Introduction
Fibrosarcoma is a malignant cancer that originates
in the connective tissue found at the ends of bones of the legs or
arms, and then spreads to the surrounding soft tissues. Soft
tissues include joint tissue, blood vessels, fat, muscles, tendons,
and fibrous tissue. Although fibrosarcoma-related morbidity is
rare, patient survival rates are low (1).
During the last few decades, epigenetics has been
one of the emerging fields in cancer research (2). Epigenetics affects the transcription
of cells, thereby regulating gene expression. Abnormal epigenetic
changes can have adverse effects on the organism. Methylation of
cytosine-phosphate-guanine (CpG) islands in the promoter region of
a gene has now been strongly linked to gene silencing.
DNA methylation is regulated by DNA
methyltransferases (DNMT1, DNMT3A and DNMT3B), which catalyze the
transfer of a methyl group from S-adenosyl-l-methionine (SAM) to
the cytosine of a CpG dinucleotide (adjacent within a single DNA
strand), immediately following replication (3).
DNMTs are classified as maintenance or de
novo methyltransferases. Maintenance DNMT1 binds methyl groups
to methylated DNA during replication, whereas de novo DNMT3A and
DNMT3B add methyl groups to CpG dinucleotides of unmethylated DNA.
Previous reports have shown that some anticancer cascades
abnormally activate the DNMT1, enzyme that maintains the DNA
methylation pattern (3).
When the levels of DNMT1 decrease, as is the case
following azacytidine or decitabine treatment, daughter strands are
less likely to undergo maintenance to restore full methylation;
thus, with each replication, CpG pairs become unmethylated,
rendering their promoter regions more accessible to transcription
factors.
DNA damage induced by 5′-azacytidine (5′-aza) is
reversible, since the drug does not influence de novo DNMT
synthesis (4–6). 5′-aza has been used clinically for
treating diverse diseases such as acute myelogenous leukemia,
hematological malignancies, gastrointestinal, lung, ovarian,
prostate, breast, and head and neck cancers, melanoma, and
malignant mesothelioma (7). Global
hypomethylation is a hallmark of cancer (8). It was believed that global
hypomethylation principally targets repetitive sequences, but some
genes involved in metastasis were also previously indicated to be
hypomethylated in cancers (9).
There are three major isoforms of cyclooxygenase
(COX), COX-1, COX-2, and COX-3. COX-1, a constitutive isoform,
plays a role in modulating physiologic activities in tissues. COX-2
is an inducible isoform of the enzyme that responds to specific
stimuli such as growth factor, hormones, endotoxins, and cytokines
(10,11). The third isoform, COX-3, an
inactive protein, is an alternative splice variant of COX-1
(12).
Overexpression of COX-2 in tumors is linked to the
over-production of the pro-inflammatory prostaglandin E2
(PGE2) (13,14). It may also trigger the acquisition
of essential cancer traits (15),
including inhibition of apoptosis (16,17),
immunosuppression (18), continued
proliferation (19), invasion
(20), angiogenesis (21,22),
and metastasis (23,24).
COX-2 is usually associated with inflammation and is
markedly upregulated in various types of cancer, as well as in
other diseases (25–28).
The methylation status of the COX-2 promoter was
shown in several cancers, and some research showed that the
transcriptional silencing of COX-2 is involved in the methylation
status of the CpG pairs of the COX-2 gene (29,30).
However, the effects of 5′-aza on COX-2 expression in human
fibrosarcoma HT-1080 cells have not been reported.
In this study, we examined whether 5′-aza regulates
COX-2 expression in human fibrosarcoma HT-1080 cells. We showed
that 5′-aza increases COX-2 expression and PGE2
production, and identified the signaling pathways involved in these
mechanisms.
Materials and methods
Reagents and chemicals
5′-azacytidine was purchased from Sigma-Aldrich (St.
Louis, MO, USA). PD 98059 was purchased from Calbiochem (San Diego,
CA, USA) and LY 294002 was purchased from Tocris (Bristol, UK).
Cell culture medium and fetal bovine serum (FBS) were obtained from
Invitrogen (Gaithersburg, MD, USA). PCR primers were purchased from
Genotek Co., (Daejeon, Korea).
Cell culture and experimental
conditions
HT1080 human fibrosarcoma cells were obtained from
the Korean Cell Line Bank (KCLB, Seoul, Korea). The cell line was
cultured in RPMI-1640 containing 10% (v/v) FBS, 50 U/ml penicillin,
and 50 μg/ml streptomycin (Sigma-Aldrich) at 37°C in a humidified
incubator containing 5% CO2. The medium was refreshed
daily over a period of 2 days. Treatment with drugs was performed
as indicated in the figure legends.
Western blot analysis
Cells were washed with PBS and lysed using RIPA
buffer containing 50 mM Tris-HCl; pH 7.4, 150 mM NaCl, 1% Nonidet
P-40, and 0.1% sodium dodecyl sulfate (SDS), supplemented with
inhibitors for proteases and phosphatases. Then the lysates were
clarified by centrifugation (1,300 rpm, 10 min, 4°C) and collected.
Proteins were size-fractionated using SDS-PAGE and transferred to a
nitrocellulose membrane (Whatman Schleicher and Schuell, Dachen,
Germany). Antibodies against DNMT-1, COX-2, pAkt, Akt, pERK, ERK,
and β-actin were used (Cell Signaling Technology, Beverly, MA,
USA). The nitrocellulose sheet was blocked with 5% non-fat dry milk
in Tris-buffered saline for 1 h. Proteins were detected using
horseradish peroxidase (HRP)-conjugated secondary antibodies,
followed by enhanced chemiluminescence (ECL). Blots were developed
using an image-Quant LAS 4000 (Amersham Biosciences Corp,
Piscataway, NJ, USA).
Reverse transcriptase-polymerase chain
reaction (RT-PCR)
HT1080 cells were collected after treatment with the
indicated drugs. Total RNA was extracted using TRIzol-reagent
(Invitrogen, Life Technologies, Carlsbad, CA, USA) according to the
manufacturer's instructions. cDNA was synthesized using
reverse-transcriptase according to the manufacturer's instructions,
and was used as the template for PCR amplification. The primer
sequences of COX-2 specific primer sets were as follows: COX-2
sense, 5′-TTC AAA TGA GAT TGT GGA AAA ATT GCT-3′; antisense, 5′-AGA
TCA TCT CTG CCT GAG TAT CTT-3′. GAPDH, sense, 5′-CGT CTT CAC CAC
CAT GGA GA-3′; antisense, 5′-CGG CCA TCA CGC CAC AGT TT-3′. GAPDH
was used as a loading control. Each sample was incubated at 95°C
for 30 sec, 55°C for 30 sec, and 72°C for 30 sec for 30 cycles.
Reaction samples were then incubated for an additional 7 min at
72°C and cooled to 4°C. PCR products were resolved on 1.2% agarose
gel.
Prostaglandin E (PGE2)
assay
Cells (2×104 cells/well) were seeded onto
96-well plates. At 24 h after treatment, conditioned medium was
harvested, and PGE2 concentrations were determined using
an ELISA assay kit according to instructions supplied by the
manufacturer (Assay Designs, Ann Arbor, MI, USA). Samples were
assayed in triplicate in each of three independent experiments.
PGE2 levels were calculated against a standard
PGE2 curve.
Immunofluorescence (IF) staining
Cells were treated for 24 h with or without 5′-aza
in the presence of inhibitor, PD 98059 or LY 294002. Cells were
harvested, fixed with ice-cold 3.5% paraformaldehyde
(Sigma-Aldrich) for 15 min, and washed with ice-cold
phosphate-buffered saline (PBS). Cells were then permeabilized with
0.1% Triton X-100 for 15 min, washed with ice-cold PBS, and stained
with antibodies against COX-2. Cell nuclei were stained with
4i,6-diamidino-2-phenylindol (DAPI, Molecular Probes/Invitrogen,
Carlsbad, CA, USA) and were observed using a fluorescence
microscope (Olympus, Tokyo, Japan) with peak excitation wavelengths
at 570 and 460 nm.
Data analysis and statistical
analyses
Values are presented as the mean ± Standard
deviation (SD) of at least three independent experiments. Data were
evaluated using the one-way ANOVA. P<0.05 was considered to
indicate statistically significant differences between values.
Results
This study aimed to determine whether 5′-aza
regulates COX-2 expression in HT1080 cells. HT1080 cells treated
with 5′-aza exhibited augmented COX-2 expression in comparison with
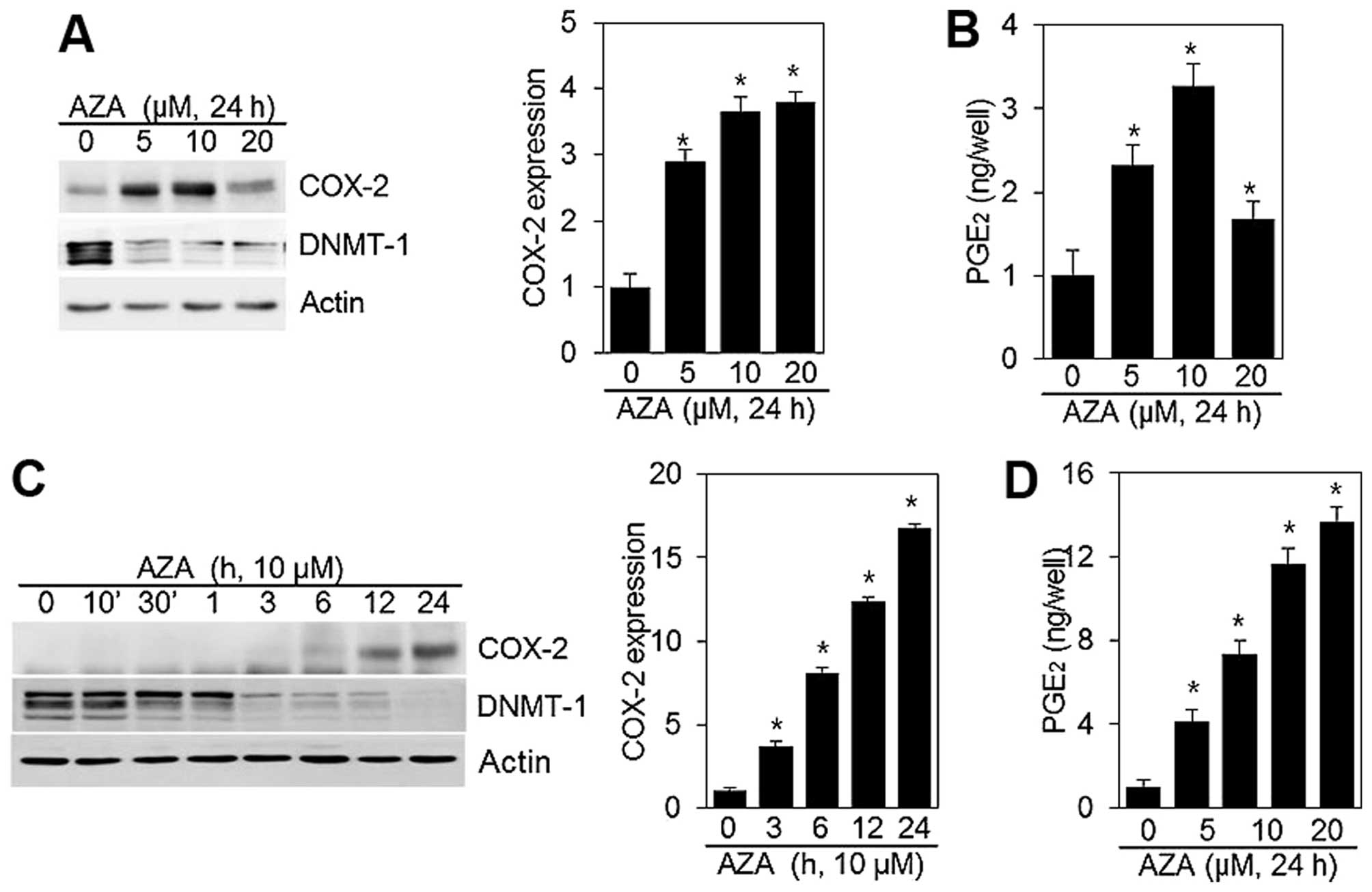
untreated cells (Fig. 1A). A
concentration-independent increase in COX-2 expression was observed
(Fig. 1A). Stimulation of cells
with 5′-aza markedly increased COX-2 expression, which was evident
within 6 h, and reached a maximum at 24 h (Fig. 1C). Densitometric evaluation of a
representative western blot experiment was performed in triplicate
(Fig. 1A and C, right panels).
To assess the effect of 5′-aza on COX-2 activity, we
quantified the production of PGE2 in HT1080 cells
untreated and treated with 5′-aza for 24 h (Fig. 1B and D). A significant increase in
PGE2 synthesis was verified in HT1080 cells treated with
5′-aza. The increase in PGE2 production and COX-2
expression induced by 5′-aza, was similar (Fig. 1B and D).
COX-2 expression is highly regulated via both
transcriptional and post-transcriptional mechanisms, depending on
its activator and the cell type (31,32).
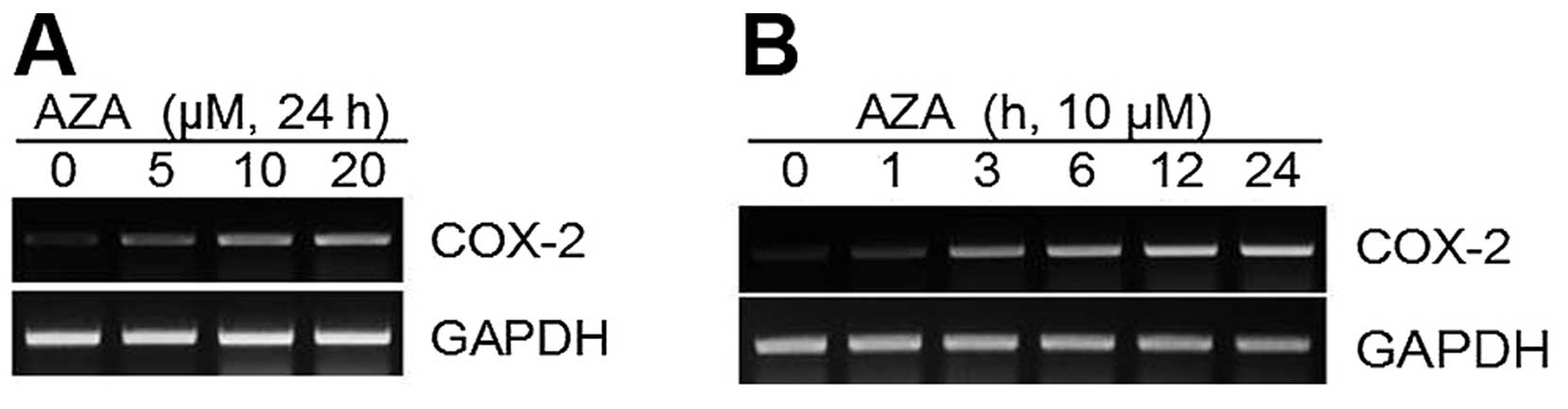
To verify the 5′-aza-induced COX-2 expression at the mRNA level, we
treated the HT1080 cells with 5′-aza. RNA was extracted from the
cells, and COX-2 mRNA levels were detected using RT-PCR. COX-2
expression increased in a dose- and time-dependent manner in cells
treated with 5′-aza (Fig. 2). To
correct differences in loading, the signal density of each COX-2
band was divided by the signal density of the GAPDH band (Fig. 2).
Several studies have shown the involvement of the
PI3K/Akt and ERK-1/2 pathways in the regulation of COX-2 expression
and PGE2 synthesis (33,34).
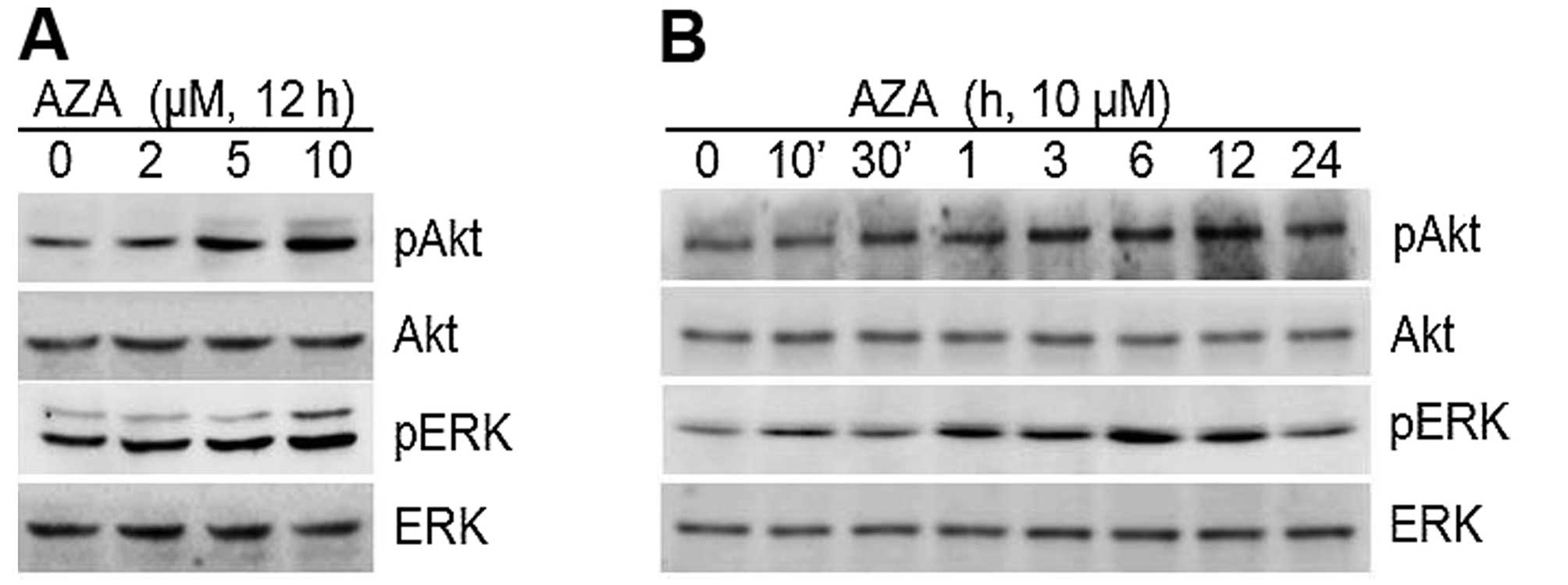
In this study, we found that 5′-aza phosphorylated
ERK-1/2 and Akt. This observation was evident within 1 h of
treatment with phosphorylation levels reaching a maximum at 12 h,
and then decreasing (Fig. 3B). The
total ERK-1/2 and Akt remained consistent through the duration of
the experiments (Fig. 3).
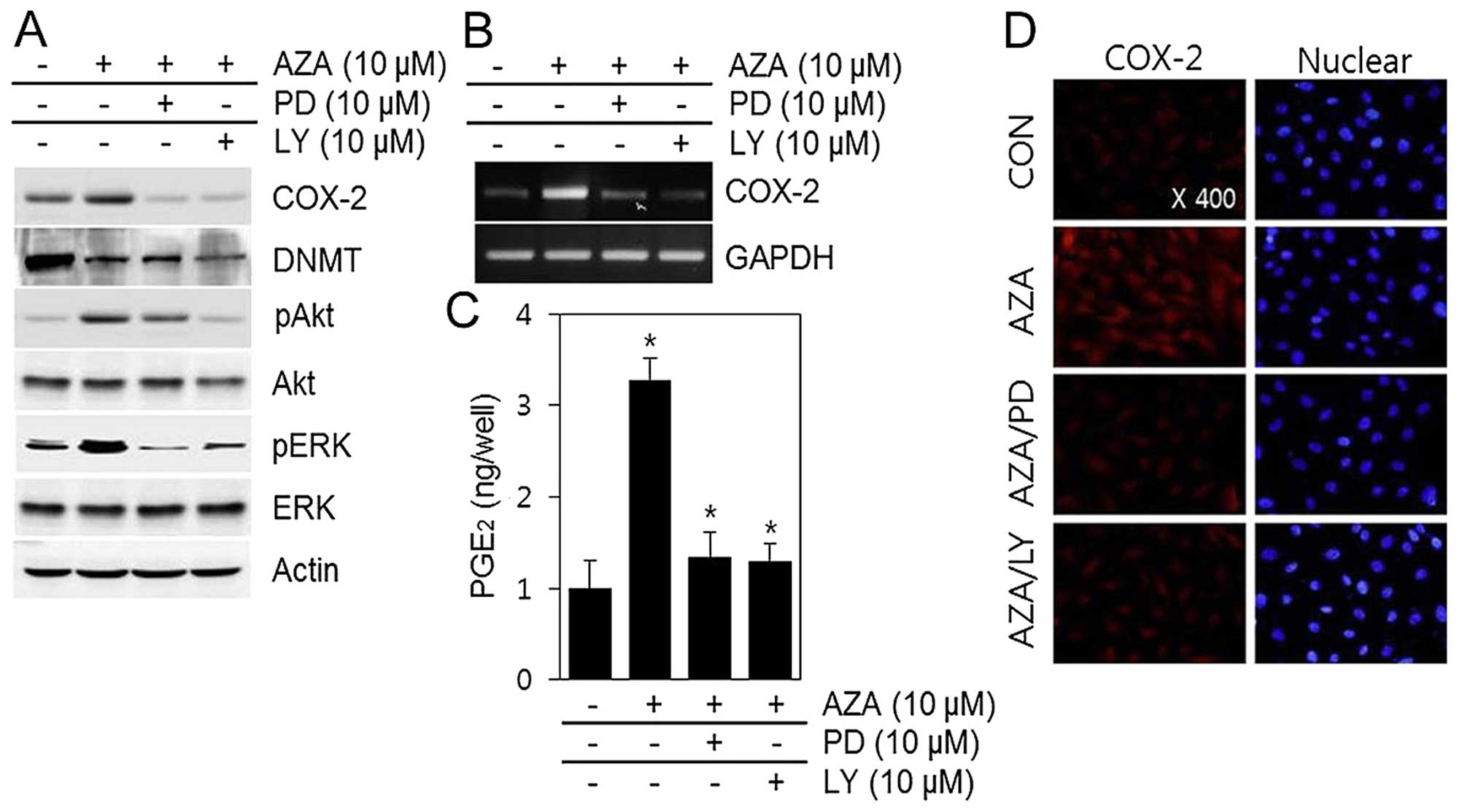
To determine whether ERK-1/2 and PI3K/Akt are
involved in 5′-aza-induced COX-2 expression and PGE2
synthesis, cells were treated with the ERK-1/2 inhibitor, PD 98059,
and the PI3K/Akt inhibitor LY 294002 (Fig. 4).
Pre-treatment with PD 98059 or LY 294002 followed by
stimulation with 5′-aza resulted in inhibition of 5′-aza-induced
effects on COX-2 mRNA expression and protein levels (Fig. 4A and B), and PGE2
synthesis (Fig. 4C).
Immunofluorescence microscopy further indicated that treatment with
5′-aza dramatically increased COX-2 expression, but these effects
were inhibited by treatment with PD 98059 and LY 294002 (Fig. 4D). Taken together, these results
indicate that the ERK-1/2 and Akt pathways participate in the
5′-aza-induced increase in COX-2 expression and PGE2
production (Fig. 4).
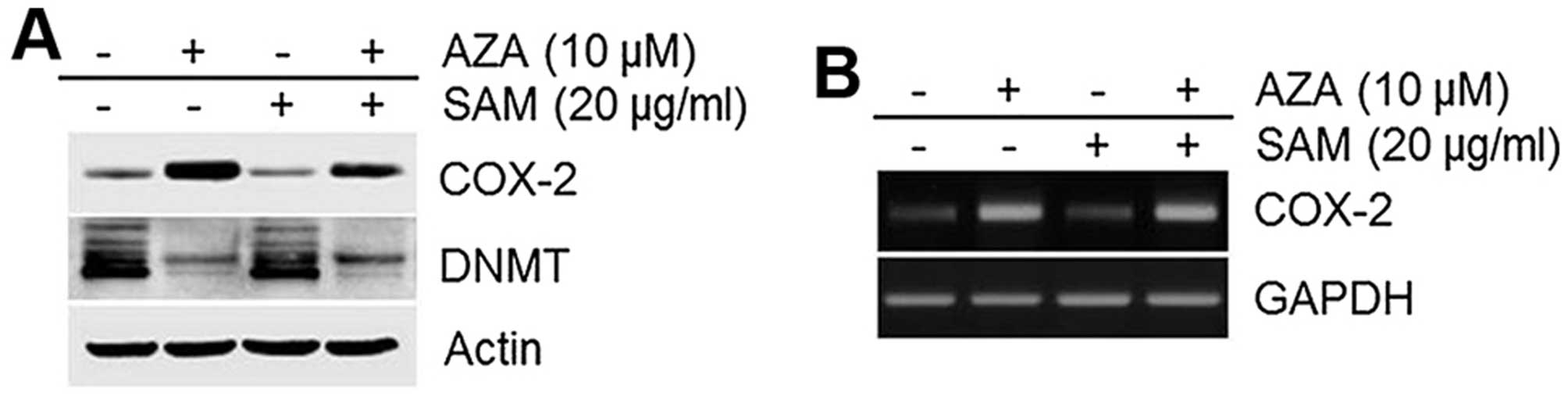
SAM is a methyl donor and an inhibitor of DNA
methylation (35). We therefore
examined and observed that SAM could inhibit the 5′-aza-induced
COX-2 expression (Fig. 5A).
Similar to COX-2 protein expression levels, COX-2 mRNA levels were
also altered by SAM (Fig. 5B).
These data demonstrate that COX-2 expression induced by 5′-aza is
regulated by alterations in DNA methylation (Fig. 5).
Discussion
COX-2 is transcriptionally modulated in normal
tissues by various factors including pro-inflammatory cytokines,
tumor necrosis factor (TNF)-α (36), interleukin (IL)-1β (37), interferon (IFN)-γ (38), and bacterial endotoxin (36).
Overexpression of COX-2 promotes cell growth, but at
the same time leads to cell cycle arrest in diverse cell types
(39,40). In other words, the upregulated
COX-2 expression may inhibit proliferation in the short-term, but
ultimately allows cancer growth in the long-term. PGE2
is a major metabolite derived from arachidonic acid and its
production is mediated by the action of COX-2. PGE2
inhibits cell growth in melanocytes (41), gastric cancer cells (42), colonic epithelial cells (43), and neuroblastoma cells (44).
In contrast, it has been reported that increased
levels of PGE2 accelerates cell motility, alters
morphology (45), and has
growth-promoting activity via the epidermal growth factor (EGF)
receptor (46).
Therefore, increased COX-2 expression and
upregulated PGE2 production lead to various responses in
carcinogenesis. Treatment with COX-2 inhibitors is known to reduce
proliferation and cause apoptosis of cancers, but our previous
results indicate that there is no relationship between COX-2
expression and proliferation of HT1080 cells (47). Although various studies on COX-2
regulation are available, the molecular basis of COX-2 expression
in cancer has not been established yet.
DNA methylation is an epigenetic mechanism of gene
silencing, with no effects on chromatin structure (48). Global hypomethylation at repetitive
sequences causes genomic instability. Aberrant hypomethylation of
CpG islands in promoter region of a gene has been implicated in the
transcriptional silencing of various genes in carcinogenesis
(48).
Studies have indicated that COX-2 promoter
methylation may be an additional regulator of COX-2 expression in
some cancer cell lines (29,49).
In this study, we therefore examined whether COX-2
expression in human HT1080 fibrosarcoma cells is regulated through
DNA methylation upon treatment with 5′-aza. We demonstrated that
COX-2 expression and PGE2 production were regulated by
the methylation status of the COX-2 promoter. Although
overexpression of COX-2 has been observed in many cancers,
concomitant increases in COX-2 expression may not be detected in
HT1080 cells. However, after treatment with 5′-aza, increased COX-2
expression became detectable, consistent with the increased COX-2
mRNA levels (Figs. 1 and 2).
These findings suggest that methylation of the COX-2
promoter may be associated with transcriptional down-regulation of
the gene. We also examined the possibility that the opposing
effects of DNA demethylation might be addressed by combining a
DNA-demethylating agent with a DNA-methylating agent. SAM is
synthesized in humans from the methyl donors present in the diet,
and is a natural compound that is a cofactor of methylation
reactions in vivo. We therefore tested whether SAM
antagonizes the effect of 5′-aza (Fig.
5). We demonstrated that SAM inhibits the global
hypomethylation caused by 5′-aza and antagonizes the effect of
5′-aza on COX-2 expression.
Promoter hypomethylation by 5′-aza upregulated COX-2
expression and PGE2 production (Figs. 1 and 2), while hypermethylation by SAM resulted
in the opposite effect (Fig.
5).
Many reports indicate that mitogen-activated protein
kinase (MAPK) and PI3K/Akt pathways are involved in regulating
COX-2 expression and PGE2 production (50,51).
We assessed the role of MAPKs and PI3K activation in 5′-aza-induced
COX-2 expression and PGE2 production (Figs. 3 and 4). 5′-aza activated ERK-1/2 and Akt, but
not p38 and c-Jun N-terminal kinase (JNK) in HT1080 cells (data not
shown). Therefore, another major finding of our study is that
5′-aza activates the ERK-1/2 and PI3K/Akt pathways (Fig. 3).
In conclusion, our data suggest that the expression
of COX-2 in HT1080 cells is regulated by DNA methylation status,
and these effects are regulated by ERK-1/2 and PI3K/Akt
pathways.
Acknowledgements
This study was supported by a grant from the
National Research Foundation of Korea
(NRF)(MEST)(NRF-2012R1A1A2043276 and 2014R1A1A3049653) and the
Korean Health Technology R&D Project, Ministry of Health and
Welfare, Republic of Korea (A120960-1201-0000300).
References
|
1
|
Papagelopoulos PJ, Galanis E, Frassica FJ,
Sim FH, Larson DR and Wold LE: Primary fibrosarcoma of bone.
outcome after primary surgical treatment. Clin Orthop Relat Res.
373:88–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baylin SB and Jones PA: A decade of
exploring the cancer epigenome - biological and translational
implications. Nat Rev Cancer. 11:726–734. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Szyf M: The role of DNA methyltransferase
1 in growth control. Front Biosci. 6:D599–D609. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stresemann C, Bokelmann I, Mahlknecht U
and Lyko F: Azacytidine causes complex DNA methylation responses in
myeloid leukemia. Mol Cancer Ther. 7:2998–3005. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Voso MT, Breccia M, Lunghi M, Poloni A,
Niscola P, Finelli C, Bari A, Musto P, Zambello R, Fianchi L, et
al: Rapid loss of response after withdrawal of treatment with
azacitidine: A case series in patients with higher-risk
myelodysplastic syndromes or chronic myelomonocytic leukemia. Eur J
Haematol. 90:345–348. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Silverman LR, Fenaux P, Mufti GJ, Santini
V, Hellström-Lindberg E, Gattermann N, Sanz G, List AF, Gore SD and
Seymour JF: Continued azacitidine therapy beyond time of first
response improves quality of response in patients with higher-risk
myelodysplastic syndromes. Cancer. 117:2697–2702. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kaminskas E, Farrell A, Abraham S, Baird
A, Hsieh LS, Lee SL, Leighton JK, Patel H, Rahman A, Sridhara R, et
al: Approval summary: Azacitidine for treatment of myelodysplastic
syndrome subtypes. Clin Cancer Res. 11:3604–3608. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Feinberg AP, Gehrke CW, Kuo KC and Ehrlich
M: Reduced genomic 5-methylcytosine content in human colonic
neoplasia. Cancer Res. 48:1159–1161. 1988.PubMed/NCBI
|
|
9
|
Shteper PJ, Zcharia E, Ashhab Y, Peretz T,
Vlodavsky I and Ben-Yehuda D: Role of promoter methylation in
regulation of the mammalian heparanase gene. Oncogene.
22:7737–7749. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu KK: Cyclooxygenase 2 induction:
Molecular mechanism and pathophysiologic roles. J Lab Clin Med.
128:242–245. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bauer AK, Dwyer-Nield LD and Malkinson AM:
High cyclo-oxygenase 1 (COX-1) and cyclooxygenase 2 (COX-2)
contents in mouse lung tumors. Carcinogenesis. 21:543–550. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nurmi JT, Puolakkainen PA and Rautonen NE:
Intron 1 retaining cyclooxygenase 1 splice variant is induced by
osmotic stress in human intestinal epithelial cells. Prostaglandins
Leukot Essent Fatty Acids. 73:343–350. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chell S, Kaidi A, Williams AC and
Paraskeva C: Mediators of PGE2 synthesis and signalling downstream
of COX-2 represent potential targets for the prevention/treatment
of colorectal cancer. Biochim Biophys Acta. 1766:104–119.
2006.PubMed/NCBI
|
|
14
|
Prescott SM and Fitzpatrick FA:
Cyclooxygenase-2 and carcinogenesis. Biochim Biophys Acta.
1470:M69–M78. 2000.PubMed/NCBI
|
|
15
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tang X, Sun YJ, Half E, Kuo MT and
Sinicrope F: Cyclooxygenase-2 overexpression inhibits death
receptor 5 expression and confers resistance to tumor necrosis
factor-related apoptosis-inducing ligand-induced apoptosis in human
colon cancer cells. Cancer Res. 62:4903–4908. 2002.PubMed/NCBI
|
|
17
|
Totzke G, Schulze-Osthoff K and Janicke
RU: Cyclooxygenase-2 (COX-2) inhibitors sensitize tumor cells
specifically to death receptor-induced apoptosis independently of
COX-2 inhibition. Oncogene. 22:8021–8030. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pockaj BA, Basu GD, Pathangey LB, Gray RJ,
Hernandez JL, Gendler SJ and Mukherjee P: Reduced T-cell and
dendritic cell function is related to cyclooxygenase-2
overexpression and pros-taglandin E2 secretion in patients with
breast cancer. Ann Surg Oncol. 11:328–339. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Castellone MD, Teramoto H, Williams BO,
Druey KM and Gutkind JS: Prostaglandin E2 promotes colon cancer
cell growth through a gs-axin-beta-catenin signaling axis. Science.
310:1504–1510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kinugasa Y, Hatori M, Ito H, Kurihara Y,
Ito D and Nagumo M: Inhibition of cyclooxygenase-2 suppresses
invasiveness of oral squamous cell carcinoma cell lines via
down-regulation of matrix metalloproteinase-2 and CD44. Clin Exp
Metastasis. 21:737–745. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gallo O, Franchi A, Magnelli L, Sardi I,
Vannacci A, Boddit V, Chiarugi V and Masini E: Cyclooxygenase-2
pathway correlates with VEGF expression in head and neck cancer.
implications for tumor angiogenesis and metastasis. Neoplasia.
3:53–61. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Masferrer JL, Leahy KM, Koki AT, Zweifel
BS, Settle SL, Woerner BM, Edwards DA, Flickinger AG, Moore RJ and
Seibert K: Antiangiogenic and antitumor activities of
cyclooxy-genase-2 inhibitors. Cancer Res. 60:1306–1311.
2000.PubMed/NCBI
|
|
23
|
Tsujii M, Kawano S and DuBois RN:
Cyclooxygenase-2 expression in human colon cancer cells increases
metastatic potential. Proc Natl Acad Sci USA. 94:3336–3340. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Byun JH, Lee MA, Roh SY, Shim BY, Hong SH,
Ko YH, Ko SJ, Woo IS, Kang JH, Hong YS, et al: Association between
cyclooxygenase-2 and matrix metalloproteinase-2 expression in
non-small cell lung cancer. Jpn J Clin Oncol. 36:263–268. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Eberhart CE, Coffey RJ, Radhika A,
Giardiello FM, Ferrenbach S and DuBois RN: Up-regulation of
cyclooxygenase 2 gene expression in human colorectal adenomas and
adenocarcinomas. Gastroenterology. 107:1183–1188. 1994.PubMed/NCBI
|
|
26
|
Ristimaki A, Honkanen N, Jankala H,
Sipponen P and Harkonen M: Expression of cyclooxygenase-2 in human
gastric carcinoma. Cancer Res. 57:1276–1280. 1997.PubMed/NCBI
|
|
27
|
Wolff H, Saukkonen K, Anttila S,
Karjalainen A, Vainio H and Ristimaki A: Expression of
cyclooxygenase-2 in human lung carcinoma. Cancer Res. 58:4997–5001.
1998.PubMed/NCBI
|
|
28
|
Liu XH and Rose DP: Differential
expression and regulation of cyclooxygenase-1 and -2 in two human
breast cancer cell lines. Cancer Res. 56:5125–5127. 1996.PubMed/NCBI
|
|
29
|
Toyota M, Shen L, Ohe-Toyota M, Hamilton
SR, Sinicrope FA and Issa JP: Aberrant methylation of the
cyclooxygenase 2 CpG island in colorectal tumors. Cancer Res.
60:4044–4048. 2000.PubMed/NCBI
|
|
30
|
Huang L, Zhang KL, Li H, Chen X-Y, Kong
Q-Y, Sun Y, Gao X, Guan H-W and Liu J: Infrequent COX-2 expression
due to promoter hypermethylation in gastric cancers in Dalian,
China. Hum Pathol. 37:1557–1567. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ramsay RG, Ciznadija D, Vanevski M and
Mantamadiotis T: Transcriptional regulation of cyclo-oxygenase
expression: Three pillars of control. Int J Immunopathol Pharmacol.
16:59–67. 2003.PubMed/NCBI
|
|
32
|
Tanabe T and Tohnai N: Cyclooxygenase
isozymes and their gene structures and expression. Prostaglandins
Other Lipid Mediat. 68–69:95–114. 2002. View Article : Google Scholar
|
|
33
|
St-Germain ME, Gagnon V, Mathieu I, Parent
S and Asselin E: Akt regulates COX-2 mRNA and protein expression in
mutated-PTEN human endometrial cancer cells. Int J Oncol.
24:1311–1324. 2004.PubMed/NCBI
|
|
34
|
Takeda K, Kanekura T and Kanzaki T:
Negative feedback regulation of phosphatidylinositol 3-kinase/akt
pathway by over-expressed cyclooxygenase-2 in human epidermal
cancer cells. J Dermatol. 31:516–523. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Detich N, Hamm S, Just G, Knox JD and Szyf
M: The methyl donor S-adenosylmethionine inhibits active
demethylation of DNA: A candidate novel mechanism for the
pharmacological effects of S-adenosylmethionine. J Biol Chem.
278:20812–20820. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Minghetti L, Walsh DT, Levi G and Perry
VH: In vivo expression of cyclooxygenase-2 in rat brain following
intraparenchymal injection of bacterial endotoxin and inflammatory
cytokines. J Neuropathol Exp Neurol. 58:1184–1191. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Giordano S, Maffe A, Williams TA,
Artigiani S, Gual P, Bardelli A, Basilico C, Michieli P and
Comoglio PM: Different point mutations in the met oncogene elicit
distinct biological properties. FASEB J. 14:399–406.
2000.PubMed/NCBI
|
|
38
|
Matsuura H, Sakaue M, Subbaramaiah K,
Kamitani H, Eling TE, Dannenberg AJ, Tanabe T, Inoue H, Arata J and
Jetten AM: Regulation of cyclooxygenase-2 by interferon gamma and
transforming growth factor alpha in normal human epidermal
keratinocytes and squamous carcinoma cells. role of
mitogen-activated protein kinases. J Biol Chem. 274:29138–29148.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
DuBois RN, Shao J, Tsujii M, Sheng H and
Beauchamp RD: G1 delay in cells overexpressing prostaglandin
endoperoxide synthase-2. Cancer Res. 56:733–737. 1996.PubMed/NCBI
|
|
40
|
Trifan OC, Smith RM, Thompson BD and Hla
T: Overexpression of cyclooxygenase-2 induces cell cycle arrest.
evidence for a prostaglandin-independent mechanism. J Biol Chem.
274:34141–34147. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Santoro MG, Philpott GW and Jaffe BM:
Inhibition of tumour growth in vivo and in vitro by prostaglandin
E. Nature. 263:777–779. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shimakura S and Boland CR: Eicosanoid
production by the human gastric cancer cell line AGS and its
relation to cell growth. Cancer Res. 52:1744–1749. 1992.PubMed/NCBI
|
|
43
|
DeRubertis FR, Craven PA and Saito R:
16,16-dimethyl prostaglandin E2 suppresses the increases in the
proliferative activity of rat colonic epithelium induced by
indomethacin and aspirin. Gastroenterology. 89:1054–1063.
1985.PubMed/NCBI
|
|
44
|
Prasad KN: Morphological differentiation
induced by prostaglandin in mouse neuroblastoma cells in culture.
Nat New Biol. 236:49–52. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sheng H, Shao J, Washington MK and DuBois
RN: Prostaglandin E2 increases growth and motility of colorectal
carcinoma cells. J Biol Chem. 276:18075–18081. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pai R, Soreghan B, Szabo IL, Pavelka M,
Baatar D and Tarnawski AS: Prostaglandin E2 transactivates EGF
receptor: A novel mechanism for promoting colon cancer growth and
gastrointestinal hypertrophy. Nat Med. 8:289–293. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gweon EJ, Jung JC and Kim SJ: Luteolin
induces inhibition of cell proliferation and COX-2 expression via
ERK pathway in human fibrosarcoma HT1080. Cancer Prev Res (Phila).
17:218–225. 2012.
|
|
48
|
Rashid A and Issa JP: CpG island
methylation in gastroenterologic neoplasia: A maturing field.
Gastroenterology. 127:1578–1588. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Akhtar M, Cheng Y, Magno RM, Ashktorab H,
Smoot DT, Meltzer SJ and Wilson KT: Promoter methylation regulates
helicobacter pylori-stimulated cyclooxygenase-2 expression in
gastric epithelial cells. Cancer Res. 61:2399–2403. 2001.PubMed/NCBI
|
|
50
|
Mercau ME, Astort F, Giordanino EF,
Martinez Calejman C, Sanchez R, Caldareri L, Repetto EM, Coso OA
and Cymeryng CB: Involvement of PI3K/akt and p38 MAPK in the
induction of COX-2 expression by bacterial lipopolysaccharide in
murine adrenocortical cells. Mol Cell Endocrinol. 384:43–51. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yu SM and Kim SJ: Production of reactive
oxygen species by withaferin A causes loss of type collagen
expression and COX-2 expression through the PI3K/akt, p38, and JNK
pathways in rabbit articular chondrocytes. Exp Cell Res.
319:2822–2834. 2013. View Article : Google Scholar : PubMed/NCBI
|