Introduction
The epithelial-mesenchymal transition (EMT) and the
reverse process, termed the mesenchymal-epithelial transition
(MET), play central roles in embryogenesis (1–3). For
example, for the period of early embryonic development, the
mesoderm generated by EMTs develops into multiple tissue types, as
well as later in development, mesodermal cells generate epithelial
organs, such as the kidney and ovary, by MET (4). Epithelial to mesenchymal transition
(EMT) is an essential process for driving plasticity during
development, yet it is an unintentional behavior of cells during
progression of the malignant tumor (5–7). In
recent years, many embryonic genes have been found to confer
malignant traits, such as motility, invasiveness, and resistance to
apoptosis, on neoplastic cells (8–16).
The EMT-associated reprogramming of pancreatic cancer cells not
only implies that fundamental changes may occur to several
regulatory networks but also that an intimate interplay exists
between them. Disturbance of a controlled epithelial balance is
triggered by altering several layers of regulation, including the
transcriptional and translational machinery, expression of
non-coding RNAs, alternative splicing and protein stability
(17–19).
SMURF2, a HECT-family ubiquitin ligase (E3), has
been implicated in diverse biological functions including TGF-β
signaling, mitotic regulation, cell polarity, motility and
chromatin modifications (20). It
appears to play complex roles in tumorigenesis. Esophageal squamous
cell carcinomas expressed high levels of SMURF2, which correlated
with poor prognosis (21). Another
study on lung adenocarcinomas and head and neck carcinomas showed a
positive correlation between SMURF2 and EGFR protein levels
(22). Yet, there are several
reports demonstrating decreased expression of SMURF2 in other types
of cancer. Protein levels of SMURF2 were found to be downregulated
in human lymphoma and breast cancer tissues relative to non-cancer
tissues (23) and its SMURF2
levels were lower in advanced tumors compared to less advanced
organ-confined tumors, suggesting association of SMURF2
downregulation with tumor progression (24). Importantly, two recent studies
using SMURF2-null mice showed that SMURF2-deficiency increased
susceptibility to spontaneous tumorigenesis in various tissues
including the liver, lung, pituitary and mammary gland (23,25).
The activity of SMURF2 to ubiquitinate and degrade RNF20, a
RING-family E3 that controls histone H2B ubiquitination and genome
stability, has been implicated for the tumor suppressive role of
SMURF2 (23).
In this study, we found that the silencing of SMURF2
promoted EMT in HPDE6c7 normal pancreas cells and overexpression of
SMURF2 inhibits TGF-β-mediated EMT in the cells. Subsequent studies
showed that SMURF2 was downregulated in pancreatic cancer tissues
and it promoted MET in pancreatic cancer cells, the expression was
negatively associated with gemcitabine-resistance, but it did not
alter cell viability, cell cycle and cell senescence. In addition,
we demonstrated that miR-15b was able to degrade SMURF2 and its
overexpression promoted EMT in pancreatic cancer and its expression
was associated with metastasis in pancreatic cancer.
Materials and methods
Pancreatic cancer tissues
Nineteen patients diagnosed with pancreatic cancer
were recruited from the Second Artillery General Hospital of PLA,
Tongji Hospital and Hubei Cancer Center. The use of human tissue
samples followed the internationally recognised guidelines as well
as local and national regulations. Informed consent was obtained
from each individual. The local Medical ethics committee approved
the experiments undertaken. Human normal pancreas HPDE6c7,
pancreatic cancer gemcitabine-sensitive cell lines HPAC, BxPC-3,
Colo357, and L3.6pl (26),
gemcitabine-resistant cell lines ASPC-1, Panc-1 and MiaPaCa-2
(26) were kindly donated by Dr
Hua Wei (University of Michigan Medical Center, MI, USA). Briefly,
cells were maintained in RPMI-1640 medium supplemented with 10%
fetal bovine serum (FBS) (Gibco, Grand Island, NY, USA) and
penicillin/streptomycin at 37°C in a humidified atmosphere with 5%
CO2.
Plasmids, pre-miR-15b/control miR,
anti-miR-15b/scramble and transfection
SMURF2-expressing plasmids/empty vectors (pcDNA3.1)
were purchased from Tiangene, Tianjin, China. The expressing
plasmids or empty vector (pcDNA3.1) used for each transfection is
10 μg. Pre-miR-15b/control miR and anti-miR-15b/scramble were
purchased from Ambion, Inc. (Ambion, Austin, TX, USA). Transfection
was performed with Lipofectamine 2000 reagent (Invitrogen, CA, USA)
according to the manufacturer's instructions.
Western blot analysis
Western blot analysis was performed as described
before (27). Briefly, after
incubation with primary antibody anti-SMURF2 (1:250; Abcam,
Cambridge, MA, USA), anti-E-cadherin (1:500; Abcam),
anti-N-cadherin (1:200; Abcam), anti-vimentin (1:500; Abcam),
anti-SNAI1 (1:500; Abcam), anti-TGFB1 (1:800; Abcam), anti-TWIST
(1:500; Abcam), anti-ZEB1 (1:500; Abcam), anti-TGFB2 (1:500;
Abcam), anti-α-SMA (1:500; Abcam) and anti-β-actin (1:500; Abcam)
overnight at 4°C, IRDye™-800 conjugated anti-rabbit secondary
antibodies (Li-COR, Biosciences, Lincoln, NE, USA) were used for 30
min at room temperature. The specific proteins were visualized by
Odyssey™ Infrared Imaging System (Gene Co., Lincoln, NE, USA).
Migration and invasion assay
For transwell migration assays, 5×104
cells were plated in the top chamber with the non-coated membrane
(24-well insert; pore size, 8 mm; BD Biosciences, San Jose, CA,
USA). For invasion assays, 1.25×105 cells were plated in
the top chamber with Matrigel-coated membrane (24-well insert; pore
size, 8 mm; BD Biosciences). In both assays, cells were plated in
medium without serum or growth factors, and medium supplemented
with serum was used as a chemoattractant in the lower chamber. The
cells were incubated for 36 h and cells that did not migrate or
invade through the pores were removed by a cotton swab. Cells on
the lower surface of the membrane were stained with the Diff-Quick
Staining Set (Dade) and counted.
Wound healing assay
Cells (6×105) were seeded onto each 35-mm
glass bottom dish (MatTek Co., Ashland, MA, USA) and cultured at
37°C with 5% CO2 for 36 h. The confluent monolayer of
cells was wounded. Mono-layer of cells were wounded with yellow
pipette tips. After washing with PBS, the cells were incubated in
fresh culture medium. The wounded areas were photographed at the
beginning (0 h) and the end (12, 24, 48 or 72 h) of the assay with
Nikon inverted microscope (ECLIPSE TE-2000U, Nikon, Japan) equipped
with a video camera (DS-U1, Nikon).
Immunofluorescence analyses
For immunofluorescence analyses, cells were plated
on glass coverslips in 6-well plates and transfected as indicated.
At 36 h after transfection, coverslips were stained with the
anti-SMURF2, anti-vimentin antibody or anti-E-cadherin antibody.
Alexa Fluor 488 goat anti-rabbit IgG antibody was used as secondary
antibody (Invitrogen). Coverslips were counterstained with DAPI
(Invitrogen-Molecular Probes, Eugene, OR, USA) for visualization of
nuclei. Microscopic analysis was performed with a confocal
laser-scanning microscope (Leica Microsystems, Bensheim, Germany).
Fluorescence intensities were measured in several viewing areas for
300 cells per coverslip and analyzed using ImageJ 1.37v software
(http://rsb.info.nih.gov/ij/index.html).
Reverse-transcription polymerase chain
reaction (RT-PCR) and quantitative real-time RT-PCR (qRT-PCR) for
SMURF2
RT-PCR and qRT-PCR were described before (27). The primer sequences for SMURF2:
forward, 5′-CGATGGCTGTTA GCAGCTTTTC-3′ and reverse,
5′-TGCCTCTGCAGGGCTT CAAAG-3′. TGFβ1: forward, 5′-CGATGGCTGTTAGCAG
CTT TTC-3′ and reverse, 5′-TGCCTCTGCAGGGCTTC AAAG-3′.
Luciferase reporter assay
The 3′ untranslated region (3′-UTR) of human SMURF2
mRNA was cloned into pRL-TK (Promega, Madison, WI, USA) using
PCR-generated fragment. Site-directed mutagenesis of the miR-15b
target-site in the SMURF2-3′-UTR was carried out using Quik
change-mutagenesis kit (Stratagene, Heidelberg, Germany), with
SMURF2-WT-luc as a template. For reporter assays, cells were
transiently transfected with WT or mutant reporter plasmids and
microRNA or anti-microRNA (as indicated in Fig. 5N–P) using Lipofectamine 2000
(Invitrogen). Reporter assays were performed 36 h post-transfection
using the Dual-luciferaseassay-system (Promega), normalized for
transfection efficiency by cotransfected Renilla-luciferase.
Real-time PCR for miRNA
Total RNA from cultured cells, with efficient
recovery of small RNAs, was isolated using the mirVana miRNA
Isolation kit (Ambion). Detection of the mature form of miRNAs was
performed using the mirVana qRT-PCR miRNA detection kit, according
to the manufacturer's instructions (Ambion). The U6 small nuclear
RNA was used as an internal control.
MTT assay
To perform the MTT assay, cells
(0.5×104/well) were plated in 96-well sterile plastic
plates and allowed to attach overnight. Cells were transfected as
indicated. After 72 h, 15 ml of MTT solution (5 mg/ml) was added to
each well and plates were incubated for 4 h. Crystalline formazan
was solubilized with 100 ml of a 10% (w/v) SDS solution.
Cell cycle analysis
Cells were starved for 24 h by deprivation of serum
to synchronize the cell cycle, and then transfected as indicated.
After transfection for 48, the cells were collected and fixed, and
then incubated with RNase A (Invitrogen). Propidium iodide (PI)
(Sigma, MO, USA) was added, followed by a 30-min incubation in the
dark. Cellular DNA content was analyzed by a FACS
(Becton-Dickinson, Franklin Lakes, NJ, USA). Data were processed
using Modfit Lt software (Version SM1120, Verity Software House,
USA).
Senescence-associated β-galactosidase
staining
Cells seeded in 6-well plates were transfected as
described previously. After 72 h, the cells were rinsed with PBS,
fixed and then incubated with freshly prepared
senescence-associated β-galactosidase (SA-β-gal) staining solution
at 37°C overnight. A total of 200 cells were counted and
percentages of SA-β-gal-positive cells calculated.
Statistical analysis
Data are presented as mean ± SEM. Student's t-test
(two-tailed) was used to compare two groups P<0.05 was
considered significant, unless otherwise indicated (χ2
test).
Results
Silencing SMURF2 promotes EMT phenotype
in HPDE6c7 normal pancreas cells
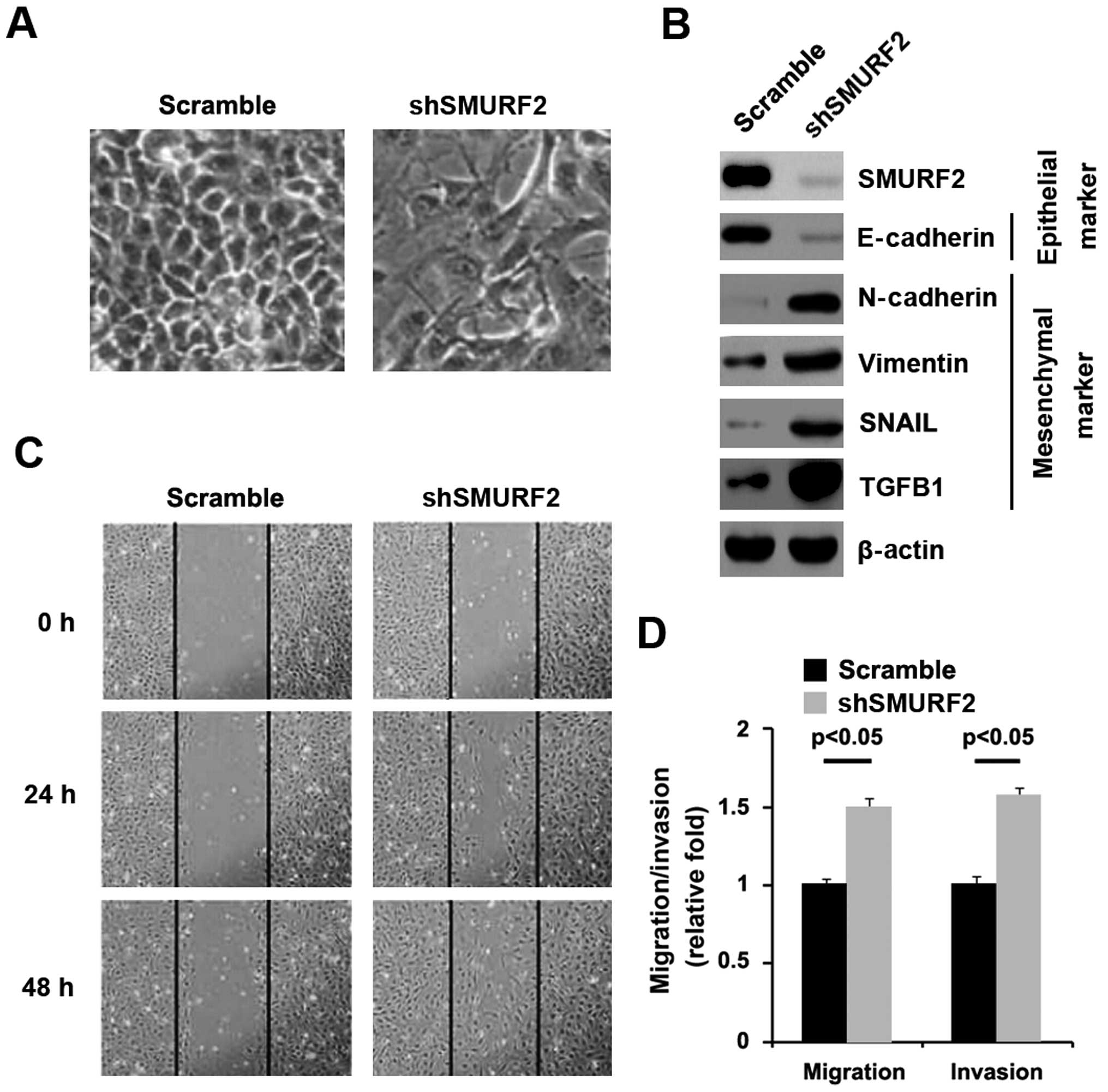
In order to identify the role of SMURF2 in normal
pancreas, we transfected HPDE6c7 cells with shSMURF2 plasmids and
then western blot analysis was performed. We found that SMURF2
protein was significantly decreased in the cells transfected with
shSMURF2 plasmids (Fig. 1B) and
its reduction caused significant changes in HPDE6c7 cell morphology
(EMT) (Fig. 1A). To further verify
that the changes in cell morphology are caused by EMT, expression
levels of epithelial and mesenchymal markers were compared in
HPDE6c7 cells transfected with shSMURF2 plasmids with the cells
transfected with scramble. The results revealed that the epithelial
markers (E-cadherin) were significantly repressed, whereas
mesenchymal markers (N-cadherin, vimentin, SNAIL and TGFB1) were
induced by silencing SMURF2 in the cells (Fig. 1B). EMT can result in increased cell
invasion and migration (28–30).
Thus, we reasoned that SMURF2 could also affect invasion and
migration in HPDE6c7 cells. To identify this reason, we performed
would healing, invasion, and migration assay. We found that
silencing SMURF2 resulted in enhanced invasion (Fig. 1D) and migration (Fig. 1C and D) in the cells.
SMURF2 inhibits TGF-β-mediated EMT
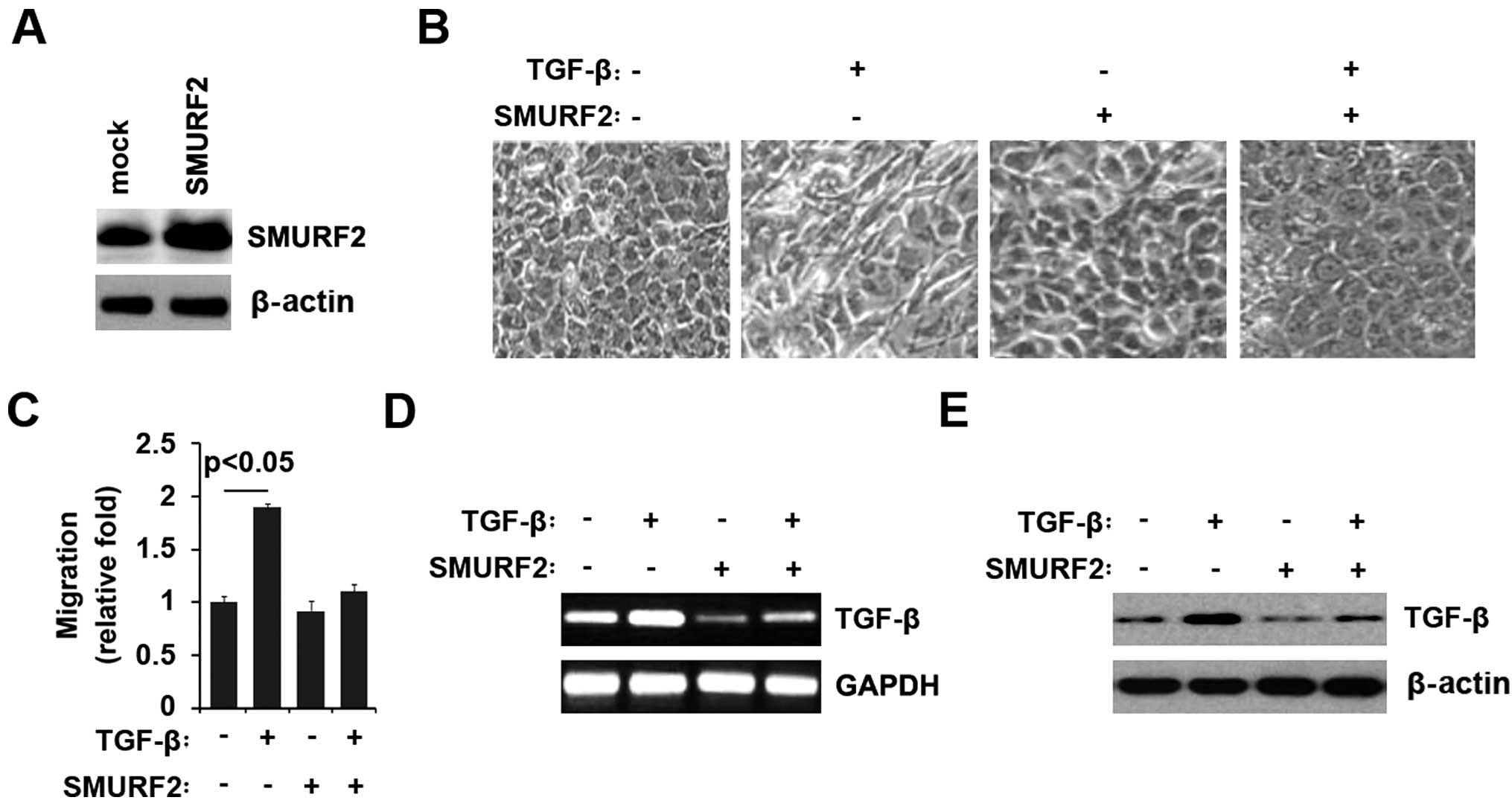
Having demonstrated that silencing SMURF2 promoted
EMT phenotype in HPDE6c7 normal pancreas cells, we reasoned that
SMURF2 overexpression could reverse EMT, namely promoting MET.
TGF-β can induce epithelial to mesenchymal transition (31). Thus, we reasoned that SMURF2 could
reverse TGF-β-induced EMT. We treated HPDE6c7 cells with TGF-β for
7 days, a standard treatment that induces EMT (32) and then western blot analysis was
performed. We found that TGF-β protein was significantly increased
in the cells (data not shown). We also performed western blot
analysis in the cells transfected with SMURF2 expressing plasmids
or empty vector. The results showed SMURF2 was significantly
increased by the expressing plasmids (Fig. 2A). Similar to previous
observations, TGF-β treatment induced EMT phenotype from a
cobblestone-like to a spindle-like morphology (Fig. 2B), accompanied by increase of
migration in the cells (Fig. 2C).
As expected, we found that SMURF2 not only reversed the change of
morphology induced by TGF-β (Fig.
2B), but also eliminated the increase of migration promoted by
TGF-β (Fig. 2C) in HPDE6c7 cells.
Next, in an attempt to identify the role of SMURF2 in regulating
TGF-β expression of HPDE6c7 cells, the cells were treated as
indicated. After treatment, TGF-β mRNA expression was detected by
RT-PCR and the results showed that TGF-β mRNA was increased by
TGF-β in the cells, but the increase was attenuated by SMURF2
(Fig. 2D). We also performed
western blot analysis to detect TGF-β protein, and found that TGF-β
was also attenuated by SMURF2 (Fig.
2E). Thus, we concluded that SMURF2 could inhibit
TGF-β-mediated EMT.
SMURF2 is downregulated in pancreatic
cancer tissues and its expression is negatively associated with
gemcitabine resistance and it promotes MET in pancreatic cancer
cells
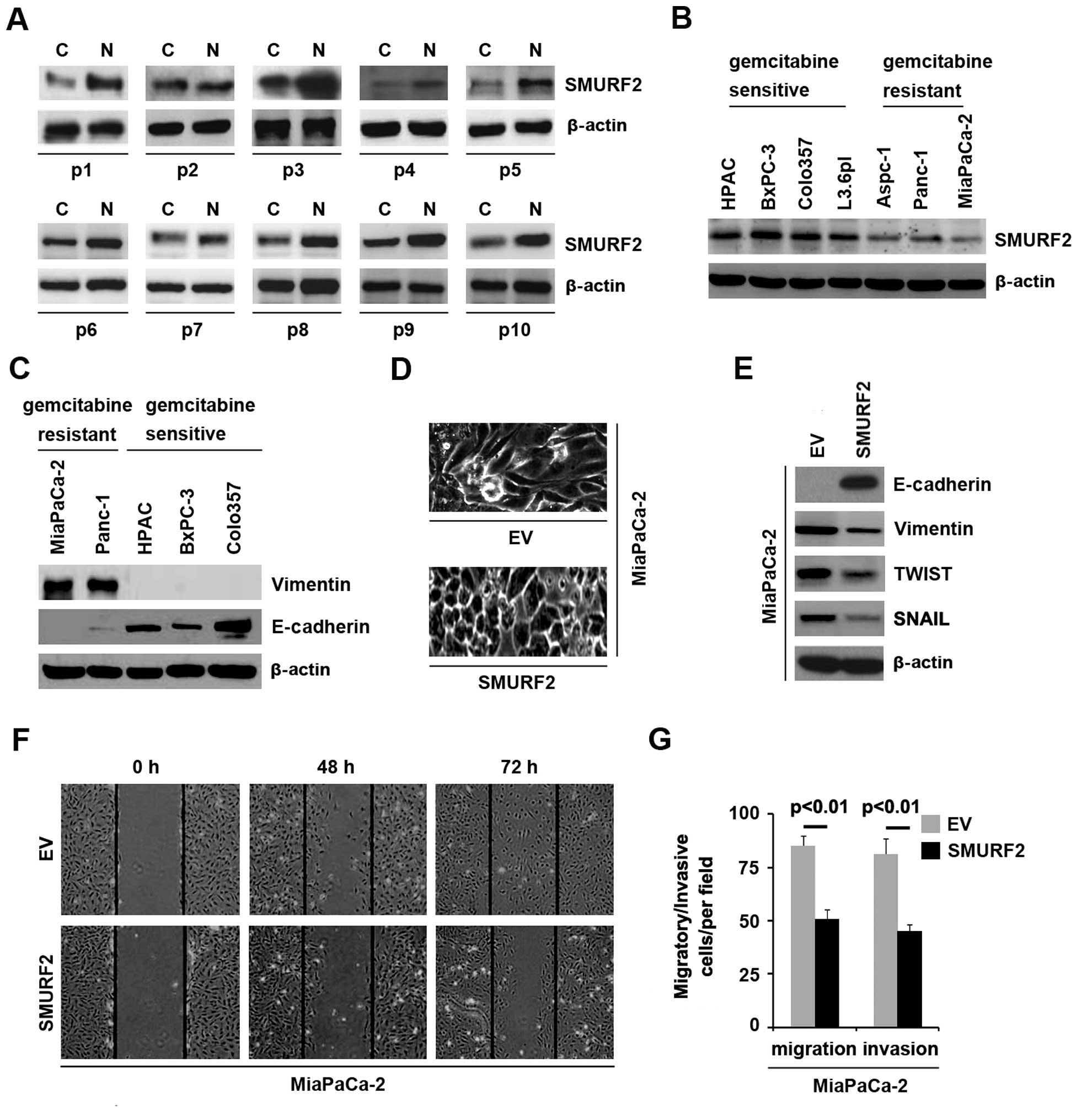
In an attempt to identify SMURF2 expression between
pancreatic cancer tissues and adjacent normal tissues, we performed
western blot analysis in cancer tissues versus normal tissues.
Protein was isolated from 10 pairs of pancreatic cancer tissues and
normal tissues (patient nos. 1–10). We found that SMURF2 protein
was significantly decreased in cancer tissues (C), compared with
adjacent normal tissues (N) (Fig.
3A). It implied that SMURF2 could be a tumor suppressive gene
in pancreatic cancer. In an attempt to identify the SMURF2 protein
expression among different pancreatic cancer cell lines, we
performed western blot analysis in pancreatic cancer cell lines
(HPAC, BxPC-3, Colo357, L3.6pl, ASPC-1, PANC-1 and MiaPaCa-2
cells). Protein isolated from the 7 cell lines was detected by
western blot analysis and the results showed that the expression of
SMURF2 was lower in gemcitabine-resistant cells (ASPC-1, PANC-1 and
MiaPaCa-2 cells) than gemcitabine-sensitive cells (HPAC, BxPC-3,
Colo357 and L3.6pl) (Fig. 3B).
Next, in order to identify whether EMT was
associated with gemcitabine-resistance, we performed western blot
analysis in gemcitabine-sensitive cells (HPAC, BxPC-3 and Colo357)
and gemcitabine-resistant cells (PANC-1 and MiaPaCa-2 cells) to
detect vimentin (mesenchymal marker) and E-cadherin (epithelial
marker). The results of western blot analysis showed that vimentin
was detected only in gemcitabine-resistant cells (PANC-1 and
MiaPaCa-2 cells) and E-cadherin was significantly elevated in
gemcitabine-sensitive cells, compared with gemcitabine-resistant
cells (Fig. 3C).
Because SMURF2 was downregulated in
gemcitabine-resistant pancreatic cancer cells and
gemcitabine-resistant pancreatic cancer cells was associated with
EMT and SMURF2 inhibited EMT in normal pancreatic cells, we
reasoned that SMURF2 was associated with EMT in pancreatic cancer.
In order to assess the role of SMURF2 in pancreatic cancer, we
transfected MiaPaCa-2 cells with SMURF2 expressing plasmids and
then western blot analysis was performed. We found that SMURF2
protein was significantly increased in the cells transfected with
SMURF2 expressing plasmids (data not shown) and its overexpression
caused significant changes in MiaPaCa-2 cells morphology (MET)
(Fig. 3D). To further verify that
the changes in cell morphology are caused by EMT, expression levels
of epithelial and mesenchymal markers were compared in MiaPaCa-2
cells transfected with SMURF2 expressing plasmids with the cells
transfected with empty vectors. The results revealed that the
epithelial marker (E-cadherin) was promoted, whereas mesenchymal
markers (vimentin, TWIST and SNAIL) were suppressed by SMURF2
overexpression in the cells (Fig.
3E). In order to identify whether SMURF2 overexpression
affected migration and invasion, we performed would healing,
migration and invasion assay in MiaPaCa-2 cells. The results showed
that SMURF2 suppressed migration (Fig.
3F and G) and invasion (Fig.
3G) in the cells. Thus, we concluded that SMURF2 is
downregulated in pancreatic cancer tissues and negatively
associated with gemcitabine resistance and it promotes MET in
pancreatic cancer cells.
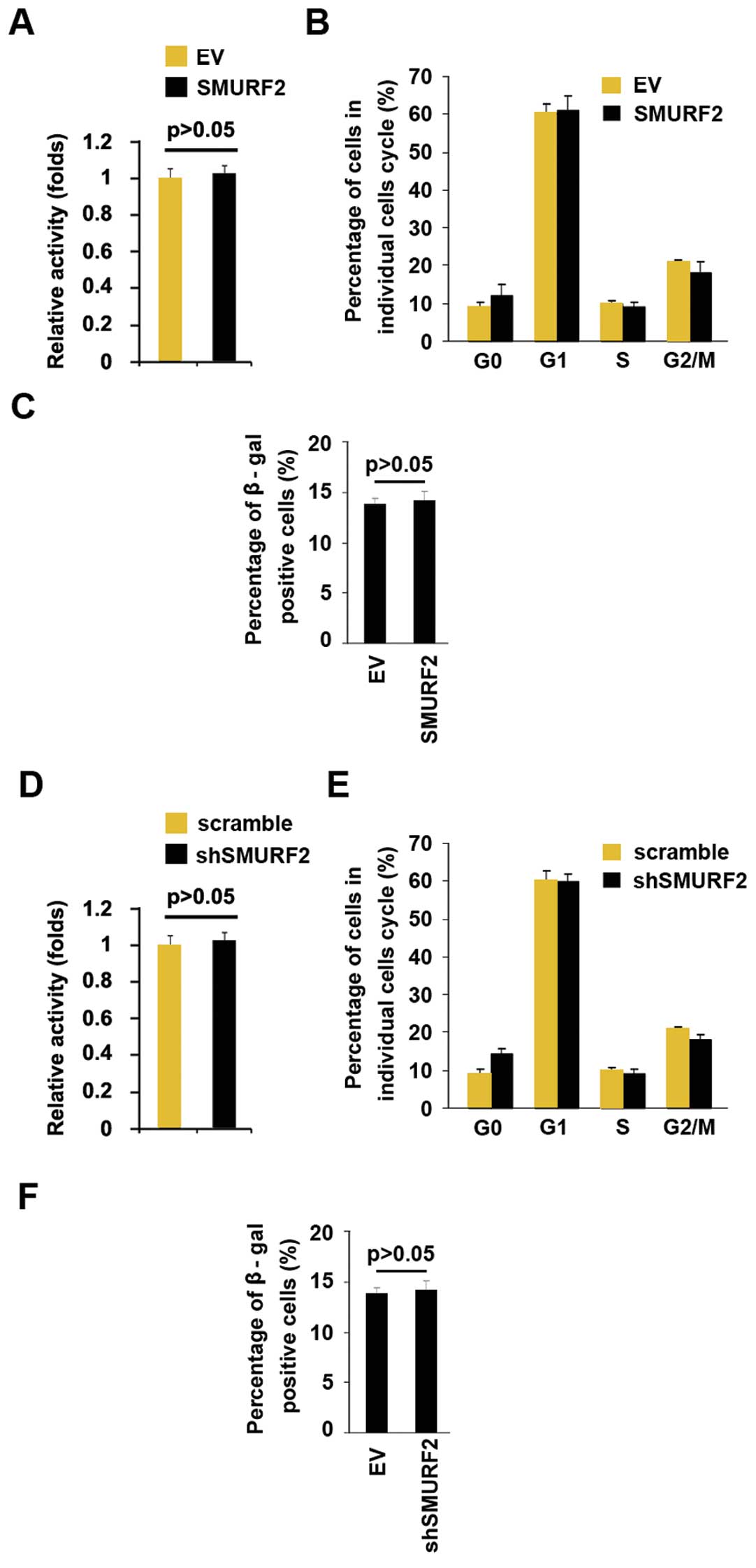
SMURF2 does not alter cell viability,
cell cycle and cell senescence
To identify whether SMURF2 could alter
proliferation, we performed an MTT assay and cell cycle analysis.
However, the result of MTT demonstrated that SMURF2 did not affect
viability of MiaPaCa-2 (Fig. 4A)
and the cell cycle was not changed by its overexpression in the
cells (Fig. 4B). Next, we
performed β-gal senescence assay, but the results showed that
SMURF2 overexpression did not alter cell senescence (Fig. 4C). Having demonstrated that SMURF2
overexpression did not alter cell viability, cell cycle and cells
senescence in MiaPaCa-2 cells, to provide further evidence that
SMURF2 was not involved in viability, cells cycle and cell
senescence of MiaPaCa-2 cells, we studied the effects of the
inhibitor of SMURF2, the shSMURF2. shSMURF2 significantly
downregulated SMURF2 expression in MiaPaCa-2 cells (data not
shown). After stable transfection, we performed MTT assay, cell
cycle analysis and β-gal senescence assay to detect cell viability,
cell cycle distribution and cell senescence of MiaPaCa-2 cells
transfected with shSMURF2 plasmids and scramble. Silencing SMURF2
did not alter viability (Fig. 4D),
cell cycle distribution (Fig. 4E)
or cell senescence (Fig. 4F).
miR-15b degrades SMURF2 in pancreatic
cancer cells
Having demonstrated that SMURF2 expression is
specifically downregulated in pancreatic cancer (Fig. 3A) and it suppressed EMT in
vitro, we investigated which mechanisms suppressed SMURF2
expression in pancreatic cancer. MicroRNAs (miRs) are a class of
small noncoding RNAs (~22 nucleotides) negatively regulating
protein-coding gene expression by targeting mRNA degradation or
translation inhibition (20,21,33).
Upregulation of specific miRNA can contribute to downregulation of
a tumor suppressive gene (34–36).
Thus we reasoned that SMURF2 was downregulated by overexpression of
specific miRNA in pancreatic cancer.
In an attempt to identify the level of miRNA
expression in pancreatic cancer MiaPaCa-2 cells (low expression of
SMURF2) and BxPC-3 cells (high expression of SMURF2) (Fig. 3B), we performed miRNA profiling in
the cell lines. RNAs isolated from the two cell lines were
hybridized to a custom miRNA microarray platform. After
hybridization, quantification, and normalization, we found that
miR-15b, miR-212 and miR-155 were significantly increased in
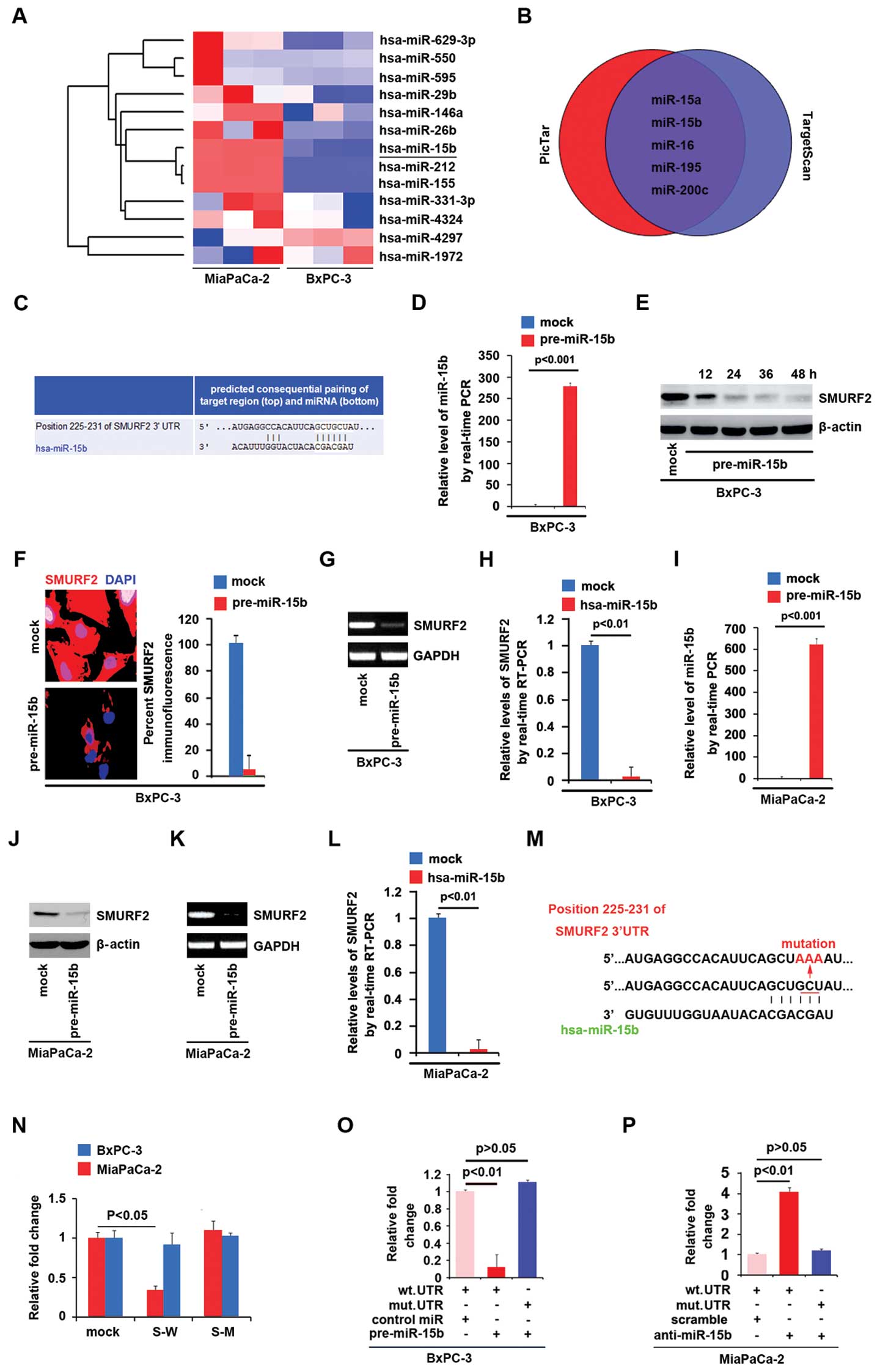
MiaPaCa-2 cells compared with BxPC-3 cells >100-fold (Fig. 5A).
As further confirmation, we used 2 commonly used
prediction algorithms - TargetScan (http://www.targetscan.org) and PicTar (http://pictar.mdc-berlin.de/) to analyze 3′-UTR of
SMURF2. The algorithms predicted that miR-15a/b, miR-16, miR-195
and miR-200c could target 3′-UTR of SMURF2 (Fig. 5B). However, we found that only
miR-15b was negatively associated with SMURF2 expression in
pancreatic cancer cells. Target sites of miR-15b on 3′-UTR of
SMURF2 are showed in Fig. 5C. We
reasoned that miR-15b could downregulate SMURF2 expression by
targeting its 3′-UTR in pancreatic cancer and that SMURF2 was
downregulated in pancreatic cancer cells due to overexpression of
miR-15b. In an attempt to identify the role of miR-15b in
regulating SMURF2 expression in pancreatic cancer cells, BxPC-3
cells were transfected with pre-miR-15b and control miR. After
transfection, miR-15b expression was detected by real-time PCR and
the results showed that miR-15b was significantly increased by
pre-miR-15b in the cells (Fig.
5D). To confirm the reason, we performed western blot analyses
in BxPC-3 cells transfected with pre-miR-15b or control miR. The
results showed that SMURF2 protein was evidently suppressed in the
cells transfected with pre-miR-15b at different time-points
(Fig. 5E). Next, we performed
immunofluorescence analysis and RT-PCR to detect SMURF2 expression
in BxPC-3 cells transfected with pre-miR-15b or control miR. The
results showed that SMURF2 protein (Fig. 5F) and mRNA (Fig. 5G) were significantly downregulated
in the cells transfected with pre-miR-15b. Consistent with the
results of RT-PCR, real-time PCR demonstrated that SMURF2 mRNA was
not reduced in BxPC-3 cells transfected with pre-miR-15b, compared
with control miR-transfected groups (Fig. 5H).
Having demonstrated that miR-15b degrades SMURF2 in
BxPC-3 cells, in order to further confirm that miR-15b degrades
SMURF2 in pancreatic cancer cells, we performed western blot
analysis, RT-PCR and real-time PCR to detect whether SMURF2 could
be suppressed by pre-miR-15b in MiaPaCa-2 cells. We showed that
miR-15b could be significantly increased by pre-miR-15b (Fig. 5I) in the cells and SMURF2 protein
(Fig. 5J) and mRNA (Fig. 5K and L) were evidently inhibited by
it.
To further demonstrate the direct regulation of
SMURF2 by miR-15b, we constructed luciferase reporters with the
targeting sequences of wild-type (SMURF2-WT-luc, S-W) and mutated
SMURF2 3′-UTRs (SMURF2-A-MUT-luc, S-M) (Fig. 5M). Both the wild-type and mutant
reporters were introduced into BxPC-3 cells and MiaPaCa-2 cells. We
found that SMURF2-WT-luc plasmids were suppressed in MiaPaCa-2
cells (miR-15b high expression).
Next, to identify that miR-15b directly targets
3′-UTR of SMURF2, luciferase reporter assay was performed in BxPC-3
cells. Luciferase reporter assay showed that the luciferase
activities of SMURF2-WT-luc plasmids were significantly suppressed
in the cells transfected with pre-miR-15b, implying that miR-15b
targeted 3′-UTR of SMURF2 mRNA (Fig.
5O). In order to further identify that miR-15b targeted 3′-UTR
of SMURF2 by the predicted sites, we mutated 3 bases in the
predicted sites (Fig. 5M). In
addition, mutant reporters were introduced into BxPC-3 cells, as
expected the luciferase activities of SMURF2-MUT-luc were not
suppressed by miR-15b in BxPC-3 cells (Fig. 5O).
We also performed luciferase reporters assay in
MiaPaCa-2 cells. First, we found that anti-miR-15b could
significantly downregulate miR-15b expression in MiaPaCa-2 cells
(data not shown) and then luciferase reporters assay showed that
the luciferase activities of SMURF2-WT-luc plasmids were
significantly promoted in the cells transfected with anti-miR-15b,
implying that miR-15b targeted 3′-UTR of SMURF2 mRNA (Fig. 5P). In order to further identify
that miR-15b targeted 3′-UTR of SMURF2 by the predicted sites in
MiaPaCa-2 cells, mutant reporters were introduced into MiaPaCa-2
cells, as expected the luciferase activities of SMURF2-MUT-luc were
not promoted by silencing miR-15b in MiaPaCa-2 cells (Fig. 5P).
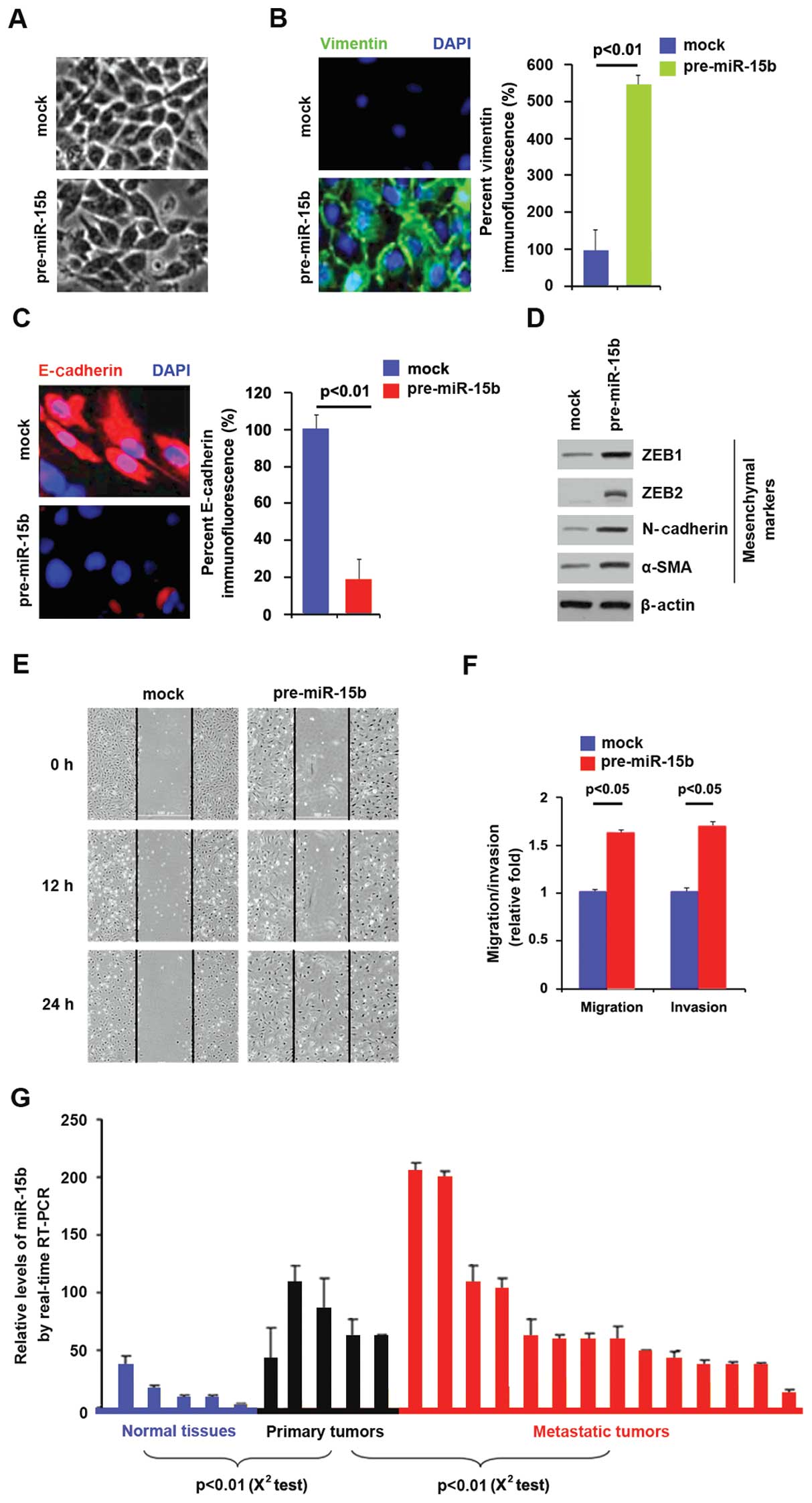
miR-15b overexpression promotes EMT in
pancreatic cancer
We had demonstrated that SMURF2 was downregulated in
pancreatic cancer tissues and it promoted MET in pancreatic cancer
cells and miR-15b degraded SMURF2 in pancreatic cancer cells.
Moreover, miR-15b was upregulated in pancreatic cancer, implying
that it is an oncogene (37).
Thus, we reasoned that contrary to SMURF2, miR-15b might promote
EMT in pancreatic cancer. miR-15b could be increased by pre-miR-15b
in BxPC-3 cells (Fig. 5D). In an
attempt to identify the role of miR-15b in regulating EMT in
pancreatic cancer, the cells were transfected with pre-miR-15b. Its
over-expression caused changes in BxPC-3 cell morphology (EMT)
(Fig. 6A). To further verify that
the change in cell morphology was caused by EMT, we performed
immunoflurescence to analyze expression of epithelial and
mesenchymal markers in pancreatic cancer cells. Expression levels
of vimentin (mesenchymal markers) and E-cadherin (epithelial
marker) were compared in BxPC-3 cells transfected with pre-miR-15b
or control miR. The results showed that vimentin was induced by
miR-15b overexpression in BxPC-3 cells (Fig. 6B), whereas E-cadherin was evidently
repressed by it (Fig. 6C). We also
performed western blot analysis and expression was analyzed of
other mesenchymal markers in the cells transfected with
pre-miR-15b. The results showed that expression of ZEB1, ZEB2,
N-cadherin and α-SMA were significantly promoted by miR-15b
(Fig. 6D). In order to identify
whether miR-15b affected migration and invasion, we performed would
healing, migration and invasion assay in BxPC-3 cells. The results
showed that miR-15b promotes migration (Fig. 6E and F) and invasion (Fig. 6F) in the cells. Having showed that
miR-15b was positively associated with invasion and migration in
vitro, we performed real-time PCR to analyze whether it was
associated with metastasis in pancreatic cancer patients.
The results demonstrated that miR-15b was increased
in primary tumors, compared with adjacent normal tissues and it was
also higher in metastatic tumors than primary tumors (Fig. 6G). All the results demonstrated
that miR-15b promoted EMT and it was positively associated with
metastasis in pancreatic cancer.
Disscussion
Pancreatic cancer ranks fourth in cancer-related
mortality, with an overall 5-year survival rate <1% and a mean
survival time of 4–6 months (38).
Late initial diagnosis, chemotherapy and radiation resistance and
early metastatic spread accounts for non-satisfactory progress in
therapy (39). There is evidence
that EMT account for drug resistance, metastasis and late
recurrence after years of dormancy (26,40–43).
Emerging evidence also suggests that the processes of EMT is
regulated by the expression status of tumor suppressive genes,
oncogenes and many microRNAs (miRNAs), which are believed to
function as key regulators of various biological and pathological
processes during tumor development and progression (37,44–46).
Here we present evidence that the expression of
SMURF2 protein is downregulated preferentially in pancreatic
cancer. The cancer-associated downregulation is consistent with the
recent study that suggested the tumor suppressive function of this
SMURF2 (47). Low expression of
SMURF2 protein was also observed in gemcitabine-resistant cell
lines, implying that its abnormal expression was associated with
drug-resistance. It is able to promote MET in normal pancreas cells
and pancreatic cancer cells. This is also consistent with the
report that EMT plays an important role in gemcitabine-resistance
(41). TGF-β functions as a
pro-metastatic factor in human cancer (48). In this study, we demonstrated that
SMURF2 inhibited TGF-β-mediated EMT in normal pancreas cells. This
further confirmed that SMURF2 is a tumor suppressive gene. In
addition, we found that miR-15b promoted EMT by degrading SMURF2 in
pancreatic cancer cells. We found that antagomirs against miR-15b
substantially increased SMURF2 levels in the glioblastoma cell
lines (unpublished data). Whether anti-miR-15b could increase
SMURF2 levels in pancreatic cancer needs further study.
Collectively, miR-15b-mediated SMURF2 regulation in
pancreatic cancer has potential basic and clinical implications. On
the one hand, miR-15b could be a powerful oncogene by promoting EMT
and regulating relevant tumor suppressor genes in pancreatic
cancer, and pharmacological inhibition of miR-15b may represent a
promising therapeutic strategy. On another hand, SMURF2 is a tumor
suppressor gene and its expression is inhibited by miR-15b in
pancreatic cancer. Further studies are clearly required.
Abbreviations:
|
EMT
|
epithelial-mesenchymal transition
|
|
MET
|
mesenchymal-epithelial transition
|
|
SMURF2
|
SMAD specific E3 ubiquitin protein
ligase 2
|
References
|
1
|
Hay ED: An overview of
epithelio-mesenchymal transformation. Acta Anat (Basel). 154:8–20.
1995. View Article : Google Scholar
|
|
2
|
Pérez-Pomares JM and Muñoz-Chápuli R:
Epithelial-mesenchymal transitions: A mesodermal cell strategy for
evolutive innovation in Metazoans. Anat Rec. 268:343–351. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Davies JA: Mesenchyme to epithelium
transition during development of the mammalian kidney tubule. Acta
Anat (Basel). 156:187–201. 1996. View Article : Google Scholar
|
|
5
|
Nieto MA: The ins and outs of the
epithelial to mesenchymal transition in health and disease. Annu
Rev Cell Dev Biol. 27:347–376. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Savagner P, Yamada KM and Thiery JP: The
zinc-finger protein slug causes desmosome dissociation, an initial
and necessary step for growth factor-induced epithelial-mesenchymal
transition. J Cell Biol. 137:1403–1419. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD
and Wang LH: Twist transcriptionally up-regulates AKT2 in breast
cancer cells leading to increased migration, invasion, and
resistance to paclitaxel. Cancer Res. 67:1979–1987. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Comijn J, Berx G, Vermassen P, Verschueren
K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D and van Roy
F: The two-handed E box binding zinc finger protein SIP1
downregulates E-cadherin and induces invasion. Mol Cell.
7:1267–1278. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hartwell KA, Muir B, Reinhardt F,
Carpenter AE, Sgroi DC and Weinberg RA: The Spemann organizer gene,
Goosecoid, promotes tumor metastasis. Proc Natl Acad Sci USA.
103:18969–18974. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mani SA, Yang J, Brooks M, Schwaninger G,
Zhou A, Miura N, Kutok JL, Hartwell K, Richardson AL and Weinberg
RA: Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis
and is associated with aggressive basal-like breast cancers. Proc
Natl Acad Sci USA. 104:10069–10074. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Oft M, Akhurst RJ and Balmain A:
Metastasis is driven by sequential elevation of H-ras and Smad2
levels. Nat Cell Biol. 4:487–494. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Peinado H, Olmeda D and Cano A: Snail, Zeb
and bHLH factors in tumour progression: An alliance against the
epithelial phenotype? Nat Rev Cancer. 7:415–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Savagner P, Kusewitt DF, Carver EA,
Magnino F, Choi C, Gridley T and Hudson LG: Developmental
transcription factor slug is required for effective
re-epithelialization by adult keratinocytes. J Cell Physiol.
202:858–866. 2005. View Article : Google Scholar
|
|
16
|
Yang J, Mani SA and Weinberg RA: Exploring
a new twist on tumor metastasis. Cancer Res. 66:4549–4552. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chang CJ, Chao CH, Xia W, Yang JY, Xiong
Y, Li CW, Yu WH, Rehman SK, Hsu JL, Lee HH, et al: p53 regulates
epithelial-mesenchymal transition and stem cell properties through
modulating miRNAs. Nat Cell Biol. 13:317–323. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gravdal K, Halvorsen OJ, Haukaas SA and
Akslen LA: A switch from E-cadherin to N-cadherin expression
indicates epithelial to mesenchymal transition and is of strong and
independent importance for the progress of prostate cancer. Clin
Cancer Res. 13:7003–7011. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hader C, Marlier A and Cantley L:
Mesenchymal-epithelial transition in epithelial response to injury:
The role of Foxc2. Oncogene. 29:1031–1040. 2010. View Article : Google Scholar :
|
|
20
|
Pasquinelli AE, Reinhart BJ, Slack F,
Martindale MQ, Kuroda MI, Maller B, Hayward DC, Ball EE, Degnan B,
Müller P, et al: Conservation of the sequence and temporal
expression of let-7 heterochronic regulatory RNA. Nature.
408:86–89. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Reinhart BJ, Slack FJ, Basson M,
Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR and Ruvkun G:
The 21-nucleotide let-7 RNA regulates developmental timing in
Caenorhabditis elegans. Nature. 403:901–906. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Esteller M: Non-coding RNAs in human
disease. Nat Rev Genet. 12:861–874. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Esquela-Kerscher A and Slack FJ: Oncomirs
- microRNAs with a role in cancer. Nat Rev Cancer. 6:259–269. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Garzon R, Calin GA and Croce CM: MicroRNAs
in cancer. Annu Rev Med. 60:167–179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Slack FJ and Weidhaas JB: MicroRNA in
cancer prognosis. N Engl J Med. 359:2720–2722. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li Y, VandenBoom TG II, Kong D, Wang Z,
Ali S, Philip PA and Sarkar FH: Upregulation of miR-200 and let-7
by natural agents leads to the reversal of
epithelial-to-mesenchymal transition in gemcitabine-resistant
pancreatic cancer cells. Cancer Res. 69:6704–6712. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liao XH, Lu DL, Wang N, Liu LY, Wang Y, Li
YQ, Yan TB, Sun XG, Hu P and Zhang TC: Estrogen receptor α mediates
proliferation of breast cancer MCF-7 cells via a
p21/PCNA/E2F1-dependent pathway. FEBS J. 281:927–942. 2014.
View Article : Google Scholar
|
|
28
|
Zuo JH, Zhu W, Li MY, Li XH, Yi H, Zeng
GQ, Wan XX, He QY, Li JH, Qu JQ, et al: Activation of EGFR promotes
squamous carcinoma SCC10A cell migration and invasion via inducing
EMT-like phenotype change and MMP-9-mediated degradation of
E-cadherin. J Cell Biochem. 112:2508–2517. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jung H, Lee KP, Park SJ, Park JH, Jang YS,
Choi SY, Jung JG, Jo K, Park DY, Yoon JH, et al: TMPRSS4 promotes
invasion, migration and metastasis of human tumor cells by
facilitating an epithelial-mesenchymal transition. Oncogene.
27:2635–2647. 2008. View Article : Google Scholar
|
|
30
|
Christiansen JJ and Rajasekaran AK:
Reassessing epithelial to mesenchymal transition as a prerequisite
for carcinoma invasion and metastasis. Cancer Res. 66:8319–8326.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shimono Y, Zabala M, Cho RW, Lobo N,
Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao E, et al:
Downregulation of miRNA-200c links breast cancer stem cells with
normal stem cells. Cell. 138:592–603. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee RC, Feinbaum RL and Ambros V: The C.
elegans heterochronic gene lin-4 encodes small RNAs with antisense
complementarity to lin-14. Cell. 75:843–854. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Meng F, Henson R, Wehbe-Janek H, Ghoshal
K, Jacob ST and Patel T: MicroRNA-21 regulates expression of the
PTEN tumor suppressor gene in human hepatocellular cancer.
Gastroenterology. 133:647–658. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhu S, Wu H, Wu F, Nie D, Sheng S and Mo
YY: MicroRNA-21 targets tumor suppressor genes in invasion and
metastasis. Cell Res. 18:350–359. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhu S, Si ML, Wu H and Mo YY: MicroRNA-21
targets the tumor suppressor gene tropomyosin 1 (TPM1). J Biol
Chem. 282:14328–14336. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee EJ, Gusev Y, Jiang J, Nuovo GJ, Lerner
MR, Frankel WL, Morgan DL, Postier RG, Brackett DJ and Schmittgen
TD: Expression profiling identifies microRNA signature in
pancreatic cancer. Int J Cancer. 120:1046–1054. 2007. View Article : Google Scholar
|
|
38
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang Z, Li Y, Ahmad A, Banerjee S, Azmi
AS, Kong D and Sarkar FH: Pancreatic cancer: Understanding and
overcoming chemoresistance. Nat Rev Gastroenterol Hepatol. 8:27–33.
2011. View Article : Google Scholar
|
|
40
|
Ellenrieder V, Hendler SF, Boeck W,
Seufferlein T, Menke A, Ruhland C, Adler G and Gress TM:
Transforming growth factor beta1 treatment leads to an
epithelial-mesenchymal trans-differentiation of pancreatic cancer
cells requiring extracellular signal-regulated kinase 2 activation.
Cancer Res. 61:4222–4228. 2001.PubMed/NCBI
|
|
41
|
Wang Z, Li Y, Kong D, Banerjee S, Ahmad A,
Azmi AS, Ali S, Abbruzzese JL, Gallick GE and Sarkar FH:
Acquisition of epithelial-mesenchymal transition phenotype of
gemcitabine-resistant pancreatic cancer cells is linked with
activation of the notch signaling pathway. Cancer Res.
69:2400–2407. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Arumugam T, Ramachandran V, Fournier KF,
Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey
DJ and Choi W: Epithelial to mesenchymal transition contributes to
drug resistance in pancreatic cancer. Cancer Res. 69:5820–5828.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sarkar FH, Li Y, Wang Z and Kong D:
Pancreatic cancer stem cells and EMT in drug resistance and
metastasis. Minerva Chir. 64:489–500. 2009.PubMed/NCBI
|
|
44
|
Tucker ON, Dannenberg AJ, Yang EK, Zhang
F, Teng L, Daly JM, Soslow RA, Masferrer JL, Woerner BM, Koki AT,
et al: Cyclooxygenase-2 expression is up-regulated in human
pancreatic cancer. Cancer Res. 59:987–990. 1999.PubMed/NCBI
|
|
45
|
Korc M, Chandrasekar B, Yamanaka Y, Friess
H, Buchier M and Beger HG: Overexpression of the epidermal growth
factor receptor in human pancreatic cancer is associated with
concomitant increases in the levels of epidermal growth factor and
transforming growth factor alpha. J Clin Invest. 90:1352–1360.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Boucher MJ, Morisset J, Vachon PH, Reed
JC, Lainé J and Rivard N: MEK/ERK signaling pathway regulates the
expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of
human pancreatic cancer cells. J Cell Biochem. 79:355–369. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu X, Gu X, Sun L, Flowers AB, Rademaker
AW, Zhou Y and Kiyokawa H: Downregulation of Smurf2, a
tumor-suppressive ubiquitin ligase, in triple-negative breast
cancers: Involvement of the RB-microRNA axis. BMC Cancer.
14:572014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Pardali K and Moustakas A: Actions of
TGF-beta as tumor suppressor and pro-metastatic factor in human
cancer. Biochim Biophys Acta. 1775:21–62. 2007.
|