Introduction
Non-Hodgkin's lymphoma (NHL) is the fifth or sixth
most common cancer in the US, and diffuse large B-cell lymphoma
(DLBCL) is the most commonly occurring lymphoma in the Western
world (1,2). Currently, the standard front-line
therapy for DLBCL is the combination of rituximab and chemotherapy
(cyclophosphamide, doxorubicin, vincristine, and prednisone)
(R-CHOP), with expected 5- and 10-year overall survival (OS) rates
of 58 and 43.5%, respectively (3).
Rituximab is a chimeric monoclonal antibody (mAb) targeting CD20.
Rituximab acts, in part, by engaging Fc receptors on immune
effector cells, such as NK and macrophages, and induces
cytotoxicity by antibody-dependent cellular cytotoxicity (ADCC). It
also activates complement-dependent cytotoxicity (CDC) and rarely
induces apoptosis (4). While
therapeutic outcomes have improved in the post-rituximab era, there
is evidence of patients exhibiting rituximab-resistance (RR). Thus,
attempts to overcome RR have been a major focus of novel
therapeutic developments. The mechanisms of resistances in
vivo are not clear. Several mechanisms underlying RR have been
postulated. These included resistance to antibody-mediated
cytotoxicity mechanisms (ADCC, CDC, and induction of apoptosis),
Fc-receptors polymorphisms, downregulation or loss of CD20
expression, altered antibody pharmacokinetics and altered molecular
signaling pathways through CD20 (5).
We have explored the potential mechanisms of
rituximab resistance by developing in vitro clones of
rituximab-resistant (RR) variants in several B-NHL cell lines and
characterized their properties. Briefly, unlike the parental
wild-type, the RR clones express CD20 but no longer respond to
treatments with rituximab or combination of rituximab and cytotoxic
drugs. Further, the RR clones overexpressed the activity of several
survival/anti-apoptotic pathways. Interference in the activity of
these hyper-activated pathways reversed resistance (6). In the hope of overcoming rituximab
resistance alternative therapies such as the use of HDAC or Bcl-2
inhibitors have also been demonstrated to enhance sensitization of
tumor cells to rituximab (7). The
efficacy of rituximab has also been shown to be augmented when used
in combination with biological agents such as interferon-α-2a
(IFN-α), specific interleukins, bortezomib and lenalidomide
(8).
An alternative strategy for the management of
patients with lymphoma has been to use biologic agents instead of
chemotherapy in relapsed and refractory lymphoma patients. Clinical
trials using rituximab alone or in combination with IFN-α have
shown that T-cells are important for the survival for lymphoma
patients (9). Preclinical studies
have suggested a synergistic activity by the combination of IFN-α
and rituximab and phase II clinical trials exploring the use of
this combination yielded promising results (10,11).
Due to the good results of this randomized phase clinical II trial,
the priming effect of INF-α on malignant B cells and immune-cells
was evaluated in a large randomized phase III trial with
preliminary promising results (12).
IFN-α is a cytokine that affects diverse biologic
functions as antiviral activity, immunomodulatory action, cell
differentiation, and cell survival or death, in a variety of cell
types (13,14). IFN-α has been employed for the
treatment of certain tumors including hairy cell leukemia, chronic
myelogenous leukemia, melanoma and renal cancer (15,16).
In some cases, the antitumor action of IFN-α has been shown to
involve the induction of apoptosis through the activation of JNK
via PKC-δ, leading to upregulation of TRAIL and activation of
Stat-1 (17).
An alternative approach to tumor immunotherapy is
the development and application of fusion proteins. Fusion proteins
have been employed to deliver cytokines, radioisotopes and toxins
for cancer therapy (18). Recent
studies have demonstrated that a fusion protein consisting of
anti-CD20 antibody and IFN-α (anti-CD20-hIFN-α) exhibited superior
activity over rituximab, IFN-α or the combination, with significant
anti-proliferative and apoptotic effects in vitro against
several B-NHL cell lines. In vivo, anti-CD20-hIFN-α showed a
significant antitumor activity against xenografts (19). Hence, we hypothesized that
anti-CD20-hIFN-α treatment may also be effective against the RR
clones.
To test the above hypothesis, the followings were
investigated: i) do the RR B-NHL clones respond to anti-CD20-hIFN-α
treatment with a decrease in both cell viability and cell recovery?
ii) Does the drug resistance of the RR clones reverse following
treatment by the combination of chemotherapeutic drugs with
anti-CD20-hIFN-α? iii) Does treatment of the RR clones with
anti-CD20-hIFN-α signal the cells and modify their
survival/anti-apoptotic pathways? and iv) Does the treatment of the
RR clones with inhibitors of gene products modified by
anti-CD20-hIFN-α result in the reversal of drug resistance? The
findings corroborated the above hypothesis. It was found that the
RR clones responded to anti-CD20-hIFN-α treatment, but not to
anti-CD20, hIFNα or combination, and treatment with
anti-CD20-hIFN-α sensitized the drug-resistant RR clones to
drug-induced apoptosis.
Materials and methods
Cell lines and reagents
The human B-NHL cell lines Ramos and 2F7 were
purchased from the ATCC (Manassas, VA, USA). Clones were developed
as previously reported (20).
Briefly, the cells were grown in the presence of step-wise
increasing concentrations of rituximab (5–20 μg/ml) for 10 weeks.
Single cells were then isolated and subjected to three consecutive
rounds of limiting dilution analyses. The cell lines were cultured
as described previously (21). All
cells used in this study were within 15 passages after
resuscitation. The cells were checked routinely by morphology and
tested for mycoplasma contamination with the
CELLshipper® Mycoplasma Detection kit
(Bionique® Testing Laboratories, Saranac Lake, NY, USA).
Rituximab was commercially obtained. CDDP was purchased from Sigma
(St. Louis, MO, USA) and was diluted in DMSO. Treanda®
(bendamustine hydrochloride) was purchased from Cephalon, Inc.,
(Teva Pharmaceutical Industries Ltd. Frazer, PA, USA).
Adriamycin® (doxorubicin) was purchased from Sun Pharma
Global FZE. The PE-labeled anti-active caspase-3 antibody and the
corresponding IgG1 isotype controls were obtained from BD
Pharmingen (San Diego, CA, USA). The following antibodies were
obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and
were directed against Bcl-XL, Bcl-6, p65, phospho-p65
(Ser 536), p38, phospho-p38 (Thr180-Tyr182), Bax, PKC-δ,
phospho-PKC-δ (Thr 505), Stat-1, phospho-Stat-1 (Tyr701 and
Ser727), and phospho-JNK (Thr183/Tyr185). The genetically
engineered anti-CD20-hIFNα was developed as described by Xuan et
al (19) and kindly provided
by Dr Sherie L. Morrison, UCLA. Human IgG (Sigma) was used as
control. IFN-α2a was purchased from Sigma-Aldrich Co. (USA), the
PKC-δ inhibitor rottlerin was obtained from Sigma-Aldrich Co.. The
p38 Map kinase inhibitor SB203580 was purchased from Cell Signaling
Technology, Inc. (USA).
Viability assay
Cell viability was assessed by either the trypan
blue dye exclusion assay by microscopy or by the XTT dye absorbance
according to the manufacturer's instructions (Roche Diagnostic
GmbH, Nonnenwald, Germany) as previously described (21). The viability of the untreated cells
was set at 100% and total cell recovery was recorded. Each
experimental condition was performed in triplicate and the SD was
calculated.
Apoptosis determination
Apoptosis was assessed in tumor cells by flow
cytometry for activated caspase-3 as previously described (21). Briefly, B-NHL cell cultures were
preincubated with various concentrations of the anti-CD20-hIFNα
fusion protein (30, 50 or 100 pM), or rituximab, rhIFN-α
(equivalent range of concentrations) or combination of rituximab
and rhIFN-α for 18 h and CDDP, Treanda or doxorubicin (10, 5 and 5
μg/ml), respectively, were added for an additional 18 h at at 37°C.
The cells were stained intracellularly for activated caspase-3 and
the samples were analyzed by flow cytometry. Population data were
acquired on a Flow Centre EPICSR XL-MCL (Coulter, Co.) with System
II software and the percentage of positive cells was recorded.
Western blot analysis for protein
expression
B-NHL cell lines were incubated with or without 100
pM of anti-CD20-hIFNα or rituximab at 37°C for 18 h. Western blot
analysis was performed as previously described (21). Briefly, cell extracts for protein
analysis were prepared by lysing 2×106 cells on ice with
cold 200 μl of radioimmunoprecipitation assay buffer [1% NP40, 0.1%
SDS, 0.5% deoxycholic acid, complete protease inhibitor cocktail
tablets (Roche Diagnostic Co.), and 1X PBS]. Lysates were
transferred to microcentrifuge tubes and sonicated in the
Sonicator, model W-220F (Heat-System Ultrasonic, Inc.) for 10 sec.
Lysates were centrifuged at 12,000 × g at 4°C for 5 min. Protein
concentration was quantified using the Bio-Rad protein assay
(Bio-Rad Laboratories). Gel loading buffer Bio-Rad (Bio-Rad
Laboratories) was added to the cell lysates at a 1:1 volume.
Samples were boiled for 5 min and were separated on 12%
SDS-polyacrylamide mini-gels and transferred to a nitrocellulose
membrane Hybond enhanced chemiluminescence (Amersham Pharmacia
Biotech) in Trans-Blot SD semidry Transfer Cell System
(Bio-Rad).
Statistical analysis
All results were expressed as the mean ± SD of data
obtained from three triplicate, independent and separate
experiments. The statistical significance of differences between
group means was determined using one-way ANOVA to compare variance.
Significant differences were considered for probabilities <5%
(p<0.05).
Results
Effects of anti-CD20-hIFN-α treatment on
the proliferation and viability of various B-NHL cell lines
The 2F7 and Ramos B-NHL cell lines and their
rituximab-resistant variants, 2F7R and Ramos R, were treated with
different concentrations of anti-CD20-hIFN-α and equimolar
concentrations of rituximab (30, 50 and 100 pM), IFN-α, or
rituximab+IFN-α or for 18 h and examined for cell recovery, cell
viability and apoptosis. Treatment with anti-CD20-hIFN-α, in
contrast to treatment with rituximab, IFN-α, or rituximab+IFN-α,
resulted in significant inhibition of cell recovery in 2F7 at a
concentration >50 pM. With 2F7R, there was a significant
inhibition of cell recovery at the concentration of 30 pM and an
augmented inhibition at 50 and 100 pM of anti-CD20-hIFN-α (Fig. 1A, upper right panel). With Ramos
cells, there was inhibition at >50 pM of anti-CD20-hIFN-α
treatment. Likewise, there was significant inhibition of Ramos R by
anti-CD20-hIFN-α at concentrations ≥50 pM (Fig. 1A, lower panels). These findings
demonstrate that, in contrast to treatments with rituximab, IFN-α
or rituximab+IFN-α, treatment with anti-CD20-hIFNα inhibited
significantly the cell recovery in both the 2F7R and Ramos R cell
lines.
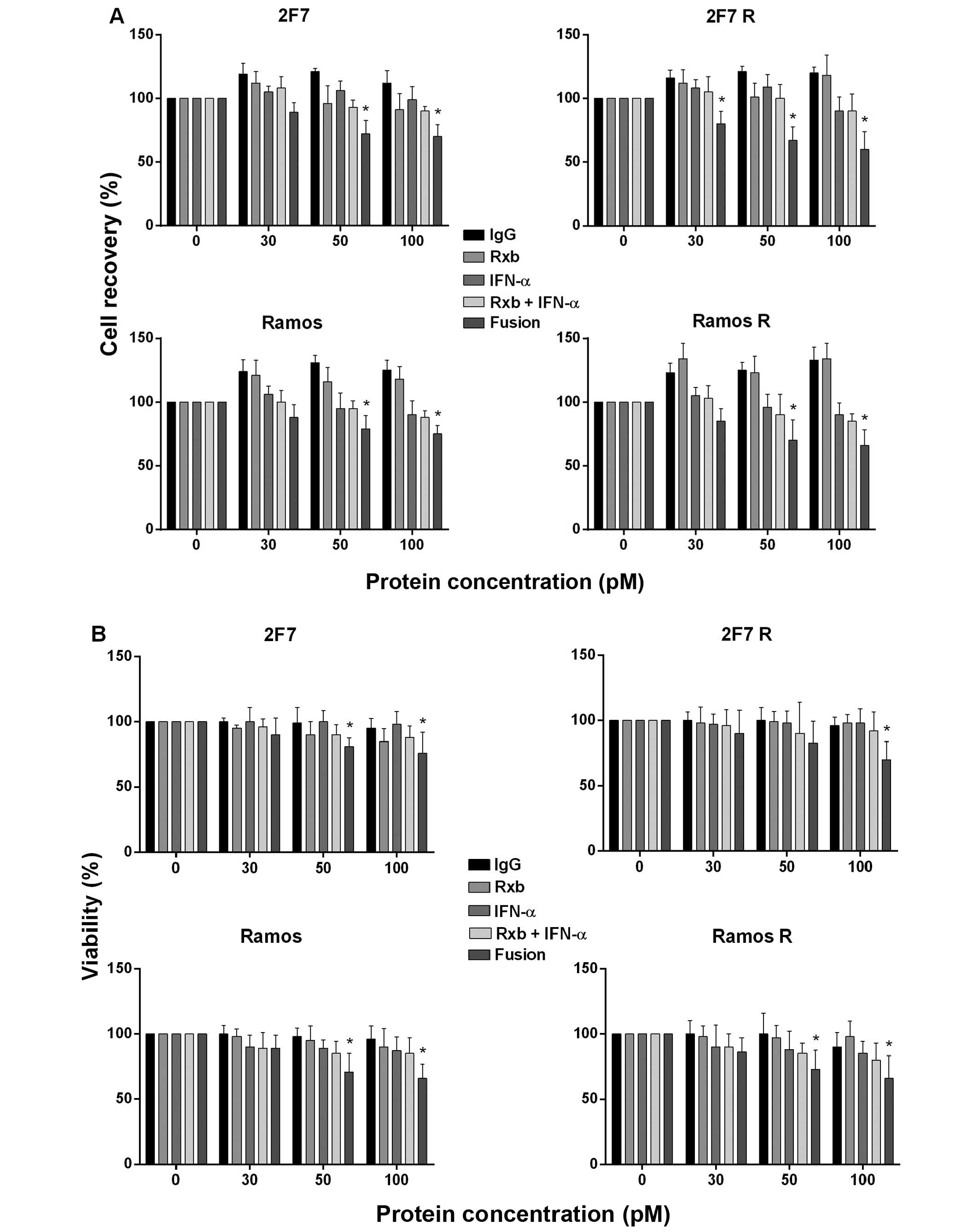 | Figure 1Anti-CD20-hIFN-α induces inhibition
of cell recovery and viability and induction of apoptosis in
rituximab-resistant (R) B-NHL cell lines. (A) The B-NHL cell lines
2F7, 2F7R, Ramos and Ramos R were treated with various
concentrations of anti-CD20-hIFN-α (30, 50 or 100 pM) or equimolar
concentrations of rituximab, rhIFN-α or the combination and
incubated for 18 h. The total cell recovery was determined by
trypan blue dye-exclusion. The B-NHL cell lines treated with normal
IgG represent 100% cells recovered. The data represent the mean±SD
from triplicate values, *p<0.05. (B) The B-NHL cell
lines 2F7, 2F7R, Ramos and Ramos R were treated with various
concentrations of anti-CD20-hIFN-α (30, 50 or 100 pM) or equimolar
concentrations of rituximab, rhIFN-α or the combination and
incubated for 18 h and cell viability was determined by the XTT
assay. B-NHL cell lines treated with normal IgG represent 100%
cells recovered. The data represent the mean±SD from triplicate
values, *p<0.05. (C) The B-NHL cell lines 2F7, 2F7 R,
Ramos and Ramos R were treated with various concentrations of
anti-CD20-hIFN-α (30, 50 or 100 pM) or equimolar concentrations of
rituximab, rhIFN-α or the combination and incubated for 18 h and
apoptosis was determined as assessed by activated caspase-3, as
described in Materials and methods. The data represent the mean±SD
from triplicate values, *p<0.05. |
The viability of the cell lines treated with the
above agents was determined microscopically by trypan blue dye
exclusion. In contrast to the treatments with rituximab, IFN-α or
rituximab+IFN-α, treatment with anti-CD20-hIFN-α induced
significant cytotoxicity in all the cell lines (2F7, 2F7R, Ramos,
and Ramos R) at concentrations ≥50 pM (Fig. 1B). These findings suggested that
the inhibition of the cell recovery shown in Fig. 1A was the result, in part, of cell
loss induced by anti-CD20-hIFN-α.
The cytotoxic activity exhibited by anti-CD20-hIFN-α
above in Fig. 1B by dye exclusion
was also examined for apoptotic activity as assessed by the
activation of procaspase-3 as described in Materials and methods.
The findings showed that treatment with anti-CD20-hIFN-α, but not
with rituximab, IFN-α or rituximab+IFN-α, induced apoptosis in all
four of the cell lines tested (Fig.
1C). With 2F7, there was significant apoptosis induction
following treatment with anti-CD20-hIFNα at 50 pM, whereas, in
2F7R, there was significant apoptosis at ≥30 pM. Also, a
significant number of cells undergoing apoptosis was recorded in
both Ramos and Ramos R cell clones treated with anti-CD20-hIFN-α at
≥30 pM.
Overall, the above findings demonstrated clearly
that the response of RR clones to treatment with anti-CD20-hIFN-α
could not be mimicked by the treatment with single agents or the
combination of anti-CD20 and hIFN-α. Further, the findings also
supported the contention that treatments with anti-CD20-hIFN-α
signalled and triggered the RR cells leading to cell death and
apoptosis. This cell signaling in the RR clones was conditioned on
the fusion protein and was not induced by the combination of
rituximab and hIFN-α.
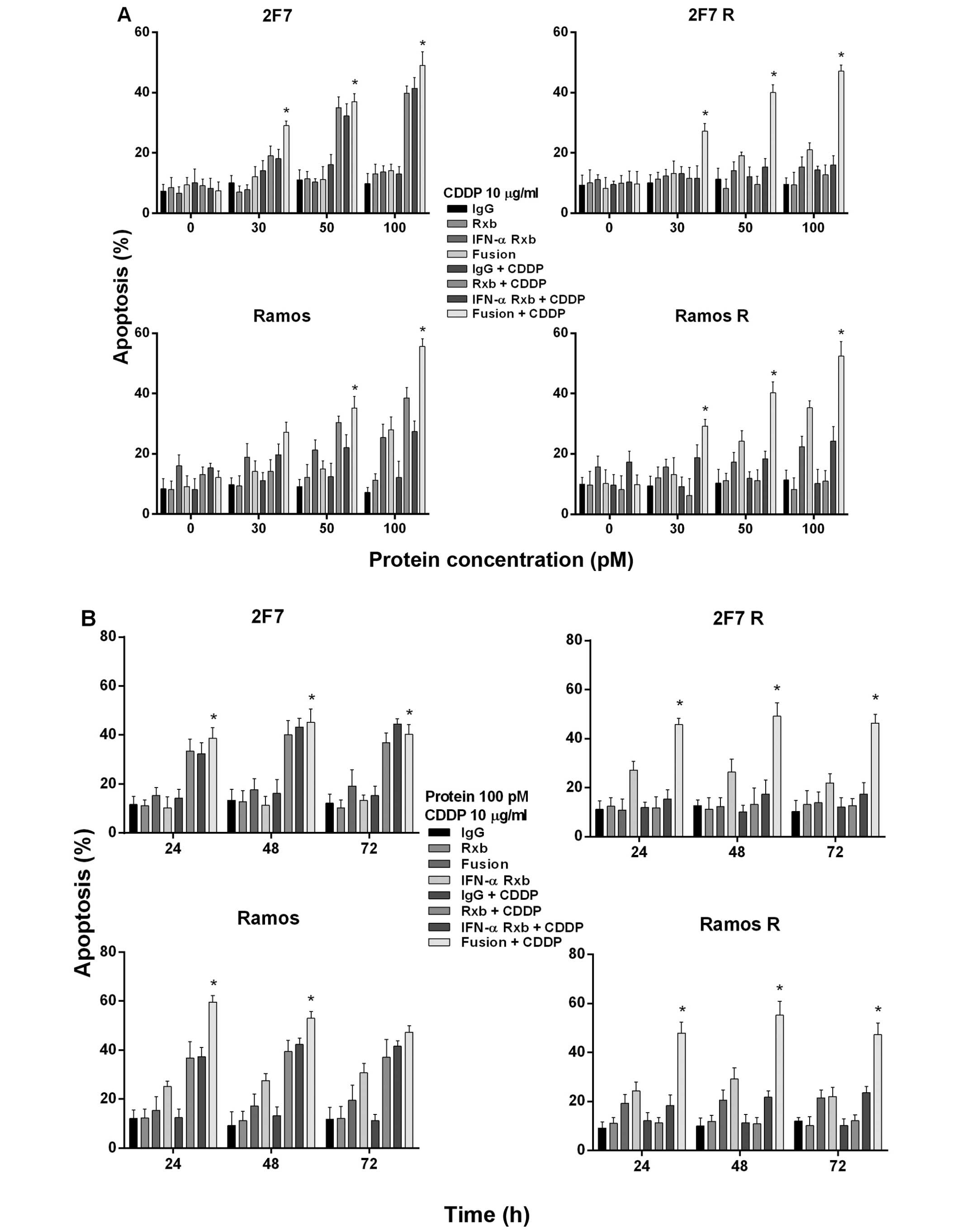
Chemosensitization of 2F7R and Ramos R
cells following treatment with anti-CD20-hIFN-α and
chemotherapeutic drugs
Previous reports have demonstrated that drug
resistance of the wild-type B-NHL cell lines, but not the
rituximab-resistant variants, can be reversed following treatment
with rituximab (22,23). These findings are corroborated here
for the 2F7, 2F7R, Ramos and Ramos R B-NHL cell lines treated with
rituximab and CDDP (Fig. 2A, upper
and bottom left panels). Noteworthy, treatment with
anti-CD20-hIFN-α and CDDP, in contrast to rituximab+CDDP, resulted
in significant induction of apoptosis in 2F7R and Ramos R at
anti-CD20-hIFN-α concentrations of ≥30 pM (Fig. 2A). The chemosensitization-induced
apoptosis by anti-CD20-hIFN-α was detected at 24, 48 and 72 h
following treatment (Fig. 2B).
These findings suggested that signaling by anti-CD20-hIFN-α on 2F7R
and Ramos R must have altered the anti-apoptotic pathways and thus,
resulting in the sensitization of the cells to CDDP apoptosis.
Similar sensitizations had been observed by the combination
treatment of rituximab+CDDP on the wild-type Ramos and 2F7 cell
lines.
In addition to CDDP, we also examined the
sensitizing activity of the anti-CD20-hIFN-α treatment on
Treanda-induced apoptosis. Treatment of the wild-type cell lines
2F7 and Ramos with rituximab+Treanda resulted in significant
apoptosis as compared to treatment with single agents (Fig. 2C, left panels). The combination of
rituximab+Treanda did not result in increased apoptosis for either
the 2F7R or Ramos R cell lines (Fig.
2C, right panels). In contrast, treatment with anti-CD20-hIFN-α
in combination with Treanda resulted in significant apoptosis in
both the 2F7R and Ramos R cell lines (Fig. 2C, right panels). Similar findings
were obtained with the combination of anti-CD20-hIFN-α and
doxorubicin (Fig. 2D). Thus,
treatment with anti-CD20-hIFN-α sensitized the RR cell lines,
findings that could not be achieved by the combination of rituximab
and chemotherapeutic agents.
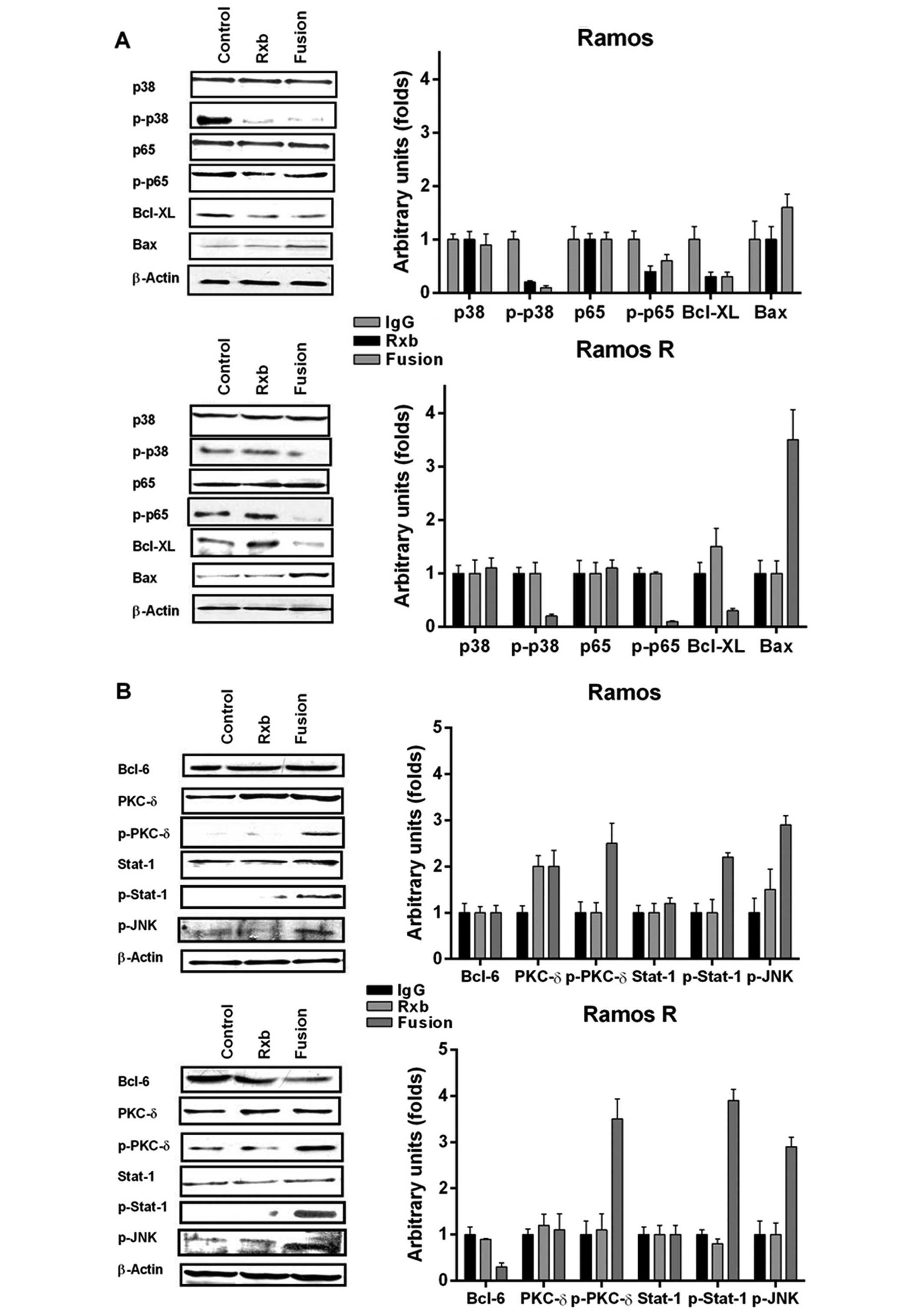
Cell signaling mediated by
anti-CD20-hIFN-α following treatment of the Ramos R and 2F7R cell
lines
Previously, we have shown that treatment of
wild-type B-NHL cell lines with rituximab resulted in the
inhibition of several survival and anti-apoptotic pathways
(23). These modifications were
responsible, in part, for the chemosensitization of the cells to
drug-induced apoptosis. Based on the present findings that
treatment of the resistant variants with anti-CD20-hIFN-α resulted
in the inhibition of cell recovery, the induction of cell apoptosis
and the sensitization to drugs, we deduced that treatment with
anti-CD20-hIFN-α must have altered cell survival pathways. We
analyzed by western blotting several proteins involved in survival
following treatment with either rituximab or anti-CD20-hIFN-α.
Treatment of Ramos with either rituximab or anti-CD20-hIFN-α
resulted in similar inhibition of p-p38, p-p65 and
Bcl-XL (Fig. 3A). In
addition, treatment with anti-CD20-hIFN-α resulted in the
upregulation of Bax. As expected, consistent with previous studies,
treatment of Ramos R with rituximab did not have any effect on the
expression of these proteins. In marked contrast, treatment of
Ramos R with anti-CD20-hIFN-α resulted in significant inhibition of
p-p38, p-p65, Bcl-XL and the induction of Bax (Fig. 3A). These findings demonstrated that
treatment with anti-CD20-hIFN-α signaled the Ramos R cells
similarly to the signaling observed following treatment of the
wild-type Ramos cells with rituximab.
In addition to the mentioned gene products, we also
examined other signaling pathways that may be induced by IFN-α and
that may have contributed to the signaling by anti-CD20-hIFN-α.
Treatment of Ramos with anti-CD20-hIFN-α resulted in the activation
of PKC-δ (p-PKC-δ) and Stat-1 but had no effect on Bcl-6, and p-JNK
(Fig. 3B). However, treatment of
Ramos R with anti-CD20-hIFN-α, but not with rituximab, resulted in
significant overexpression of p-PKC-δ and p-Stat-1 (Fig. 3B) and inhibition of Bcl-6
expression: there was no effect on p-JNK. Treatment of Ramos or
Ramos R with hIFN-α induced p-PKC-δ (Fig. 3C), suggesting that hIFN-α in the
fusion protein contributed to the activation of p-PKC-δ.
The above findings demonstrated that treatment of
the Ramos R cell line with anti-CD20-hIFNα impacted pathways
already observed following treatment of wild-type Ramos with
rituximab alone as well as by IFN-α alone. Thus, rituximab and IFN-
α in the anti-CD20-hIFN-α fusion protein each contributed to the
signaling observed in Ramos R.
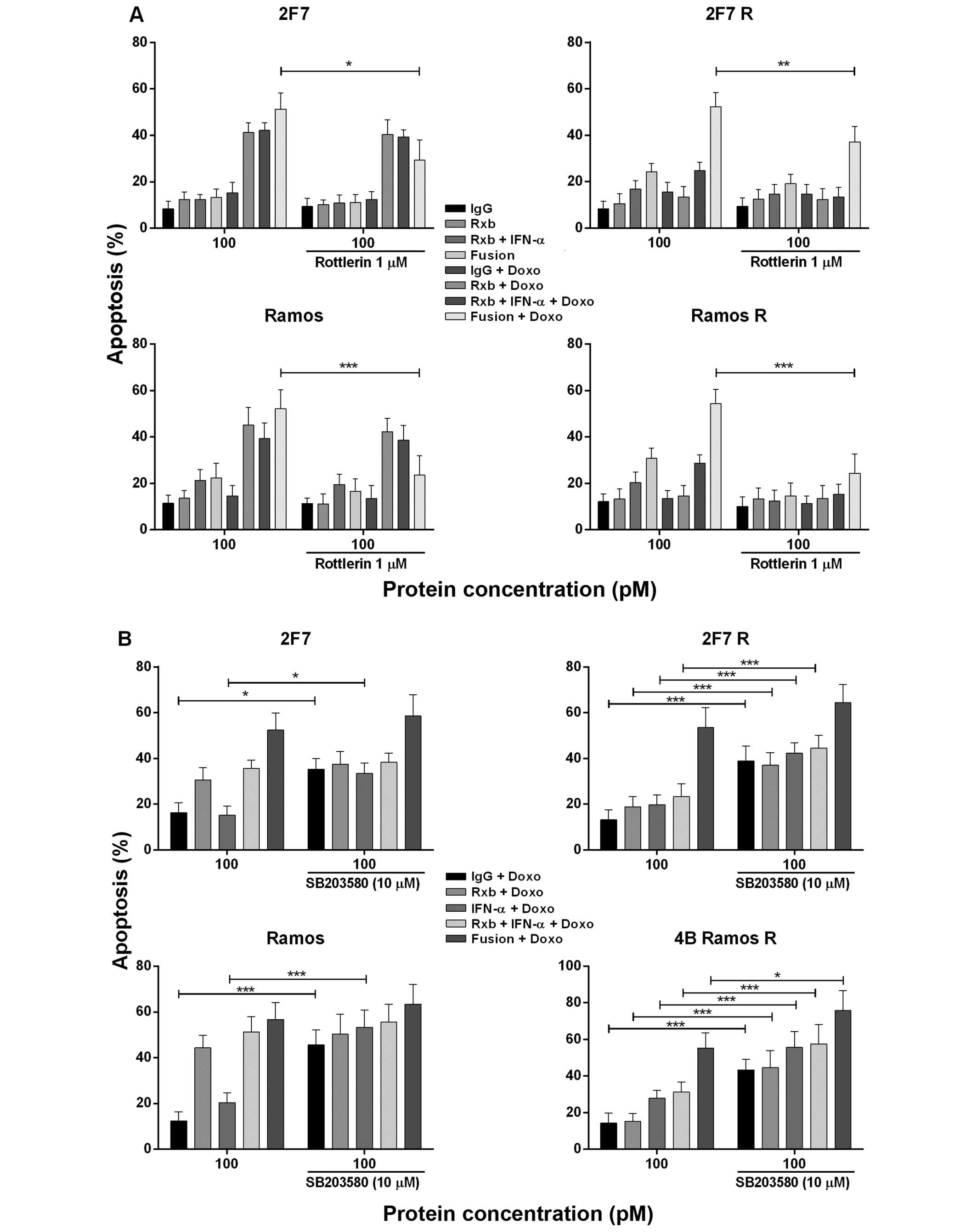
Roles of the p-p38 and PKC-δ by
anti-CD20-hIFN-α in chemosensitization to CDDP and doxorubicin
a) Effect of the PKC-δ inhibitor
Rotterin on the sensitization of 2F7 R and Ramos R cell lines by
anti-CD20-hIFN-α to CDDP-induced apoptosis
Since treatment of Ramos R with anti-CD20-hIFN-α
induced p-pKC-δ (Fig. 3B) we
examined the role of p-pKC-δ induction on chemosensitization by
anti-CD20-hIFN-α. Treatment with the PKC inhibitor rottlerin
significantly inhibited the chemosensitization induced by
anti-CD20-hIFN-α in 2F7 (p<0.005), Ramos (p<0.001), and Ramos
R (p<0.001) (Fig. 4A). PKC-δ is
activated by IFN-α and there was also significant inhibition by any
combination containing IFN-α. These findings suggest the
participation of IFN-α in anti-CD20-hIFNα induced
chemosensitization of Ramos R cells.
b) Effect of p38 MAPK inhibition by
anti-CD20-hIFN-α in chemosensitization
We have reported that treatment of B-NHL cells with
rituximab inhibited p-p38 MAPK activity and sensitized the cells to
drug apoptosis (24). Treatment of
Ramos R with anti-CD20-hIFN-α, but not with rituximab alone,
inhibited p-p38 MAPK (Fig. 3A).
Thus, we examined the role of anti-CD20-hIFNα-induced inhibition of
p-p38 MAPK in chemosensitization of 2F7 R and Ramos R to
doxorubicin-induced apoptosis. Treatment with the p-p38 MAPK
inhibitor SB203580 was found to significantly augment the apoptosis
induced by the combination of anti-CD20-hIFN-α and doxorubicin in
all four cell lines (Fig. 4B).
There was also augmentation of apoptosis by doxorubicin alone and
doxorubicin+IFN-α + rituximab. The augmented apoptosis by treatment
with anti-CD20-hIFN-α and doxorubicin suggested that it is
regulated, in part, by p-p38 MAPK and that inhibition p-p38 MAPK by
anti-CD20-hIFNα in 2F7 R and Ramos R participated in the
chemosensitization observed.
Discussion
The present standard therapy for B-NHL is anti-CD20
mAb, rituximab, plus CHOP. Although it has been shown that the
combination treatment with chemotherapy and rituximab improved the
remissions and the overall survival in indolent B cell lymphomas
(25), the majority of patients
remains incurable and new therapeutic approaches are needed. The
present study reports, for the first time, the novel finding
demonstrating that treatment by anti-CD20-hIFN-α of
rituximab-resistant (RR) B-NHL clones results in the inhibition of
cell proliferation, induction of apoptosis and sensitization to
drug-induced apoptosis. These findings are reminiscent of our
previous studies that demonstrated that intracellular inhibitors of
survival pathways sensitized the RR B-NHL clones to apoptosis by
various chemotherapeutic drugs. The observed response of the RR
clones to anti-CD20-hIFN-α was specific to the fusion protein as
neither anti-CD20, hIFN-α nor the combination was effective. The
anti-CD20-hIFN-α-mediated antitumor effects on the RR clones
resulted from the independent cell signaling pathways triggered by
the fusion protein whereby both anti-CD20 and hIFN-α contributed to
the antitumor activity. These findings provide a new potential
therapeutic application of anti-CD20 IFN-α for the treatment of
patients who are initially unresponsive or become refractory to
treatment with rituximab monotherapy or combination of rituximab
with chemotherapy.
As previously observed, the anti-CD20-hIFN-α fusion
protein was shown to exert greater anti-proliferative and cytotoxic
effects on rituximab sensitive lines compared with treatment with
either single agent alone or with the combination of anti-CD20 and
IFN-α (19). Of interest, the
level of apoptosis achieved by anti-CD20-hIFN-α on the RR clones
was higher than that achieved on the parental wild-type cells.
Previously, we have reported that the potent activity of the fusion
protein against human lymphoma cells is dependent on targeting
IFN-α to the IFN-α receptor on the tumor cell surface. In addition,
we have shown that the RR clones have high levels of IFN-αR
compared to the wild-type parental cell lines (19).
We have reported that treatment of sensitive, but
drug-resistant B-NHL cell lines, with rituximab were sensitized to
various chemotherapeutic drugs and synergy was achieved (20). We now demonstrate that the
combination of anti-CD20-hIFN-α and chemotherapeutic drugs [CDDP,
doxorubicin and bendamustine (Treanda)] resulted in reversal of
rituximab and drug resistance of RR clones to apoptosis.
Examination of the intracellular pathways, that may
be implicated in the sensitization, we showed that the treatment of
RR clones with anti-CD20-hIFN-α resulted in the inhibition of both
the p38 MAPK and NF-κB pathways. These findings are reminiscent to
those previously observed following treatment of wild-type cells
with rituximab (20,23,24).
These findings suggested that treatment of RR cells with
anti-CD20-hIFN-α resulted in the recovery of the cell signaling
mediated by rituximab in the parental wild-type cells: this
recovery, however, required that anti-CD20 is physically linked to
IFN-α.
The interaction of IFN-α with its receptor results
in the phosphorylation of receptor-associated janus kinases (JAK1
and Tyk2) and leading to the activation of signal transducer
activators of transcription (STAT) (26,27)
and in B lymphoma cells it induced the activation of JNK1 via PKC-δ
(17). In addition, recent results
identified type I IFNs as the first group of cytokines that can
downregulate Bcl-6 expression directly in germinal center (GC) B
cells (28). In B cells, Bcl-6
modulates both the activation and apoptosis, in addition to
controlling DNA-damage sensing and response (29). The phosphorylation of Bcl-6 protein
induces its subsequent degradation by the ubiquitin-proteasome
pathway (30,31). Inhibition of Bcl-6 arrests the
proliferation and induces apoptosis in B-NHL cell lines (32), through the regulation of MAPK and
NF-κB pathways; thus, this link between the inhibition of Bcl-6 and
NF-κB can explain, in part, the inhibition of NF-κB mediated by the
fusion protein.
We also explored the role IFN-α in cell signaling by
the fusion protein. We observed induction of p-PKC-δ in both the
wild-type and the RR cells by the fusion protein, but not by
rituximab. Treatment with the PKC inhibitor rottlerin reversed the
chemosensitizing effects of anti-CD20-hIFN-α, findings that are
consistent with p-PKC-δ playing a role in the reversal of
resistance. Clearly, IFN-α signals the cells by activation of
p-JNK, however, in the present findings, there was no effect by the
fusion protein on this activity in either the wild-type or the RR
clones. In addition, treatment with anti-CD20-hIFN-α resulted in
the inhibition of Bcl-XL and Bcl-6 and the induction of
Bax, gene products that regulate apoptosis and that might play a
direct role in the chemosensitization observed by the treatment of
the RR clones with anti-CD20-hIFN-α.
In addition, the role of p38 MAPK inhibition by
anti-CD20-hIFN-α on chemosensitization was corroborated by the use
of the p38 inhibitor, SB203580, which mimicked anti-CD20-hIFN-α in
the reversal of drug resistance in the RR clones. Therefore, the
anti-CD20-hIFN-α-mediated inhibition of p38 MAPK and NF-κB and
target genes such as Bcl-XL, Bcl-6, p-Stat-1 and p-PKC-δ
appear to play a role in the reversal of drug resistance.
CD20 is a membrane-associated non-glycosylated
phosphoprotein expressed on the surface of all mature B-cells, and
it plays a key role in the development and differentiation of
B-cells into plasma cells. The natural CD20 ligand is still
unknown, its function is suspected to be similar to a calcium
channel in the cell membrane (33). Recently, it was suggested that CD20
may play a central role in the generation of the T-cell-independent
antibody response (34). In
addition, recent data suggest that CD20-Ab or rituximab potentiates
B lymphocytes for the production of interferon (35). In some studies, an additive or
synergistic activity of INF with rituximab has been reported in the
treatment of lymphomas (10). This
relationship between CD20 and IFN may contribute to the efficacy of
the anti-CD20-hIFN-α fusion protein. Alternatively, crosslinking
with CD20 may prevent the internalization or downregulation of the
IFN receptors, resulting in a more prolonged and effective
IFN-α-induced signal. The mechanism by which anti-CD20-hIFN-α may
be acting on the RR clones is not completely understood.
CD20 is constitutively associated to lipid rafts and
this association depends on cholesterol and a short
membrane-proximal cytoplasmic sequence (36). The presence of a dynamic interplay
between the neutral glycosphingolipid CD77 and CD20 in B cell
lymphomas has been reported (37).
Cross-linking of CD77 with SLT-1 induces colocalization with BCR
and CD20 in Ramos cells resulting in a regulation of Lyn kinase
activity and an increase of the accessibility of the monoclonal Ab
to CD20 (38). The above findings
could indicate the possible interaction at the cell surface of CD20
and CD77 and this interaction can modulate the accessibility and
the CD20 molecular signaling-mediated antibodies. CD77 has been
implicated to play a role in IFN-α signal transduction (39). The roles of CD77 in IFN and CD20
signaling may be mediated through interactions between CD77, the
intracellular domains of CD20 and the IFNR-1 of the IFN-α receptor
as observed on other proteins, such as CD19 (40). This observation suggests a role for
the IFNR/CD77/CD20 interactions on the cytotoxic effect of the
anti-CD20-hIFN-α on B-NHL cells. Anti-CD20-hIFN-α is more potent
and effective than either IFN-α or anti-CD20 alone or their
combination, suggesting that the cross-linking between the IFN
receptor and CD20 signaling can potentiate its effect, probably
mediated by CD77 interactions. We and others have previously
reported that targeting IFN-α to CD20 on B-cell lymphomas resulted
in high potency and efficacy in vitro and in vivo
models (19,41,42).
The anti-CD20-IFN-α fusion protein induced the
activation of PKC-δ, which is involved, in part, in the
chemosensitization as shown here in the RR B-NHL cells treated with
the PKC-δ inhibitor rottlerin. Furthermore, since PKC-δ was
hyper-phosphorylated in Ramos R cells, its inhibition only
partially decreased the drug-induced apoptosis suggesting that
PKC-δ activation alone is not sufficient for the antiproliferative
and proapoptotic actions of the anti-CD20-IFN-α fusion protein.
Related studies have demonstrated that PKC-δ has multiple targets
in response to apoptotic stimuli, including IFN-α (43–46).
For example, it has been shown that PKC-δ mediated the activation
of caspase-3 (45) and activation
of Bax (47). Our results show
that the treatment with the anti-CD20-IFNα fusion protein induced
high expression of Bax in Ramos R cells. The classical type 1 IFNs
pathway included JAK/STAT activation (26). For instance, IFN-α induced
prolonged JNK1 activation (48)
with subsequent Stat-1 Ser 727 phosphorylation at least through
PKC-δ signaling (17,49). Stat-1 activation favors the
induction of apoptosis (50). We
analyzed the activation of JNK/Stat-1 pathway after fusion protein
treatment and the activation of these proteins was observed,
further suggesting that the classical JNK/Stat-1 activation plays a
role in the action of the anti-CD20-IFN-α against RR B-NHL
cells.
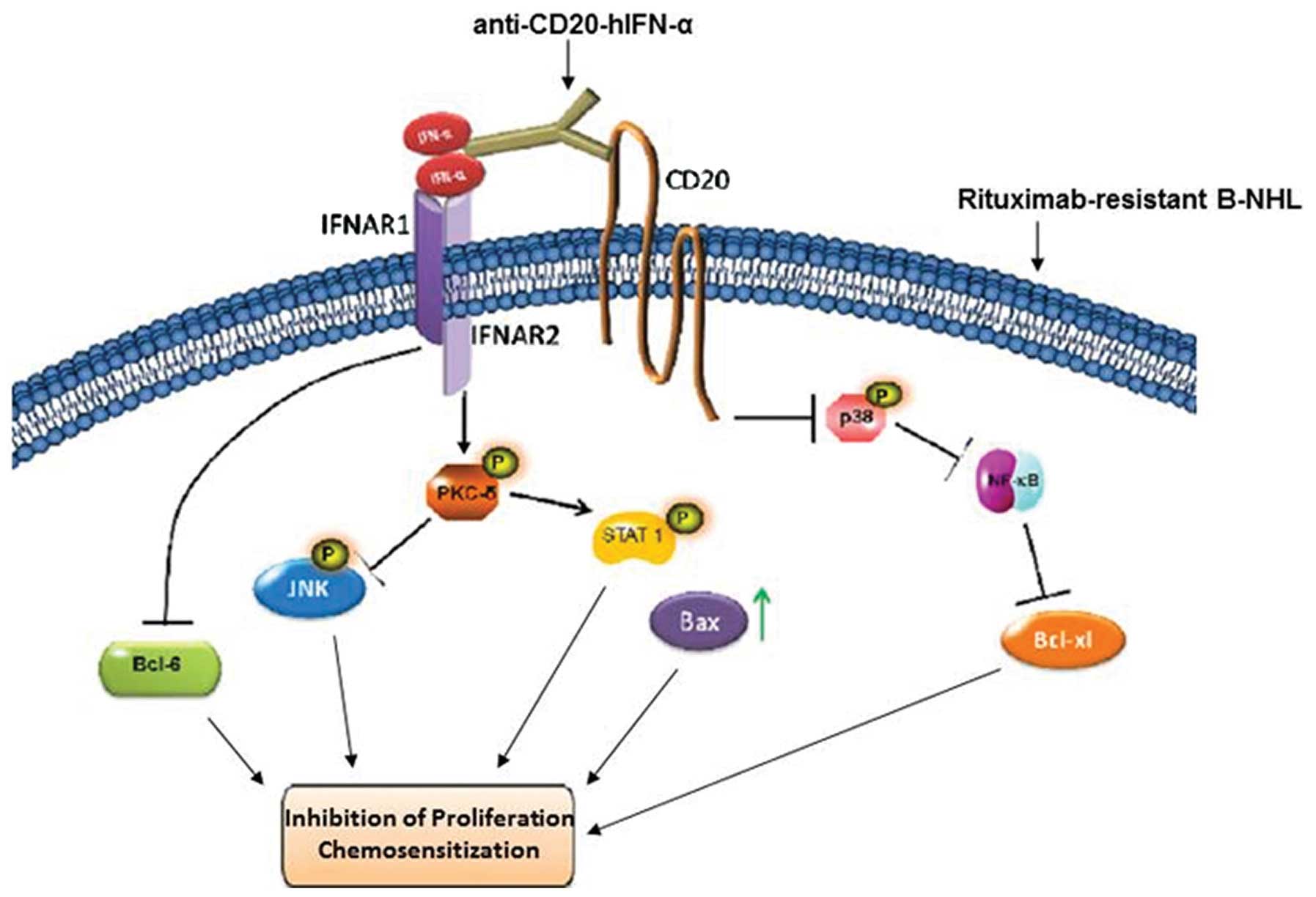
Clearly, the present findings (schematically
diagrammed in Fig. 5) using RR
B-NHL cell lines need to be validated with tumor derived RR B-NHL
cells in both untreated patients and patients resistant to
treatment. In addition, the findings need also to be validated
in vivo on the antitumor effect of anti-CD20 in mice bearing
RR tumor xenografts as monotherapy and in combination with drug
therapy. The findings suggest new therapeutic options for the
treatment of refractory B-NHL cells or CD20-mediated diseases that
no longer respond to rituximab and its combination with
chemotherapeutic drugs. Such an application clearly would be
targeted and possibly less toxic overall.
Acknowledgements
This study was supported in part by academic support
from the Grant FIS/IMSS/PROT/G13/1191 from the IMSS R-2013-785-029
(M.I.V.), CONACYT (275373) (G.G.V.), Jonsson Comprehensive Cancer
Center (M.I.V.), UCLA AIDS Institute (M.I.V.), and Fogarty
International Center Fellowship (D43 TW00013-14) (M.I.V. and
S.H.-Y.). The authors acknowledge the technical assistance of
Andrea Garcia-Olin in the experiments and the assistance of Melissa
Cao in the preparation of the manuscript. This study was also
supported by various donors (B.B.) and by the Johnson Comprehensive
Cancer Center at UCLA (B.B.).
References
|
1
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: The impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zelenetz AD, Abramson JS, Advani RH,
Andreadis CB, Byrd JC, Czuczman MS, Fayad L, Forero A, Glenn MJ,
Gockerman JP, et al: NCCN Clinical Practice Guidelines in Oncology:
non-Hodgkin's lymphomas. J Natl Compr Canc Netw. 8:288–334.
2010.PubMed/NCBI
|
|
3
|
Coiffier B, Thieblemont C, Van Den Neste
E, Lepeu G, Plantier I, Castaigne S, Lefort S, Marit G, Macro M,
Sebban C, et al: Long-term outcome of patients in the LNH-98.5
trial, the first randomized study comparing rituximab-CHOP to
standard CHOP chemotherapy in DLBCL patients: A study by the Groupe
d'Etudes des Lymphomes de l'Adulte. Blood. 116:2040–2045. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Glennie MJ, French RR, Cragg MS and Taylor
RP: Mechanisms of killing by anti-CD20 monoclonal antibodies. Mol
Immunol. 44:3823–3837. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rezvani AR and Maloney DG: Rituximab
resistance. Best Pract Res Clin Haematol. 24:203–216. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vega MI, Martinez-Paniagua M, Jazirehi AR,
Huerta-Yepez S, Umezawa K, Martinez-Maza O and Bonavida B: The
NF-kappaB inhibitors (bortezomib and DHMEQ) sensitise
rituximab-resistant AIDS-B-non-Hodgkin lymphoma to apoptosis by
various chemotherapeutic drugs. Leuk Lymphoma. 49:1982–1994. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shimizu R, Kikuchi J, Wada T, Ozawa K,
Kano Y and Furukawa Y: HDAC inhibitors augment cytotoxic activity
of rituximab by upregulating CD20 expression on lymphoma cells.
Leukemia. 24:1760–1768. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kimby E: Biological therapy doublets:
Pairing rituximab with interferon, lenalidomide, and other
biological agents in patients with follicular lymphoma. Curr
Hematol Malig Rep. 7:221–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wahlin BE, Sundström C, Holte H, Hagberg
H, Erlanson M, Nilsson-Ehle H, Lindén O, Nordström M, Ostenstad B,
Geisler CH, et al: T cells in tumors and blood predict outcome in
follicular lymphoma treated with rituximab. Clin Cancer Res.
17:4136–4144. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Davis TA, Maloney DG, Grillo-López AJ,
White CA, Williams ME, Weiner GJ, Dowden S and Levy R: Combination
immunotherapy of relapsed or refractory low-grade or follicular
non-Hodgkin's lymphoma with rituximab and inter-feron-alpha-2a.
Clin Cancer Res. 6:2644–2652. 2000.PubMed/NCBI
|
|
11
|
Sacchi S, Federico M, Vitolo U, Boccomini
C, Vallisa D, Baldini L, Petrini M, Rupoli S, Di Raimondo F, Merli
F, et al: GISL: Clinical activity and safety of combination
immunotherapy with IFN-alpha 2a and Rituximab in patients with
relapsed low grade non-Hodgkin's lymphoma. Haematologica.
86:951–958. 2001.PubMed/NCBI
|
|
12
|
Kimby E, Jurlander J, Geisler C, Hagberg
H, Holte H, Lehtinen T, Ostenstad B, Hansen M, Osterborg A, Lindén
O, et al; Nordic Lymphoma Group. Long-term molecular remissions in
patients with indolent lymphoma treated with rituximab as a single
agent or in combination with interferon alpha-2a: A randomized
phase II study from the Nordic Lymphoma Group. Leuk Lymphoma.
49:102–112. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chawla-Sarkar M, Lindner DJ, Liu YF,
Williams BR, Sen GC, Silverman RH and Borden EC: Apoptosis and
interferons: Role of interferon-stimulated genes as mediators of
apoptosis. Apoptosis. 8:237–249. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hayashida M, Hoshika A, Kanetaka Y, Yanase
N and Mizuguchi J: IFN-alpha sensitizes daudi B lymphoma cells to
anti-IgM induced loss of mitochondrial membrane potential through
activation of c-Jun NH(2)-terminal kinase. J Interferon Cytokine
Res. 26:421–429. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gutterman JU: Cytokine therapeutics:
Lessons from interferon alpha. Proc Natl Acad Sci USA.
91:1198–1205. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Spielberger RT, Mick R, Ratain MJ and
Golomb HM: Interferon treatment for hairy cell leukemia. An update
on a cohort of 69 patients treated from 1983 to 1986. Leuk
Lymphoma. 14(Suppl 1): 89–93. 1994.PubMed/NCBI
|
|
17
|
Yanase N, Hayashida M, Kanetaka-Naka Y,
Hoshika A and Mizuguchi J: PKC-δ mediates interferon-α-induced
apoptosis through c-Jun NH(2)-terminal kinase activation. BMC Cell
Biol. 13:7–15. 2012. View Article : Google Scholar
|
|
18
|
Schrama D, Reisfeld RA and Becker JC:
Antibody targeted drugs as cancer therapeutics. Nat Rev Drug
Discov. 5:147–159. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xuan C, Steward KK, Timmerman JM and
Morrison SL: Targeted delivery of interferon-alpha via fusion to
anti-CD20 results in potent antitumor activity against B-cell
lymphoma. Blood. 115:2864–2871. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jazirehi AR, Vega MI and Bonavida B:
Development of rituximab-resistant lymphoma clones with altered
cell signaling and cross-resistance to chemotherapy. Cancer Res.
67:1270–1281. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vega MI, Huerta-Yepez S, Martinez-Paniagua
M, Martinez-Miguel B, Hernandez-Pando R, González-Bonilla CR, Chinn
P, Hanna N, Hariharan K, Jazirehi AR, et al: Rituximab-mediated
cell signaling and chemo/immunosensitization of drug-resistant
B-NHL is independent of its Fc functions. Clin Cancer Res.
15:6582–6594. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alas S, Emmanouilides C and Bonavida B:
Inhibition of interleukin 10 by rituximab results in
down-regulation of bcl-2 and sensitization of B-cell non-Hodgkin's
lymphoma to apoptosis. Clin Cancer Res. 7:709–723. 2001.PubMed/NCBI
|
|
23
|
Bonavida B: Rituximab-induced inhibition
of antiapoptotic cell survival pathways: Implications in
chemo/immunoresistance, rituximab unresponsiveness, prognostic and
novel therapeutic interventions. Oncogene. 26:3629–3636. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vega MI, Huerta-Yepaz S, Garban H,
Jazirehi A, Emmanouilides C and Bonavida B: Rituximab inhibits p38
MAPK activity in 2F7 B NHL and decreases IL-10 transcription:
Pivotal role of p38 MAPK in drug resistance. Oncogene.
23:3530–3540. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Czuczman MS and Gregory SA: The future of
CD20 monoclonal antibody therapy in B-cell malignancies. Leuk
Lymphoma. 51:983–994. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kotenko SV and Pestka S: Jak-Stat signal
transduction pathway through the eyes of cytokine class II receptor
complexes. Oncogene. 19:2557–2565. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Stark GR, Kerr IM, Williams BR, Silverman
RH and Schreiber RD: How cells respond to interferons. Annu Rev
Biochem. 67:227–264. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Salamon D, Adori M, He M, Bönelt P,
Severinson E, Kis LL, Wu L, Ujvari D, Leveau B, Nagy N, et al: Type
I interferons directly down-regulate BCL-6 in primary and
transformed germinal center B cells: Differential regulation in B
cell lines derived from endemic or sporadic Burkitt's lymphoma.
Cytokine. 57:360–371. 2012. View Article : Google Scholar
|
|
29
|
Basso K, Saito M, Sumazin P, Margolin AA,
Wang K, Lim WK, Kitagawa Y, Schneider C, Alvarez MJ, Califano A, et
al: Integrated biochemical and computational approach identifies
BCL6 direct target genes controlling multiple pathways in normal
germinal center B cells. Blood. 115:975–984. 2010. View Article : Google Scholar :
|
|
30
|
Niu H, Ye BH and Dalla-Favera R: Antigen
receptor signaling induces MAP kinase-mediated phosphorylation and
degradation of the BCL-6 transcription factor. Genes Dev.
12:1953–1961. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Phan RT, Saito M, Kitagawa Y, Means AR and
Dalla-Favera R: Genotoxic stress regulates expression of the
proto-oncogene Bcl6 in germinal center B cells. Nat Immunol.
8:1132–1139. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Basso K and Dalla-Favera R: Roles of BCL6
in normal and transformed germinal center B cells. Immunol Rev.
247:172–183. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cragg MS, Walshe CA, Ivanov AO and Glennie
MJ: The biology of CD20 and its potential as a target for mAb
therapy. Curr Dir Autoimmun. 8:140–174. 2005. View Article : Google Scholar
|
|
34
|
Kuijpers TW, Bende RJ, Baars PA, Grummels
A, Derks IA, Dolman KM, Beaumont T, Tedder TF, van Noesel CJ,
Eldering E, et al: CD20 deficiency in humans results in impaired T
cell-independent antibody responses. J Clin Invest. 120:214–222.
2010. View Article : Google Scholar :
|
|
35
|
Xu D, Staedman A and Zhang L: CD20
antibody primes B lymphocytes for type I interferon production.
PLoS One. 8:e679002013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Polyak MJ, Tailor SH and Deans JP:
Identification of a cytoplasmic region of CD20 required for its
redistribution to a detergent-insoluble membrane compartment. J
Immunol. 161:3242–3248. 1998.PubMed/NCBI
|
|
37
|
Jarvis RM, Chamba A, Holder MJ, Challa A,
Smith DC, Hodgkin MN, Lord JM and Gordon J: Dynamic interplay
between the neutral glycosphingolipid CD77/Gb3 and the therapeutic
antibody target CD20 within the lipid bilayer of model B lymphoma
cells. Biochem Biophys Res Commun. 355:944–949. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Holder MJ, Chamba A, Hardie DL, Deans JP
and Gordon J: Improved access to CD20 following B cell receptor
cross-linking at Burkitt's lymphoma cell surfaces. Leuk Res.
28:1197–1202. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Khine AA and Lingwood CA: Functional
significance of globotriaosyl ceramide in interferon-alpha(2)/type
1 interferon receptor-mediated antiviral activity. J Cell Physiol.
182:97–108. 2000. View Article : Google Scholar
|
|
40
|
Maloney MD, Binnington-Boyd B and Lingwood
CA: Globotriaosyl ceramide modulates interferon-alpha-induced
growth inhibition and CD19 expression in Burkitt's lymphoma cells.
Glycoconj J. 16:821–828. 1999. View Article : Google Scholar
|
|
41
|
Rossi EA, Goldenberg DM, Cardillo TM,
Stein R and Chang CH: CD20-targeted tetrameric interferon-alpha, a
novel and potent immunocytokine for the therapy of B-cell
lymphomas. Blood. 114:3864–3871. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rossi EA, Rossi DL, Stein R, Goldenberg DM
and Chang CH: A bispecific antibody-IFNalpha2b immunocytokine
targeting CD20 and HLA-DR is highly toxic to human lymphoma and
multiple myeloma cells. Cancer Res. 70:7600–7609. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Brodie C and Blumberg PM: Regulation of
cell apoptosis by protein kinase c delta. Apoptosis. 8:19–27. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jackson DN and Foster DA: The enigmatic
protein kinase Cdelta: Complex roles in cell proliferation and
survival. FASEB J. 18:627–636. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Reyland ME: Protein kinase Cdelta and
apoptosis. Biochem Soc Trans. 35:1001–1004. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Saijo K, Mecklenbräuker I, Schmedt C and
Tarakhovsky A: B cell immunity regulated by the protein kinase C
family. Ann NY Acad Sci. 987:125–134. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yanase N, Ohshima K, Ikegami H and
Mizuguchi J: Cytochrome c release, mitochondrial membrane
depolarization, caspase-3 activation, and Bax-alpha cleavage during
IFN-alpha-induced apoptosis in Daudi B lymphoma cells. J Interferon
Cytokine Res. 20:1121–1129. 2000. View Article : Google Scholar
|
|
48
|
Yanase N, Hata K, Shimo K, Hayashida M,
Evers BM and Mizuguchi J: Requirement of c-Jun NH2-terminal kinase
activation in interferon-alpha-induced apoptosis through
upregulation of tumor necrosis factor-related apoptosis-inducing
ligand (TRAIL) in Daudi B lymphoma cells. Exp Cell Res. 310:10–21.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kaur S, Parmar S, Smith J, Katsoulidis E,
Li Y, Sassano A, Majchrzak B, Uddin S, Tallman MS and Fish EN: Role
of protein kinase C-delta (PKC-delta) in the generation of the
effects of IFN-alpha in chronic myelogenous leukemia cells. Exp
Hematol. 33:550–557. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kumar A, Commane M, Flickinger TW, Horvath
CM and Stark GR: Defective TNF-alpha-induced apoptosis in
STAT1-null cells due to low constitutive levels of caspases.
Science. 278:1630–1632. 1997. View Article : Google Scholar : PubMed/NCBI
|