Introduction
Colorectal cancer is the third leading cause of
cancer-related deaths in the USA and has one of the highest rates
of mortality among cancers worldwide. In addition, the incidence of
colorectal cancer is increasing dramatically in Asian countries
including South Korea (1–6). In the USA alone, a total of 136,000
new colorectal cancer cases were diagnosed in 2014, and the
American Cancer Society projects that more than 50,310 patients
will die from this disease during 2014. Despite continual efforts
to understand the biological mechanisms of colon cancer
progression, the primary mechanisms underlying colon cancer
pathogenesis remain largely unclear.
Wnt proteins were first identified as oncogenes in
mouse breast cancers (7,8), and downstream targets of Wnt
signaling are highly activated in human colon cancers (9–11).
Dysregulation of the Wnt/β-catenin signaling pathway plays a key
role in colon carcinogenesis (11). Likewise, both abnormal activation
of the canonical Wnt/β-catenin pathway and upregulation of the
β-catenin/T-cell factor (TCF) response to transcriptional signaling
play critical roles in early colorectal carcinogenesis (12,13).
Taken together, it is clear that abnormal regulation of the
Wnt/β-catenin signaling has an important role in colon
carcinogenesis.
3,3′-Diindolylmethane (DIM) is a natural compound
derived from cruciferous vegetables. Studies from our laboratory
and others have shown that DIM has inhibitory effects on cancer
cell growth and induces apoptotic cancer cell death (4,14–19).
Although several studies have shown that DIM has anti-proliferative
effects on many types of cancer (19), the biological mechanisms by which
DIM induces apoptosis in colon cancer cells have not been fully
elucidated. However, the inhibitory effects of DIM on colon cancer
cells may be due to altered patterns of gene expression. To clarify
the changes in gene expression patterns of DIM on colon cancer, we
performed a microarray experiment, seeking to identify important
changes in gene expression patterns following DIM treatment of
colon cancer cell lines. In this study, we generated gene
expression data from the HCT116 human colon cancer cell line.
Statistical analyses on gene expression data revealed that the
levels of a total of 1,424 genes were significantly altered, many
of which were associated with regulation of cell growth, cell
cycle, and apoptosis. Noteworthy, the Wnt signaling pathway network
including β-catenin and c-Myc was significantly downregulated by
DIM treatment in colon cancer cells. Therefore, our results suggest
for the first time that DIM suppresses the proliferation of colon
cancer cells via inactivation of β-catenin and targeting the
Wnt/β-catenin signaling pathway by DIM may represent a new approach
for the treatment and prevention of colon cancer.
Materials and methods
Cell lines, culture conditions and
reagents
The DLD-1 and HCT116 cell lines were obtained from
the University of Texas, M.D. Anderson Cancer Center (Houston, TX,
USA). The DLD-1 cell line was cultured in RPMI-1640 medium (Gibco,
Grand Island, NY, USA) and the HCT116 cell line was cultured in
DMEM F12 medium (Gibco). The media contained 10% fetal bovine serum
(Gibco), 100 mg/ml streptomycin, and 100 IU/ml penicillin. Cells
were maintained under standard conditions at 37°C in a 5%
CO2 humidified atmosphere. DIM was purchased from LKT
Laboratories (St. Paul, MN, USA). Antibodies were purchased from
the following commercial sources: Cyclin D1, p-β-catenin, β-catenin
(Cell Signaling Technology, Inc., Beverly, MA, USA); c-Myc, SOX4,
GAPDH (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), and
c-FOS (Cell Signaling Technology Inc., Danvers, MA, USA).
Cell viability assay
Cell proliferation was assessed as previously
described (4). Briefly, DLD-1 and
HCT116 cells were seeded in 96-well plates and allowed to grow
overnight. The following day, cells were treated with varying doses
of DIM. After 72 h, 50 μl of MTT
[3-(4,5-dimethylthiazol-2,5-di-phenyl) tetrazolium bromide; 2
mg/ml] was added to each well. After incubation for 3 h at 37°C,
the media were removed and DMSO was added to solubilize the
formazan crystals for 30 min. Finally, the absorbance at 570 nm was
determined on an Epoch microplate spectrophotometer (BioTeck,
Winnoski, VT, USA).
Colony formation assay
Colony formation assay was performed as previously
described (20). Briefly, cells
(5×104 cells) were seeded into 6-well plates and
incubated for 2 weeks until the colonies were large enough to be
clearly discerned. The cells were fixed with methanol and stained
with crystal violet, and the number of colonies was counted
manually under a microscope and photographed.
Microarray experiment
The microarray experiment was performed as
previously described (4). Briefly,
a mirVana™ miRNA isolation labeling kit (Ambion Inc., TX, USA) was
used according to the manufacturer's protocol to isolate total RNA
from colon cancer cells. An Illumina Human-12 BeadChip V.4
microarray (Illumina, San Diego, CA, USA) was used for sample
hybridization. Gene expression data were extracted using Genome
Studio (Illumina). Microarray analysis was performed by the Shared
Research Equipment Assistance Program of the Korea Basic Science
Institute, MEST.
Microarray data analysis
Data were normalized using the quantile
normalization method with the linear models for microarray data
(LIMMA) package in the R statistical environment (21,22).
The class comparison tool, part of BRB Array Tools suite
(Biometrics Research Branch, National Cancer Institute, MD, USA),
was used to perform multiple comparisons of t-statistics with
estimation of false discovery rate (FDR) and gene expression
differences were considered statistically significant at P-value
<0.001. Cluster and Treeview programs were used to generate heat
maps of gene expression (23).
Gene networks were analyzed using Ingenuity Pathway Analysis
software (Ingenuity Systems Inc., CA, USA). Kaplan-Meier plots and
the log-rank test were used to estimate patient prognosis.
Real-time PCR
Real-time reverse transcription PCR analysis was
used to quantify levels of gene expression as previously described
(24). Briefly, Total RNA (1 μg)
from each sample was reverse-transcribed using the PrimeScript™ RT
reagent kit (Takara Bio Inc, Otsu, Shiga, Japan) according the
manufacturer's protocol. The PCR program was initiated at 95°C for
30 sec and 95°C for 15 sec followed by 40 cycles and 60°C for 1
min. Primer sequences were as follows: β-catenin sense:
5′-GAGCCTGCCATCTGTGCTCT-3′ and β-catenin anti-sense:
5′-ACGCAAAGGTGCATGATTTG-3′; c-Myc sense:
5′-CAGCTGCTTAGACGCTGGATT-3′ and c-Myc antisense:
5′-GTAGAAATACGGCTGCACCGA-3′; cyclin D1 sense:
5′-AGGAACAGAAGTGCGAGGAGG-3′ and cyclin D1 anti-sense:
5′-GGATGGAGTTGTCGGTGTAGATG-3′; c-FOS sense:
5′-GAGGACCTTATCTGTGCGTGAA-3′ and c-FOS antisense:
5′-TGAGTCCACACATGGATGCTT-3′; SOX4 sense: 5′-CCCCCTGGGCCTGTACG-3′
and SOX-4 antisense: 5′-CCGGGCTCGAAGTTAAAATC-3′; GAPDH sense:
5′-GTCTCCTCTGACTTCAACAGCG-3′ and GAPDH anti-sense:
5′-ACCACCCTGTTGCTGTAGCCAA-3′.
Western blot analysis
DLD-1 and HCT116 colon cancer cells were plated and
allowed to attach for 24 h. DIM was added to cell cultures at the
indicated concentrations and incubated for 72 h. Cell lysates were
prepared by suspending the cells in lysis buffer (Intron
Biotechnology, Seoul, South Korea) and the resulting lysates were
subjected to routine western blotting analysis as previously
described (4,14).
Cell cycle analysis
Cell cycle analysis was performed by analyzing total
DNA content with a FACstar flow cytometer (Becton-Dickinson, San
Jose, CA, USA) using Becton Dickinson software (Lysis II and
CellFit) as previously described (25).
Other statistical analysis
Statistically significant differences between
experimental groups and controls were determined by one-way ANOVA
and comparisons between groups were made with Student's t-test.
Results are expressed as the mean ± SEM. P-values <0.05 were
considered to indicate statistical significance.
Results
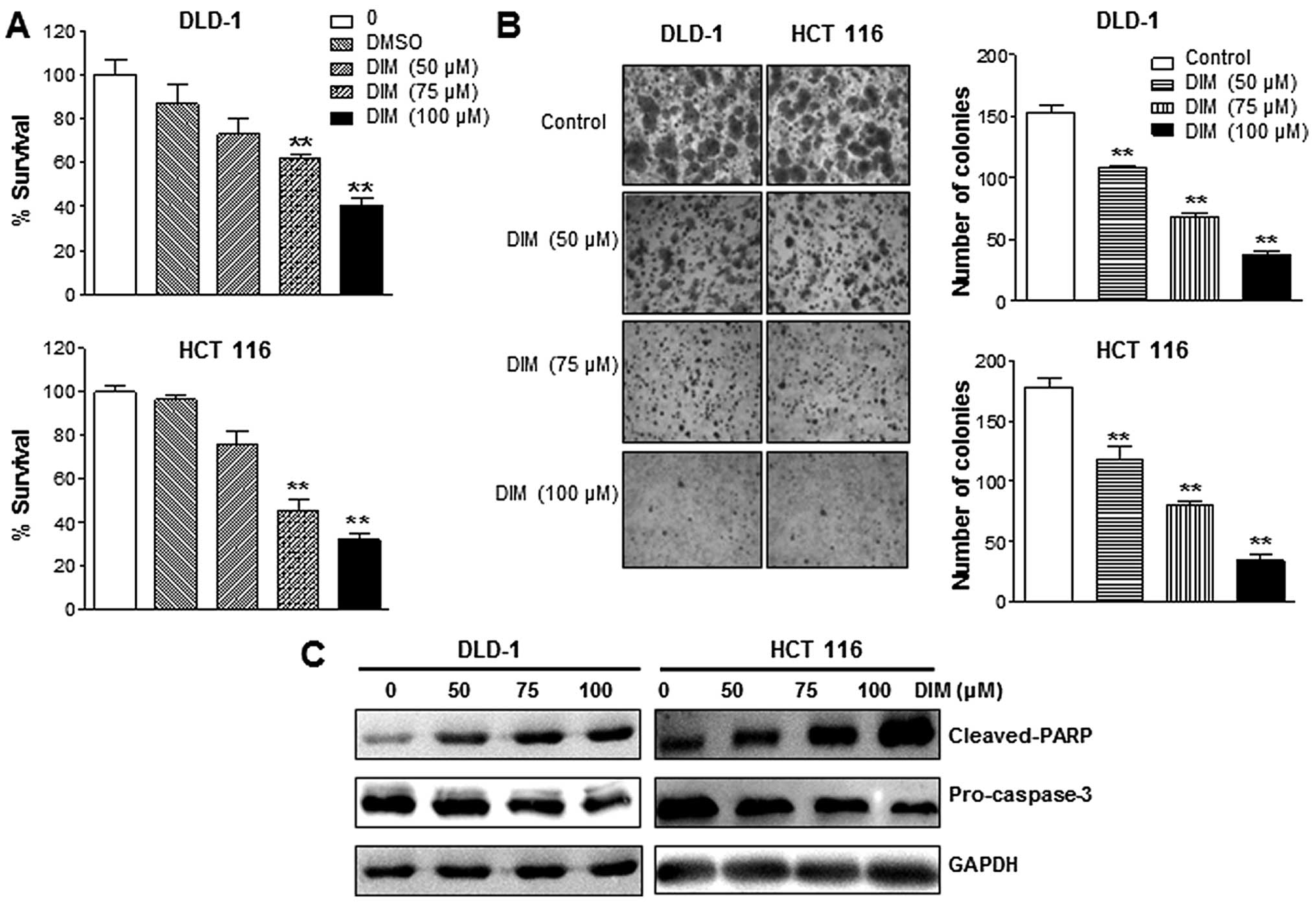
DIM inhibits the growth of colon cancer
cells
The effects of DIM on human colon cell growth were
examined using DLD-1 and HCT116 cells following treatment at
various concentrations for 72 h. As shown in Fig. 1, DIM inhibited cell proliferation
of DLD-1 and HCT116 cells in a dose-dependent manner (Fig. 1A). Treatment of DIM resulted in a
significant inhibition of colony formation in DLD-1 and HCT116
cells in a dose-dependent manner (Fig.
1B). We also evaluated cell viability by quantifying levels of
apoptotic proteins. We found that the DIM treatment resulted in a
significant induction of apoptosis in DLD-1 and HCT116 cells. As
shown in Fig. 1C, DIM
significantly increased cleaved-PARP protein levels in a
dose-dependent manner. Likewise, levels of pro-caspase-3 were
significantly decreased by DIM treatment (Fig. 1C). Taken together, these data
indicated that the DIM inhibits proliferation of human colon cancer
cells.
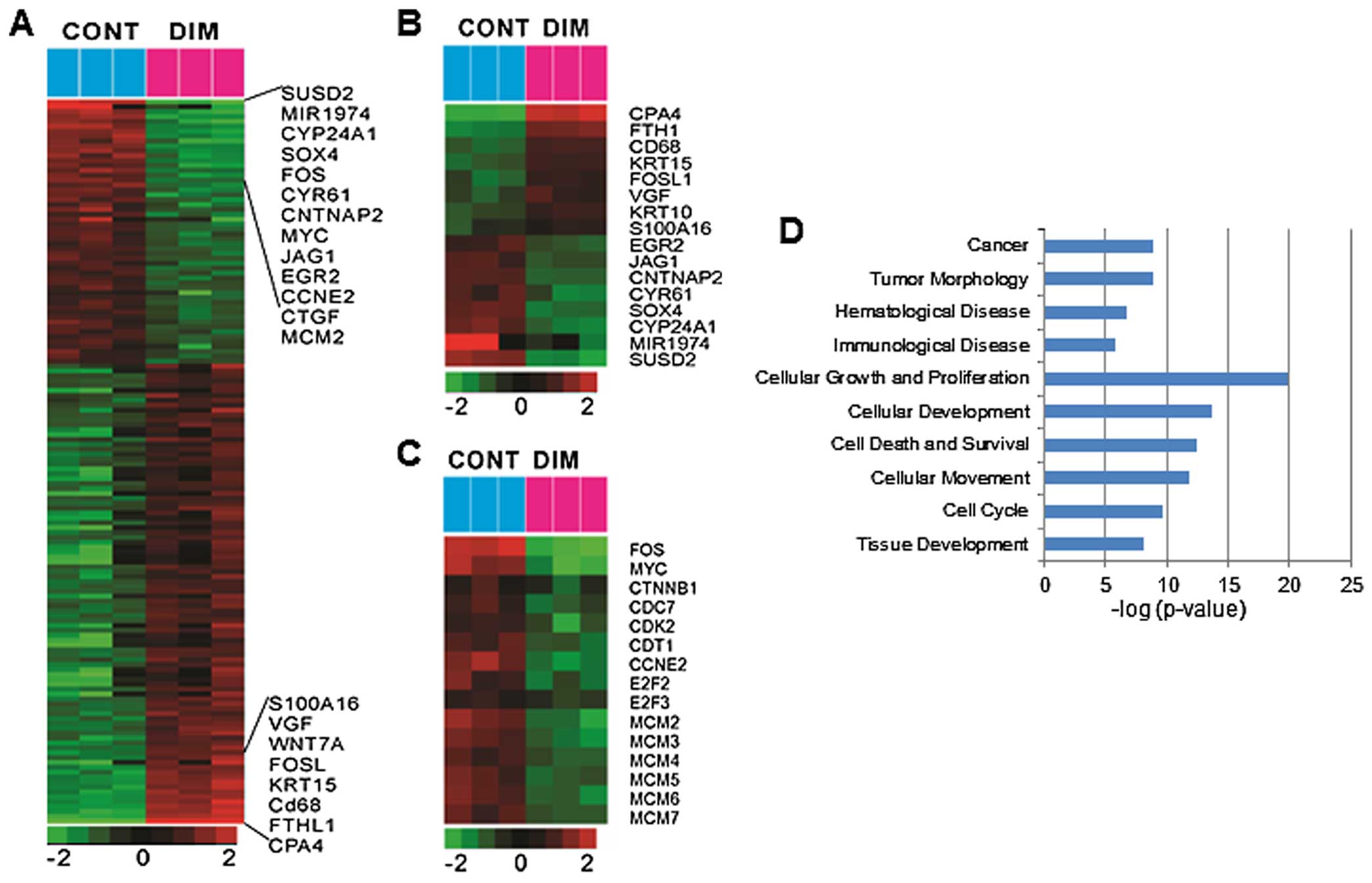
Microarray analysis
To further investigate the effects of DIM on colon
cancer cells, we performed gene expression profiling using an
Illumina bead array platform. We identified a total of 1,424 genes
significantly associated with the effect of DIM in HCT116 cells
(Fig. 2A). Specifically, a total
of 692 genes were significantly upregulated by DIM treatment while
731 genes were downregulated. Among these genes, s100A16, VGF,
FOSL, KRT15, CD68, FTHL1, CPA4 were the most highly upregulated
while SUSD2, MIR1974, CYP24A1, SOX4, FOS, CYR61, CNTNAP2, MYC,
JAG1, EGR2, CCNE2, CTGF, MCM2 were the most significantly
downregulated (Fig. 2B). The most
significantly altered pathway was that of cell cycle control of
chromosomal replication (CDC7, CDK2, CDT1, MCM2, MCM3, MCM4, MCM5,
MCM6, MCM7, E2F2 and E2F3), which was significantly downregulated
(Fig. 2C). Several oncogenes (Myc,
SOX4, CTGF, MCM2 and FOS) and cell cycle related genes (cyclin D
and cyclin E) were significantly downregulated by DIM treatment in
HCT116 cells (Fig. 2C). Gene set
enrichment analysis also revealed highly presentation of cellular
growth and proliferation involved in cell cycle regulation by DIM
treatment (Fig. 2D). We next tried
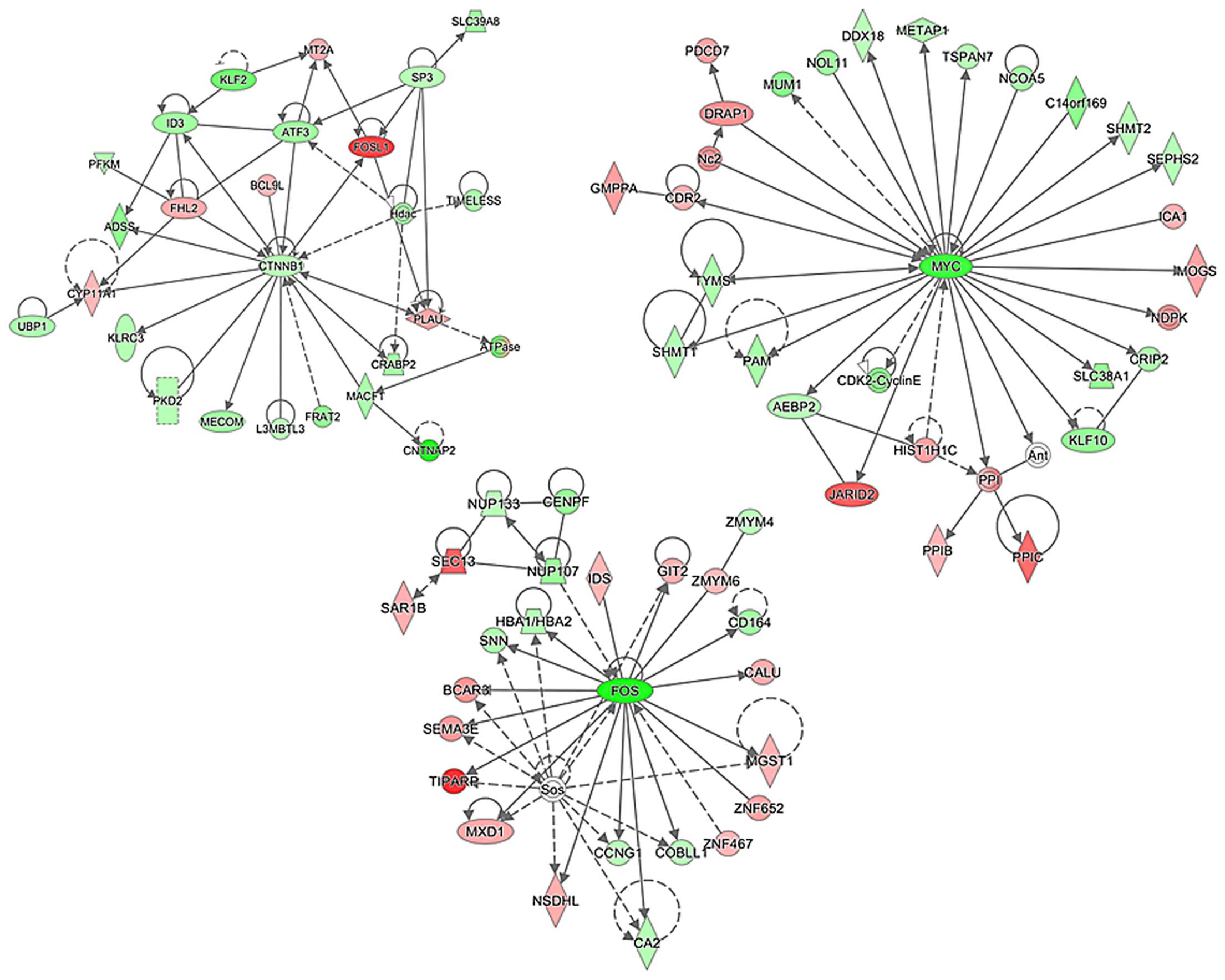
to identify the gene networks potentially involved in the DIM
effects on colon cancer cells using Ingenuity™ Pathway Analysis
(IPA). Our initial analysis identified a number of putative
networks, and functional connectivity of the top network revealed
significant downregulation of β-catenin (CTNNB1), Myc, and FOS
genes (Fig. 3), suggesting
suppression of β-catenin (CTNNB1), Myc and FOS activity by DIM
treatment in colon cancer cells. Importantly, altered expression of
these genes by DIM treatment affects other genes that are
subsequently responsible for controlling colon cancer cell
growth.
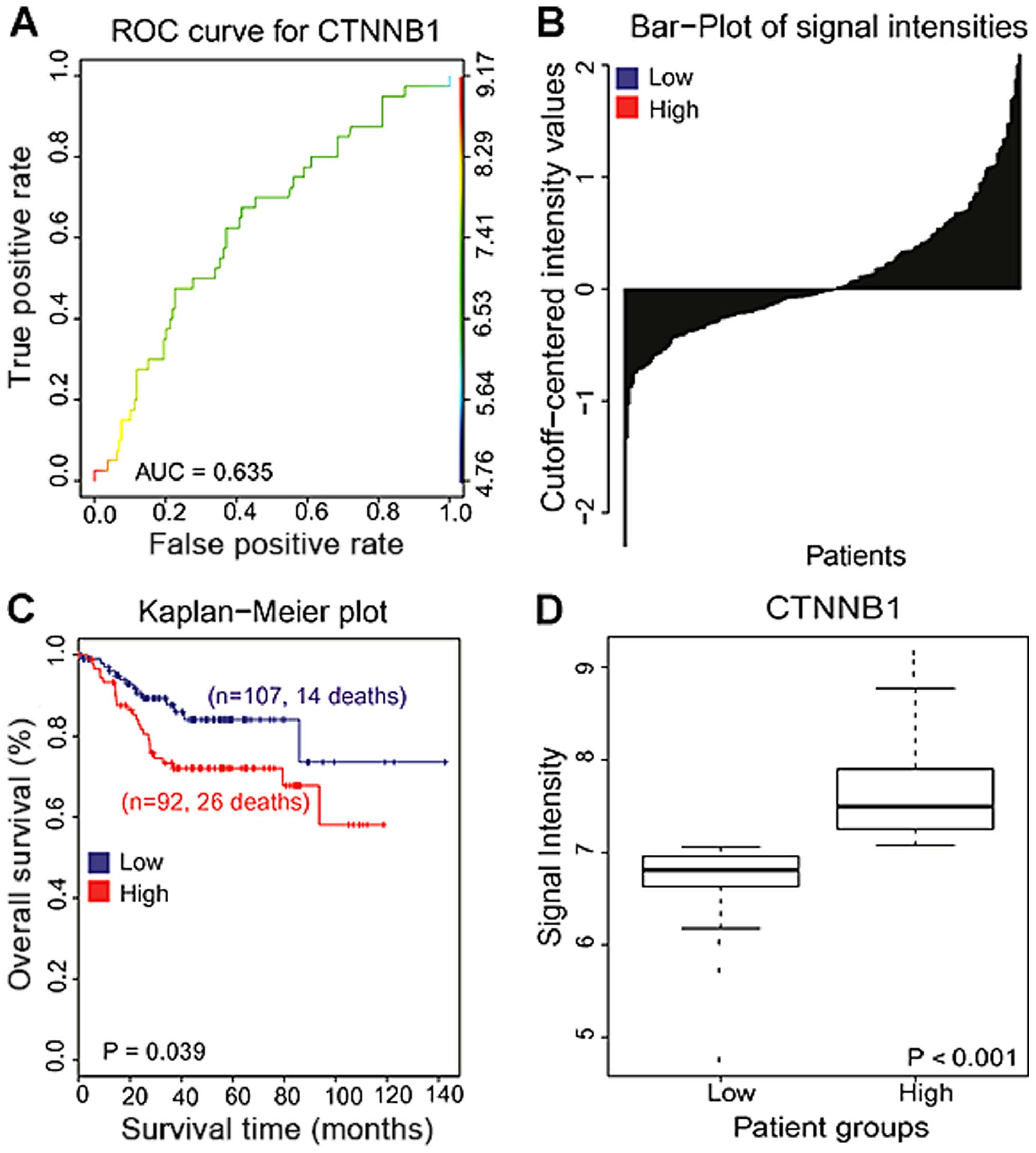
Activation of β-catenin is associated
with poor prognosis in colon cancer patients
Given that Wnt proteins are regarded as oncogenes
and activating mutations frequently convert the downstream target
of Wnt signaling, β-catenin, to an oncogene in human colon cancers,
we next examined the association between the expression of
β-catenin and clinical data of colon cancer patients. Publically
available data from 226 colon cancer patients were downloaded from
the National Center for Biotechnology Information GEO (GSE 14333).
To estimate the β-catenin expression signature responsible for the
clinical overall survival, we used receiver operating
characteristic (ROC) curves from censored overall survival (OS)
data using nearest neighbor estimation methods with a cut-off value
of 7.057 intensities and under the curve (AUC) were calculated
(AUC=0.635) (26–28). Associations between β-catenin and
the patient survival rate were assessed using Kaplan-Meier plots
and log-rank tests. Analysis of Kaplan-Meir plots revealed that
patients with high expression of β-catenin (CTNNB1) had poorer
overall survival compared with those of patients with low
expression of β-catenin (CTNNB1), suggesting that activation of
β-catenin (CTNNB1) is significantly associated with prognosis in
colon cancer patients (P=0.039, Fig.
4). When comparing the microarray gene expression data, the
expression levels of β-catenin (CTNNB1) were significantly
different between low and high expression of β-catenin with colon
cancer patients (P<0.001, Fig.
4).
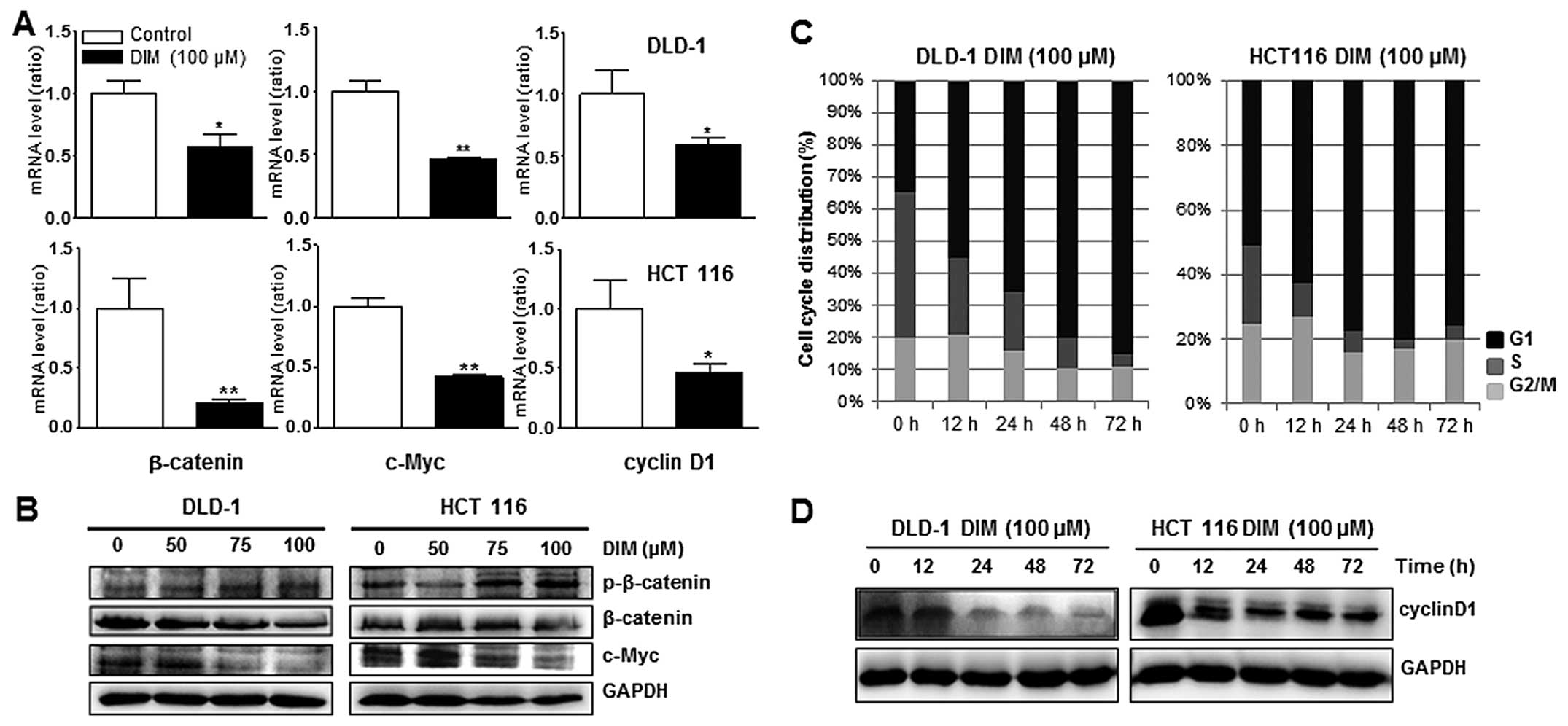
Downregulation of β-catenin and c-Myc
expression by DIM treatment in DLD-1 and HCT116 cells
Since β-catenin can control expression of c-Myc and
cyclin D1 and induce cell proliferation in colon cancer cells, we
next tested whether β-catenin and c-Myc genes could be altered by
DIM treatment in DLD-1 and HCT116 cells using qPCR and western blot
analysis. qPCR showed that β-catenin and c-Myc were significantly
decreased in both cell lines by treatment with 100 μM DIM (Fig. 5A). Protein levels of β-catenin and
c-Myc were also downregulated, while that of p-β-catenin, the
inactive form of β-catenin, was upregulated in a dose-dependent
manner after DIM treatment (Fig.
5B). These results suggested that DIM inhibited activation of
β-catenin and c-Myc in colon cancer cells, indicating that DIM
induces colon cancer cell death by inactivation of the Wnt
signaling pathway, which normally promotes proliferation of cells
in the colon.
DIM induces cell cycle arrest in DLD-1
and HCT116 cells
FACs analysis was performed to determine whether DIM
regulates cell cycle progression in human colon cancer cells. DIM
treatment resulted in a significant, time-dependent increase in the
proportion of the cell population in the G1 phase in both DLD-1 and
HCT116 human colon cancer cells, suggesting that DIM induced G1
cell cycle arrest (Fig. 5C). We
also found that protein level of cyclin D1 were downregulated in a
time-dependent manner after DIM treatment in DLD-1 and HCT116 cells
(Fig. 5D). Taken together, these
results indicated that DIM induced G1 phase arrest in human colon
cancer cells through downregulation of cyclin D1.
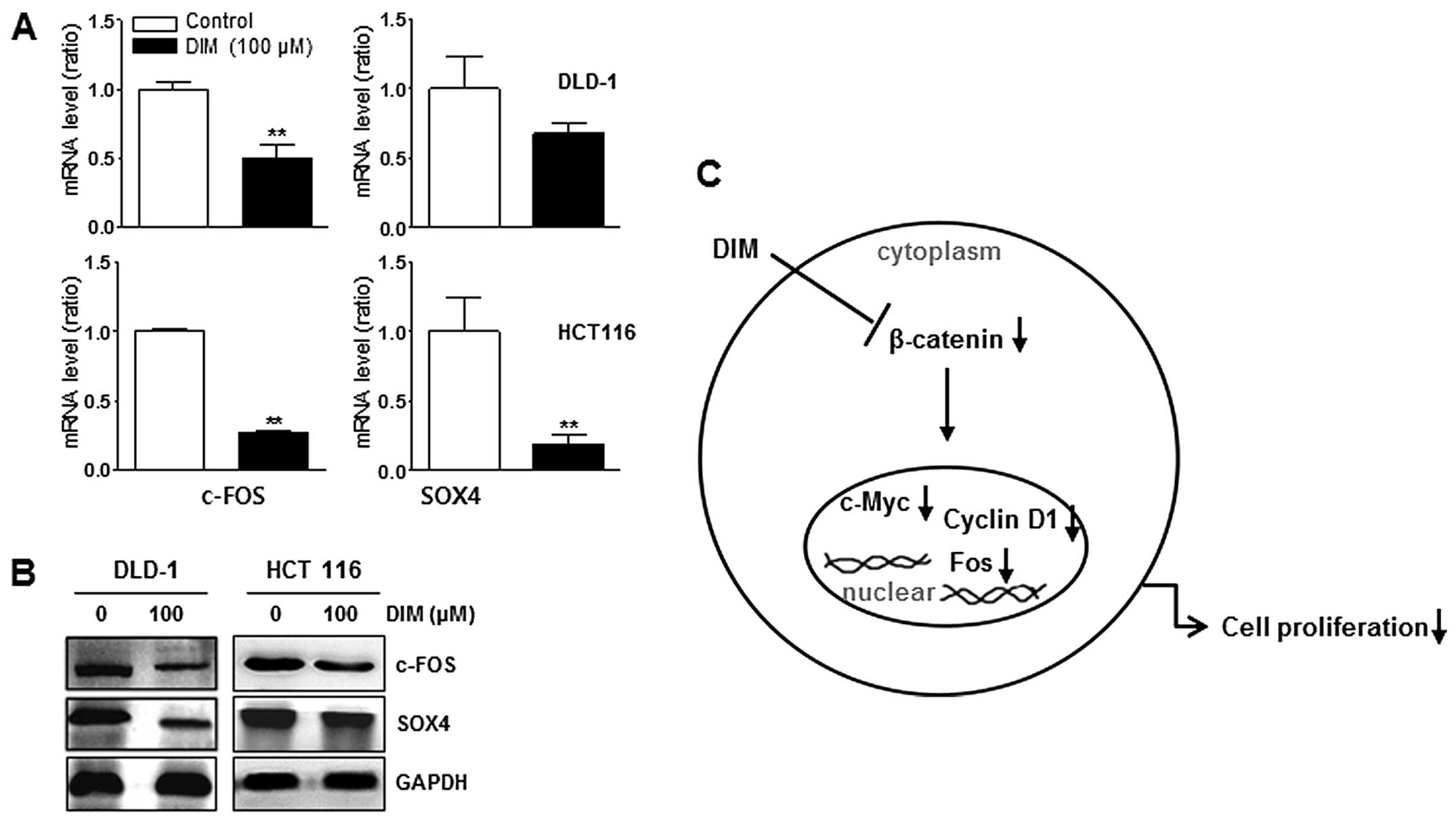
Downregulation of FOS and SOX4 expression
by DIM treatment in DLD-1 and HCT116 cells
Protein and mRNA levels of c-FOS and SOX-4 were
significantly decreased in both DLD-1 and HCT116 cell lines by
treatment with 100 μM DIM (Fig. 6A and
B). These results suggested that DIM inhibited activation of
c-FOS and SOX-4 in colon cancer cells, suggesting that DIM also
suppressed oncoproteins in colon cancer cells.
Discussion
In the present study, we profiled gene expression
following DIM treatment of colon cancer cells. We found that DIM
significantly inhibited proliferation of colon cancer cells
mediated by altered expression of genes involved in cell cycle
progression and Wnt signaling. Our results showed for the first
time that β-catenin and c-Myc, which are major genes involved in
colon carcinogenesis, are significantly downregulated by DIM
treatment in colon cancer cells.
Gene expression profiling is a powerful tool that is
useful for uncovering the molecular mechanisms underlying cellular
functions in human cancers. Since DIM is a nontoxic, natural
compound isolated from cruciferous vegetables, its anti-cancer
effects have been actively explored by many researchers for cancer
prevention and treatment (19,29–31).
Wide ranging pleiotropic anti-tumor signals elicited by DIM have
been previously shown to be orchestrated through the Ah receptor,
Akt and NFkB pathways impinging on cell cycle arrest and altering
angiogenesis, invasion, and metastasis of a variety of cancer cells
(19,29,31–34).
DIM has been reported to induce apoptosis in colon cancer cells by
several mechanisms involving cell cycle arrest, inactivation of
Akt, induction of proteasomal degradation of class I histone
deacetylases, and downregulation of survivin (4,35–37).
However, a comprehensive biological mechanism by which DIM inhibits
colon cancer cell growth and induces apoptosis remains elusive.
We used gene expression profiling to determine how
exposure to DIM alters the transcriptome of colon cancer cells. We
found that DIM significantly altered the expression of more than
1000 genes. The top canonical pathway modified by DIM treatment in
colon cancer cells was cell cycle control of chromosomal
replication. Specifically, we observed significantly decreased
expression of numerous genes involved in cell cycle control of
chromosomal replication (CDC7, CDK2, CDT1, MCM2, MCM3, MCM4, MCM5,
MCM6, MCM7, E2F2 and E2F3). Gene set enrichment analysis also
showed high presentation of cellular growth and proliferation
involved in cell cycle regulation by DIM treatment. In addition,
several oncogenes (Myc, SOX4, CTGF, β-catenin and FOS) were
significantly downregulated by DIM treatment. IPA showed that DIM
significantly altered putative gene networks surrounding β-catenin
(CTNNB1), Myc and FOS. Specifically, the expression of many
downstream target genes of β-catenin (CTNNB1) and Myc were
downregulated, as were β-catenin and Myc themselves, suggesting
that transcriptional activity of β-catenin and Myc was suppressed
by DIM. This down-regulation might be responsible for the
inhibition of colon cancer cell growth. Alterations in gene
expression profiles by DIM exposure of breast cancer cells have
been reported (38); however, to
the best of our knowledge, this is the first report of a gene
expression profiling study on colon cancer cells treated with
DIM.
Since the downstream target of Wnt signaling,
β-catenin, is an oncogene in human colon cancer, we investigated
the relationship between the expression of β-catenin and clinical
outcomes in colon cancer. Publically available data from 226 colon
cancer patients was used to determine whether there is an
association between β-catenin activation and patient survival.
Kaplan-Meir plot analysis and log-rank test analysis showed that
patients with high expression levels of β-catenin had poorer
overall survival when compared with patients who had low levels of
β-catenin expression, suggesting that β-catenin is significantly
associated with prognosis in colon cancer patients. Our results
strongly indicated that the Wnt/β-catenin signaling pathway plays a
crucial role in carcinogenesis and progression of colon cancer.
The β-catenin and Tcf genes regulate expression of
c-Myc and cyclin D1 and regulate cell proliferation (39,40).
In the present study, we found that DIM significantly suppressed
the mRNA expression levels of β-catenin and c-Myc in two different
colon cancer cell lines (DLD-1 and HCT116). We also found that
protein levels of β-catenin and c-Myc were significantly
downregulated after DIM treatment while level of phosphorylated
β-catenin, the inactive form of β-catenin that is degraded by a
ubiquitin-proteasome mechanism, was increased. We also showed that
DIM induced a significant increase in the proportion of the cell
population in the G1 phase of the cell cycle in a time-dependent
manner in both DLD-1 and HCT116 cells. We confirmed that protein
levels of c-Myc, FOS, and Cyclin D1 were also inhibited in a
time-dependent manner after DIM treatment in DLD-1 and HCT116
cells. Together, these findings suggested that DIM induced G1 phase
arrest in human colon cancer cells by altering the expression of
c-Myc, FOS and cyclin D1, which may be mediated through suppression
of β-catenin. Therefore, the anti-proliferative effects of DIM in
colon cancer cells may be associated with inhibition of β-catenin,
which in turn may induce cell cycle arrest by regulating expression
of cyclin D1, FOS and c-Myc (Fig.
6C). However, further studies are needed to elucidate how DIM
specifically regulates target genes downstream of β-catenin in the
cytosol and nucleus.
In conclusion, to the best of our knowledge, this is
the first study to show that DIM inhibits the Wnt/β-catenin pathway
in colon cancer cells. We believe that targeting Wnt/β-catenin
signaling with DIM may be an attractive strategy for the prevention
and treatment of colon cancer.
Acknowledgements
This study was supported by the Mid-career
Researcher Program through NRF grant (NRF-2013R1A2A2A04008115)
funded by the Korea government (MEST) and was supported by a
National Research Foundation of Korea (NRF) grant funded by the
Korean government (MISP, no. 2008-0062279).
References
|
1
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 59:225–249.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Center MM, Ward E and Thun MJ:
Cancer occurrence. Methods Mol Biol. 471:3–29. 2009. View Article : Google Scholar
|
|
3
|
DeSantis CE, Lin CC, Mariotto AB, Siegel
RL, Stein KD, Kramer JL, Alteri R, Robbins AS and Jemal A: Cancer
treatment and survivorship statistics, 2014. CA Cancer J Clin.
64:252–271. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li XJ, Leem SH, Park MH and Kim SM:
Regulation of YAP through an Akt-dependent process by 3,
3′-diindolylmethane in human colon cancer cells. Int J Oncol.
43:1992–1998. 2013.PubMed/NCBI
|
|
5
|
Cho KH, Park S, Lee KS, Jang SI, Yoo KB,
Kim JH and Park EC: A single measure of cancer burden in Korea from
1999 to 2010. Asian Pac J Cancer Prev. 14:5249–5255. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Park JH, Lee KS and Choi KS: Burden of
cancer in Korea during 2000–2020. Cancer Epidemiol. 37:353–359.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huguet EL, McMahon JA, McMahon AP,
Bicknell R and Harris AL: Differential expression of human Wnt
genes 2, 3, 4, and 7B in human breast cell lines and normal and
disease states of human breast tissue. Cancer Res. 54:2615–2621.
1994.PubMed/NCBI
|
|
8
|
Weber-Hall SJ, Phippard DJ, Niemeyer CC
and Dale TC: Developmental and hormonal regulation of Wnt gene
expression in the mouse mammary gland. Differentiation. 57:205–214.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moon RT and Miller JR: The APC tumor
suppressor protein in development and cancer. Trends Genet.
13:256–258. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takahashi M, Fukuda K, Sugimura T and
Wakabayashi K: Beta-catenin is frequently mutated and demonstrates
altered cellular location in azoxymethane-induced rat colon tumors.
Cancer Res. 58:42–46. 1998.PubMed/NCBI
|
|
11
|
Bienz M and Clevers H: Linking colorectal
cancer to Wnt signaling. Cell. 103:311–320. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Luu HH, Zhang R, Haydon RC, Rayburn E,
Kang Q, Si W, Park JK, Wang H, Peng Y, Jiang W, et al:
Wnt/beta-catenin signaling pathway as a novel cancer drug target.
Curr Cancer Drug Targets. 4:653–671. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mishra L, Shetty K, Tang Y, Stuart A and
Byers SW: The role of TGF-beta and Wnt signaling in
gastrointestinal stem cells and cancer. Oncogene. 24:5775–5789.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li XJ, Park ES, Park MH and Kim SM:
3,3′-Diindolylmethane suppresses the growth of gastric cancer cells
via activation of the Hippo signaling pathway. Oncol Rep.
30:2419–2426. 2013.PubMed/NCBI
|
|
15
|
Kim SJ, Lee JS and Kim SM:
3,3′-Diindolylmethane suppresses growth of human esophageal
squamous cancer cells by G1 cell cycle arrest. Oncol Rep.
27:1669–1673. 2012.PubMed/NCBI
|
|
16
|
Rahman KM, Li Y and Sarkar FH:
Inactivation of akt and NF-kappaB play important roles during
indole-3-carbinol-induced apoptosis in breast cancer cells. Nutr
Cancer. 48:84–94. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hong C, Kim HA, Firestone GL and Bjeldanes
LF: 3,3′-Diindolylmethane (DIM) induces a G(1) cell cycle arrest in
human breast cancer cells that is accompanied by Sp1-mediated
activation of p21(WAF1/CIP1) expression. Carcinogenesis.
23:1297–1305. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hong C, Firestone GL and Bjeldanes LF:
Bcl-2 family-mediated apoptotic effects of 3,3′-diindolylmethane
(DIM) in human breast cancer cells. Biochem Pharmacol.
63:1085–1097. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Banerjee S, Kong D, Wang Z, Bao B, Hillman
GG and Sarkar FH: Attenuation of multi-targeted
proliferation-linked signaling by 3,3′-diindolylmethane (DIM): From
bench to clinic. Mutat Res. 728:47–66. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ye S, Lee KB, Park MH, Lee JS and Kim SM:
p63 regulates growth of esophageal squamous carcinoma cells via the
Akt signaling pathway. Int J Oncol. 44:2153–2159. 2014.PubMed/NCBI
|
|
21
|
Bolstad BM, Irizarry RA, Astrand M and
Speed TP: A comparison of normalization methods for high density
oligo-nucleotide array data based on variance and bias.
Bioinformatics. 19:185–193. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim SM, Park YY, Park ES, Cho JY, Izzo JG,
Zhang D, Kim SB, Lee JH, Bhutani MS, Swisher SG, et al: Prognostic
biomarkers for esophageal adenocarcinoma identified by analysis of
tumor transcriptome. PLoS One. 5:e150742010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Eisen MB, Spellman PT, Brown PO and
Botstein D: Cluster analysis and display of genome-wide expression
patterns. Proc Natl Acad Sci USA. 95:14863–14868. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jin H, Park MH and Kim SM:
3,3′-Diindolylmethane potentiates paclitaxel-induced antitumor
effects on gastric cancer cells through the Akt/FOXM1 signaling
cascade. Oncol Rep. 33:2031–2036. 2015.PubMed/NCBI
|
|
25
|
Kim M, Kim M, Lee S, Kuninaka S, Saya H,
Lee H, Lee S and Lim DS: cAMP/PKA signalling reinforces the
LATS-YAP pathway to fully suppress YAP in response to actin
cytoskeletal changes. EMBO J. 32:1543–1555. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cho JY, Lim JY, Cheong JH, Park YY, Yoon
SL, Kim SM, Kim SB, Kim H, Hong SW, Park YN, et al: Gene expression
signature-based prognostic risk score in gastric cancer. Clin
Cancer Res. 17:1850–1857. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Heagerty PJ and Zheng Y: Survival model
predictive accuracy and ROC curves. Biometrics. 61:92–105. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim SM, Leem SH, Chu IS, Park YY, Kim SC,
Kim SB, Park ES, Lim JY, Heo J, Kim YJ, et al: Sixty-five
gene-based risk score classifier predicts overall survival in
hepatocellular carcinoma. Hepatology. 55:1443–1452. 2012.
View Article : Google Scholar
|
|
29
|
Nicastro HL, Firestone GL and Bjeldanes
LF: 3,3′-diindolyl-methane rapidly and selectively inhibits
hepatocyte growth factor/c-Met signaling in breast cancer cells. J
Nutr Biochem. 24:1882–1888. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen C, Chen SM, Xu B, Chen Z, Wang F, Ren
J, Xu Y, Wang Y, Xiao BK and Tao ZZ: In vivo and in vitro study on
the role of 3,3′-diindolylmethane in treatment and prevention of
nasopharyngeal carcinoma. Carcinogenesis. 34:1815–1821. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ahmad A, Ali S, Ahmed A, Ali AS, Raz A,
Sakr WA and Rahman KM: 3, 3′-Diindolylmethane enhances the
effectiveness of herceptin against HER-2/neu-expressing breast
cancer cells. PLoS One. 8:e546572013. View Article : Google Scholar
|
|
32
|
Li Y, Kong D, Ahmad A, Bao B and Sarkar
FH: Targeting bone remodeling by isoflavone and
3,3′-diindolylmethane in the context of prostate cancer bone
metastasis. PLoS One. 7:e330112012. View Article : Google Scholar
|
|
33
|
Gao N, Cheng S, Budhraja A, Liu EH, Chen
J, Chen D, Yang Z, Luo J, Shi X and Zhang Z: 3,3′-Diindolylmethane
exhibits anti-leukemic activity in vitro and in vivo through an
Akt-dependent process. PLoS One. 7:e317832012. View Article : Google Scholar
|
|
34
|
Weng JR, Bai LY, Chiu CF, Wang YC and Tsai
MH: The dietary phytochemical 3,3′-diindolylmethane induces G2/M
arrest and apoptosis in oral squamous cell carcinoma by modulating
Akt-NF-κB, MAPK, and p53 signaling. Chem Biol Interact.
195:224–230. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li Y, Li X and Guo B: Chemopreventive
agent 3,3′-diindolyl-methane selectively induces proteasomal
degradation of class I histone deacetylases. Cancer Res.
70:646–654. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Choi HJ, Lim Y and Park JH: Induction of
G1 and G2/M cell cycle arrests by the dietary compound
3,3′-diindolylmethane in HT-29 human colon cancer cells. BMC
Gastroenterol. 9:392009. View Article : Google Scholar
|
|
37
|
Bhatnagar N, Li X, Chen Y, Zhou X, Garrett
SH and Guo B: 3,3′-diindolylmethane enhances the efficacy of
butyrate in colon cancer prevention through down-regulation of
survivin. Cancer Prev Res (Phila). 2:581–589. 2009. View Article : Google Scholar
|
|
38
|
Rahman KW, Li Y, Wang Z, Sarkar SH and
Sarkar FH: Gene expression profiling revealed survivin as a target
of 3,3′-diindolylmethane-induced cell growth inhibition and
apoptosis in breast cancer cells. Cancer Res. 66:4952–4960. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tetsu O and McCormick F: Beta-catenin
regulates expression of cyclin D1 in colon carcinoma cells. Nature.
398:422–426. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
40
|
He TC, Sparks AB, Rago C, Hermeking H,
Zawel L, da Costa LT, Morin PJ, Vogelstein B and Kinzler KW:
Identification of c-MYC as a target of the APC pathway. Science.
281:1509–1512. 1998. View Article : Google Scholar : PubMed/NCBI
|