Introduction
Metastatic melanoma is generally an incurable
neoplasm. Few patients achieve meaningful clinical responses to
conventional cytotoxic chemotherapy. In recent years, there have
been significant developments in immunotherapy and molecular
targeted therapy for the treatment of metastatic melanoma.
Antibodies targeting immune inhibitory checkpoints, specifically
CTLA4 and PD1/PD-L1 have produced meaningful improvements in
melanoma survival rates (1).
Inhibitors targeting BRAF produce frequent, but temporary responses
in patients with V600E mutations (2). Alternative strategies to target the
BRAF signalling are also been tested including MEK inhibitors, and
both BRAF and MEK inhibitors are now approved as single agent
therapies for metastatic melanoma. Furthermore, combining the BRAF
and MEK inhibitors may further improve response rates (3).
These new therapies have resulted in improvements
the 5-year survival rates for patients with metastatic melanoma but
there is a need for new targets and therapies to further improve
survival rates. The aim of the present study was to identify novel
therapeutic targets by screening a library of 160 protein kinase
inhibitors, including cyclin dependent kinase (CDK) inhibitors.
Protein kinases play central roles in the regulation
of cell proliferation, differentiation and apoptosis. Deregulated
kinases are often found to be oncogenic and can be central to the
survival of cancer cells (4,5).
Progression through the cell cycle is dependent on CDKs, a family
of serine/threonine kinases that consist of a catalytic subunit
known as CDK and a regulatory subunit known as a cyclin. Many of
the genes involved in cell cycle progression are frequently mutated
in human cancers and deregulated CDK activity can be considered a
hallmark of malignancy (6–9).
The tumour suppressor p16INK4a, encoded by the
CDKN2A gene, is inactivated by mutation, deletion or promoter
methylation in 30–70% of melanomas (10). A 2010 study by Jonsson et al
(11) demonstrated p16INK4a
mutation, promoter methylation or lack of expression occurred in
16, 25 and 82% of melanoma metastases, respectively. The p16INK4a
protein binds to CDK4/6 and inhibits interaction with D-type
cyclins, which would otherwise stimulate passage through the G1
phase of the cell cycle. The frequent loss of p16INK4a in melanomas
suggests that CDK4 activity may be unchecked in melanoma and may
play a role in promoting uncontrolled proliferation of melanoma
cells. Furthermore, mutation or overexpression of CDK4, combined
with amplification of cyclin D1, has been implicated in resistance
to BRAF inhibition in V600E-mutated melanoma cells, and
amplification of cyclin D1 is detected in ~17% of BRAF
V600E-mutated human metastatic melanomas (12).
The druggable nature of kinases has sparked
considerable interest in pursuing CDKs as novel targets in
anticancer drug development. Selective inhibition of CDKs may limit
the progression of a tumour cell through the cell cycle and
facilitate the induction of apoptosis (6,13).
Materials and methods
Cells and reagents
Malme-3M, Sk-Mel-2, Sk-Mel-5, Sk-Mel-28, M14 and
Lox-IMVI melanoma cell lines were obtained from the Department of
Developmental Therapeutics, National Cancer Institute (Bethesda,
MD, USA). WM-115 and WM-266-4 melanoma cell lines were obtained
from the European Association Culture Collection (UK). Malme-3M,
Sk-Mel-2, Sk-Mel-5, Sk-Mel-28, M14 and Lox-IMVI cell lines were
maintained at 37°C with 5% CO2 in RPMI-1640 medium
(Sigma-Aldrich, Co. Wicklow, Ireland) with 10% fetal calf serum
(FCS; Lonza, Tewkesbury, UK). WM-115 and WM-266-4 were maintained
at 37°C with 5% CO2 in minimal essential medium (MEM;
Sigma-Aldrich) with 10% FCS (BioWhittaker, Walkersville, MD, USA),
2 mM L-glutamine (Life Technologies, Dublin, Ireland), 1 mM
non-essential amino acids (Life Technologies) and 1 mM sodium
pyruvate (Life Technologies). Stock solutions of fascaplysin (Merck
Millipore, Watford, UK) (10 mM), PLX4032 (Sequoia Research Products
Ltd., Pangbourne, UK) (10 mM), elacridar (Sigma-Aldrich) (10 mM)
and temozolomide (Sigma-Aldrich) (103 mM) were prepared in dimethyl
sulfoxide (DMSO) PD0332991 (provided by Pfizer, Peapack, NJ, USA)
(10 mM) was prepared in ultrapure water.
InhibitorSelect™ 384-well protein kinase
inhibitor library I
The InhibitorSelect protein kinase inhibitor library
I (Merck Millipore) was supplied with 160 protein kinase inhibitors
in a 384-well plate at a volume of 25 μl and a concentration of 10
mM in DMSO and were stored at −80°C. Stock solutions (1 mM) were
prepared by dilution in DMSO, and stored at −20°C. Initial
screening of the 160 protein kinase inhibitors was performed at 1
μM concentration on the Sk-Mel-2 and Sk-Mel-28 cell lines.
Cells/well (1×103) were seeded in 96-well plates. Plates
were incubated overnight at 37°C followed by addition of drugs at
the appropriate concentrations and incubated for a further 5 days
until wells were 80–90% confiuent. At completion of the assay the
colorimetric acid phosphatase assay was used to determine cell
viability.
Proliferation assays and acid phosphatase
assay
All cells lines were seeded at 1×103
cells/well in 96-well plates except for Malme-3M and WM-115 which
were seeded at 2×103 cells/well. Plates were incubated
overnight at 37°C followed by addition of drug at the appropriate
concentrations and incubated for a further 5 days until wells were
80–90% confluent. All media were removed and the wells were washed
once with phosphate-buffered saline (PBS; Sigma-Aldrich).
Paranitrophenol phosphate substrate (7.25 mM; Sigma-Aldrich) in 0.1
M sodium acetate buffer with 0.1% Triton-X (Sigma-Aldrich) pH 5.5
was added to each well and incubated at 37°C for 2 h. To stop the
reaction 50 μl of 1 M NaOH was added and the absorbance was read at
405 nM (reference, 620 nM).
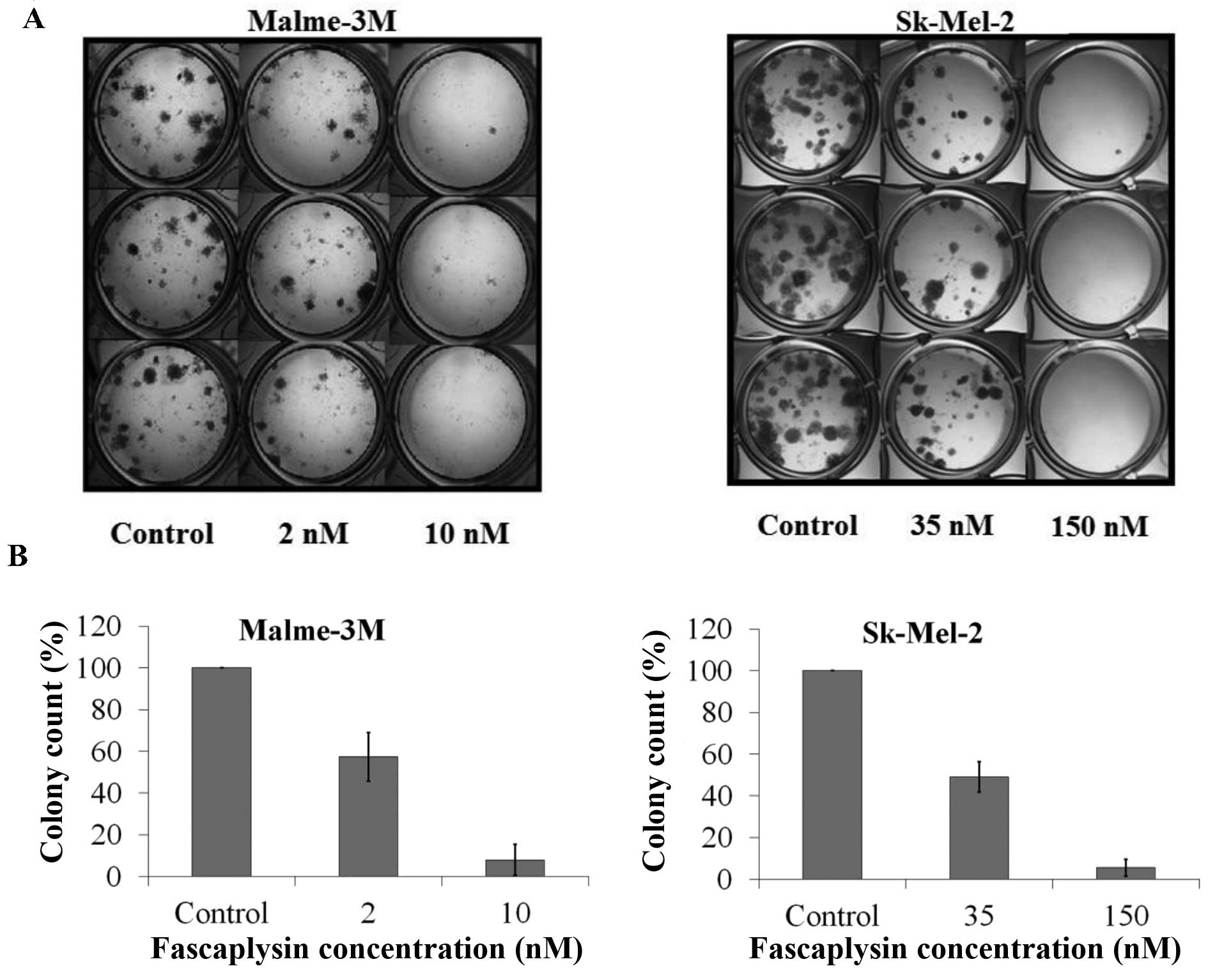
Clonogenic assays
Malme-3M were seeded at 600 cells/well and Sk-Mel-2
were seeded at 125 cells/well in a 24-well plate. The cells were
incubated overnight at 37°C. Media were removed and the drugs were
added at the appropriate concentrations. Drug containing medium was
removed after 4 days and replaced with drug-free media. The medium
was replenished every 4/5 days thereafter. Malme-3M cells were
incubated for 17 days and Sk-Mel-2 cells were incubated for 14
days. When ready to stain, media were removed and the cells washed
gently with PBS twice. The cells were then fixed in cold Methacare
solution (4°C, 75% v/v methanol, 25% v/v acetic acid) for 30 min.
The Methacare solution was removed and the cells were washed twice
with PBS before staining with 1% crystal violet (Cruinn
Diagnostics, Dublin, Ireland) for 1 h. The cells were then rinsed
with water and left to dry. Stained colonies were counted by
eye.
Cell cycle assays
Malme-3M cells (2.5×104 per well) were
seeded in 24-well plates and incubated overnight at 37°C.
Fascaplysin was then added at the appropriate concentration and the
plates incubated at 37°C for 48 h. Media was collected and the
wells washed once with PBS. Cells were trypsinised and added to the
media collected for each sample. Cells were centrifuged at 300 × g
for 5 min and the media aspirated. The cell pellet was resuspended
in 150 μl PBS and transferred to a round bottomed 96-well plate.
The plate was centrifuged at 300 × g for 5 min and the supernatant
aspirated leaving ~15 μl in each well. The remaining volume was
used to resuspend the cells and 200 μl of ice cold 70% ethanol was
added. The plates were then stored at −20°C for 2 h. After fixing,
the cells were spun at 450 × g for 5 min, the supernatant removed,
washed with 200 μl of PBS and spun again at 450 × g. The PBS was
then removed and 200 μl of Guava cell cycle reagent (Merck
Millipore) was added to each well. The cells were mixed by
pipetting and stored at room temperature shielded from the light
for 30 min. Cells were analysed on the Guava easyCyte (Merck
Millipore) and the data were analysed using ModFit LT software
(Verity Software House, Topsham, ME, USA).
Terminal DNA transferase-mediated dUTP
nick end labelling (TUNEL) assay
The TUNEL assays were performed using the Guava
TUNEL kit for fiow cytometry (Merck Millipore), according to the
manufacturer's protocol. Briefiy, 2.5×104 cells were
seeded per well in 24-well plates and incubated overnight at 37°C,
followed by addition of drug at the appropriate concentrations.
After 48 h, media were collected and the wells washed once with
PBS. Cells were trypsinised and added to the media collected for
each sample. Cells were centrifuged at 300 × g for 5 min and the
medium was aspirated. The pellet was re-suspended in 150 μl of PBS
and transferred to a round bottomed 96-well plate. A total of 50 μl
of 4% paraformaldehyde (Sigma-Aldrich) made up in PBS was added to
the wells and mixed. Cells were incubated at 4°C for 60 min. The
plate was centrifuged at 300 × g for 5 min and the supernatant
aspirated leaving ~15 μl in each well. The remaining volume was
used to resuspend the cells and 200 μl of ice cold 70% ethanol
(Sigma-Aldrich) was added to the cells. The plates were then stored
at −20°C for 2 h. After fixation, the cells including positive and
negative controls were spun at 300 × g for 5 min. Cells were washed
a further 2 times at 300 × g with wash buffer. The wash buffer was
aspirated and 25 μl of DNA labelling mix was added to each well and
the cells mixed. The plates were covered with parafilm and
incubated for 60 min at 37°C. Rinsing buffer (200 μl) was then
added to each well and the plates spun at 300 × g for 5 min. The
supernatant was aspirated and 50 μl of anti-BrdU staining mix added
to each well, with the plate stored in the dark, at room
temperature for 30 min. At the end of the incubation 150 μl of
rinsing buffer was added to each well. Cells were analysed on the
Guava EasyCyte (Merck Millipore). Positive and negative controls
supplied with the kit were performed with each assay.
Protein extraction and western
blotting
RIPA buffer (500 μl; Sigma-Aldrich) with 1× protease
inhibitors, 2 mM phenylmethanesulphonylfiouride (PMSF) and 1 mM
sodium orthovanadate (Sigma-Aldrich) was added to cells and
incubated on ice for 20 min. Following centrifugation at 16,000 × g
for 10 min at 4°C the resulting lysate was stored at −80°C. Protein
quantification was performed using the bicinchoninic acid (BCA)
assay (Life Technologies). Protein (40 μg) in sample buffer [3 mM
Tris HCl; 5% sodium dodecyl sulphate (SDS); 12.5%
β-mercaptoethanol; 29% glycerol; 0.1% bromophenol blue] was heated
to 95°C for 5 min and proteins were separated on 10% pre-cast
Tris-glycine gels (Lonza). The resolved proteins were then
transferred to nitrocellulose membranes (Life Technologies) using
the iBlot transfer system (Life Technologies). The membrane was
blocked with 5% milk powder (Bio-Rad Laboratories, Hempstead, UK)
in 0.1% PBS-Tween at room temperature for 1 h, then incubated
overnight at 4°C in primary antibody with 0.1% PBS-Tween in 5% milk
powder. Primary antibodies used were anti-CDK4 (1:1,000; Santa Cruz
Biotechnology, Heidelberg, Germany), anti-cyclin D1 (1:1,000; Cell
Signaling Technology, Leiden, The Netherlands), anti-Rb (1:1,000;
Cell Signaling Technology), anti-phospho-Rb (1:3,000; Cell
Signaling Technology), and anti-α-tubulin (1:1,000; Sigma-Aldrich).
The membrane was washed 3 times with 0.5% PBS-Tween and then
incubated at room temperature with anti-mouse (Sigma-Aldrich) or
anti-rabbit (Sigma-Aldrich) secondary antibody in 5% milk powder
with 0.5% PBS-Tween for 1 h. The membrane was washed three times
with 0.5% PBS-Tween followed by one wash with PBS alone. Detection
was performed using Luminol (Santa Cruz Biotechnology) or ECL™
Advance western blotting detection kit (GE Healthcare,
Buckinghamshire, UK).
Statistical analysis
IC50 values and combination index (CI)
values were calculated using CalcuSyn software (Biosoft, Cambridge,
UK). The Student's t-test was used to determine the significance of
the effects of fascaplysin and PD0332991 on the levels of cell
cycle arrest and apoptosis induction, where a P-value of <0.05
was deemed significant. The Pearson's correlation was used to
examine the relationship between expression of CDK4, cyclin D1, Rb
and phospho-Rb by western blotting and sensitivity to fascaplysin
and PD0332991.
Results
Melanoma cells are sensitive to cyclin
dependent kinase inhibitors
Of the 160 protein kinase inhibitors tested 29
achieved ≥50% growth inhibition in the Sk-Mel-2 cell line when
tested at 1 μM concentration. Twenty compounds achieved ≥50% growth
inhibition in the Sk-Mel-28 cell line when tested at the same dose.
The 20 compounds that achieved ≥50% growth inhibition in the
Sk-Mel-28 cell line achieved similar levels of inhibition in the
Sk-Mel-2 cell line (Table I). CDK
inhibitors represented 6 of the 20 compounds. Four CDK1/2
inhibitors were identified, achieving between 55.3–99.8% growth
inhibition in Sk-Mel-28 cells and 62.9–99.6% growth inhibition in
Sk-Mel-2 cells. Two CDK4 inhibitors, CDK4 inhibitor III and
fascaplysin, achieved 72.6 and 99.2% growth inhibition in Sk-Mel-28
cells and 53.9 and 98.0% growth inhibition in Sk-Mel-2 cells,
respectively.
 | Table IPercentage growth inhibition (±
standard deviation) by the 20 most active compounds tested at 1 μM
in Sk-Mel-2 and Sk-Mel-28 melanoma cell lines. |
Table I
Percentage growth inhibition (±
standard deviation) by the 20 most active compounds tested at 1 μM
in Sk-Mel-2 and Sk-Mel-28 melanoma cell lines.
| % Growth
inhibition |
|---|
|
|
|---|
| Sk-Mel-28 | Sk-Mel-2 |
|---|
| Akt inhibitor
IV | 86.3±6.4 | 95.3±1.7 |
| Alsterpaullone,
2-Cyanotheyl | 65.6±13.0 | 64.5±4.1 |
| Aurora kinase/CDK
inhibitor | 55.3±21.8 | 62.9±5.6 |
| CDK 1/2 inhibitor
III | 99.9±0.0 | 98.0±1.0 |
| CDK 4 inhibitor
III | 72.6±22.8 | 53.9±8.5 |
| Cdk/crk
inhibitor | 99.8±0.0 | 99.6±0.2 |
| EGFR inhibitor | 87.7±5.7 | 94.3±0.3 |
| Fascaplysin,
synthetic | 99.2±0.6 | 98.0±1.0 |
| Herbimycon A,
streptomyces sp. | 73.9±15.3 | 89.6±3.8 |
| IC261 | 87.5±7.0 | 93.8±1.2 |
| JNK inhibitor | 91.5±1.3 | 95.9±1.0 |
| K-252a,
Nocardiopsis sp. | 93.3±0.6 | 92.8±3.3 |
| MK2a inhibitor | 92.6±1.9 | 93.9±1.9 |
| PDGF receptor
tyrosine kinase inhibitor IV | 89.9±1.0 | 95.3±1.4 |
| PDK1/AKT/Flt dual
pathway inhibitor | 99.9±0.0 | 99.8±0.3 |
| PKR inhibitor | 68.0±2.4 | 83.7±3.3 |
| PI-103 | 89.0±1.3 | 79.2±6.7 |
| Rapamycin | 66.8±5.0 | 65.5±5.9 |
| Staurosporine,
streptomyces sp. | 99.2±0.3 | 99.6±0.4 |
| TGF-β RI inhibitor
III | 75.5±3.5 | 74.5±8.2 |
Fascaplysin inhibits the growth of
melanoma cells
The anti-proliferative effect of synthetic
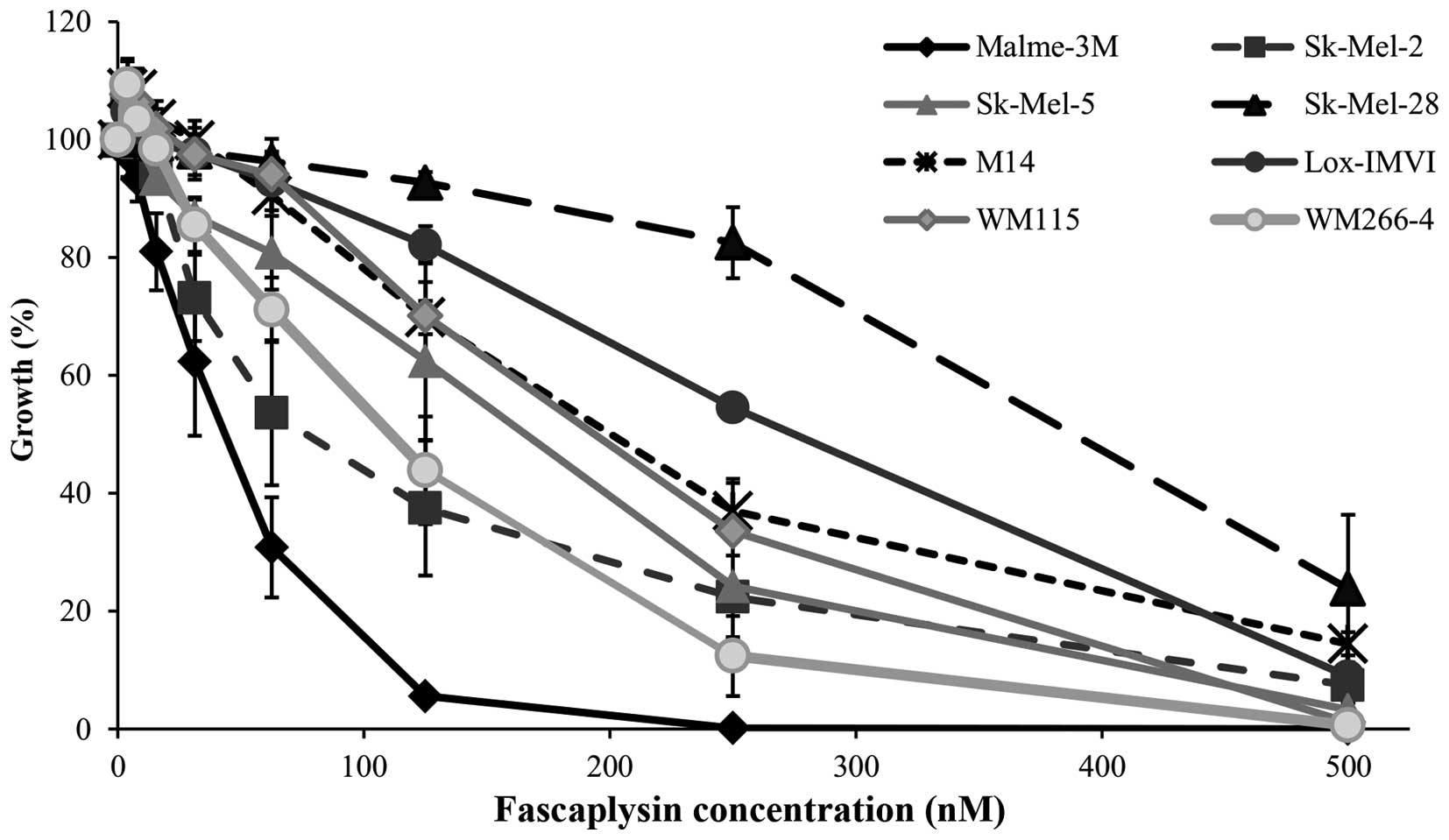
fascaplysin was further investigated in a panel of 8 melanoma cell
lines. All cell lines were sensitive to fascaplysin at nanomolar
concentrations, with IC50 values ranging from 32.9 to
221.3 nM (Fig. 1 and Table II). The NRAS mutated Sk-Mel-2
cells and the most sensitive BRAF mutated cell line Malme-3M were
selected for further testing. Fascaplysin significantly inhibited
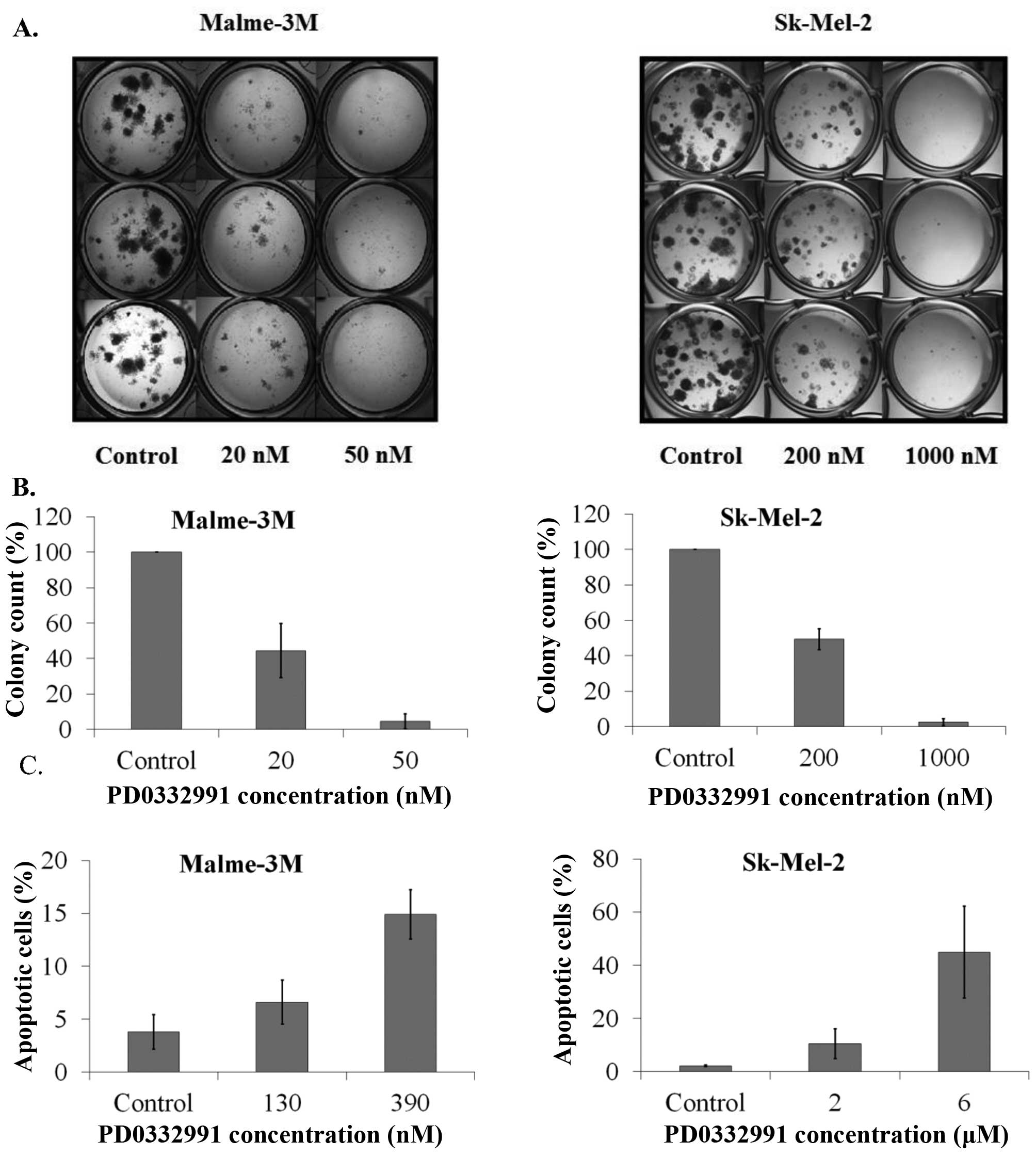
the clonogenic growth of both Malme-3M and Sk-Mel-2 cells (Fig. 2).
| Table IIIC50 concentrations of
fascaplysin (nM) and PD0332991 (μM) in a panel of melanoma cell
lines. |
Table II
IC50 concentrations of
fascaplysin (nM) and PD0332991 (μM) in a panel of melanoma cell
lines.
| Mutation
status | Fascaplysin
(nM) | PD0332991 (μM) |
|---|
| Sk-Mel-2 | NRAS | 74.5±6.1 | 2.14±0.24 |
| Malme-3M | BRAF | 32.9±1.7 | 0.14±0.01 |
| Sk-Mel-5 | BRAF | 93.0±11.1 | 0.50±0.05 |
| Sk-Mel-28 | BRAF | 220.0±19.1 | 1.83±0.18 |
| M14 | BRAF | 178.7±23.4 | 2.29±0.28 |
| Lox-IMVI | BRAF | 221.3±18.4 | 0.30±0.03 |
| WM115 | BRAF | 166.6±25.9 | 1.00±0.11 |
| WM266-4 | BRAF | 87.5±13.5 | 0.13±0.01 |
Fascaplysin induces apoptosis in melanoma
cell lines
To determine if the anti-proliferative effects of
fascaplysin were cytostatic or cytocidal, we performed cell cycle
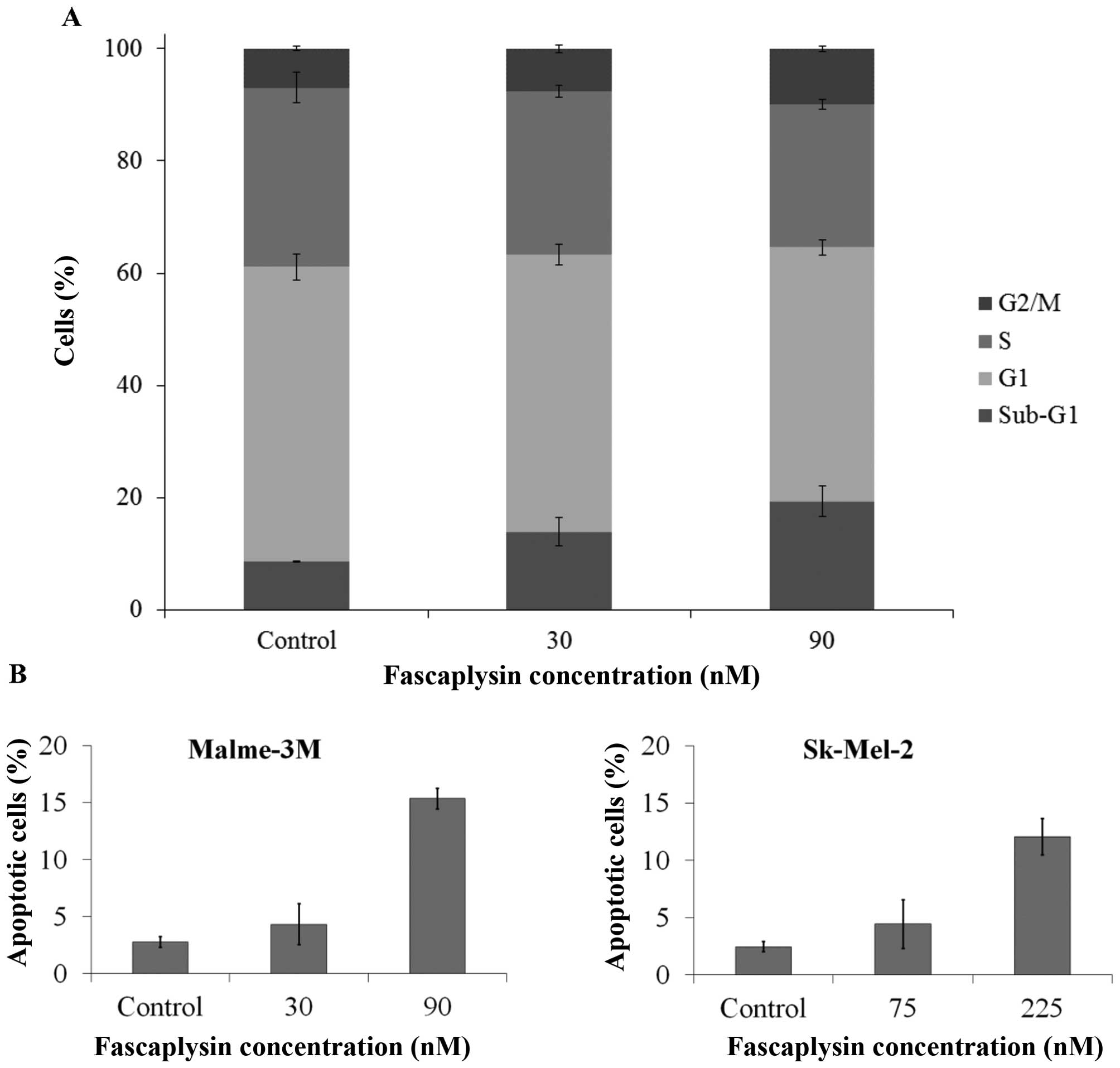
analysis. In Malme-3M cells, 48-h treatment with fascaplysin
induced a significant increase in the subG1 fraction (Fig. 3A), to 19.3±2.7% in response to 90
nM fascaplysin (P=0.020) compared with a subG1 fraction of 8.6±0.1%
for control. To confirm the increase in the subG1 fraction was due
to apoptosis induction, we used the TUNEL assay. Treatment with
fascaplysin caused a small but significant increase in apoptosis in
both Malme-3M cells (90 nM, 15.3±0.9%; P=0.0002) and Sk-Mel-2 cells
(225 nM, 12.1±1.6%; P=0.006) (Fig.
3B).
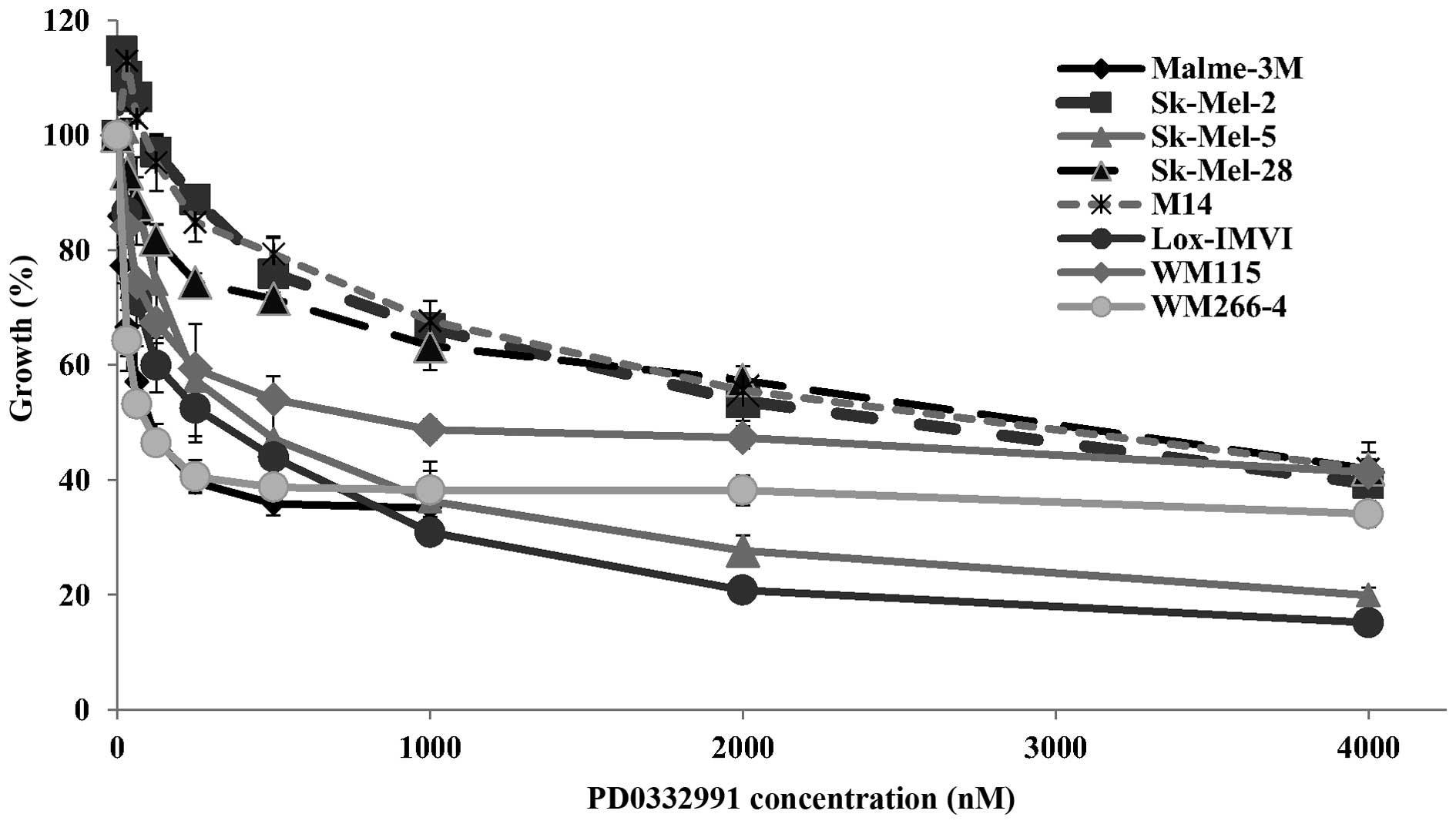
Evaluation of PD0332991 activity in
melanoma cells
As fascaplysin, a laboratory grade CDK4 inhibitor,
showed significant activity in the melanoma cell lines, we tested a
therapeutic CDK4/6 inhibitor PD0332991 in the panel of melanoma
cell lines. Surprisingly, although PD0332991 is a more potent
inhibitor of CDK4 (IC50=0.011 μM compared to 0.35 μM for
fascaplysin), the melanoma cell lines were generally less sensitive
to PD0332991 than to fascaplysin with IC50 values
ranging from 0.13 to 2.29 μM (Table
II and Fig. 4). The Sk-Mel-2,
Sk-Mel-28, M14 and WM-115 cell lines showed relative resistance to
PD0332991 with IC50 values >1 μM. Notably, the
WM-266-4 cell line, derived from a metastatic tumour, was
sensitive, while the WM-115 cell line, derived from the primary
tumour from the same patient, was resistant to PD0332991.
PD0332991 also inhibited clonogenic growth and
induced apoptosis in Malme-3M and Sk-Mel-2 cells, albeit at higher
concentrations than fascaplysin (Fig.
5A and B). In the sensitive Malme-3M cells, 48-h treatment with
PD0332991 (390 nM) induced apoptosis in 14.9±2.3% of cells
(P=0.004). PD0332991 induced apoptosis in 44.9±17.2% of Sk-Mel-2
cells following 48-h treatment at a dose of 6 μM (P=0.05) (Fig. 5C).
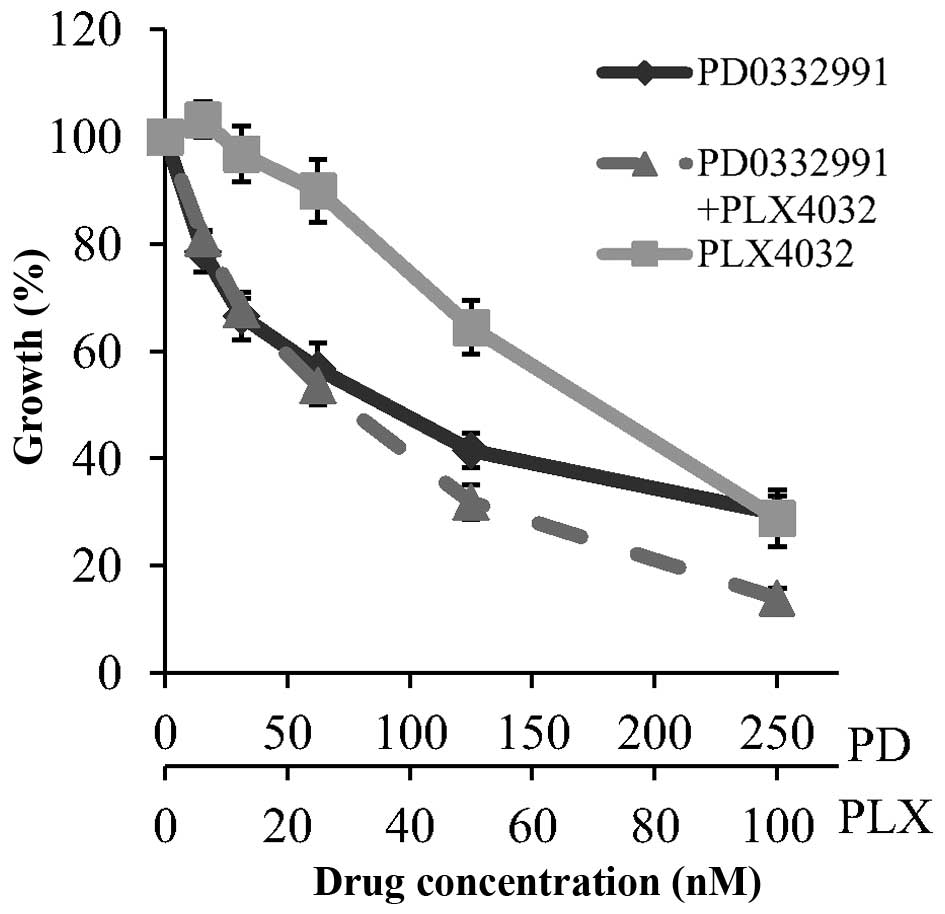
Drug combination assays were performed to examine
the effect of PD0332991 combined with temozolomide and the BRAF
inhibitor PLX4032. Concurrent treatment with PD0332991 and
temozolomide did not improve response compared to PD0332991 alone
(data not shown). However, the combination of PD0332991 and PLX4032
was found to be additive, with a combination index (at
ED50) of 1.02±0.09, in the Malme-3M cell line (Fig. 6).
CDK4 expression correlates with response
to PD0332991
To determine if the differential response to
PD0332991 in the melanoma cell lines is due to drug effiux we
tested the effect of an inhibitor of the drug effiux pumps
P-glycoprotein and breast cancer resistance protein (BCRP), in two
cell lines which displayed resistance to PD0332991, SK-Mel-2 and
WM115. Elacridar (GF120918, 0.5 μM) did not enhance response to
PD0332991 in either of the cell lines (data not shown).
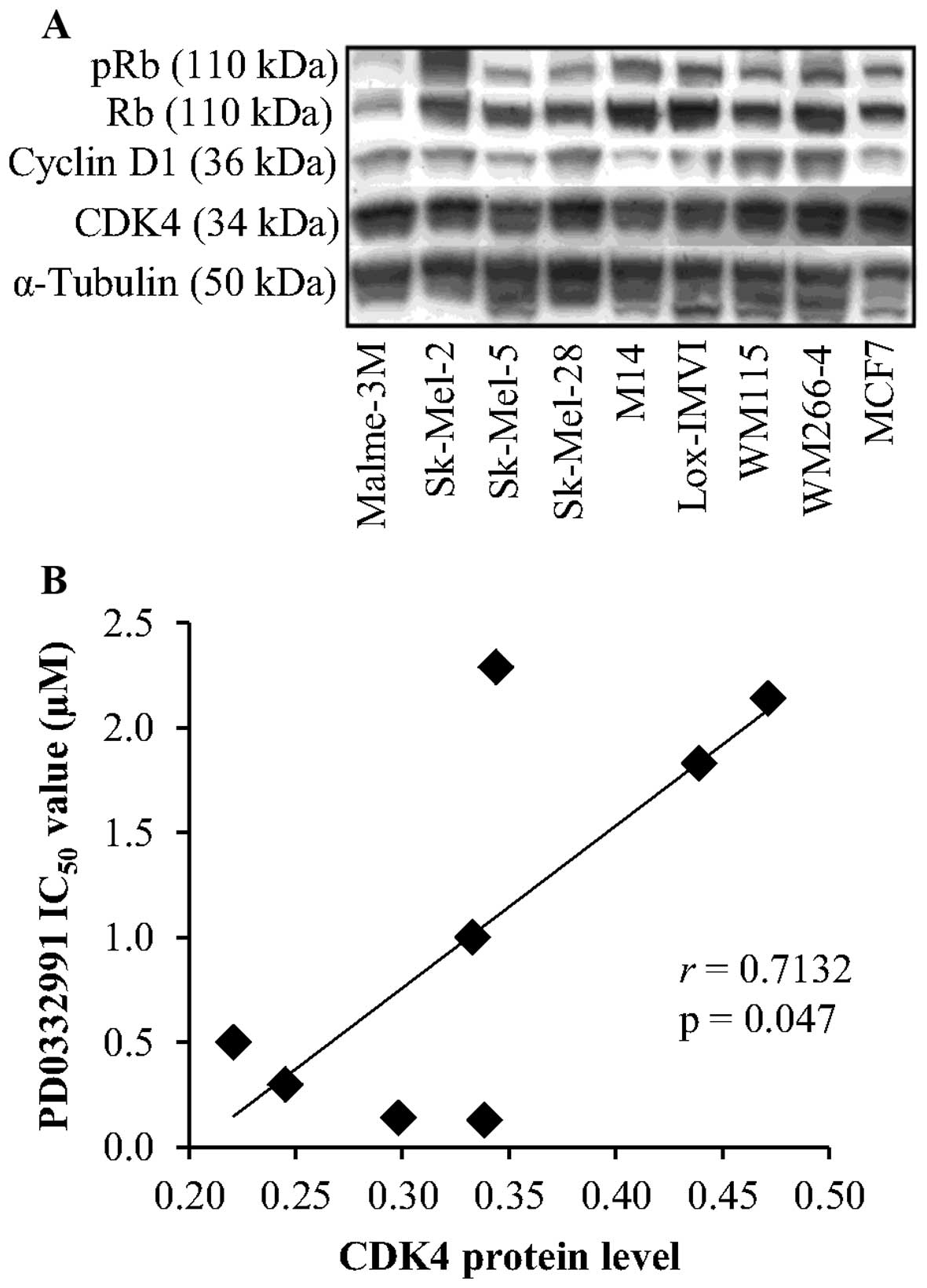
CDK4, cyclin D1, Rb and phospho-Rb protein levels
were evaluated by western blotting. CDK4 protein was detected at
high levels in the panel of 8 melanoma cell lines (Fig. 7A). No correlation was observed
between expression of CDK4 and sensitivity to fascaplysin (r=0.036,
P=0.933). In contrast, high levels of CDK4 expression were
significantly associated with reduced sensitivity to PD0332991,
that is higher IC50 values (r=0.713, P=0.047) (Fig. 7B). No significant correlation was
observed between sensitivity to either fascaplysin or PD0332991 and
cyclin D1, Rb and phospho-Rb levels.
Discussion
To identify novel targets for treatment, we screened
a library of 160 protein kinase inhibitors in 2 melanoma cell
lines. For a disease representative approach, we selected 2
melanoma cell lines with genetic mutations common to melanoma. The
Sk-Mel-2 cell line carries an NRAS mutation, NRAS is mutated in 18%
of melanomas (range, 0–50%) (14).
The Sk-Mel-28 cell line is BRAF mutated, BRAF is mutated in 41% of
melanomas (range 22–72%) (14).
NRAS and BRAF mutations are believed to be mutually exclusive in
melanoma (15).
Six of the 20 most effective compounds were CDK
inhibitors. This result is not surprising considering the role of
CDKs in cell cycle control. Progression through the mammalian cell
cycle is a tightly regulated process. Along with their associated
cyclins, CDKs are master regulators of cell proliferation. More
than 11 CDKs have been identified, with CDKs 1–4 and 6 directly
involved in driving the cell cycle. Deregulated CDK activity
results in uncontrolled proliferation and represents a hallmark of
malignancy (16).
Activating mutations, or amplification of CDK4 have
been described in both familial and sporadic cases of melanoma
(17). Intact p16INK4a inhibits
CDK4 activity and loss of p16, which occurs frequently in melanoma,
results in abnormal CDK4 activity (18). Amplification of cyclin D, which
binds to CDK4, can drive cell cycle progression, and has been
observed in a subset of melanomas (19). These factors provide evidence that
CDK4 plays an important role in melanoma progression. Thus, we
chose to further evaluate CDK4 inhibition using fascaplysin, a
synthetic selective CDK4 inhibitor.
Fascaplysin was originally isolated as a compound
showing antimicrobial activity from the sponge
Fascaplysinopsis sp (20).
It is a selective inhibitor of CDK4 (IC50=0.35 μM) and
CDK6 (IC50=3.4 μM) and not selective for the other CDKs
or other kinases (21,22). Fascaplysin, or derivatives of
fascaplysin, have shown direct antitumour activity, by inducing
apoptosis, and anti-angiogenic effects in preclinical tumour
models, including sarcoma (23)
and a number of the National Cancer Institute panel of cell lines
(24).
All melanoma cell lines tested were sensitive to
fascaplysin at concentrations in the nanomolar range. Notably,
WM-266-4 cells were significantly more sensitive to growth
inhibition by fascaplysin than WM-115 cells. WM-115 is a primary
melanoma cell line, while WM-266-4 is a metastatic cell line,
derived from the same patient (25). The different sensitivity may be
related to the faster proliferation rate of the metastatic cell
line WM-266-4 (26). The other
cell lines used in this study are all derived from metastatic
tumours, and all show similar sensitivity to fascaplysin as the
WM-266-4 cell line, with the exception of Lox-IMVI which shows
intermediate sensitivity. The lower sensitivity in the primary
WM-115 cells, suggests that primary melanomas might be less
sensitive to CDK4 inhibition than metastatic tumours but this would
require further testing in a larger panel of cell lines derived
from primary and metastatic tumours.
Consistent with previous studies showing that
fascaplysin can induce apoptosis in vitro and in vivo
(23,27) we showed that fascaplysin increases
the subG1 fraction and apoptosis induction in melanoma cells.
Although fascaplysin has not been tested in cancer
patients, several multi-target CDK inhibitors are currently being
evaluated in clinical trials (5).
PD0332991 is a highly specific inhibitor of CDK4
(IC50=0.011 μM) and CDK6 (IC50=0.016 μM)
(28) and is currently in phase II
trials in several solid tumours. One phase II study is testing
PD0332991 in refractory solid tumours including stage IV melanoma
(NCT01037790).
Four of the 8 melanoma cell lines tested were
sensitive to PD0332991. Loss of retinoblastoma protein (Rb) has
been correlated with resistance to PD0332991 (29), however, all of the melanoma cell
lines tested expressed detectable levels of Rb. Analysis of the
mutational status of the panel of cell lines using the COSMIC
database (http://cancer.sanger.ac.uk/cancergenome/projects/cell_lines/)
and the Cancer Cell Line Encyclopedia (http://www.broadinstitute.org/ccle), suggest that
mutations in CDK4 or p16INK4A do not predict response or
resistance, as only one of the melanoma cell lines (SK-MEL-28) has
a mutation in CDK4 and there are no reported mutations in p16INK4A
in these cell lines. Sanchez-Martinez et al (30) previously reported that PD0332991 is
a substrate for the drug effiux pumps P-glycoprotein (P-gp) and
breast cancer resistance protein (BCRP). Therefore, we tested
elacridar, an inhibitor of P-gp and BCRP, but it had no impact on
response to PD0332991 in the cell lines tested, suggesting that
P-gp and BCRP do not play a role in resistance to PD0332991 in the
melanoma cell lines. However, P-gp expression has been reported in
~50% of primary melanomas and 74% of metastatic melanomas (31), and thus may impact on sensitivity
to PD0332991 in melanoma patients.
Using semi-quantitative western blotting, we found
that lower CDK4 protein levels correlated with sensitivity to
PD0332991 in the panel of 8 cell lines. A recent study showed that
37 out of 47 melanoma cell lines were sensitive to PD0332991 in
vitro (GI50 <1 μM). Sensitivity to PD0332991 did
not correlate with CDK4 mRNA expression in that study, but low
levels of CDKN2A mRNA, or mutation or loss of CDKN2A correlated
with sensitivity (32). Further
evaluation of CDK4 protein levels in a larger panel of cell lines,
and ultimately in tumour samples from melanoma patients treated
with PD0332991, would be required to definitively determine the
clinical relevance of CDK4 protein as a predictive biomarker for
PD0332991 response.
The reason for the differential sensitivity to
fascaplysin and PD0332991 is not fully understood. It may be due to
the reported ability of fascaplysin to bind to and intercalate into
DNA (33).
Although combinations of either fascaplysin or
PD0332991 with temozolomide, a derivative of dacarbazine commonly
used to treat melanoma, did not show enhanced anti-proliferative
effects, combined treatment with PD0332991 and the therapeutic BRAF
inhibitor PLX4032 was additive in the BRAF mutated cell line
tested. A recent study by Jalili et al (34) showed that dual inhibition of CDK2
and CDK4 enhanced response to BRAF and MEK inhibitors in melanoma
cells in vitro and in vivo. Thus, combining CDK4
inhibition with BRAF or MEK targeted therapies may provide greater
therapeutic benefit than combinations with chemotherapy.
In summary, using kinase inhibitor screening, we
showed that melanoma cells are particularly sensitive to CDK
inhibition in vitro. Further testing of the therapeutic CDK4
inhibitor PD0332991 showed that 4 out of 8 melanoma cell lines
tested are sensitive to CDK4 inhibition and that CDK4 inhibition
induces apoptosis in melanoma cells. Our results suggest that CDK4
inhibition represents a promising approach for the treatment of
metastatic melanoma.
Acknowledgements
The present research was supported by the Cancer
Clinical Research Trust.
References
|
1
|
Bhatia S, Tykodi SS, Lee SM and Thompson
JA: Systemic therapy of metastatic melanoma: On the road to cure.
Oncology (Williston Park). 29:126–135. 2015.
|
|
2
|
Dummer R and Flaherty KT: Resistance
patterns with tyrosine kinase inhibitors in melanoma: New insights.
Curr Opin Oncol. 24:150–154. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Long GV, Stroyakovskiy D, Gogas H,
Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A,
Grob JJ, et al: Combined BRAF and MEK inhibition versus BRAF
inhibition alone in melanoma. N Engl J Med. 371:1877–1888. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hunter T and Cooper JA: Protein-tyrosine
kinases. Annu Rev Biochem. 54:897–930. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cicenas J and Valius M: The CDK inhibitors
in cancer research and therapy. J Cancer Res Clin Oncol.
137:1409–1418. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Diaz-Padilla I, Siu LL and Duran I:
Cyclin-dependent kinase inhibitors as potential targeted anticancer
agents. Invest New Drugs. 27:586–594. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Malumbres M and Barbacid M: Cell cycle
kinases in cancer. Curr Opin Genet Dev. 17:60–65. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lapenna S and Giordano A: Cell cycle
kinases as therapeutic targets for cancer. Nat Rev Drug Discov.
8:547–566. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sharpless E and Chin L: The INK4a/ARF
locus and melanoma. Oncogene. 22:3092–3098. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jonsson A, Tuominen R, Grafström E,
Hansson J and Egyhazi S: High frequency of p16(INK4A) promoter
methylation in NRAS-mutated cutaneous melanoma. J Invest Dermatol.
130:2809–2817. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Smalley KS, Lioni M, Dalla Palma M, Xiao
M, Desai B, Egyhazi S, Hansson J, Wu H, King AJ, Van Belle P, et
al: Increased cyclin D1 expression can mediate BRAF inhibitor
resistance in BRAF V600E-mutated melanomas. Mol Cancer Ther.
7:2876–2883. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sausville EA: Complexities in the
development of cyclin-dependent kinase inhibitor drugs. Trends Mol
Med. 8(Suppl): S32–S37. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee JH, Choi JW and Kim YS: Frequencies of
BRAF and NRAS mutations are different in histological types and
sites of origin of cutaneous melanoma: A meta-analysis. Br J
Dermatol. 164:776–784. 2011. View Article : Google Scholar
|
|
15
|
Meier F, Schittek B, Busch S, Garbe C,
Smalley K, Satyamoorthy K, Li G and Herlyn M: The RAS/RAF/MEK/ERK
and PI3K/AKT signaling pathways present molecular targets for the
effective treatment of advanced melanoma. Front Biosci.
10:2986–3001. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Harper JW and Adams PD: Cyclin-dependent
kinases. Chem Rev. 101:2511–2526. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Walker GJ, Flores JF, Glendening JM, Lin
AH, Markl ID and Fountain JW: Virtually 100% of melanoma cell lines
harbor alterations at the DNA level within CDKN2A, CDKN2B, or one
of their downstream targets. Genes Chromosomes Cancer. 22:157–163.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ibrahim N and Haluska FG: Molecular
pathogenesis of cutaneous melanocytic neoplasms. Annu Rev Pathol.
4:551–579. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Curtin JA, Fridlyand J, Kageshita T, Patel
HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Bröcker EB, LeBoit PE, et
al: Distinct sets of genetic alterations in melanoma. N Engl J Med.
353:2135–2147. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Roll DM, Ireland CM, Lu HSM and Clardy J:
Fascaplysin, an unusual antimicrobial pigment from the marine
sponge Fascaplysinopsis Sp. J Org Chem. 53:3276–3278. 1988.
View Article : Google Scholar
|
|
21
|
Soni R, Muller L, Furet P, Schoepfer J,
Stephan C, Zumstein-Mecker S, Fretz H and Chaudhuri B: Inhibition
of cyclin-dependent kinase 4 (Cdk4) by fascaplysin, a marine
natural product. Biochem Biophys Res Commun. 275:877–884. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shafiq MI, Steinbrecher T and Schmid R:
Fascaplysin as a specific inhibitor for CDK4: Insights from
molecular modelling. PLoS One. 7:e426122012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yan X, Chen H, Lu X, Wang F, Xu W, Jin H
and Zhu P: Fascaplysin exert anti-tumor effects through apoptotic
and anti-angiogenesis pathways in sarcoma mice model. Eur J Pharm
Sci. 43:251–259. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Segraves NL, Robinson SJ, Garcia D, Said
SA, Fu X, Schmitz FJ, Pietraszkiewicz H, Valeriote FA and Crews P:
Comparison of fascaplysin and related alkaloids: A study of
structures, cytotoxicities, and sources. J Nat Prod. 67:783–792.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McArthur AG, Young RJ, Sheppard KE, Mar V,
Waldeck K, Fox SB, Kelleher FC, Liu W, Dobrovic A, Pearson R, et
al: Clinical significance of genomic alterations of the
CDK4-pathway and sensitivity to the CDK4 inhibitor PD0332991 in
melanoma. J Clin Oncol. 30:85202012.
|
|
26
|
Herlyn M, Balaban G, Bennicelli J, Guerry
D IV, Halaban R, Herlyn D, Elder DE, Maul GG, Steplewski Z, Nowell
PC, et al: Primary melanoma cells of the vertical growth phase:
Similarities to metastatic cells. J Natl Cancer Inst. 74:283–289.
1985.PubMed/NCBI
|
|
27
|
Lu XL, Zheng YL, Chen HM, Yan XJ, Wang F
and Xu WF: Anti-proliferation of human cervical cancer HeLa cell
line by fascaplysin through apoptosis induction. Yao Xue Xue Bao.
44:980–986. 2009.(In Chinese).
|
|
28
|
Fry DW, Harvey PJ, Keller PR, Elliott WL,
Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, et
al: Specific inhibition of cyclin-dependent kinase 4/6 by PD
0332991 and associated antitumor activity in human tumor
xenografts. Mol Cancer Ther. 3:1427–1438. 2004.PubMed/NCBI
|
|
29
|
Finn RS, Dering J, Conklin D, Kalous O,
Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, et al: PD
0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially
inhibits proliferation of luminal estrogen receptor-positive human
breast cancer cell lines in vitro. Breast Cancer Res. 11:R772009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sanchez-Martinez C, Gelbert L, Shannon H,
De Dios A, Staton BA, Ajamie RT, Sawada G, Wishart GN and Raub TJ:
LY2835219, a potent oral inhibitor of the cyclin-dependent kinases
4 and 6 (CDK4/6) that crosses the blood-brain barrier and
demonstrates in vivo activity against intracranial human brain
tumor xenografts. Mol Cancer Ther. 10:B2342011. View Article : Google Scholar
|
|
31
|
Walsh N, Kennedy S, Larkin AM,
Tryfonopoulos D, Eustace AJ, Mahgoub T, Conway C, Oglesby I,
Collins D, Ballot J, et al: Membrane transport proteins in human
melanoma: Associations with tumour aggressiveness and metastasis.
Br J Cancer. 102:1157–1162. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Young RJ, Waldeck K, Martin C, Foo JH,
Cameron DP, Kirby L, Do H, Mitchell C, Cullinane C, Liu W, et al:
Loss of CDKN2A expression is a frequent event in primary invasive
melanoma and correlates with sensitivity to the CDK4/6 inhibitor
PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res.
27:590–600. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hörmann A, Chaudhuri B and Fretz H: DNA
binding properties of the marine sponge pigment fascaplysin. Bioorg
Med Chem. 9:917–921. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jalili A, Wagner C, Pashenkov M, Pathria
G, Mertz KD, Widlund HR, Lupien M, Brunet JP, Golub TR, Stingl G,
et al: Dual suppression of the cyclin-dependent kinase inhibitors
CDKN2C and CDKN1A in human melanoma. J Natl Cancer Inst.
104:1673–1679. 2012. View Article : Google Scholar : PubMed/NCBI
|