Introduction
Neuroblastoma is a malignant tumor consisting of
neural crest derived undifferentiated neuroectodermal cells. These
tumors are biologically heterogeneous, with cell populations
differing in their genetic programs, maturation stage and malignant
potential (1,2). As neuroblastoma cells seem to have
the capacity to differentiate spontaneously in vivo and
in vitro (3), their
heterogeneity could affect treatment outcome. Recent studies have
provided a link between increased metastatic potential and
drug-resistant phenotypes, indicating that in addition to the
development of drug resistance, chemotherapy of tumors may cause
changes in their biological characteristics (4,5).
Unfortunately, little improvement in therapeutic options in high
risk neuroblastoma has been made in the last decade, requiring a
need for the development of new therapies.
Recently, we suggested a novel treatment for
neuroblastomas, utilizing a drug targeting DNA, the plant alkaloid
ellipticine. This compound and its derivatives act as potent
anticancer agents via a combined mechanism involving cell cycle
arrest and induction of apoptosis. Cell death induced by
ellipticine has been shown to engage a p53-dependent pathway, cell
cycle arrest, interaction with several kinases and induction of the
mitochondrial pathway of apoptotic cell death. Cell cycle arrest
was shown to result from DNA damage caused by a variety of tumor
chemotherapeutic agents; this is also the case for ellipticines.
Formation of covalent DNA adducts after ellipticine enzymatic
activation with cytochrome P450 (CYP) and peroxidase enzymes is one
of the most important mechanisms of its pharmacological action
(summarized in refs. 6,7). We found that exposure of human
neuroblastoma IMR-32, UKF-NB-3 and UKF-NB-4 cell lines to this
agent resulted in strong inhibition of cell growth, followed by
induction of apoptosis (6–11). These effects were associated with
formation of two major covalent ellipticine-derived DNA adducts,
identical to those formed by the CYP- and peroxidase-mediated
ellipticine metabolites, 13-hydroxy- and 12-hydroxyellipticine
(6,7,12–16).
The levels of covalent ellipticine-derived DNA
adducts correlated with ellipticine toxicity in IMR-32 and UKF-NB-4
cell lines. Cells of both lines accumulated in S phase, suggesting
that ellipticine-DNA adducts interfere with DNA replication. We
therefore concluded that formation of ellipticine-DNA adducts was
the predominant DNA-damaging mechanism of ellipticine action,
resulting in its high cytotoxicity to these neuroblastoma cells
(6–8,11).
Nevertheless, this drug is unfortunately capable of
inducing resistance in neuroblastoma cells. Exposure of UKF-NB-4
cells to increasing concentrations of ellipticine resulted in a
resistant line assigned as UKF-NB-4ELLI (8,17).
In the UKF-NB-4ELLI cells, lower accumulation of this
drug was found in the nuclei after treatment of these cells with
ellipticine than in the parental line (17), which consequently leads to lower
levels of DNA adducts and decreased ellipticine toxicity in these
cells. Ellipticine resistance in neuroblastoma is caused by a
combination of overexpression of Bcl-2, efflux or degradation of
the drug, downregulation of topoisomerases and the upregulation of
vacuolar (V)-ATPase (17). The
mechanism of V-ATPase contribution to induction of resistance to
ellipticine in the UKF-NB-4ELLI cell line was
investigated in this study.
Vacuolar V-ATPase is the multi-subunit membrane
protein complex (18) responsible
for the acidification of some intracellular compartments such as
trans-Golgi network, endosomes, lysosomes and secretory granules.
The V-ATPase-dependent acidification of Golgi complex is essential
for the synthesis and delivery of the lysosomal hydrolases from
endoplasmic reticulum/Golgi to lysosomes (19–21).
The acidic microenvironment caused by changes in the pH gradient
between the intracellular and the extracellular compartments as
well as the pH gradient between the cytoplasm and the intracellular
organelles can be significantly involved in the mechanism of drug
resistance (22,23). These changes in pH lead to
neutralization of weakly basic drugs by the acidic tumor
microenvironment or the sequestration of drugs into lysosomal
vesicles (22–26). Whether these mechanisms and if so,
which of them are responsible for V-ATPase-dependent development of
resistance of UKF-NB-4 cells to ellipticine need to be answered.
Therefore, this feature was studied.
Materials and methods
Cell lines and cell culture
UKF-NB-4 neuroblastoma cell line, established from
recurrent bone marrow metastases of high risk neuroblastoma, was a
gift of Professor J. Cinatl (J.W. Goethe University, Frankfurt,
Germany). The ellipticine-resistant cell line, designated
UKF-NB-4ELLI, was established by exposing UKF-NB-4 cells
to increasing concentrations from 0.2 to 2.5 μM ellipticine over 36
months. The drug resistance of UKF-NB-4ELLI cells to
ellipticine was verified using the MTT test (17). Each cell line was cultivated in
Iscove's modified Dulbecco's medium (IMDM) supplemented with 10%
(v/v) fetal bovine serum (both from Life Technologies, Carlsbad,
CA, USA), maintained at 37°C and 5% CO2. The medium for
UKF-NB-4ELLI cells was the same, but contained 2.5 μM
ellipticine (8). Resistance of
UKF-NB-4ELLI cells to ellipticine caused by changes in
expression of several genes and chromosome modifications (detailed
in ref. 17) is maintained during
more than four passages of these cells without ellipticine
(17). Before experiments,
UKF-NB-4ELLI cells were cultivated for at least one
passage without ellipticine, in order to remove ellipticine from
these cells. Ellipticine, chloroquine, wortmannin and bafilomycin A
were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Electron microscopy
UKF-NB-4 and UKF-NB-4ELLI cells
(5×105) were grown on glass 60 mm dishes either
untreated or treated with 5 μM ellipticine and 100 nM bafilomycin A
as well as combination of 5 μM ellipticine and 100 nM bafilomycin A
for 1 h at 37°C. In the case of a combined treatment, bafilomycin A
was added to the incubations 20 min before adding ellipticine.
Cells were mechanically re-suspended, washed, centrifuged and fixed
with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer pH 7.4
for 90 min. Samples were centrifuged (16,000 × g for 5 min) and
pellets were postfixed for 60 min with 2% OsO4 in 0.1 M
sodium cacodylate buffer pH 7.4, dehydrated in graded series of
alcohol and embedded in a Durcupan-Epon mixture. Ultrathin sections
were prepared on Leica EM UC6 ultramicrotome (Leica Microsystems,
Vienna, Austria) contrasted with uranyl acetate and lead citrate
and examined by a JEM 1011 transmission electron microscope (Jeol,
Tokyo, Japan).
Fluorescence microscopy
Acidic vesicular organelle stained, UKF-NB-4 cells
(5×105) were grown on 35 mm glass bottom culture dishes
(In Vitro Scientific, Sunnyvale, CA, USA) for 24 h before adding
the compounds. Cells were treated either with 5 μM ellipticine
alone or in combination with either 100 nM bafilomycin A or 25 μM
chloroquine for 1 h at 37°C, then incubated with 75 nM
LysoTracker® Red (Life Technologies) for 30 min. After
washing with Hanks' balanced salt solution (Sigma-Aldrich), cells
were observed with a laser-scanning confocal microscope, Olympus
FV1000 (Olympus, Tokyo, Japan). For excitation of the
LysoTracker® Red, laser with an excitation wavelength of
559 nM was used; emitted light was collected in the range of
570–670 nM. For excitation of green-ellipticine fluorescence,
solid-state laser with an excitation wavelength of 473 nM was used
and emitted light was collected in the range of 485–545 nM. All
images were recorded with a ×40 objective using a zoom factor of ×2
and the Olympus FluoView FV1000 system. Each fluorescence channel
was scanned individually (sequential scanning). Fluorescent
channels were pseudocolored with RGB values corresponding to each
of the fluorophore emission spectral profiles.
Western blot analysis of V-ATPase
(ATP6V0D1 membrane domain) protein expression
In order to analyze V-ATPase (ATP6V0D1 membrane
domain) protein expression, western blotting was used. UKF-NB-4 and
UKF-NB-4ELLI cell (1.5×106) pellets were
re-suspended in 25 mM Tris-HCl buffer pH 7.6 containing 150 mM
NaCl, 1% detergent Igepal® CA-630 (Sigma Chemical Co.,
St. Louis, MO, USA), 1% sodium deoxycholate and 0.1% sodium dodecyl
sulfate (SDS) and with solution of Complete™ (Roche, Basel,
Switzerland) at concentrations described by the provider. The
samples were incubated for 30 min on ice and thereafter centrifuged
for 20 min at 20,000 × g and 4°C. Supernatants were used for
additional analysis. Protein concentrations were assessed using the
DC protein assay (Bio-Rad, Hercules, CA, USA) according to Lowry
method. Proteins (15 μg) were electrophoretically separated using
4–20% TGX precast gel (100 mA). After migration, proteins were
transferred to a nitrocellulose membrane and incubated with 5%
non-fat milk to block non-specific binding. The membranes were then
exposed to specific anti-ATP6V0D1 mouse monoclonal antibody (1:500;
Abcam, Cambridge, UK). Membranes were washed and exposed to
peroxidase-conjugated anti-IgG secondary antibodies (1:2,000;
Bio-Rad) and the antigen-antibody complex was visualized by
enhanced chemiluminescence detection system according to the
manufacturer's instructions (Immun-Star HRP Substrate; Bio-Rad),
using X-ray film (MEDIX XBU; Foma, Hradec Kralove, Czech Republic).
Antibody against actin (1:1,000; Sigma-Aldrich) was used as loading
control.
Determination of apoptosis by Annexin
V/DAPI labeling
UKF-NB-4 and UKF-NB-4ELLI cells
(5×105) were seeded in 35-mm culture dishes overnight.
Bafilomycin A, chloroquine and/or ellipticine in the
above-mentioned concentrations were added to dishes and the cells
were incubated for 24 h. The cells were collected by trypsinization
and washed with phosphate-buffered saline (PBS). Annexin V staining
was accomplished by following producer's instructions (Exbio,
Vestec, Czech Republic). The cells were re-suspended in Annexin V
binding buffer (Exbio), then Annexin V-Dy647 was added and samples
were incubated for 15 min in the dark at ambient temperature. DAPI
(2.5 μg/ml) was added just before analysis. Cells were analyzed
using LSR II Flow Cytometer (BD Bioscience, San Jose, CA, USA).
3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS) assay
The IC50 values of ellipticine were
determined by CellTiter 96® AQueous One Solution Cell
Proliferation Assay (Promega, Fitchburg, WI, USA) in a 96-well
plate. For a dose-response curve, cells were seeded in 100 μl of
medium with 5×103 cells/well with 100 nM bafilomycin A,
25 μM chloroquine or 100 nM wortmanin, and 20 min later
ellipticine, in serial dilutions, was added. After three days of
incubation at 37°C in 5% CO2, 7 μl of MTS solution per
well was added and the plates were incubated for 2 h. The
absorbance at 490 nM was measured for each well by multiwell ELISA
reader VersaMax (Molecular Devices, Sunnyvale, CA, USA). Each value
is the mean of 8 wells. The mean absorbance of medium controls was
the background and was subtracted. The IC50 values were
calculated from three independent experiments using the linear
regression of the dose-log response curves by SoftMaxPro
software.
Western blot analysis of autophagosomal
marker proteins LC3-I and LC3-II
To induce autophagy, UKF-NB-4 and
UKF-NB-4ELLI cells were starved in Hanks' balanced salt
solution (Sigma-Aldrich) for 4 h at 37°C with or without the
inhibitors of autophagy, wortmannin (0.1 μM), chloroquine (25 μM)
or bafilomycin A (100 nM). Subsequently cells were collected and
lysed in a Laemmli sample buffer (Sigma-Aldrich) and were subjected
to immunoanalysis. Protein concentrations were assessed using a DC
protein assay kit (Bio-Rad) according to manufacturer's
instructions. Sample protein (50 μg) was subjected to
SDS-polyacrylamide gel electrophoresis. After migration, proteins
were transferred to nitrocellulose membranes and incubated with 5%
non-fat milk (Bio-Rad). The membranes were exposed to anti-LC3
(microtubule-associated protein 1A/1B-light chain 3) antibody
diluted 1:400 (Novus Biologicals, Littleton, CO, USA) overnight at
4°C. Membranes were then washed three times with PBS/Tween-20 and
exposed to horseradish peroxidase-conjugated goat anti-rabbit
anti-IgG (H+L) secondary antibodies (Bio-Rad). The antigen-antibody
complex was visualized using chemiluminescence by Immun-Star HRP
Substrate kit (Bio-Rad). Antibodies against actin (1:1,000;
Sigma-Aldrich) were used as loading control.
DNA isolation from neuroblastoma cells
and 32P-postlabeling of ellipticine-DNA adducts
Neuroblastoma cell lines were seeded 24 h prior to
treatment at a density of 5×105 cells/ml in two 75
cm2 culture flasks in a total volume of 20 ml of IMDM.
After 24 h incubations with 5 μM ellipticine in IMDM, the cells
were harvested after trypsinizing by centrifugation at 2000 × g for
3 min and two washing steps with 5 ml of PBS yielded a cell pellet,
which was stored at −80°C until DNA isolation. An analogous
procedure was used to evaluate the effect of treatment of
neuroblastoma cells with bafilomycin A or chloroquine before adding
ellipticine. Cells were treated with 100 nM bafilomycin A or 25 μM
chloroquine for 20 min before adding ellipticine. DNA from
neuroblastoma cells treated with 5 μM ellipticine in the presence
or absence of 100 nM bafilomycin A and/or 25 μM chloroquine for 24
h was isolated by the phenol-chloroform extraction as described
(8,9,13,27,28).
The 32P-postlabeling of nucleotides using nuclease P1
enrichment, found previously to be appropriate to detect and
quantify ellipticine-derived DNA adducts formed in vitro
(12,13,27–30)
and in vivo (6,7,31–33)
was used.
Statistical analysis
Data are expressed as mean ± standard deviations
(SD). Student's t-test (two-tailed) was used for statistical
analysis. P-values <0.05 were considered statistically
significant, and are indicated in the figures as
*P<0.05, **P<0.01 and
***P<0.001.
Results
Ellipticine induces cytoplasmic
vacuolization in neuroblastoma cells and accumulates in these
vacuoles
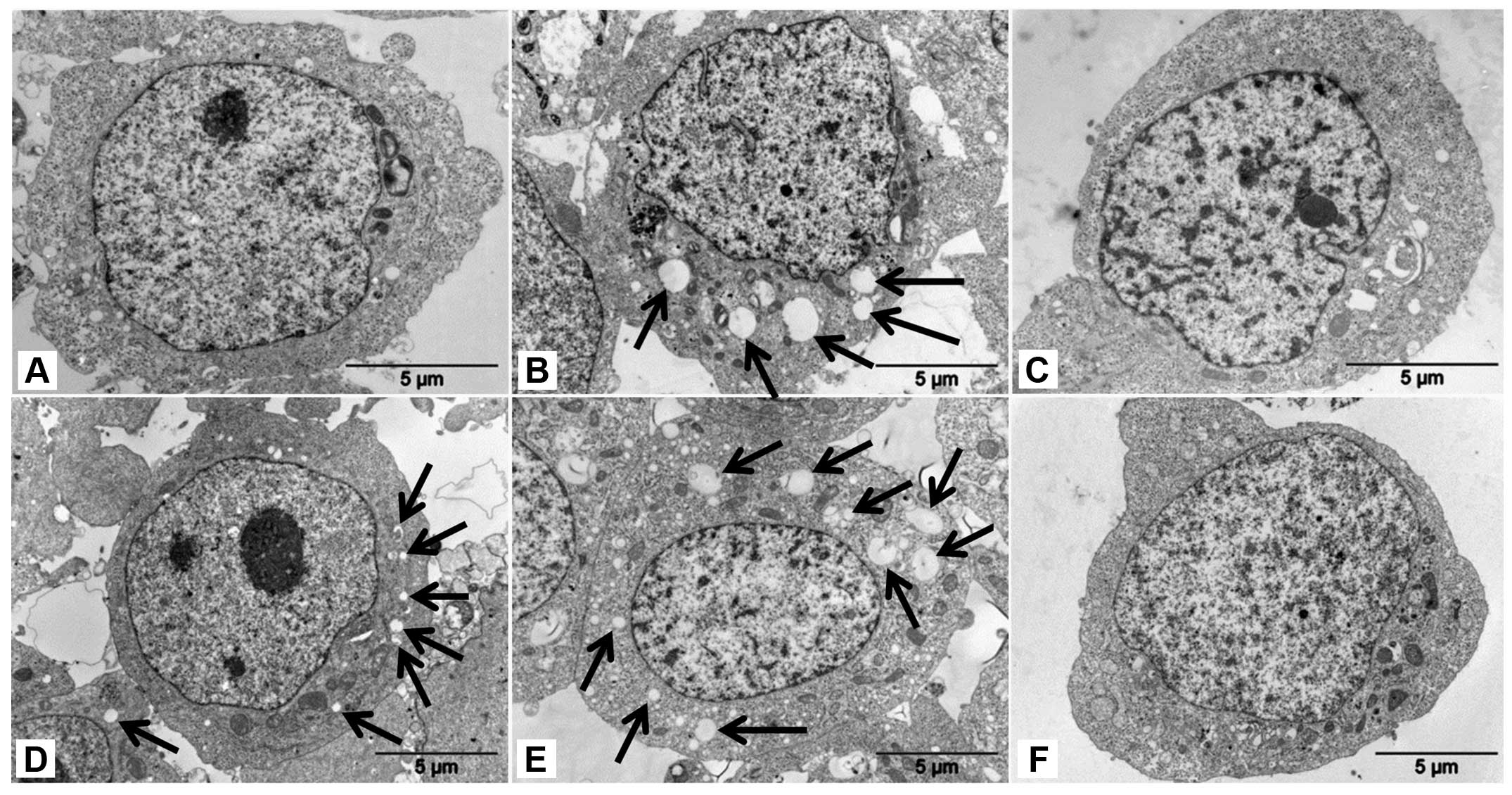
Treatment of neuroblastoma UKF-NB-4 cells, both
sensitive and resistant (UKF-NB-4ELLI) to ellipticine,
with ellipticine at concentrations that are toxic to these cells (5
μM) induced extensive cytoplasmic vacuolization in these cells
(vacuoles are indicated by arrows in Fig. 1B and E). The higher number of these
vacuoles was generated in UKF-NB-4ELLI cells resistant
to ellipticine than in the parent UKF-NB-4 cell line. The vacuolar
vesicles of a small size were also present in the
UKF-NB-4ELLI cell line prepared by cultivation of
UKF-NB-4 cells with increasing concentrations of ellipticine (from
0.2 to 2.5 μM) over 36 months (17) (vesicles are indicated by arrows in
Fig. 1D). The vacuoles were
already detectable 30 min after adding the ellipticine (data not
shown). This ellipticine-mediated cytoplasmic vacuolization seems
to be a general phenomenon, because it appears also in the
neuroblastoma cell lines SK-N-AS and UKF-NB-3 (data not shown).
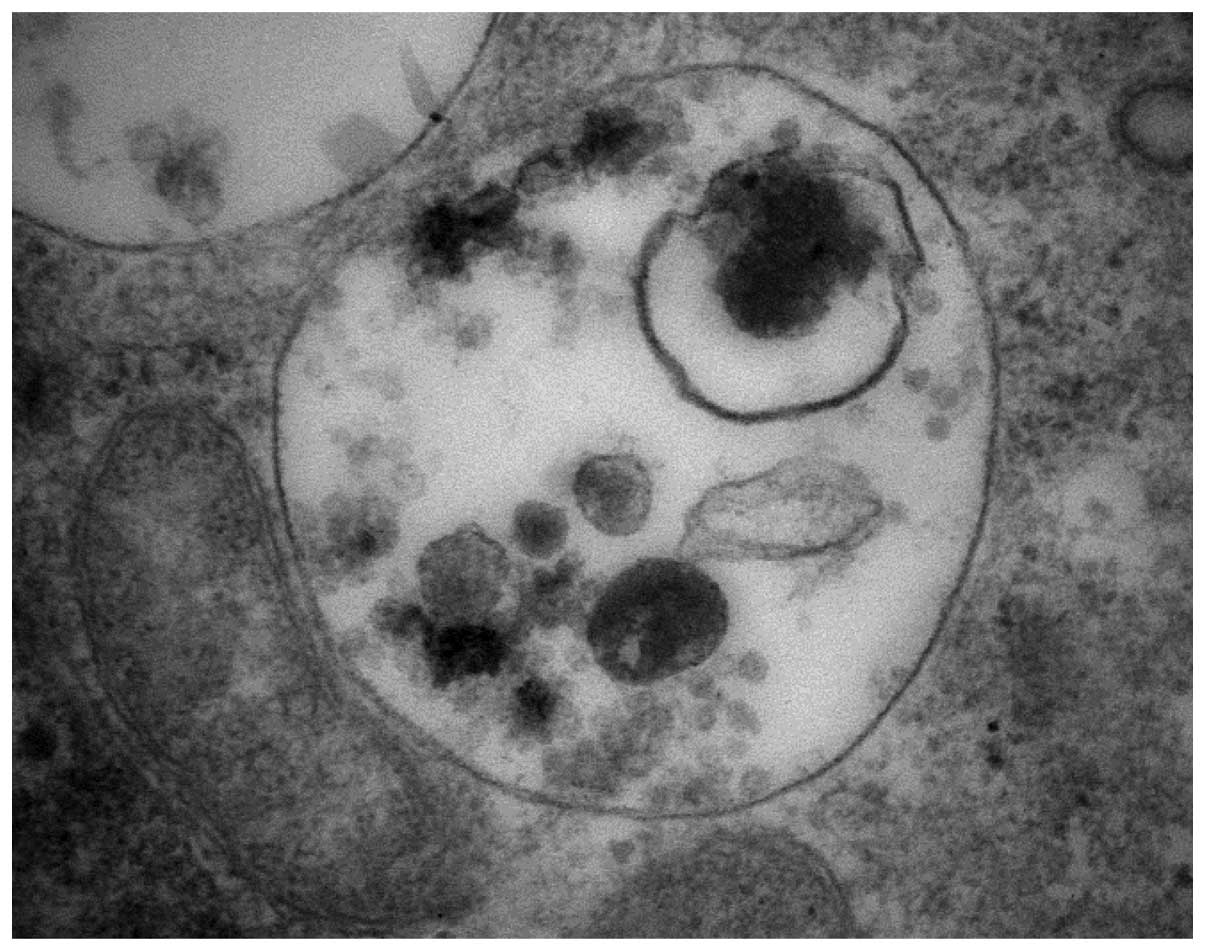
Under the electron microscope, ellipticine-induced vacuoles were
found to be electron-lucent and to contain some heterogeneous
material (the darker structures in a vacuole shown in Fig. 2). They, however, lacked any
detectable content of cytoplasmic material (organelles) and were
lined by a single membrane (Fig.
2), ruling out autophagy. Nevertheless, in order to
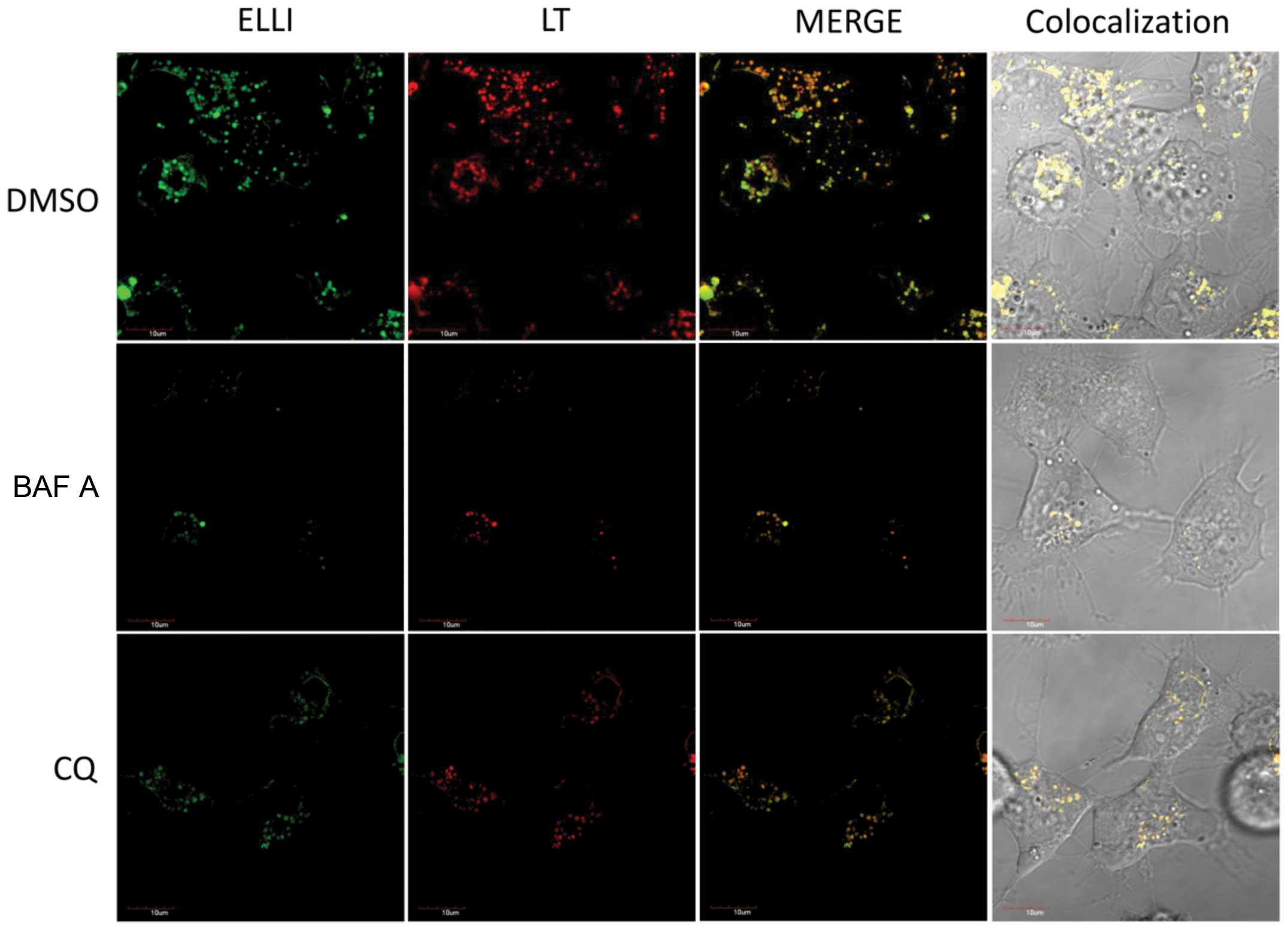
characterize the vacuoles further, we used confocal microscopy of
cells stained with two specific lysosomal markers,
lysosomal-associated membrane protein 1 (LAMP1) (34) and a lysosomal marker selective for
acidic compartments, LysoTracker® Red (35). Unfortunately, the use of LAMP1 as a
marker failed in our experiments, because LAMP1 could not be
applied simultaneously with ellipticine. The anti-LAMP1 is namely
used on fixed cells and fixation interferes with ellipticine
detection (data not shown). The results found using confocal
microscopy of cells stained with LysoTracker® Red
(Fig. 3) and the finding that the
ellipticine-induced vacuoles are single membrane vesicles (Figs. 1 and 2) suggested that these vacuoles are
lysosomes.
The green fluorescence of ellipticine (excitation,
440 nM; emission, 520 nm) (10)
allowed the detection of its intracellular localization. At
physiological pH, ellipticine exists in both protonated (charged)
and unprotonated (uncharged) forms (7). As shown in Fig. 3, the UKF-NB-4 cells exposed to
ellipticine contained ellipticine-specific green fluorescent
vesicles where ellipticine is accumulated. Some of the vesicles
where ellipticine was present colocalized with a lysosomal marker
LysoTracker® Red (Fig.
3). Hence, ellipticine as a protonated chemical is trapped in
these vesicles formed in the cells. This may be caused by the pKa
value of this compound and the pH gradient between cytoplasm and
acidic vacuoles developed by ellipticine. Namely, ellipticine has a
pKa of ~6, and can be protonated in a weakly acidic environment
(7,36,37).
The trapping of ellipticine in these acidic vesicles is followed by
osmotic swelling and dilatation (Fig.
1).
A contribution of V-ATPase to ellipticine-induced
vacuolation and ellipticine sequestration into these vacuoles was
investigated with its specific inhibitor, bafilomycin A (38,39)
and the lysosomotropic drug chloroquine, the agent that enters
selectively the lysosomes and inhibits enzymes for which the acidic
pH is crucial (40).
Ellipticine-induced vacuolation and intravesicular
ellipticine-associated fluorescence were abolished by co-treatment
of tested neuroblastoma cells with bafilomycin A and chloroquine
(Figs. 1 and 3). These results suggest that ellipticine
is responsible for the V-ATPase-mediated formation of cytoplasmic
vacuoles (i.e. lysosomes) in these neuroblastoma cells, and that is
able to be sequestrated into these acidic compartments.
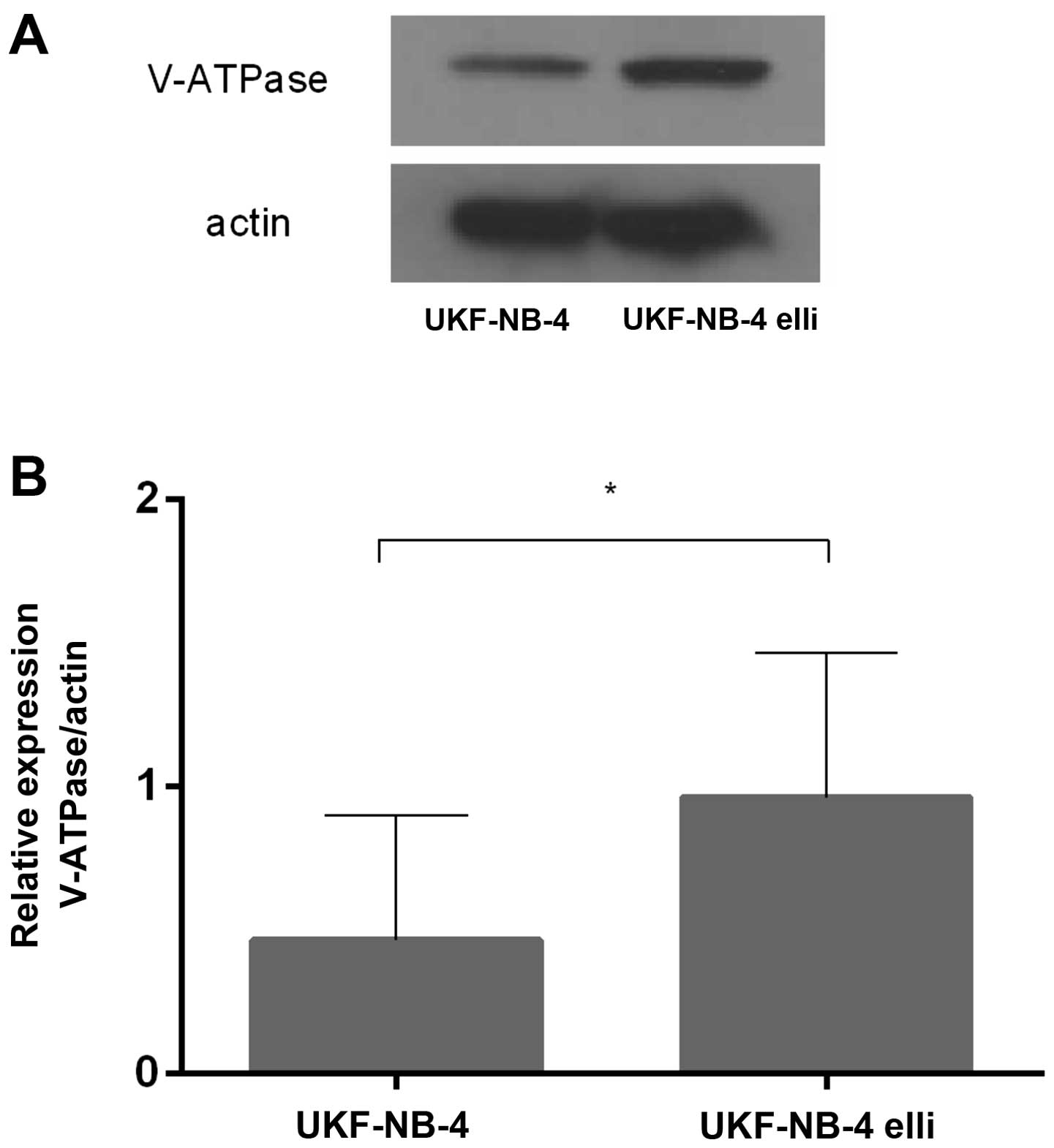
Expression of V-ATPase in the ellipticine
sensitive and resistant UKF-NB-4 cells
Because of the suspected role of upregulation of the
V-ATPase gene in induction of resistance of UKF-NB-4 cells
to ellipticine (17), we further
investigated expression of this enzyme both in the ellipticine
sensitive and resistant UKF-NB-4 cells. Using western blot
analyses, expression of a protein product of ATP6V0D1, the
gene of the V-ATPase membrane domain, which is upregulated in
several drug-resistant cell lines including UKF-NB-4ELLI
(17,38–43),
was measured in the tested cells. As shown in Fig. 4, the V-ATPase (ATP6V0D) protein
levels were 2.3-times higher in the resistant
UKF-NB-4ELLI cell line than in its parental sensitive
line. These results are in agreement with previous finding which
demonstrated upregulation of the ATP6V0D1 gene in
ellipticine-resistant neuroblastoma cells (17), and point out its importance for
acquiring resistance to ellipticine.
Treatment of neuroblastoma cells with
bafilomycin A or chloroquine increases the cytotoxic effects of
ellipticine and decreases their resistance to ellipticine
The UKF-NB-4 and UKF-NB-4ELLI cell lines
were treated with either ellipticine alone or after pretreatment
with bafilomycin A or chloro-quine. The cytotoxic effects of
ellipticine to neuroblastoma cells in the presence or absence of
these inhibitors were analyzed by two methods: i) by detection of
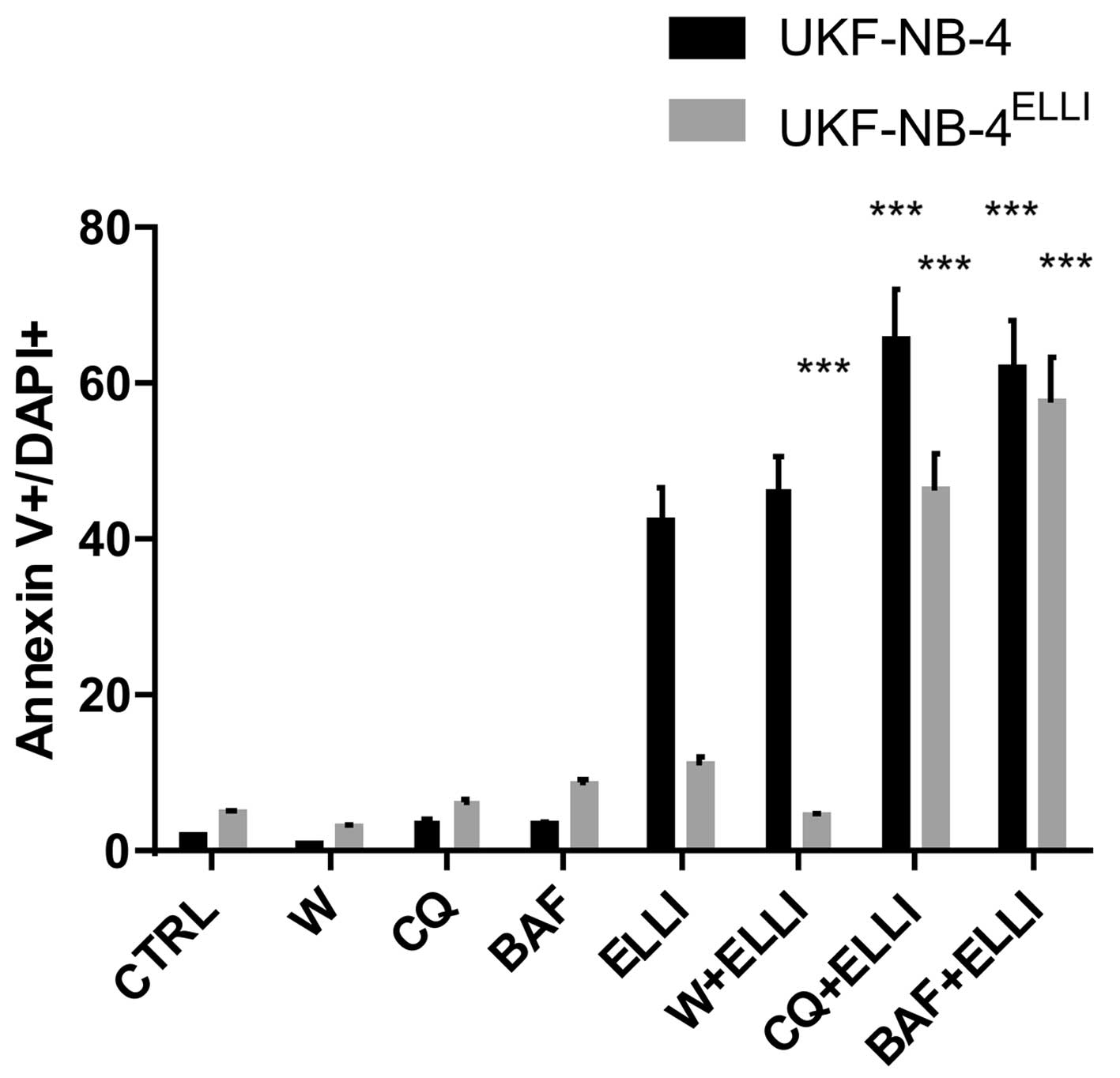
apoptosis in the cells using Annexin V/DAPI labeling (Fig. 5) and ii) by MTS assay (Table I). Treatment of neuroblastoma cells
with bafilomycin A or chloroquine did not induce apoptosis in these
cells (Fig. 5). However,
pretreatment of the cells with these compounds enhanced markedly
the ellipticine-mediated apoptosis induction in both the sensitive
and ellipticine-resistant neuroblastoma cells and decreased the
resistance of UKF-NB-4ELLI cells to ellipticine
(Fig. 5). In addition,
pretreatment of cells with bafilomycin A and/or chloroquine was
able to reduce the values of IC50 both in the
ellipticine-sensitive and ellipticine-resistant cell lines to the
lower IC50 values (Table
I). These results demonstrate that a decrease in sensitivity of
neuroblastoma cells to ellipticine is indeed caused by the potency
of this drug to induce the formation of acidified vesicles having
the lysosomal character in these cells, which additionally trapped
the protonated ellipticine, thereby decreasing its cytotoxic
effects. They also strongly support the suggestion that these
processes participated in ellipticine-induced resistance of
UKF-NB-4 cells.
 | Table IThe effect of bafilomycin A,
chloroquine and wortmannin on the IC50 values for
ellipticine in ellipticine-sensitive UKF-NB-4 and
ellipticine-resistant UKF-NB-4ELLI neuroblastoma cell
lines. |
Table I
The effect of bafilomycin A,
chloroquine and wortmannin on the IC50 values for
ellipticine in ellipticine-sensitive UKF-NB-4 and
ellipticine-resistant UKF-NB-4ELLI neuroblastoma cell
lines.
| IC50 for
ellipticine (μM) |
|---|
|
|
|---|
| Compound | UKF-NB-4 cells |
UKF-NB-4ELLI cells |
|---|
| Ellipticine | 0.86±0.007 |
1.42±0.004c |
| Ellipticine + 100
nM bafilomycin A |
0.21±0.006a |
0.69±0.014a,c |
| Ellipticine + 25 μM
chloroquine |
0.19±0.010a |
0.35±0.012a,c |
| Ellipticine + 100
nM wortmannin |
1.02±0.005b |
1.39±0.014c |
Nevertheless, it should be noted that bafilomycin A
and chloroquine act not only as the inhibitors of lysosomal
proteases, but that they can also partially prevent maturation of
autophagic vacuoles. They, namely, also inhibit fusion between
autophagosomes and lysosomes, because they are inhibitors of the
late phase of autophagy (40).
Hence, their augmented effects may be caused also by authophagy
inhibition. Here, we examined this possibility, namely, whether
their potentiating effect on ellipticine-mediated cytotoxicity to
neuroblastoma cells is related to autophagy inhibition. For such a
study, we used the inhibitor of phosphatidylinositol 3-kinase
(PI3K) wortmannin (44,45), since, as an inhibitor of this
enzyme (44,45), it dictates the autophagy
development in cells (46). In
contrast to bafilomycin A and chloroquine, wortmannin had no effect
on induction of apoptosis in neuro-blastoma cells exposed to
ellipticine (Fig. 5). It did not
reduce the value of IC50 for ellipticine in these cells
(Table I). These findings
demonstrate that the bafilomycin A- and chloroquine-mediated
increase in cytotoxicity and induction of apoptosis caused by
ellipticine determined in this study are not related to
autophagy.
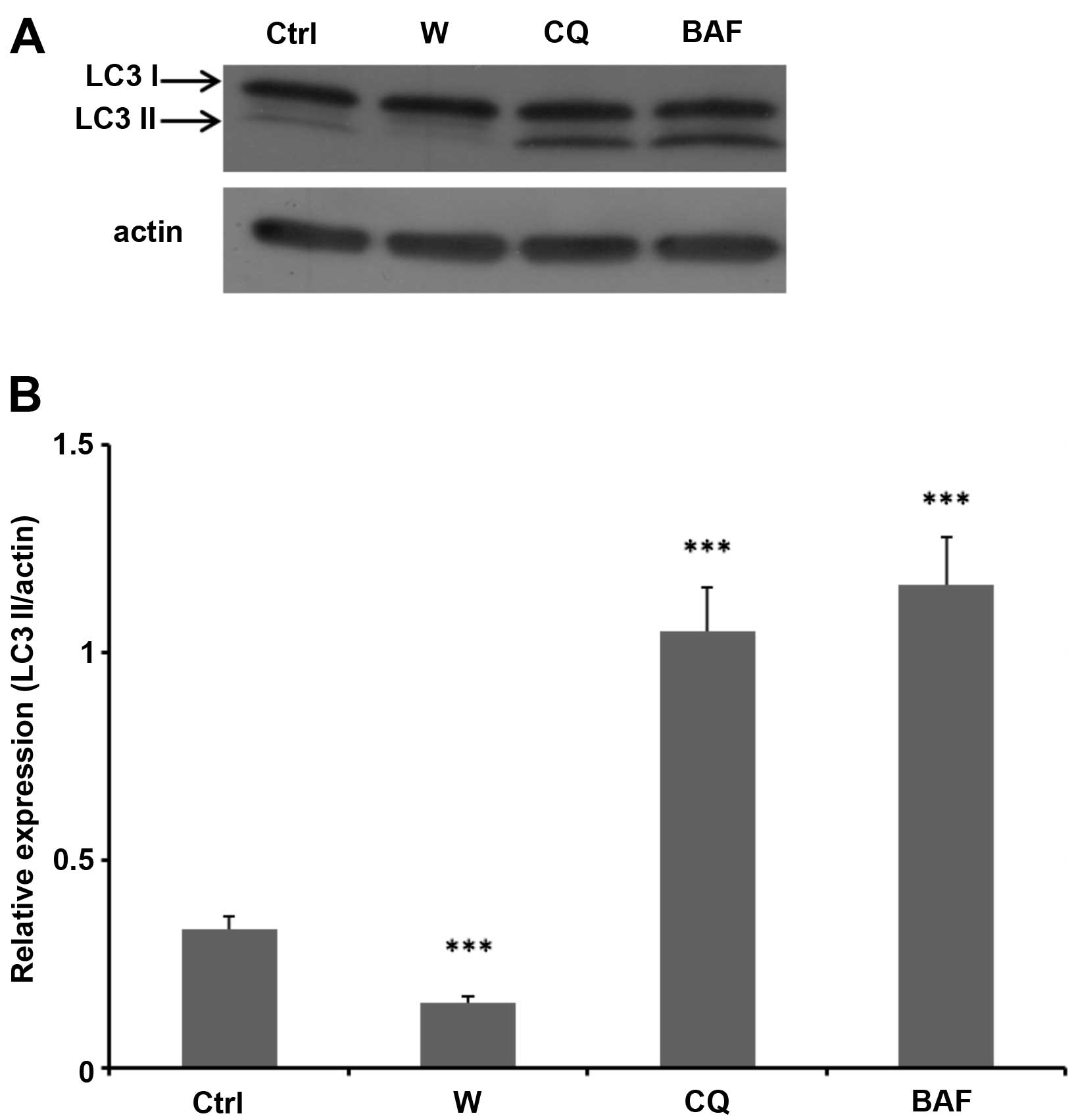
Effectiveness of autophagy inhibitors in tested
lines was also investigated by examining the expression of an
autophagosomal marker protein LC3-II (Fig. 6), the protein that is highly
expressed in both membranes of autophagosomes. Lysosomal turnover
of the autophagosomal marker LC3-II namely reflects autophagic
activity, and therefore determination of levels of LC3-II is
considered as a method suitable to monitor the autophagy process
(47). In our experiments,
autophagy in neuroblastoma cells was induced by their starvation
and proved by expression of LC3-II in these cells (Fig. 6). High expression of LC-II in these
cells were also induced by bafilomycin A and chloroquine (Fig. 6) because both these compounds as
inhibitors of proteolytic processes in the lysosomes (38–40)
increased lysosomal pH that consequently led to decreased activity
of lysosomal proteases. These processes blocked lysosomal
degradation and rescued intact LC3-II in neuroblastoma cells
(Fig. 6). In contrast, wortmannin
as a blocker of autophagosome formation decreased the expression of
LC3-II induced by starvation (Fig.
6). This finding again suggests that the increase in
ellipticine-mediated cytotoxicity and induction of apoptosis by
ellipticine due to bafilomycin A and chloroquine in neuroblastoma
cells are not related to autophagy.
Treatment of neuroblastoma cells with
bafilomycin A and chloroquine prior to ellipticine increases the
formation of covalent ellipticine-derived DNA adducts
Since formation of covalent DNA adducts of
ellipticine is one of the major modes of ellipticine action in
various cancer cells including neuro-blastoma (6–9,11,12,28,29),
we investigated whether treatment of UKF-NB-4 and
UKF-NB-4ELLI cells with bafilomycin A or chloroquine
prior to ellipticine changes DNA adduct levels. Two major DNA
adducts identical to those formed by the ellipticine metabolites,
13-hydroxy- and 12-hydroxyellipticine, with deoxyguanosine in DNA
(13,15), and two minor DNA adducts of unknown
structures were detected in neuroblastoma cells treated with
ellipticine. The levels of the ellipticine-DNA adducts were lower
in a resistant cell line (Fig. 7
and Table II), as it has already
been found in our previous study (8). However, treatment with either
bafilomycin A or chloroquine prior to ellipticine significantly
increased levels of ellipticine-DNA adducts in both cell lines
(Fig. 7 and Table II). This corresponded to enhanced
cytotoxic effects of ellipticine on these cells (Fig. 5). These results indicate that
bafilomycin A- and chloroquine-mediated inhibition of ellipticine
sequestration into vacuoles led to higher concentrations of
ellipticine in cytoplasm and nuclei to be activated to species
forming covalent DNA adducts.
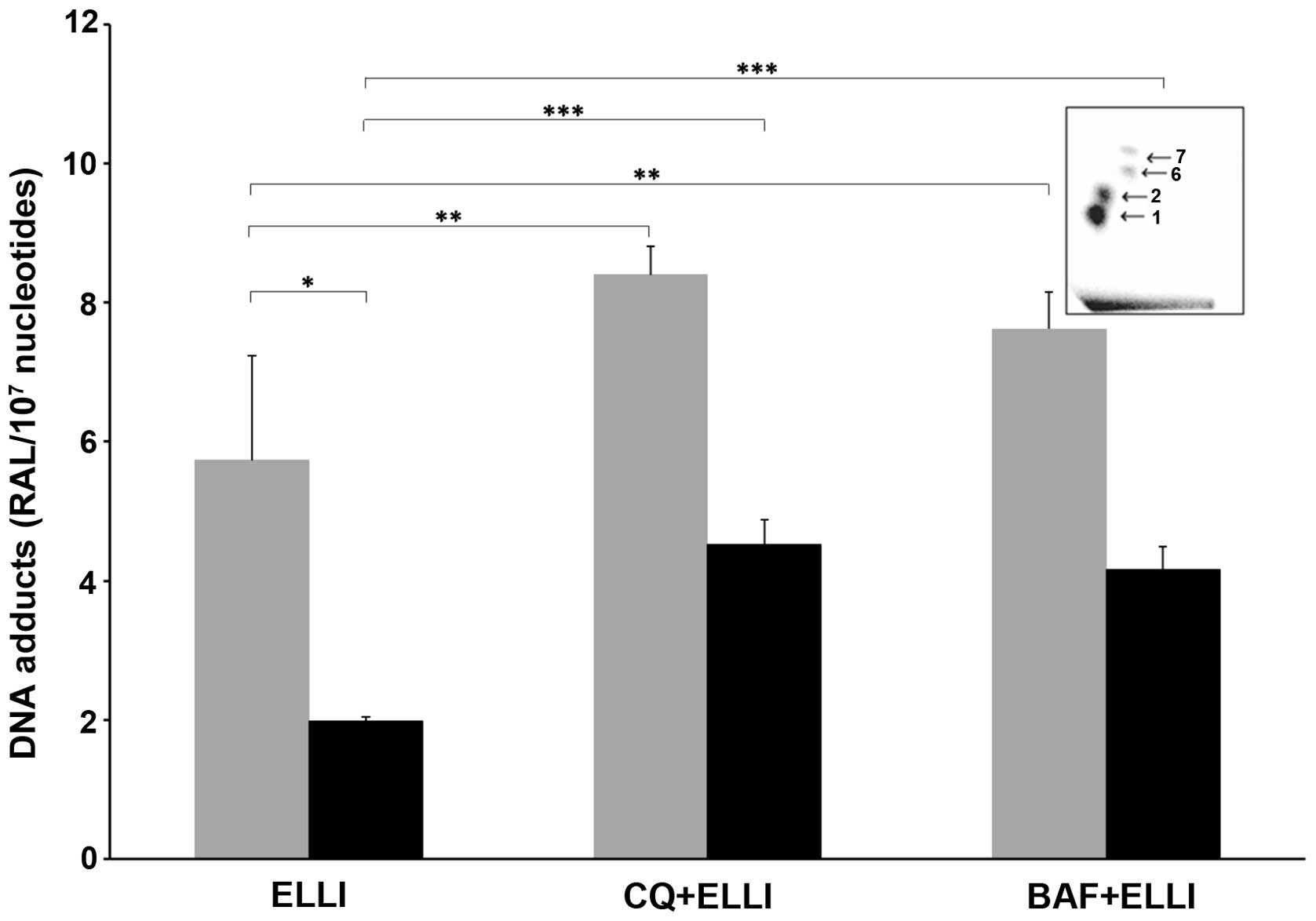 | Figure 7Levels of covalent DNA adducts (sum
of adducts 1, 2, 6 and 7 shown in insert) formed in UKF-NB-4 (grey
columns) and UKF-NB-4ELLI (black columns) neuroblastoma
cells after 24 h treatment with ellipticine (ELLI, 5 μM) either
without pretreatment or pretreatment with bafilomycin A (BAF, 100
nM) or chloroquine (CQ, 25 μM) (the cells were pretreated with
bafilomycin A and/or chloroquine for 20 min before adding
ellipticine and further incubated 24 h). The data represent means
of total levels of ellipticine-DNA adducts and standard deviations
determined from three independent experiments. Values of relative
adduct labeling (RAL) are expressed as adducts/107
normal nucleotides. Asterisks represent statistical significance as
calculated by Student's t-test (*P<0.05,
**P<0.01 and ***P<0.001). Insert, an
autoradiographic profile of ellipticine-DNA adducts formed in
UKF-NB-4 cells determined by 32P-postlabeling. The
adduct spots 1 and 2 are formed in deoxyguanosine residues of DNA
by the ellipticine metabolites, 13-hydroxy- and
12-hydroxyellipticine (6,7,13,15). |
| Table IIDNA adduct formation by ellipticine
in UKF-NB-4 and UKF-NB-4ELLI cell lines. |
Table II
DNA adduct formation by ellipticine
in UKF-NB-4 and UKF-NB-4ELLI cell lines.
| RAL (mean ±
SD/107 nucleotides)a |
|---|
|
|
|---|
| Cells | Adduct 1b | Adduct 2b | Adduct 6b | Adduct 7b | Total |
|---|
| UKF-NB-4 |
| ELLI | 2.92±0.85 | 1.75±0.78 | 0.55±0.03 | 0.51±0.005 | 5.73±1.51 |
| BAF + ELLI | 4.3±0.67c | 2.03±0.12d | 0.69±0.03c | 0.60±0.03c | 7.62±0.41c |
| CQ + ELLI | 4.08±0.72c | 2.64±0.14e | 0.91±0.04e | 0.77±0.4e | 8.4±0.53c |
|
UKF-NB-4ELLI |
| ELLI | 1.02±0.01g | 0.89±0.01g | 0.5±0.01 | 0.03±0.01g | 1.99±0.06g |
| BAF + ELLI | 1.69±0.21c,g | 1.65±0.07e,f | 0.6±0.03c | 0.22±0.01e,g | 4.16±0.36e,f |
| CQ + ELLI | 1.78±0.1e,g | 1.82±0.11e,f | 0.51±0.03f | 0.41±0.02e,g | 4.52±0.33e,g |
Discussion
The results found in this study demonstrate for the
first time that sequestration of anticancer drug ellipticine into
the subcellular compartments (i.e. lysosomes) of UKF-NB-4
neuroblastoma cells is one of the mechanisms contributing to the
development of ellipticine-resistance in these cells. Such
processes finally result in a decrease in ellipticine cytotoxic
effects (8,17). We demonstrated that this resistance
is, among other mechanisms, dependent on upregulation of the
V-ATPase gene (17).
Indeed, here we found that the V-ATPase protein expression is
enhanced in the ellipticine-resistant UKF-NB-4ELLI cell
line.
Since V-ATPase is the major enzyme responsible for
the acidification of subcellular compartments, it acidifies newly
formed cytoplasmic vacuolar vesicles by pumping protons across the
membranes (19–21). This process is a necessary step for
additional sequestration of the protonated form of ellipticine
within these organelles. Finally, this sequestration results in
lower cytoplasmic concentrations of ellipticine, less nuclear
accumulation (17) and lower DNA
damage by ellipticine (Table II
and Fig. 7) and therefore also
lower toxic effects to these cells (Table I and Fig. 5) and our previous study (8). The formation of covalent
ellipticine-derived DNA adducts, which was found to be lower in
ellipticine-resistant UKF-NB-4ELLI cells, was increased
by the inhibitor of V-ATPase, bafilomycin A, and/or the
lysosomotropic drug chloroquine that blocks formation of lysosomes
(48). In concordance to these
results, exposure of the tested cells to bafilomycin A and
chloroquine enhanced markedly the cytotoxicity of ellipticine on
these cells and decreased resistance of UKF-NB-4ELLI to
ellipticine.
Based on these results, we can conclude that the
decrease in ellipticine-mediated cytotoxicity on UKF-NB-4 cells as
well as in induction of resistance to ellipticine in the
ellipticine-resistant UKF-NB-4ELLI cell line is
associated with vacuolar trapping of this drug, which may be
abolished by bafilomycin A or by chloroquine. Therefore,
therapeutic implications could be derived from this study. In
principle, the components of the endocytic/lysosomal pathway could
be molecular targets for a combination therapy of neuroblastoma
with chemotherapeutic drugs and probably also for that of other
cancers.
Acknowledgements
This study was supported by GACR (grants
P301/10/0356 and 14-8344S), Charles University in Prague
(UNCE204025/2012) and by the Ministry of Health of the Czech
Republic for conceptual development of research organization
00064203 (University Hospital Motol, Prague, Czech Republic).
References
|
1
|
Brodeur GM: Neuroblastoma: Biological
insights into a clinical enigma. Nat Rev Cancer. 3:203–216. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schwab M, Westermann F and Hero Band
Berthold F: Neuroblastoma: Biology and molecular and chromosomal
pathology. Lancet Oncol. 4:472–480. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Morgenstern BZ, Krivoshik AP, Rodriguez V
and Anderson PM: Wilms' tumor and neuroblastoma. Acta Paediatr
(Suppl). 93:78–85. 2004. View Article : Google Scholar
|
|
4
|
Kotchetkov R, Driever PH, Cinatl J,
Michaelis M, Karaskova J, Blaheta R, Squire JA, Von Deimling A,
Moog J and Cinatl J Jr: Increased malignant behavior in
neuroblastoma cells with acquired multi-drug resistance does not
depend on P-gp expression. Int J Oncol. 27:1029–1037.
2005.PubMed/NCBI
|
|
5
|
Michaelis M, Klassert D, Barth S, Suhan T,
Breitling R, Mayer B, Hinsch N, Doerr HW, Cinatl J and Cinatl J Jr:
Chemoresistance acquisition induces a global shift of expression of
aniogenesis-associated genes and increased pro-angogenic activity
in neuroblastoma cells. Mol Cancer. 8:802009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stiborová M, Rupertová M and Frei E:
Cytochrome P450- and peroxidase-mediated oxidation of anticancer
alkaloid ellipticine dictates its anti-tumor efficiency. Biochim
Biophys Acta. 1814:175–185. 2011. View Article : Google Scholar
|
|
7
|
Stiborová M and Frei E: Ellipticines as
DNA-targeted chemo-therapeutics. Curr Med Chem. 21:575–591. 2014.
View Article : Google Scholar
|
|
8
|
Poljaková J, Eckschlager T, Hrabeta J,
Hrebacková J, Smutný S, Frei E, Martínek V, Kizek R and Stiborová
M: The mechanism of cytotoxicity and DNA adduct formation by the
anticancer drug ellipticine in human neuroblastoma cells. Biochem
Pharmacol. 77:1466–1479. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Poljakova J, Hrebackova J, Dvorakova M,
Moserova M, Eckschlager T, Hrabeta J, Göttlicherova M, Kopejtkova
B, Frei E, Kizek R, et al: Anticancer agent ellipticine combined
with histone deacetylase inhibitors, valproic acid and trichostatin
A, is an effective DNA damage strategy in human neuroblastoma.
Neuro Endocrinol Lett. 32(Suppl 1): 101–116. 2011.PubMed/NCBI
|
|
10
|
Stiborová M, Eckschlager T, Poljaková J,
Hrabĕta J, Adam V, Kizek R and Frei E: The synergistic effects of
DNA-targeted chemotherapeutics and histone deacetylase inhibitors
as therapeutic strategies for cancer treatment. Curr Med Chem.
19:4218–4238. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stiborova M, Poljakova J, Mrizova I, et
al: Expression levels of enzymes metabolizing an anticancer drug
ellipticine determined by electromigration assays influence its
cytotoxicity to cancer cells - a comparative study. Int J
Electrochem Sci. 9:5675–5689. 2014.
|
|
12
|
Stiborová M, Bieler CA, Wiessler M and
Frei E: The anticancer agent ellipticine on activation by
cytochrome P450 forms covalent DNA adducts. Biochem Pharmacol.
62:1675–1684. 2001. View Article : Google Scholar
|
|
13
|
Stiborová M, Sejbal J, Borek-Dohalská L,
Aimová D, Poljaková J, Forsterová K, Rupertová M, Wiesner J,
Hudecek J, Wiessler M, et al: The anticancer drug ellipticine forms
covalent DNA adducts, mediated by human cytochromes P450, through
metabolism to 13-hydroxyellipticine and ellipticine N2-oxide.
Cancer Res. 64:8374–8380. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stiborova M, Rupertova M, Schmeiser HH and
Frei E: Molecular mechanisms of antineoplastic action of an
anticancer drug ellipticine. Biomed Pap Med Fac Univ Palacky
Olomouc Czech Repub. 150:13–23. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stiborová M, Poljaková J, Ryslavá H,
Dracínský M, Eckschlager T and Frei E: Mammalian peroxidases
activate anticancer drug ellipticine to intermediates forming
deoxyguanosine adducts in DNA identical to those found in vivo and
generated from 12-hydroxyellipticine and 13-hydroxyellipticine. Int
J Cancer. 120:243–251. 2007. View Article : Google Scholar
|
|
16
|
Kizek R, Adam V, Hrabeta J, Eckschlager T,
Smutny S, Burda JV, Frei E and Stiborova M: Anthracyclines and
ellipticines as DNA-damaging anticancer drugs: Recent advances.
Pharmacol Ther. 133:26–39. 2012. View Article : Google Scholar
|
|
17
|
Procházka P, Libra A, Zemanová Z,
Hřebačková J, Poljaková J, Hrabĕta J, Bunček M, Stiborová M and
Eckschlager T: Mechanisms of ellipticine-mediated resistance in
UKF-NB-4 neuroblastoma cells. Cancer Sci. 103:334–341. 2012.
View Article : Google Scholar
|
|
18
|
Marshansky V, Rubinstein JL and Grüber G:
Eukaryotic V-ATPase: Novel structural findings and functional
insights. Biochim Biophys Acta. 1837:857–879. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Maxfield FR and McGraw TE: Endocytic
recycling. Nat Rev Mol Cell Biol. 5:121–132. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Goldman SDB, Funk RS, Rajewski RA and
Krise JP: Mechanisms of amine accumulation in, and egress from,
lysosomes. Bioanalysis. 1:1445–1459. 2009. View Article : Google Scholar
|
|
21
|
Coutinho MF, Prata MJ and Alves S:
Mannose-6-phosphate pathway: A review on its role in lysosomal
function and dysfunction. Mol Genet Metab. 105:542–550. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Simon S, Roy D and Schindler M:
Intracellular pH and the control of multidrug resistance. Proc Natl
Acad Sci USA. 91:1128–1132. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mahoney BP, Raghunand N, Baggett B and
Gillies RJ: Tumor acidity, ion trapping and chemotherapeutics. I
Acid pH affects the distribution of chemotherapeutic agents in
vitro. Biochem Pharmacol. 66:1207–1218. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chapuy B, Koch R, Radunski U, Corsham S,
Cheong N, Inagaki N, Ban N, Wenzel D, Reinhardt D, Zapf A, et al:
Intracellular ABC transporter A3 confers multidrug resistance in
leukemia cells by lysosomal drug sequestration. Leukemia.
22:1576–1586. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Spugnini EP, Citro G and Fais S: Proton
pump inhibitors as anti vacuolar-ATPases drugs: A novel anticancer
strategy. J Exp Clin Cancer Res. 29:442010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yamagishi T, Sahni S, Sharp DM, Arvind A,
Jansson PJ and Richardson DR: P-glycoprotein mediates drug
resistance via a novel mechanism involving lysosomal sequestration.
J Biol Chem. 288:31761–31771. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Frei E, Bieler CA, Arlt VM, Wiessler M and
Stiborová M: Covalent binding of the anticancer drug ellipticine to
DNA in V79 cells transfected with human cytochrome P450 enzymes.
Biochem Pharmacol. 64:289–295. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Poljaková J, Frei E, Gomez JE, Aimová D,
Eckschlager T, Hrabeta J and Stiborová M: DNA adduct formation by
the anticancer drug ellipticine in human leukemia HL-60 and
CCRF-CEM cells. Cancer Lett. 252:270–279. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Martinkova E, Dontenwill M, Frei E and
Stiborova M: Cytotoxicity of and DNA adduct formation by
ellipticine in human U87MG glioblastoma cancer cells. Neuro
Endocrinol Lett. 30(Suppl 1): 60–66. 2009.PubMed/NCBI
|
|
30
|
Martinkova E, Maglott A, Leger DY, Bonnet
D, Stiborova M, Takeda K, Martin S and Dontenwill M: alpha5beta1
integrin antagonists reduce chemotherapy-induced premature
senescence and facilitate apoptosis in human glioblastoma cells.
Int J Cancer. 127:1240–1248. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Stiborová M, Breuer A, Aimová D,
Stiborová-Rupertová M, Wiessler M and Frei E: DNA adduct formation
by the anticancer drug ellipticine in rats determined by
32P postlabeling. Int J Cancer. 107:885–890. 2003.
View Article : Google Scholar
|
|
32
|
Stiborová M, Rupertová M, Aimová D,
Ryslavá H and Frei E: Formation and persistence of DNA adducts of
anticancer drug ellipticine in rats. Toxicology. 236:50–60. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Stiborová M, Arlt VM, Henderson CJ, Wolf
CR, Kotrbová V, Moserová M, Hudecek J, Phillips DH and Frei E: Role
of hepatic cytochromes P450 in bioactivation of the anticancer drug
ellipticine: studies with the hepatic NADPH:cytochrome P450
reductase null mouse. Toxicol Appl Pharmacol. 226:318–327. 2008.
View Article : Google Scholar
|
|
34
|
Hunziker W and Geuze HJ: Intracellular
trafficking of lysosomal membrane proteins. Bioessays. 18:379–389.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Oeste CL, Seco E, Patton WF, Boya P and
Pérez-Sala D: Interactions between autophagic and endo-lysosomal
markers in endothelial cells. Histochem Cell Biol. 139:659–670.
2013. View Article : Google Scholar
|
|
36
|
Garbett NC and Graves DE: Extending
nature's leads: The anti-cancer agent ellipticine. Curr Med Chem
Anticancer Agents. 4:149–172. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wu Y, Sadatmousavi P, Wang R, Lu S, Yuan
YF and Chen P: Self-assembling peptide-based nanoparticles enhance
anticancer effect of ellipticine in vitro and in vivo. Int J
Nanomed. 7:3221–3233. 2012.
|
|
38
|
Bowman EJ, Siebers A and Altendorf K:
Bafilomycins: A class of inhibitors of membrane ATPases from
microorganisms, animal cells, and plant cells. Proc Natl Acad Sci
USA. 85:7972–7976. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huss M and Wieczorek H: Inhibitors of
V-ATPases: Old and new players. J Exp Biol. 212:341–346. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shacka JJ, Klocke BJ, Shibata M, Uchiyama
Y, Datta G, Schmidt RE and Roth KA: Bafilomycin A1 inhibits
chloroquine-induced death of cerebellar granule neurons. Mol
Pharmacol. 69:1125–1136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Martínez-Zaguilán R, Raghunand N, Lynch
RM, Bellamy W, Martinez GM, Rojas B, Smith D, Dalton WS and Gillies
RJ: pH and drug resistance. I Functional expression of plasmalemmal
V-type H+-ATPase in drug-resistant human breast
carcinoma cell lines. Biochem Pharmacol. 57:1037–1046. 1999.
View Article : Google Scholar
|
|
42
|
Murakami T, Shibuya I, Ise T, Chen ZS,
Akiyama S, Nakagawa M, Izumi H, Nakamura T, Matsuo K, Yamada Y, et
al: Elevated expression of vacuolar proton pump genes and cellular
PH in cisplatin resistance. Int J Cancer. 93:869–874. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Huang L, Lu Q, Han Y, Li Z, Zhang Z and Li
X: ABCG2/V-ATPase was associated with the drug resistance and tumor
metastasis of esophageal squamous cancer cells. Diagn Pathol.
7:1802012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wymann MP, Bulgarelli-Leva G, Zvelebil MJ,
Pirola L, Vanhaesebroeck B, Waterfield MD and Panayotou G:
Wortmannin inactivates phosphoinositide 3-kinase by covalent
modification of Lys-802, a residue involved in the phosphate
transfer reaction. Mol Cell Biol. 16:1722–1733. 1996.PubMed/NCBI
|
|
45
|
Arcaro A and Wymann MP: Wortmannin is a
potent phospha-tidylinositol 3-kinase inhibitor: The role of
phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses.
Biochem J. 296:297–301. 1993. View Article : Google Scholar
|
|
46
|
Blommaart EF, Krause U, Schellens JP,
Vreeling-Sindelárová H and Meijer AJ: The phosphatidylinositol
3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in
isolated rat hepatocytes. Eur J Biochem. 243:240–246. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tanida I, Ueno T and Kominami E: LC3 and
sutophagy. Methods Mol Biol. 445:77–88. 2008. View Article : Google Scholar
|
|
48
|
Solomon VR and Lee H: Chloroquine and its
analogs: A new promise of an old drug for effective and safe cancer
therapies. Eur J Pharmacol. 625:220–233. 2009. View Article : Google Scholar : PubMed/NCBI
|