,
Ijichi H, et al: Therapeutic effect of c-Jun N-terminal kinase
inhibition on pancreatic cancer. Cancer Sci. 104:337–344. 2013.
View Article : Google Scholar
|
|
61
|
Chen P, O’Neal JF, Ebelt ND, Cantrell MA,
Mitra S, Nasrazadani A, Vandenbroek TL, Heasley LE and Van Den Berg
CL: Jnk2 effects on tumor development, genetic instability and
replicative stress in an oncogene-driven mouse mammary tumor model.
PLoS One. 5:e104432010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Das M, Garlick DS, Greiner DL and Davis
RJ: The role of JNK in the development of hepatocellular carcinoma.
Genes Dev. 25:634–645. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kennedy NJ, Sluss HK, Jones SN, Bar-Sagi
D, Flavell RA and Davis RJ: Suppression of Ras-stimulated
transformation by the JNK signal transduction pathway. Genes Dev.
17:629–637. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Hübner A, Mulholland DJ, Standen CL,
Karasarides M, Cavanagh-Kyros J, Barrett T, Chi H, Greiner DL,
Tournier C, Sawyers CL, et al: JNK and PTEN cooperatively control
the development of invasive adenocarcinoma of the prostate. Proc
Natl Acad Sci USA. 109:12046–12051. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ying J, Li H, Cui Y, Wong AH, Langford C
and Tao Q: Epigenetic disruption of two proapoptotic genes
MAPK10/JNK3 and PTPN13/FAP-1 in multiple lymphomas and carcinomas
through hypermethylation of a common bidirectional promoter.
Leukemia. 20:1173–1175. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Yoshida S, Fukino K, Harada H, Nagai H,
Imoto I, Inazawa J, Takahashi H, Teramoto A and Emi M: The c-Jun
NH2-terminal kinase3 (JNK3) gene: Genomic structure, chromosomal
assignment, and loss of expression in brain tumors. J Hum Genet.
46:182–187. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Jurewicz A, Matysiak M, Tybor K and Selmaj
K: TNF-induced death of adult human oligodendrocytes is mediated by
c-jun NH2-terminal kinase-3. Brain. 126:1358–1370. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Dajas-Bailador F, Bantounas I, Jones EV
and Whitmarsh AJ: Regulation of axon growth by the JIP1-AKT axis. J
Cell Sci. 127:230–239. 2014. View Article : Google Scholar :
|
|
69
|
Kim AH, Sasaki T and Chao MV:
JNK-interacting protein 1 promotes Akt1 activation. J Biol Chem.
278:29830–29836. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Pan J, Pei DS, Yin XH, Hui L and Zhang GY:
Involvement of oxidative stress in the rapid Akt1 regulating a JNK
scaffold during ischemia in rat hippocampus. Neurosci Lett.
392:47–51. 2006. View Article : Google Scholar
|
|
71
|
Kim AH, Yano H, Cho H, Meyer D, Monks B,
Margolis B, Birnbaum MJ and Chao MV: Akt1 regulates a JNK scaffold
during excitotoxic apoptosis. Neuron. 35:697–709. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Cerezo A, Martínez-A C, Lanzarot D,
Fischer S, Franke TF and Rebollo A: Role of Akt and c-Jun
N-terminal kinase 2 in apoptosis induced by interleukin-4
deprivation. Mol Biol Cell. 9:3107–3118. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Levresse V, Butterfield L, Zentrich E and
Heasley LE: Akt negatively regulates the cJun N-terminal kinase
pathway in PC12 cells. J Neurosci Res. 62:799–808. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Okubo Y, Blakesley VA, Stannard B, Gutkind
S and Le Roith D: Insulin-like growth factor-I inhibits the
stress-activated protein kinase/c-Jun N-terminal kinase. J Biol
Chem. 273:25961–25966. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wang Q, Zhang QG, Wu DN, Yin XH and Zhang
GY: Neuroprotection of selenite against ischemic brain injury
through negatively regulating early activation of ASK1/JNK cascade
via activation of PI3K/AKT pathway. Acta Pharmacol Sin. 28:19–27.
2007. View Article : Google Scholar
|
|
76
|
Kim AH, Khursigara G, Sun X, Franke TF and
Chao MV: Akt phosphorylates and negatively regulates apoptosis
signal-regulating kinase 1. Mol Cell Biol. 21:893–901. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Aikin R, Maysinger D and Rosenberg L:
Cross-talk between phosphatidylinositol 3-kinase/AKT and c-jun
NH2-terminal kinase mediates survival of isolated human islets.
Endocrinology. 145:4522–4531. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Xie D, Gore C, Zhou J, Pong RC, Zhang H,
Yu L, Vessella RL, Min W and Hsieh JT: DAB2IP coordinates both
PI3K-Akt and ASK1 pathways for cell survival and apoptosis. Proc
Natl Acad Sci USA. 106:19878–19883. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Park HS, Kim MS, Huh SH, Park J, Chung J,
Kang SS and Choi EJ: Akt (protein kinase B) negatively regulates
SEK1 by means of protein phosphorylation. J Biol Chem.
277:2573–2578. 2002. View Article : Google Scholar
|
|
80
|
Murakami T, Takagi H, Suzuma K, Suzuma I,
Ohashi H, Watanabe D, Ojima T, Suganami E, Kurimoto M, Kaneto H, et
al: Angiopoietin-1 attenuates H2O2-induced
SEK1/JNK phosphorylation through the phosphatidylinositol
3-kinase/Akt pathway in vascular endothelial cells. J Biol Chem.
280:31841–31849. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Barthwal MK, Sathyanarayana P, Kundu CN,
Rana B, Pradeep A, Sharma C, Woodgett JR and Rana A: Negative
regulation of mixed lineage kinase 3 by protein kinase B/AKT leads
to cell survival. J Biol Chem. 278:3897–3902. 2003. View Article : Google Scholar
|
|
82
|
Wen XR, Li C, Zong YY, Yu CZ, Xu J, Han D
and Zhang GY: Dual inhibitory roles of geldanamycin on the c-Jun
NH2-terminal kinase 3 signal pathway through suppressing the
expression of mixed-lineage kinase 3 and attenuating the activation
of apoptosis signal-regulating kinase 1 via facilitating the
activation of Akt in ischemic brain injury. Neuroscience.
156:483–497. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Song JJ and Lee YJ: Dissociation of Akt1
from its negative regulator JIP1 is mediated through the
ASK1-MEK-JNK signal transduction pathway during metabolic oxidative
stress: A negative feedback loop. J Cell Biol. 170:61–72. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Logan SK, Falasca M, Hu P and Schlessinger
J: Phosphatidylinositol 3-kinase mediates epidermal growth
factor-induced activation of the c-Jun N-terminal kinase signaling
pathway. Mol Cell Biol. 17:5784–5790. 1997.PubMed/NCBI
|
|
85
|
Vivanco I, Palaskas N, Tran C, Finn SP,
Getz G, Kennedy NJ, Jiao J, Rose J, Xie W, Loda M, et al:
Identification of the JNK signaling pathway as a functional target
of the tumor suppressor PTEN. Cancer Cell. 11:555–569. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Cui J, Han SY, Wang C, Su W, Harshyne L,
Holgado-Madruga M and Wong AJ: c-Jun NH(2)-terminal kinase 2alpha2
promotes the tumorigenicity of human glioblastoma cells. Cancer
Res. 66:10024–10031. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Tsuiki H, Tnani M, Okamoto I, Kenyon LC,
Emlet DR, Holgado-Madruga M, Lanham IS, Joynes CJ, Vo KT and Wong
AJ: Constitutively active forms of c-Jun NH2-terminal kinase are
expressed in primary glial tumors. Cancer Res. 63:250–255.
2003.PubMed/NCBI
|
|
88
|
Nitta RT, Del Vecchio CA, Chu AH, Mitra
SS, Godwin AK and Wong AJ: The role of the c-Jun N-terminal kinase
2-α-isoform in non-small cell lung carcinoma tumorigenesis.
Oncogene. 30:234–244. 2011. View Article : Google Scholar
|
|
89
|
Antonyak MA, Kenyon LC, Godwin AK, James
DC, Emlet DR, Okamoto I, Tnani M, Holgado-Madruga M, Moscatello DK
and Wong AJ: Elevated JNK activation contributes to the
pathogenesis of human brain tumors. Oncogene. 21:5038–5046. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Bost F, McKay R, Bost M, Potapova O, Dean
NM and Mercola D: The Jun kinase 2 isoform is preferentially
required for epidermal growth factor-induced transformation of
human A549 lung carcinoma cells. Mol Cell Biol. 19:1938–1949.
1999.PubMed/NCBI
|
|
91
|
Rong Y, Belozerov VE, Tucker-Burden C,
Chen G, Durden DL, Olson JJ, Van Meir EG, Mackman N and Brat DJ:
Epidermal growth factor receptor and PTEN modulate tissue factor
expression in glioblastoma through JunD/activator protein-1
transcriptional activity. Cancer Res. 69:2540–2549. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Bonavia R, Inda MM, Vandenberg S, Cheng
SY, Nagane M, Hadwiger P, Tan P, Sah DW, Cavenee WK and Furnari FB:
EGFRvIII promotes glioma angiogenesis and growth through the NF-κB,
interleukin-8 pathway. Oncogene. 31:4054–4066. 2012. View Article : Google Scholar
|
|
93
|
Gu J, Tamura M and Yamada KM: Tumor
suppressor PTEN inhibits integrin- and growth factor-mediated
mitogen-activated protein (MAP) kinase signaling pathways. J Cell
Biol. 143:1375–1383. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Hettinger K, Vikhanskaya F, Poh MK, Lee
MK, de Belle I, Zhang JT, Reddy SA and Sabapathy K: c-Jun promotes
cellular survival by suppression of PTEN. Cell Death Differ.
14:218–229. 2007. View Article : Google Scholar
|