Introduction
The phosphatidylinositol 3-kinase
(PI3K)/AKT/mammalian target of rapamycin (mTOR) axis lies
downstream of receptor tyrosine kinases (RTKs) and plays a key role
in regulating critical cellular processes such as growth,
metabolism, size, motility and survival (1,2).
This signalling network is reported to be the most commonly altered
pathway in human cancers and activation can occur through several
different mechanisms. In breast cancer these include,
overexpression/amplification of HER2 (ErbB2/neu), activating
mutations in PIK3CA, the gene encoding the α catalytic subunit of
PI3K, mutation or amplification of AKT and loss of the lipid
phosphatase, phosphatase and tensin homolog (PTEN) (3). Several preclinical studies have
suggested that activation of the pathway, either through PIK3CA
mutations or PTEN loss, can predict sensitivity to a range of PI3K
pathway inhibitors, targeting different nodes on the signalling
axis (4–7). Data are also emerging from the clinic
to suggest that patients with PIK3CA mutations, treated with
PI3K/AKT/mTOR inhibitors, demonstrate a higher response rate than
patients without mutations (8,9).
Despite the initial activity of PI3K pathway
inhibitors, resistance is likely to occur, as has been seen with
other targeted therapies in the clinic (10,11).
Normal activation of PI3K signalling is regulated by feedback
inhibition of upstream components of the pathway such as IRS1 and
RTKs (reviewed in ref. 12). It is
becoming increasingly evident that PI3K pathway inhibitors relieve
this feedback, which may therefore limit their anti-tumour
activity. For example, treatment with rapamycin, the mTORC1
inhibitor, inhibits the phosphorylation and activation of p70S6K
and disrupts the negative feedback loop to IRS-1 and Grb10
(13–15). This leads to activation of
insulin-like growth factor-1 receptor (IGF-1R) and
re-phosphorylation and activation of AKT. Furthermore, additional
feedback mechanisms, involving FOXO transcription factors, have
subsequently been identified in HER2-amplified and ER+
breast cancer cells (16–18). Inhibition of AKT results in the
activation of FOXO-dependent gene transcription of multiple RTKs,
including IGF-1R and HER2/HER3, leading to reactivation of PI3K
signalling as well as activation of compensatory pathways such as
ERK1/ERK2. These data suggest that targeting the PI3K pathway in
combination with anti-HER2 agents would be superior to monotherapy
treatment. This is further supported by the fact that
hyperactivation of the PI3K pathway, either through mutations in
PIK3Ca or loss of PTEN, has been associated with resistance to
HER2-targeted therapies such as trastuzumab and lapatanib (19–22).
Consistent with these observations, we and others have demonstrated
enhanced anti-tumour efficacy in preclinical models of
HER2-amplified breast cancer by combining PI3K pathway inhibitors
with trastuzumab or lapatinib (7,16,23–26).
In this study, we sought to extend these findings,
to demonstrate the mechanism of enhanced activity in combination
and also to determine whether a similar effect could be observed
with the novel inhibitor AZD8931, which displays equipotent
activity against EGFR, ErbB2 (HER2) and ErbB3 (HER3) signalling
(27). We show that AZD5363 in
combination with AZD8931 causes profound shutdown of the PI3K
signalling pathway as well as concomitant inhibition of the ERK
pathway. This resulted in decreased proliferation and enhanced cell
death in a range of HER2-amplified breast cancer lines in
vitro and tumour regression in vivo. These data indicate
that combining AZD5363 with a HER2 agent represents an attractive
therapeutic strategy in the clinic for the treatment of
HER2-amplified breast cancer.
Materials and methods
Cell lines and treatments
All cell lines were maintained at 37°C and 5%
CO2 in a humidified atmosphere and grown in RPMI-1640
growth media supplemented with 10% FBS and 2 mmol/l glutamine. The
BT474c cell line was subcloned from the human breast cell line
BT474, provided by Drs J. Albanell and J. Baselga, Laboratorio
Ricerca Oncologica, Barcelona, Spain. BT20 cells were purchased
from the European Collection of Cell Cultures, SUM52PE were from
Asterand and the remaining lines were all from the American Type
Culture Collection. The identity of all cell lines was confirmed
using short tandem repeat analysis as previously described
(7). AZD5363, AZD8931 and AZD6244
were dissolved in DMSO to a concentration of 10 mmol/l and stored
under nitrogen.
Determination of cell growth
Cells were seeded in 384-well black, clear bottomed
plates (Greiner Bio-One, Stonehouse, UK) at 1,000–2,000 cells per
well, cultured for 18–24 h and treated with increasing
concentrations of AZD5363 and AZD8931 (0.03–3 μmol/l) in a 6×6
dosing matrix. After 5 days of treatment, live cell number was
determined using a Sytox Green endpoint as previously described
(7). Briefly, Sytox Green nucleic
acid dye (Invitrogen) diluted in TBS-EDTA buffer was added to cells
at a final concentration of 0.13 μmol/l and the number of dead
cells detected using an Accumen Explorer (TTP Labtech, Melbourn,
UK). Cells were then permeabilised by the overnight addition of
saponin (0.03% final concentration, diluted in TBS-EDTA buffer) and
a total cell count measured. The live cell count was then
determined by subtracting the number of dead cells per well from
the total number of cells. Pre-dose measurements were made to
indicate the number of live cells at the start of the experiment
and thus an indication of whether the treatment regimen had
resulted in cell death. The data are presented as % growth using
the NCI formulas as follows: [(Ti-Tz)/(C-Tz)] ×100 for values for
which Ti≥Tz and [(Ti-Tz)/Tz] ×100 for concentrations for which
Ti<Tz where, Tz represents the number of live cells at time
zero, C represents the control growth and Ti represents the number
of live cells in the presence of each drug regimen. This formula
gives a growth percentage from −100% to +100%. Negative scores are
for cell killing and positive scores are for anti-proliferation.
Experiments were performed in triplicate.
Analysis of combination activity
Two dimensional dose response matrix and curve
fitting were processed in the combination extension of Genedata
Screener12™ (Genedata, Basel, Switzerland). To enable this, %
growth values were converted to a positive scale using a modified
NCI formula as follows: [1-(Ti-Tz)/(C-Tz)] ×100 for values for
which Ti≥Tz and [1-(Ti-Tz)/Tz] ×100 for concentrations for which
Ti<Tz. This gave a scale of 0–200% growth inhibition where
0–100% is for anti-proliferation and 100–200% is for cell killing.
Combination activity (synergism) was calculated using the Loewe
dose-additivity model as previously described (28,29).
This model of additivity provides a null-reference that is
predicted by the expected response if the two agents were the same
drug. The 3-dimensional model surface, predicted from the two
single-agent response curves, is subtracted from the
experimentally-derived 3-dimensional dose effect surface to
generate a difference volume. This excess matrix volume can be
integrated to generate a synergy score. A synergy score cut-off
>5 was used to identify combinations of interest in the initial
high-throughput screen.
Western blot assay
Cells were grown in 60-mm dishes and treated with
AZD5363, AZD8931 or a combination of the two agents, for the
indicated concentrations and times. Cells were washed once with 1
ml ice-cold PBS and then lysed for 15 min on ice in buffer
consisting of 25 mmol/l Tris-HCl, 3 mmol/l EDTA, 3 mmol/l EGTA, 50
mmol/l NaF, 2 mmol/l sodium orthovanadate, 0.27 mol/l sucrose, 10
mmol/l B-glycerophosphate, 5 mmol/l pyrophosphate and 0.5% TX-100
and supplemented with protease and phosphatase inhibitors (Sigma).
Protein lysates were cleared by microcentrifugation at 13,000 rpm
for 10 min at 4°C and the supernatants aliquoted and stored at
−80°C. Protein concentration was determined using the Pierce BCA
Protein Assay kit (Thermo Scientific). Equivalent amounts of
protein (12 μg) from each sample were resolved by SDS-PAGE and
transferred by electroblotting onto nitrocellulose membranes
(Invitrogen, Carlsbad, CA, USA).
Membranes were then blocked and immunoblotted with
the following antibodies overnight at 4°C: HER3, p-HER3 (Y1197),
pHER3 (Y1289), HER2, p-HER2 (Tyr1221/1222), AKT, p-AKT (Ser473),
p-S6 (Ser235/236), 4EBP1, p-4EBP1 (Ser65), p-FOXO1/3A (Thr24/32),
ERK, p-ERK (Thr202/Tyr204), p-NDRG1 (T346), PARP, Cleaved caspase
3, (all from Cell Signaling Technology), Vinculin (Sigma) and
p-PRAS40 (Thr246; Invitrogen). After washing three times in
PBS-Tween the membranes were then incubated for 1 h at room
temperature with anti-mouse or anti-rabbit horseradish peroxidase
(HRP)-conjugated secondary antibodies (1:2000; Cell Signaling
Technology). Antibody-protein complexes were visualised by
chemiluminescence using SuperSignal West Dura Chemiluminescent
Substrate (Thermo Scientific) and images captured and quantified
using the ChemiGenius system (Syngene, Cambridge, UK)
Xenograft studies
Mice were bred, maintained and treated in accordance
with Institutional guidelines, as previously described (7). HCC1954 cells were harvested from T225
cm tissue culture flasks and a single cell suspension of >90%
viability was injected into the left flank of female nude mice
(nu/nu:Alpk) in a volume of ~0.1 ml in 50% matrigel. When mean
tumour sizes reached ~0.3 cm3, the mice were randomized
into control and treatment groups. Compounds were dosed by oral
gavage in a suspension formulation of
hydroxypropylmethylcellulose/Tween-80. The control group received
vehicle alone, twice daily by oral gavage. Tumour volumes (measured
by caliper), animal body weight and tumour condition were recorded
twice weekly for the duration of the study. The tumour volume was
calculated (taking length to be the longest diameter across the
tumour and width to be the corresponding perpendicular diameter)
using the formula: (length × width) × √(length × width) × (π/6).
Growth inhibition from the start of treatment was assessed by
comparison of the differences in tumour volume between control and
treated groups. Since the variance in mean tumour volume data
increases proportionally with volume (and is therefore
disproportionate between groups), data were log-transformed to
remove any size dependency before statistical evaluation.
Statistical significance was evaluated using a one-tailed,
two-sample t-test. At the end of the study mice were sacrificed by
CO2 euthanasia. The tumours were harvested and
snap-frozen in liquid nitrogen for analysis of protein expression
by ELISA.
ELISA
Frozen tumours were homogenized using FastPrep
methodology with FastPrep lysis matrix A (MP Biomedicals, Santa
Anna, CA, USA). Lysates were generated in buffer consisting of 25
mmol/l Tris-HCl, 3 mmol/l EDTA, 3 mmol/l EGTA, 50 mmol/l NaF, 2
mmol/l sodium orthovanadate, 0.27 mol/l sucrose, 10 mmol/l
B-glycerophosphate, 5 mmol/l pyrophosphate and 0.5% TX-100 and
supplemented with protease and phosphatase inhibitors (Sigma).
Total and phospho-EGFR, ErbB2 (HER2) and ErbB3 (HER3) were measured
by solid phase sandwich ELISA according to the manufacturer’s
protocol (R&D Systems, Abingdon, UK).
Results
Combining AZD5363 with the EGFR/HER2/HER3
signalling inhibitor AZD8931, results in synergistic growth
inhibition in HER2-amplified breast cancer lines
A panel of 23 breast cancer cell lines, representing
different segments of breast cancer, was screened to identify cell
lines in which AZD8931 synergizes with AZD5363 to inhibit
proliferation. The combination was evaluated in a 6×6 dose matrix
format, which allows the drug combination activity to be analysed
over a wide concentration range. Using a quantitative synergy
score, based on the Loewe model of additivity (28), we demonstrate that combination
activity is more frequently observed in the HER2-amplified cell
lines (Table I). By defining a
relative synergy cut-off (synergy score, >5) we found that 6/7
HER2-amplified lines tested show a synergistic interaction between
AZD5363 and AZD8931. Interestingly, two triple negative breast
cancer lines, MDAMB468 and BT20, which are reported to have
amplified EGFR (30) also showed a
synergistic interaction.
 | Table ISynergy scores for the combination of
AZD5363 plus AZD8931 across a panel of breast cancer cell lines
representing the different segments of breast cancer. |
Table I
Synergy scores for the combination of
AZD5363 plus AZD8931 across a panel of breast cancer cell lines
representing the different segments of breast cancer.
| Cell line | Synergy score | HER2 | ER | PIK3CA mutation |
|---|
| CAMA1 | 4.7 | − | + | WT |
| KPL1 | 0.9 | − | + | E545K |
| MCF7 | 1.7 | − | + | E545K |
| MDAMB134V1 | 0.0 | − | + | WT |
| T47D | 2.5 | − | + | H1047R |
| BT474c | 9.8 | + | + | K111N |
| HCC1419 | 8.3 | + | + | WT |
| MDAMB361 | 12.9 | + | + | E545K |
| HCC1569 | 0.7 | + | − | WT |
| HCC1954 | 7.5 | + | − | H1047R |
| KPL4 | 15.0 | + | − | H1047R |
| SKBR3 | 10.2 | + | − | WT |
| MDAMB453 | 4.3 | − | − | H1047R |
| BT20 | 8.5 | − | − | H1047R |
| EVSAT | 0.3 | − | − | WT |
| HCC1187 | 0.3 | − | − | WT |
| HCC1395 | 1.7 | − | − | WT |
| HCC70 | 4.0 | − | − | WT |
| MDAMB157 | 2.1 | − | − | WT |
| MDAMB415 | 3.4 | − | − | WT |
| MDAMB436 | 2.3 | − | − | WT |
| MDAMB468 | 12.5 | − | − | WT |
| SUM52PE | 4.3 | − | − | WT |
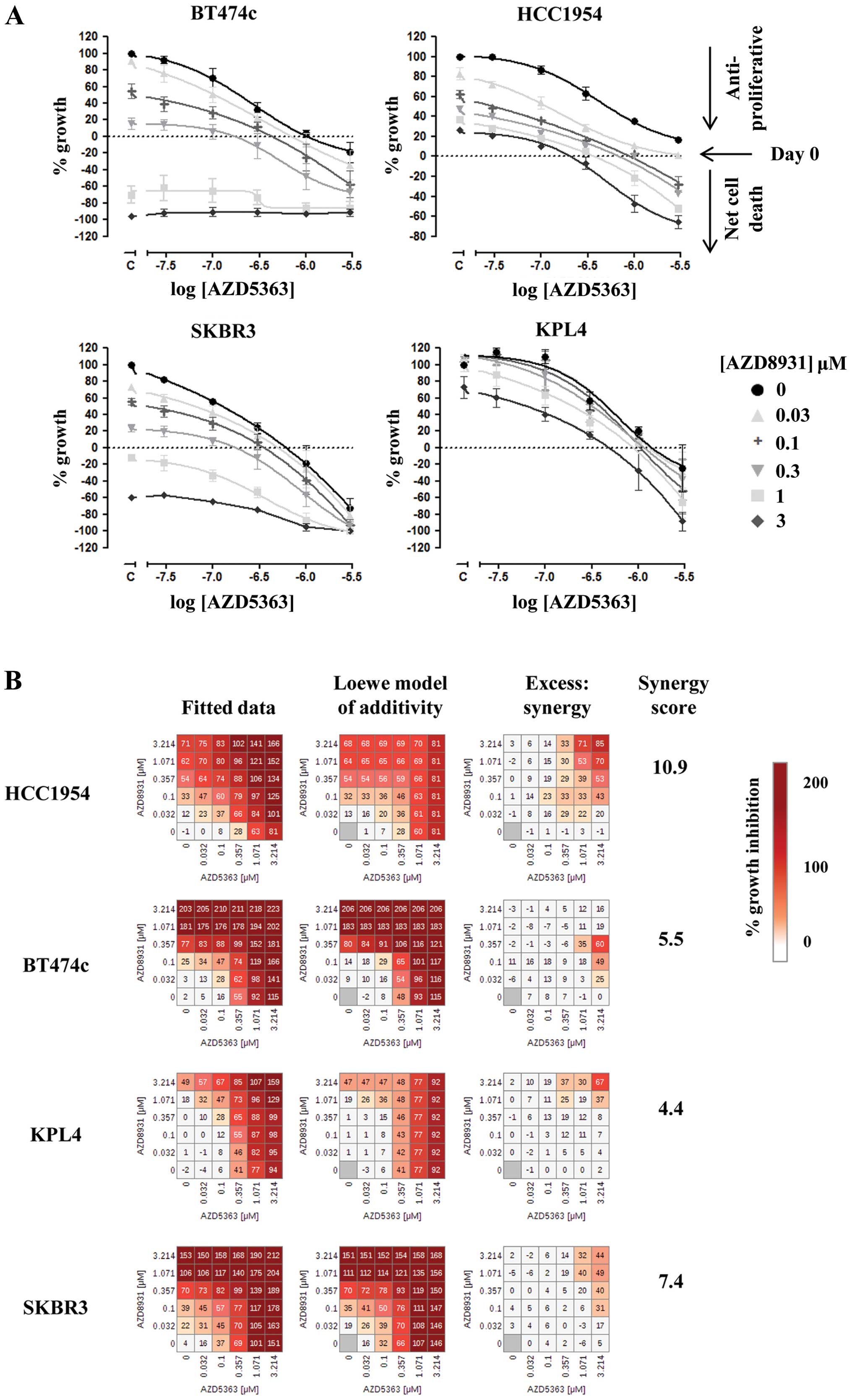
To confirm the results of the high-throughput screen
and further evaluate the effect of combining AZD5363 and AZD8931 we
selected a mini-panel of four HER2-amplified breast cancer cell
lines and assessed synergistic growth inhibition using the 5-day
sytox green assay to measure live cell number. Analysis of the
growth curves demonstrated that AZD5363 exerted monotherapy
anti-proliferative activity in all of the HER2-amplified lines, as
previously described (7) but only
induced cell death at concentrations ≤1 μM in two of these lines,
BT474c and SKBR3. However, addition of AZD8931 was able to increase
the anti-proliferative effects of AZD5363 and cause enhanced cell
death over either agent alone in all four of the cell lines tested
(Fig. 1A). Analysis of synergism,
as described in Materials and methods, suggested that the greatest
synergy was observed in HCC1954 cells for this combination
(Fig. 1B), with synergy scores of
10.9, 11.4 and 7.8 being generated across the three separate
experiments. Mean synergy scores for BT474c, KPL4 and SKBR3 were
4.0, 4.8 and 8.3, respectively.
AKT inhibition by AZD5363 leads to
induction of phospho and total HER2/3 levels
Previous studies have demonstrated that inhibition
of the PI3K/AKT/mTOR pathway leads to the activation of
compensatory pathways through feedback induction of receptor
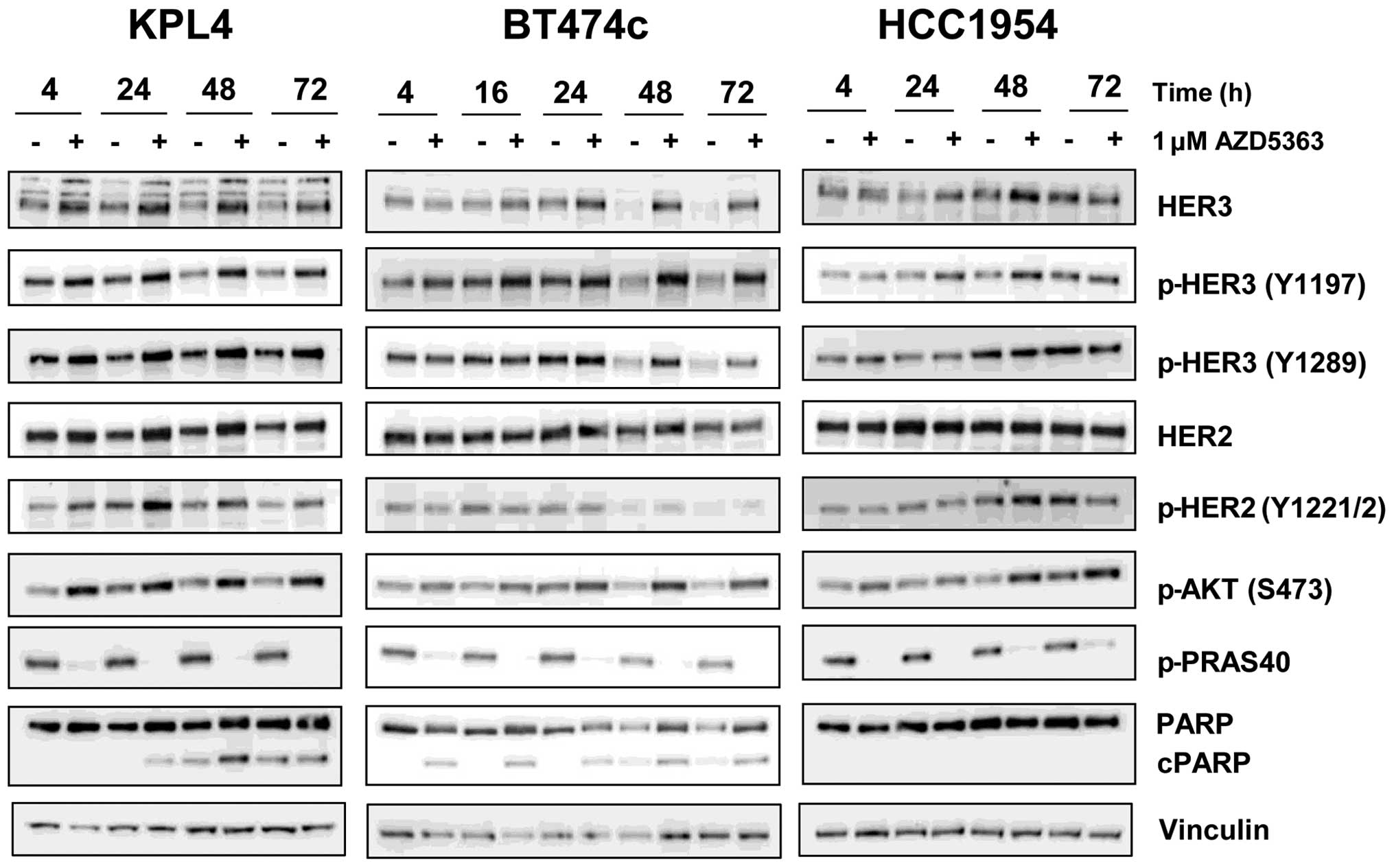
tyrosine kinases such as HER2 and HER3. To determine whether
AZD5363 resulted in similar feedback induction we analysed phospho
and total HER2 and HER3 levels, in three of the HER2-amplified
lines that had shown good combination effects, following exposure
to 1 μM AZD5363 for up to 72 h. This concentration is clinically
relevant, as a steady state Cmin of ~1 μM is achieved
throughout the dosing period at the clinically tolerated dosing
schedule of 480 mg bid (31). As
expected, we observed reproducible, time-dependent increases in
phospho and total levels of HER3, and to a lesser extent HER2, in
all three of the cell lines examined (Fig. 2).
In addition, pAKT levels were seen to increase. This
was not unexpected as it has been reported that binding of an
inhibitor to the ATP site of AKT can result in hyperphosphorylation
of the kinase in the absence of any pathway feedback effects
(32). In contrast, the levels of
phospho-PRAS40, a direct substrate of AKT, were markedly decreased,
confirming inhibition of AKT activity by AZD5363 at 1 μM. It was
unclear why the levels of HER3 appeared to decrease in DMSO-treated
BT474c cells over time, but was perhaps due to the cells
approaching confluence at the later time points. However, all
subsequent experiments assessing total and phosphorylated endpoints
were performed at 24 h, when HER3 levels remained constant.
Analysis of cleaved PARP levels suggested that whilst 1 μM AZD5363
could induce apoptosis in BT474c and KPL4 cells it was unable to
induce cell death as a monotherapy in the HCC1954 cell line. This
is consistent with data from the growth curve analysis; in BT474c
and KPL4 cultures the cell number fell below the initial seeding
density at concentrations of approximately 1 μM, suggesting potent
anti-proliferative effects and some cell death, whereas in HCC1954
cultures, 20% growth was still observed even at concentrations as
high as 3 μM (Fig. 1A).
AZD8931 limits RTK feedback induced by
AZD5363 and results in more prominent shutdown of downstream
signalling pathways and induction of apoptosis
To determine whether AZD8931 could limit the RTK
feedback induced by AZD5363, we assessed the phosphorylation status
of HER2 and HER3 as well as several downstream signalling molecules
by western blotting in HCC1954 and BT474c cells. In HCC1954 cells,
AZD5363 caused a dose-dependent increase in HER2/HER3
phosphorylation at 24 h which was prevented by the addition of 1 μM
AZD8931. Phosphorylation of HER2 (Y1221/2) and HER3 (Y1289) was
essentially undetectable by western blotting in the presence of
AZD8931 (Fig. 3A).
A similar result was obtained in BT474c cells with
0.3 μM AZD8931. This lower concentration was used due to the
potency of AZD8931 at inducing cell death in this cell line.
Likewise, AZD8931 limited the increase in phospho-AKT which occurs
as a result of treatment with AZD5363, returning phospho-AKT back
to control levels in both cell lines. This subsequently resulted in
more pronounced inhibition of the PI3K/AKT/mTOR signalling pathway
as evidenced by a further reduction in PRAS40, S6 and 4EBP1
phosphorylation over either agent alone. PI3K inhibition has also
been shown to result in compensatory activation of the ERK
signalling pathway, probably at least in part as a consequence of
HER2 activation (24). We
therefore decided to assess whether AZD5363 increased ERK
phosphorylation in BT474c and HCC1954 cells and if so, could this
be prevented by the addition of AZD8931. Twenty-four hour exposure
to AZD5363 increased phosphorylated ERK in both cell lines. This
induction was completely abrogated by the combined treatment with
AZD8931. To further investigate whether the induction of
phospho-ERK might in part be responsible for limiting the
anti-proliferative activity of AZD5363 in HER2-amplified cell lines
we tested whether the addition of AZD6244, a MEK1/2 inhibitor, was
also able to synergise with AZD5363. Good synergy was observed in
several breast cell lines, and this tended to correlate with cell
lines which had shown good synergy with AZD8931 (Fig. 3B).
The data in Fig. 1
suggested that combining AZD5363 with AZD8931 caused greater cell
death than either agent alone. To determine whether this was due to
enhanced apoptosis we analysed cleaved PARP levels by western
blotting and demonstrated a greater induction of cleaved PARP in
the combination treatment (Fig.
3A). This was particularly evident in the BT474c cells,
occurring across the entire concentration range of AZD5363, whereas
in HCC1954 cells, induction of cleaved PARP was only really
observed at the top concentration of 5 μM.
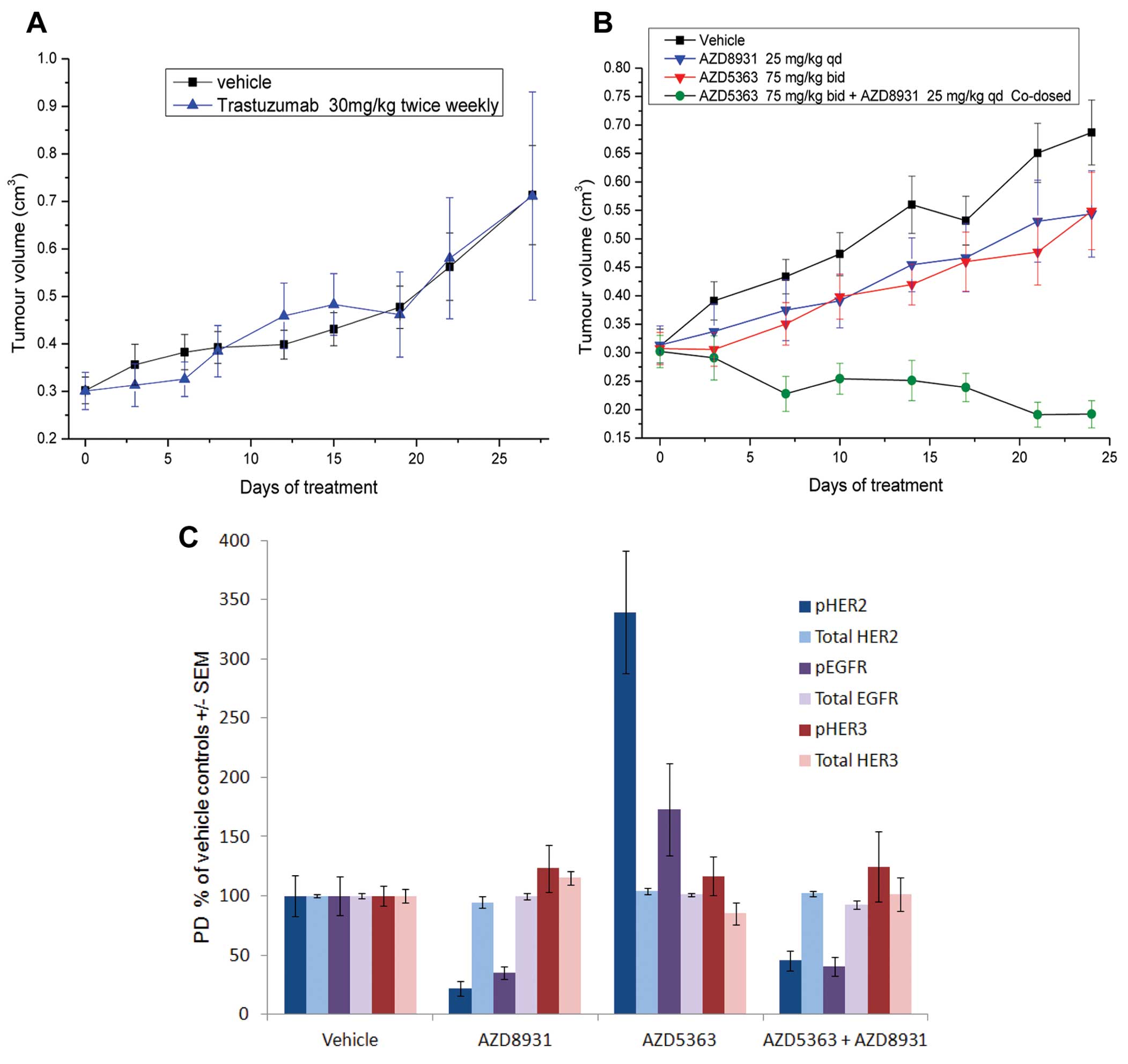
Anti-tumour activity of AZD5363 and
AZD8931 in the HCC1954 xenograft model
To determine whether the enhanced anti-proliferative
activity translated in vivo we investigated the effects of
the combination in the HCC1954 xenograft model, a HER2-amplified,
PIK3CA mutant cell line which is resistant to Trastuzumab therapy
(Fig. 4A). Monotherapy AZD5363 (75
mg/kg bid) and AZD8931 (25 mg/kg qd) inhibited tumour growth by 42%
(p=0.05) and 39% (p=0.07) respectively, compared with vehicle
controls (Fig. 4B). In contrast,
the combination of AZD5363 and AZD8931 was well tolerated and
caused pronounced tumour regression which was sustained for the
duration of the dosing period (130% inhibition compared with
vehicle controls; p<0.0001 compared with controls and
monotherapy groups). To evaluate whether AZD5363-induced feedback
to HER receptors was also observed in vivo we analysed
phospho and total EGFR, HER2 and HER3 levels at the end of
anti-tumour study. As expected, AZD8931 inhibited the
phosphorylation of HER2 and EGFR, whereas these were increased by
AZD5363 monotherapy treatment. The induction of pHER2 and pEGFR by
AZD5363 was ameliorated when dosed in combination with AZD8931
(Fig. 4C). There was no
significant effect of either agent on phospho HER3 levels in
vivo.
Discussion
Numerous PI3K/AKT/mTOR pathway inhibitors, including
AZD5363, have progressed into the clinic and are currently being
tested in phase I/II clinical trials. To date, modest clinical
activity has been observed for these agents when dosed as a
monotherapy. Furthermore, data emerging from both preclinical and
clinical studies with other targeted therapies suggests that
resistance is likely to occur following prolonged treatment.
Therefore, a pressing need exists to develop rational combination
hypotheses to enhance the therapeutic response and prevent or delay
the development of resistance. Relief of feedback to receptor
tyrosine kinases has been increasingly demonstrated to be a
potential resistance mechanism for inhibitors of the PI3K
signalling pathway and in breast cancer this frequently occurs
through increased expression and phosphorylation of the HER family
of receptors, particularly HER2 and HER3 (16,24–26).
Likewise, resistance to trastuzumab (Herceptin) has been associated
with increased PI3K signalling, either through hyper-activating
mutations in PIK3CA or loss of the tumour suppressor PTEN (19–22).
Combined inhibition of PI3K/AKT/mTOR signalling with HER2/HER3
signalling inhibitors is therefore an attractive therapeutic
strategy for the treatment of breast cancer.
In this study, we demonstrated that the activity of
AZD5363 could be enhanced by the addition of AZD8931, as shown by
synergistic growth inhibition in vitro and enhanced
anti-tumour activity in vivo. Using a panel of breast cell
lines, representing different segments of disease, we showed that
this combination activity was mainly observed in cell lines
containing amplified HER2 or in the case of two triple negative
lines, amplified EGFR. This is consistent with AZD8931 being a pan
EGFR/HER2/HER3 signalling inhibitor.
AKT inhibition resulted in feedback upregulation and
phosphorylation of HER3 and to a lesser extent HER2, in
vitro. This is consistent with previous studies where
upregulation of cell surface receptors, including HER3, have been
observed in response to a range of PI3K/AKT/mTOR inhibitors
(16,18,25).
In those studies the activation of FOXO transcription factors, as a
direct result of AKT inhibition, was shown to be responsible for
the increased expression of RTKs. Given that AZD5363 potently
inhibits FOXO phosphorylation and induces translocation of FOXO3a
to the nucleus in BT474c cells (Fig.
3) and (7) it is likely that
this is the mechanism of HER3 induction in these cells. Indeed, Fox
et al demonstrated that knockdown of FOXO3a abrogated
AZD5363-mediated induction of IGF-1R, IGF-I and IGF-II mRNA in
their ER+ models of long-term estrogen deprivation
(18). Surprisingly, whilst
AZD5363 induced the phosphorylation of HER2 and EGFR in
vivo, the levels of phosphorylated HER3 remained similar to
control treated samples. This may suggest that different feedback
mechanisms are activated in vivo, with receptor activation
being heavily dependent on the microenvironment and source of
appropriate ligand.
In all cases, addition of AZD8931 prevented the
induction of phospho HER2/3 and led to a more prominent suppression
of the downstream markers of PI3K pathway signalling. As well as
reactivating the inhibited pathway, loss of negative feedback has
also been shown to activate parallel signalling networks. For
example, inhibition of PI3K/AKT/mTOR with BEZ235 resulted in
compensatory activation of ERK signalling which was shown to occur
via activation of HER family receptors (24). We observed a similar effect
following inhibition of AKT with AZD5363. Twenty-four hour exposure
to AZD5363 resulted in a marked increase of phospho ERK in BT474c
and HCC1954 cells and this induction was completely prevented by
the addition of AZD8931. Furthermore, the MEK1/2 inhibitor,
AZD6244, was also able to synergise with AZD5363 in a range of
HER2-amplified cell lines, suggesting that activation of ERK
signalling in response to AZD5363 treatment may serve as a
potential mechanism to limit its efficacy in HER2-amplified breast
cancer lines.
Trastuzumab has had a major impact on the treatment
of HER2-amplified metastatic breast cancer. However, not all
HER2-amplified patients respond to treatment and patients that
initially respond eventually develop resistance. Numerous
mechanisms of resistance have been identified including
hyper-activation of the PI3K signalling pathway, expression of
p95HER2 receptor, a truncated form of HER2 which lacks the
extracellular ligand-binding domain and dimerisation with other
RTKs (33). Several clinical
trials, combining PI3K/mTOR inhibitors with HER2 agents in
trastuzumab refactory patients have been initiated (www.clinicaltrial.gov; NCT00736970, NCT01471847,
NCT00317720, NCT01283789). We have previously demonstrated that
AZD5363 enhances the efficacy of both trastuzumab and lapatinib in
the KPL4 xenograft model which is only partially sensitive to the
HER2 agents as monotherapy (7).
Herein, we further show that the combination of AZD5363 and AZD8931
was highly synergistic in HCC1954 cells, a model which expresses
high levels of p95HER2 and is resistant to trastuzumab therapy. It
is therefore suggested that the combination of AZD5363 with an
inhibitor of HER2/HER3 signalling, such as AZD8931, is worth
pursuing as a possible treatment option for patients with amplified
HER2 who have previously progressed on HER2-directed therapy. The
potential application of AZD5363 in the neoadjuvant setting, where
patients fail to show a pathologic complete response (pCR) on
HER2-directed therapy alone, is also worthy of further
evaluation.
Acknowledgements
AZD5363 was discovered by AstraZeneca subsequent to
a collaboration with Astex Therapeutics (and its collaboration with
the Institute of Cancer Research and Cancer Research Technology
Limited).
References
|
1
|
Altomare DA and Testa JR: Perturbations of
the AKT signaling pathway in human cancer. Oncogene. 24:7455–7464.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bellacosa A, Kumar CC, Di Cristofano A and
Testa JR: Activation of AKT kinases in cancer: Implications for
therapeutic targeting. Adv Cancer Res. 94:29–86. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Engelman JA: Targeting PI3K signalling in
cancer: Opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Serra V, Markman B, Scaltriti M, et al:
NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and
inhibits the growth of cancer cells with activating PI3K mutations.
Cancer Res. 68:8022–8030. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ihle NT, Lemos R Jr, Wipf P, Yacoub A,
Mitchell C, Siwak D, Mills GB, Dent P, Kirkpatrick DL and Powis G:
Mutations in the phosphatidylinositol-3-kinase pathway predict for
antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is
a dominant predictor for resistance. Cancer Res. 69:143–150. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Di Nicolantonio F, Arena S, Tabernero J,
et al: Deregulation of the PI3K and KRAS signaling pathways in
human cancer cells determines their response to everolimus. J Clin
Invest. 120:2858–2866. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Davies BR, Greenwood H, Dudley P, et al:
Preclinical pharmacology of AZD5363, an inhibitor of AKT:
Pharmacodynamics, antitumor activity, and correlation of
monotherapy activity with genetic background. Mol Cancer Ther.
11:873–887. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Janku F, Tsimberidou AM, Garrido-Laguna I,
et al: PIK3CA mutations in patients with advanced cancers treated
with PI3K/AKT/mTOR axis inhibitors. Mol Cancer Ther. 10:558–565.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Janku F, Hong DS, Fu S, et al: Assessing
PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR
inhibitors. Cell Rep. 6:377–387. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bucheit AD and Davies MA: Emerging
insights into resistance to BRAF inhibitors in melanoma. Biochem
Pharmacol. 87:381–389. 2014. View Article : Google Scholar
|
|
11
|
Tartarone A, Lazzari C, Lerose R,
Conteduca V, Improta G, Zupa A, Bulotta A, Aieta M and Gregorc V:
Mechanisms of resistance to EGFR tyrosine kinase inhibitors
gefitinib/erlotinib and to ALK inhibitor crizotinib. Lung Cancer.
81:328–336. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chandarlapaty S: Negative feedback and
adaptive resistance to the targeted therapy of cancer. Cancer
Discov. 2:311–319. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Haruta T, Uno T, Kawahara J, Takano A,
Egawa K, Sharma PM, Olefsky JM and Kobayashi M: A
rapamycin-sensitive pathway down-regulates insulin signaling via
phosphorylation and proteasomal degradation of insulin receptor
substrate-1. Mol Endocrinol. 14:783–794. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
O’Reilly KE, Rojo F, She QB, et al: mTOR
inhibition induces upstream receptor tyrosine kinase signaling and
activates Akt. Cancer Res. 66:1500–1508. 2006. View Article : Google Scholar
|
|
15
|
Yu Y, Yoon SO, Poulogiannis G, et al:
Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate
that negatively regulates insulin signaling. Science.
332:1322–1326. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chandarlapaty S, Sawai A, Scaltriti M,
Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK,
Baselga J and Rosen N: AKT inhibition relieves feedback suppression
of receptor tyrosine kinase expression and activity. Cancer Cell.
19:58–71. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Muranen T, Selfors LM, Worster DT,
Iwanicki MP, Song L, Morales FC, Gao S, Mills GB and Brugge JS:
Inhibition of PI3K/mTOR leads to adaptive resistance in
matrix-attached cancer cells. Cancer Cell. 21:227–239. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fox EM, Kuba MG, Miller TW, Davies BR and
Arteaga CL: Autocrine IGF-I/insulin receptor axis compensates for
inhibition of AKT in ER-positive breast cancer cells with
resistance to estrogen deprivation. Breast Cancer Res. 15:R552013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nagata Y, Lan KH, Zhou X, et al: PTEN
activation contributes to tumor inhibition by trastuzumab, and loss
of PTEN predicts trastuzumab resistance in patients. Cancer Cell.
6:117–127. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Berns K, Horlings HM, Hennessy BT, et al:
A functional genetic approach identifies the PI3K pathway as a
major determinant of trastuzumab resistance in breast cancer.
Cancer Cell. 12:395–402. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Eichhorn PJ, Gili M, Scaltriti M, et al:
Phosphatidylinositol 3-kinase hyperactivation results in lapatinib
resistance that is reversed by the mTOR/phosphatidylinositol
3-kinase inhibitor NVP-BEZ235. Cancer Res. 68:9221–9230. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chandarlapaty S, Sakr RA, Giri D, et al:
Frequent mutational activation of the PI3K-AKT pathway in
trastuzumab-resistant breast cancer. Clin Cancer Res. 18:6784–6791.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Miller TW, Forbes JT, Shah C, et al:
Inhibition of mammalian target of rapamycin is required for optimal
antitumor effect of HER2 inhibitors against HER2-overexpressing
cancer cells. Clin Cancer Res. 15:7266–7276. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Serra V, Scaltriti M, Prudkin L, et al:
PI3K inhibition results in enhanced HER signaling and acquired ERK
dependency in HER2-overexpressing breast cancer. Oncogene.
30:2547–2557. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chakrabarty A, Sánchez V, Kuba MG,
Rinehart C and Arteaga CL: Feedback upregulation of HER3 (ErbB3)
expression and activity attenuates antitumor effect of PI3K
inhibitors. Proc Natl Acad Sci USA. 109:2718–2723. 2012. View Article : Google Scholar :
|
|
26
|
García-García C, Ibrahim YH, Serra V, et
al: Dual mTORC1/2 and HER2 blockade results in antitumor activity
in preclinical models of breast cancer resistant to anti-HER2
therapy. Clin Cancer Res. 18:2603–2612. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hickinson DM, Klinowska T, Speake G, et
al: AZD8931, an equipotent, reversible inhibitor of signaling by
epidermal growth factor receptor, ERBB2 (HER2), and ERBB3: A unique
agent for simultaneous ERBB receptor blockade in cancer. Clin
Cancer Res. 16:1159–1169. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lehár J, Krueger AS, Avery W, et al:
Synergistic drug combinations tend to improve therapeutically
relevant selectivity. Nat Biotechnol. 27:659–666. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rickles RJ, Tam WF, Giordano TP III, et
al: Adenosine A2A and beta-2 adrenergic receptor agonists: Novel
selective and synergistic multiple myeloma targets discovered
through systematic combination screening. Mol Cancer Ther.
11:1432–1442. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Garnett MJ, Edelman EJ, Heidorn SJ, et al:
Systematic identification of genomic markers of drug sensitivity in
cancer cells. Nature. 483:570–575. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Elvin P, Palmer A, Womack C, et al:
Pharmacodynamic activity of the AKT inhibitor AZD5363 in patients
with advanced solid tumors. J Clin Oncol. 32(Suppl 5):
25412014.
|
|
32
|
Okuzumi T, Fiedler D, Zhang C, Gray DC,
Aizenstein B, Hoffman R and Shokat KM: Inhibitor hijacking of Akt
activation. Nat Chem Biol. 5:484–493. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rexer BN and Arteaga CL: Intrinsic and
acquired resistance to HER2-targeted therapies in HER2
gene-amplified breast cancer: Mechanisms and clinical implications.
Crit Rev Oncog. 17:1–16. 2012. View Article : Google Scholar : PubMed/NCBI
|