Introduction
Breast cancer is the most frequently diagnosed
neoplasm in women. In the majority of cases, hormonal manipulation
by chemical or surgical oophorectomy, or by pharmacological
intervention with anti-estrogens such as tamoxifen, remains one of
the most effective approaches in the treatment of estrogen receptor
(ER)-positive breast malignancies (1,2).
Both de novo and acquired resistance to endocrine based
therapies results from actual or functional loss of ER and is
paralleled by cellular transition from an epithelial to a
mesenchymal phenotype. Commonly referred to as EMT, this is
associated with enhanced proliferative and invasive capacity and
results in poor clinical outcome.
Voltage-gated sodium channels (VGSCs) are
heteromeric membrane protein complexes containing a single
pore-forming α subunit and one or more smaller auxiliary β subunits
(3–7). They are classically responsible for
initiation and propogation of action potential in excitable cells
(8). They effect rapid
Na+ influx coincident with efflux of intracellular
K+. In mammals, ten genes encoding VGSCα have been
described, nine of which constitute one family with designations of
Nav1.1 to Nav1.9, and Nax (4,6,9–14).
These isoforms are encoded by the genes SCN1A to
SCN11A. VGSCα forms an ion pore that differs in its
tetrodotoxin (TTX) sensitivity; being sensitive to either nanomolar
(Nav1.1 to Nav1.4, Nav1.6 and
Nav1.7) or micromolar (Nav1.5,
Nav1.8 and Nav1.1) concentrations of the
toxin (4,9,12,15).
VGSCβ are also members of the immunoglobulin superfamily of cell
adhesion molecules (CAMs), and responsible for regulating channel
gating (16). Four subunits of
VGSCβ (β1-β4 encoded by the genes SCN1B to SCN4B)
have been identified in mammals; β1, β1A/B, and β3 are
non-covalently bound to the α subunit, while β2 and β4 are linked
by disulfide bonds. VGSCβ are multifunctional molecules; they boost
channel kinetics, transfer voltage-dependence and expand channel
expression in the cell membrane (17). These subunits also promote cell
adhesion in vitro, both in the presence and absence of the α
subunit (7). Thus they are
expressed in non-excitable cells such as glia, human endothelial
cells and T-lymphocytes (10,11).
In addition, they have been found to be overexpressed in various
forms of tumors, promoting adhesion, galvanotaxis, motility and
invasion, and are therefore associated with poor clinical prognosis
(9,11,18–21).
For example, in prostate cancer cells, enhanced expression of VGSCα
subunit was observed in the highly metastatic cell line
MAT-LyLu/PC-3 compared with the poorly metastatic cell line
AT-2/LNCaP (18), and tetrodotoxin
treatment resulted in significant inhibition of PC3 cell invasion
in vitro (20).
In breast cancer, the α subunit gene (SCN5A)
and nNav1.5 protein were found to be upregulated in the
highly metastatic de novo resistant MDA-MB-231 cells, in
contrast to the weakly metastatic MCF-7 (3,22),
and are involved in the enhancement of extracellular matrix (ECM)
degradation (23), in part through
activation of acidic cysteine cathepsins B and S (24).
We have established several endocrine-resistant
breast cancer cell lines by shRNA-mediated silencing of ER. These
have all undergone EMT, and acquired enhanced proliferative and
invasive capacity towards various serum components, insulin-like
growth factor-1 and epidermal growth factor (EGF) (25–27).
Since VGSC expression/activity was shown to be enhanced in highly
metastatic cancer cells, we were interested to test if this channel
shows enhanced expression level and activity in our acquired form
of endocrine-resistant breast cancer cells (pII), and if their
invasive behavior is correlated with blockade of VGSC activity. In
the present study, we examined the involvment of VGSCs in these
cells, with respect to functions related to tumor progression,
either by inhibiting channel activity with pharmacological agents
(phenytoin and tetrodotoxin) or through siRNA-mediated reduction of
Nav1.5 channels. We show for the first time a
pro-invasive effect of VGSCs in breast cancer cells with acquired
endocrine resistance, modulated in part through enhancement of
proteases (cathepsin E and kallikrein 10) and MMP (such as MMP-7)
activity.
Materials and methods
Cell lines
MCF-7 breast cancer cells were obtained from the
American Type Culture Collection (VA, USA). pII cell line (ER
silenced) was established in our laboratory by transfection of
MCF-7 with ER directed shRNA plasmid as described previously
(25,27). For routine culture, all cell lines
were maintained as monolayers in advanced Dulbecco's minimum
essential medium (DMEM) containing phenol red and supplemented with
5% fetal bovine serum (FBS), 600 μg/ml L-glutamine, 100 U/ml
penicillin, 100 μg/ml streptomycin and 6 ml/500 100X non-essential
amino acids (all from Invitrogen, CA, USA), and grown at 37°C in an
incubator gassed with an atmosphere of 5% CO2 and
maintained at 95% humidity.
Drugs, reagents and antibodies
5,5-Diphenylhydantoin sodium salt (PHT; Sigma, USA)
was prepared by dissolution in NaOH and stored at −20°C.
Tetrodotoxin (TTX; Tocris, UK) was prepared by dissolution in
physiological saline solution at pH 7.4, and stored at −80°C. Stock
solutions (10 mM) were diluted with PBS to give final
concentrations of 100 nM, 1, 10, 50 and 100 μM. EGF powder (Sigma)
was re-suspended in 0.1% BSA at 0.1 mg/ml, and stored in aliquots
at −20°C. This stock solution was freshly diluted with sterile PBS
to give final concentrations of 10, 50 and 100 ng/ml. Phallotoxin
and goat anti-rabbit IgG were obtained from Alexa.
Anti-Nav1.5 anti-body (ab56240) was obtained from Abcam,
UK. P-ERK1/2, P-Akt, actin, and anti-HRP-conjugated secondary
antibodies were obtained from Cell Signaling, USA.
Electrophysiology
Membrane currents were recorded from cells grown on
glass coverslips using the whole-cell patch clamp technique, as
described previously (3,28). A Multiclamp 700B amplifier was used
to make recordings in voltage clamp mode, compensating for series
resistance by 40–60%. A Digidata 1440A interface (Molecular
Devices) was used to digitize currents, which were low-pass
filtered at 10 kHz, sampled at 50 kHz and then analyzed using
pClamp 10.4 software. Linear leak currents were subtracted using a
P/6 protocol (29).
MTT assay
Approximately 104 cells were seeded into
triplicate wells of 12-well plates and allowed to attach overnight.
Either vehicle only (control) or VGSC inhibitors PHT or TTX (100
nM-100 μM) were then added to the cells. Growth was assessed after
4 days of incubation. Briefly, 1 ml of MTT
[3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide]
reagent (Promega, USA) (0.5 mg/ml) was added to each well and
plates incubated at 37°C for 30 min followed by the addition of 1
ml acidic isopropanol and vigorous re-suspension of the converted
blue dye. Absorbance of the suspension was measured at 595 nm with
background subtraction at 650 nm.
Cell motility wound healing assay
pII cells were cultured in 12-well plates to 80–90%
confluency. A scratch was created in the cell monolayer using a
sterile p100 yellow pipette tip and an image of the scratched area
was captured immediately (0 h). The media was then replaced with
vehicle (control), phenytoin, or TTX at concentrations of 100 nM,
1, 10, 50 and 100 μM diluted in DMEM. Cells were cultured at
37°C/5% CO2. After 24 h, an image was captured of the
same scratched area. The width of the scratch at 24 h was
calculated as a percentage of the width at 0 h; a minimum of 3
areas along the scratch were measured.
Agarose invasion assay
Ultra-pure agarose (Invitrogen) was melted in PBS,
supplemented with DMEM containing 5% FBS, and allowed to solidify
in individual wells of 6-well dishes at room temperature. Once set,
cells (4×104) that had been exposed to various
concentrations of PHT or TTX, or vehicle (control), were loaded
into wells in the agarose formed as previously described (26). Plates were incubated at 37°C in 5%
CO2 humidified atmosphere. After 24 h, cells that had
penetrated into the agarose were manually counted by visual
microscopic examination. Random cell invasion was determined as the
total number of cells which moved in both lateral directions out of
the well. In another experimental setup, cells were treated with
EGF (10 ng/ml) in the presence or absence of various concentrations
of PHT or TTX and invasion determined. In this latter case, the
agarose was mixed with insulin transferrin selenium (ITS) instead
of 5% FBS since serum itself has invasive stimulatory
components.
Cultrex BME cell invasion assay
pII cell invasion was also assessed by the
Cultrex® 24-well BME cell invasion assay obtained from
Trevigen (USA) according to the manufacturer's instructions. In
brief, the invasion chamber was coated with 100 μl of 1X basement
membrane extract (BME) solution and incubated overnight at 37°C.
pII cells serum-starved over-night at 37°C/5% CO2, were
re-suspended at 106 cells/ml in DMEM (control) or DMEM
containing various doses of PHT (1, 10, 50 and 100 μM), and 100 μl
of suspension was loaded into the upper chamber. The lower chamber
was loaded with 500 μl DMEM supplemented with 10% FBS, as a
chemoattractant. Cells were incubated at 37°C, 5% CO2
and allowed to migrate from the top chamber to the bottom. After 48
h, liquid from both top and bottom chambers was removed by
aspiration and chambers gently washed with 1X cell wash buffer,
provided by the supplier. Calcein-AM/cell dissociation solution
complex was added to the bottom chamber and left for 1 h at 37°C/5%
CO2. Cells internalize Calcein-AM and intracellular
esterases cleave acetomethylester (AM) moiety generating
fluorescent free calcein. Invading cells were determined by
recording the fluorescence emission using a microplate reader with
a filter set of excitation/emission = 485/535 nm (Cultrex,
2008).
Confocal microscopy
MCF-7 and pII cells grown overnight at 37°C, 5%
CO2 in 8-well glass chambered slides (Lab-Tek, USA) were
either left untreated or exposed for 30 min to EGF (50 ng/ml) then
fixed with 3.7% paraformaldehyde and stained with phallotoxin
(green fluorescence) to visualize F-actin, Nav1.5
antibody (red fluorescence), and DAPI (blue fluorescence) to
visualize the nuclei, and examined by confocal microscopy using a
Carl Zeiss LSM 700 microscope.
Western blotting
pII cells were cultured in 6-well plates to 80%
confluence and then serum-starved overnight before addition of
either vehicle or EGF (50 ng/ml). After 30-min exposure cells were
harvested by scraping into 300 μl of lysis buffer containing 50 mM
HEPES, 50 mM NaCl, 5 mM EDTA 1% Triton X-100, 100 μg/ml PMSF, 10
μg/ml aprotinin, and 10 μg/ml leupeptin. Protein was determined by
the standard Bradford assay and 6 μg were mixed with an equal
volume of 2X SDS and heated at 90°C for 10 min. Lysates were loaded
onto a 10% SDS-polyacrylamide gel and electrophoresed at 150 V for
1 h. Proteins were transferred to a nitrocellulose membrane and
blocked with 2% BSA for 1 h before being incubated overnight at 4°C
with either total or pAkt antibody (1/600 dilution), pERK1/2
antibody (1/1,000 dilution), Nav1.5 antibody (1/100
dilution), or actin antibody (1/1,000 dilution) prepared in 2% BSA.
The membrane was washed and incubated with anti-HRP-conjugated
secondary antibody (1/500 dilution) for 1 h, developed with Super
Signal ECL and visualized with Kodak X-ray film.
Matrix metalloproteinase activity
The general activity of MMPs was determined using a
kit from Abcam (cat no. ab112146) according to the manufacturer's
protocol. pII cells were seeded into 6-well plates and allowed to
grow to 80% confluence. Cells were serum starved overnight, and
then either left untreated or exposed to 100 μM phenytoin for 1 h
followed by EGF stimulation (100 ng/ml) for 30 min. Then, 25 μl of
the media was removed and added to 25 μl of 2 mM APMA working
solution and incubated for 15 min at 25°C, followed by addition of
50 μl of green substrate solution. MMP activity was measured at
10-min intervals for 1 h, at 37°C by recording fluorescence
emission using a microplate reader with a filter set of
excitation/emission = 485/535 nm.
Proteome profiler analysis
The relative change in 35 human proteases was
detected using Proteome Profiler™ human protease array kit (cat no.
ARY021B, R&D Systems, Inc., Minneapolis, MN, USA) following the
manufacturer's protocol. Briefly, pII cells were cultured in 6-well
plates until reaching 80–90% confluency, then serum-starved
overnight, and either left untreated (UT, control) or exposed to 50
μM phenytoin for 1 h followed by EGF stimulation (100 ng/ml) for 30
min. Cell lysate was harvested by scraping into 300 μl of lysis
buffer containing 50 mM HEPES, 50 mM NaCl, 5 mM EDTA 1% Triton
X-100, 100 μg/ml PMSF, 10 μg/ml aprotinin, and 10 μg/ml leupeptin.
Protein was determined by the standard Bradford assay.
Nitrocellulose membranes with duplicate spots of selected capture
antibodies were incubated in 2 ml of array buffer 6 (works as
blocking buffer) in 4-well multi-dishes on a rocking platform for 1
h. For each membrane, protein samples (200 μg) were incubated at
room temperature with 15 μl of protease detection cocktail for 1 h,
before adding onto the membrane, and incubated overnight at 4°C.
The membranes were then washed with 1X wash buffer and incubated
with streptavidin HRP for 30 min. Following another wash, the
membranes were incubated with chemi-reagent mix and exposed for
10–20 min. Spot intensity was quantified using a densitometer and
the average of duplicate spots on the membrane was normalized with
the average negative control spots according to the manufacturer's
protocol.
siRNA transfection
pII cells were plated in 12-well plates in complete
DMEM and incubated for 18 h at 37°C, 5% CO2.
Transfection was performed using 25 pmol of Na+ CP type
Vα siRNA (h) obtained from Santa Cruz Biotechnology (cat no.
sc-42640). Solution A was prepared following the manufacturer's
protocol by diluting 1 μl Stemfect RNA transfection reagent
(Stemgent, cat no. 00-0069) into 24 μl buffer. Solution B was made
by diluting 2.5 μl of siRNA transfection reagent into 22.5 μl of
buffer. Solutions A and B were then mixed, incubated for 15 min,
and added dropwise to the cells. After 48–72 h cells were harvested
and RNA extracted for determination of SCN5A expression by
SYBR-Green real-time quantitative PCR as described below.
RNA extraction
RNA was extracted from transfected pII cells and
purified using the RNeasy kit (Qiagen, USA) following the
manufacturer's protocol. The concentration and yield of RNA was
determined spectroscopically using the Nano-Drop (Pharmacia) and
integrity checked by agarose gel electrophoresis.
Quantitative real-time PCR
RNA was converted to cDNA using a High-Capacity cDNA
Reverse Transcription kit from Applied Biosystems. Quantitative PCR
was performed in the ABI 7500 FAST thermocycler in a reaction
volume of 20 μl using the SYBER green master mix from Invitrogen).
Primers for SCN5A gene (forward primer
5′-CACGCGTTCACTTTCCTTC-3′, reverse primer
5′-CATCAGCCAGCTTCTTCACA-3′; 208-bp product) and β-actin were
synthesized in the HSC Research Core Facility, Kuwait
University.
Statistical analysis
Means of various groups were compared using the
Sudent's t-test. Differences were considered significant at
p≤0.05.
Results
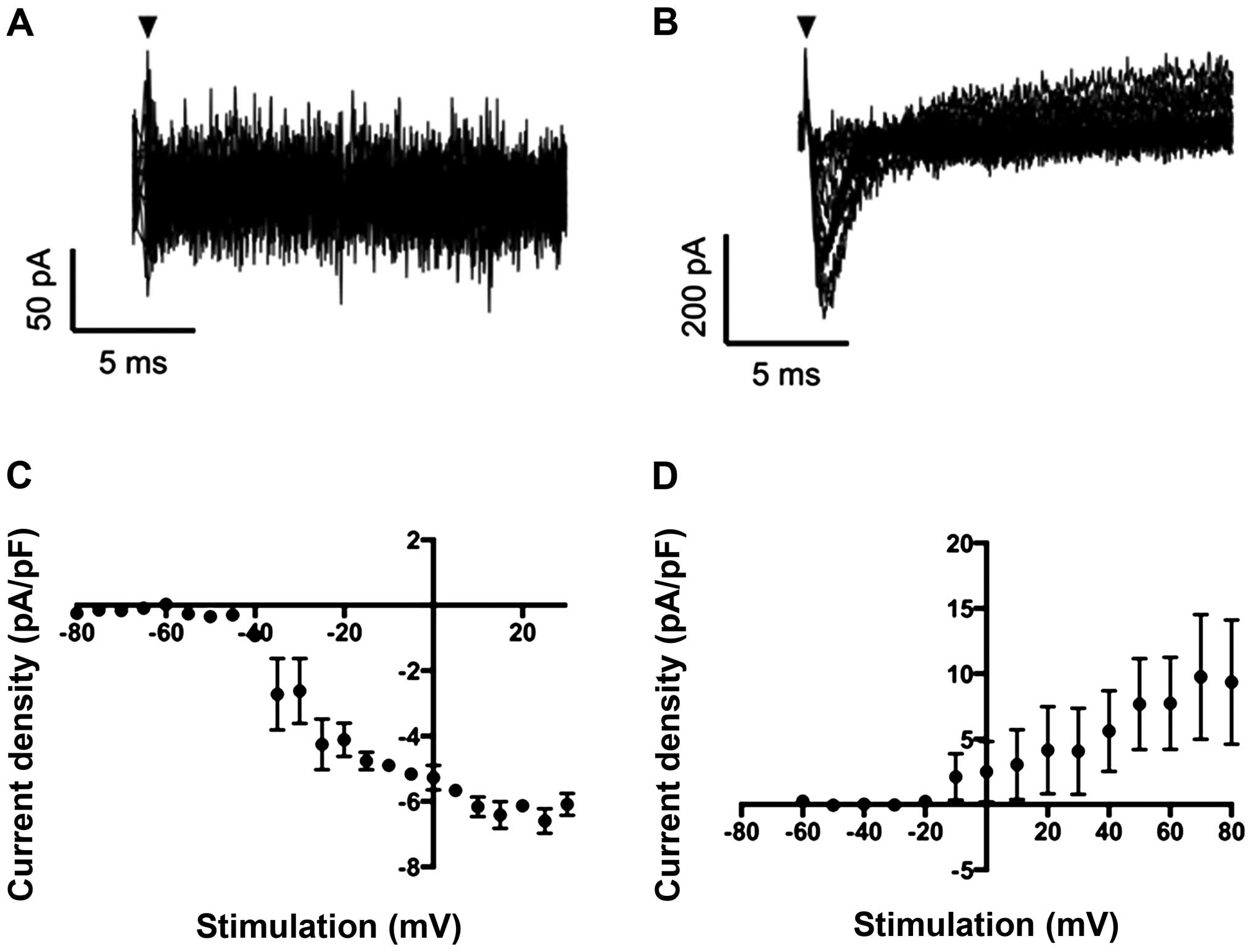
VGSC current in pII and MCF-7 cells
Whole-cell patch clamp recording revealed that VGSC
currents were absent in MCF-7 cells (n=8 recordings), consistent
with previous reports (Fig. 1A)
(22). Interestingly, however, ER
silencing in pII cells resulted in the upregulation of a fast
inward Na+ current in 2 of 5 cells recorded (Fig. 1B–D).
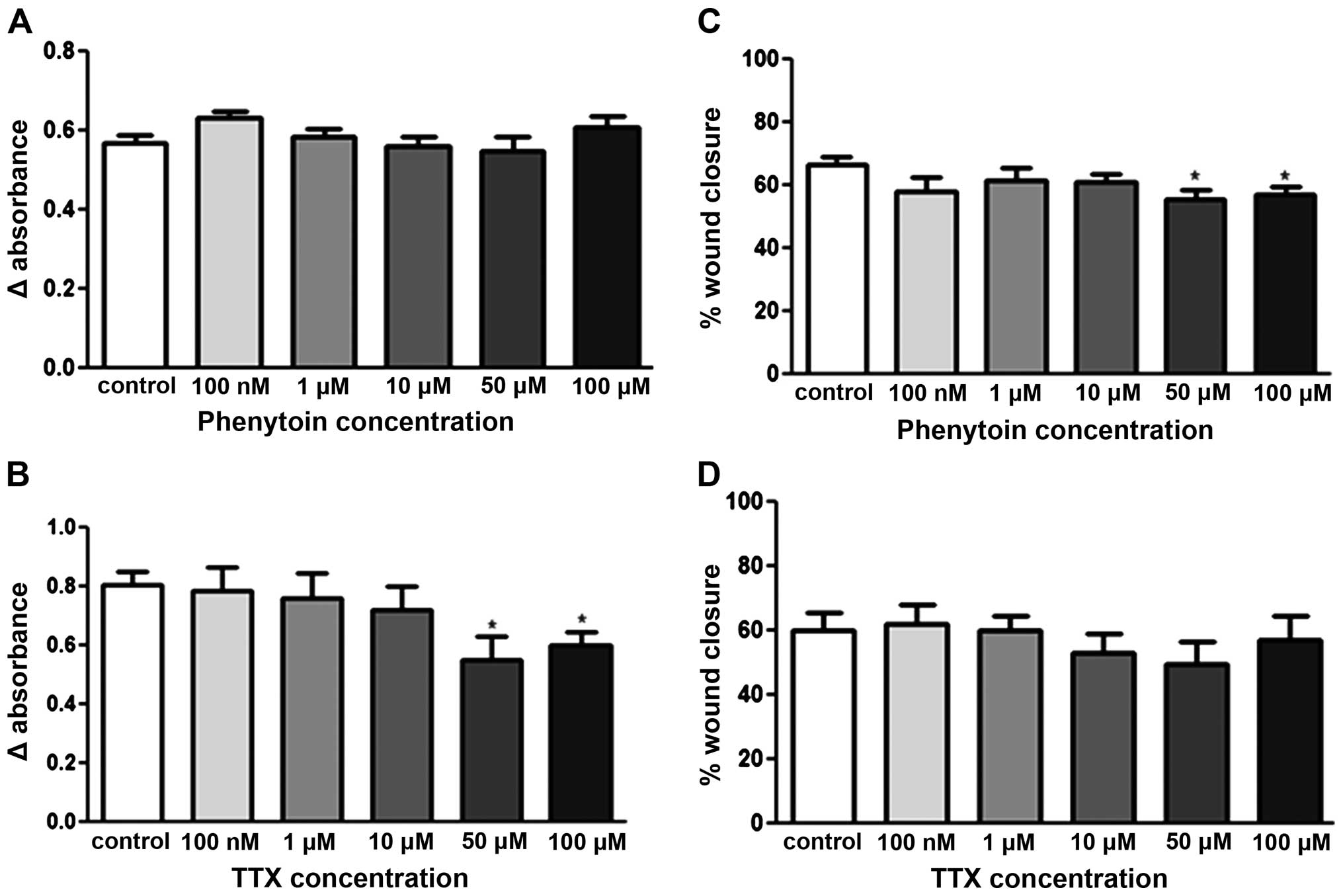
Effect of phenytoin and TTX on pII cell
proliferation and motility
The effect of various doses (100 nM-100 μM) of PHT
and TTX on pII cell proliferation was assessed using the MTT assay.
As shown in Fig. 2A, phenytoin had
no effect, while TTX exhibited a small inhibitory effect (25–30%)
when used at the higher concentrations (50–100 μM; Fig. 2B). On the other hand, TTX had no
effect on cell motility whereas phenytoin had a small (8–10%)
inhibitory effect at 50–100 μM (Fig.
2C and D).
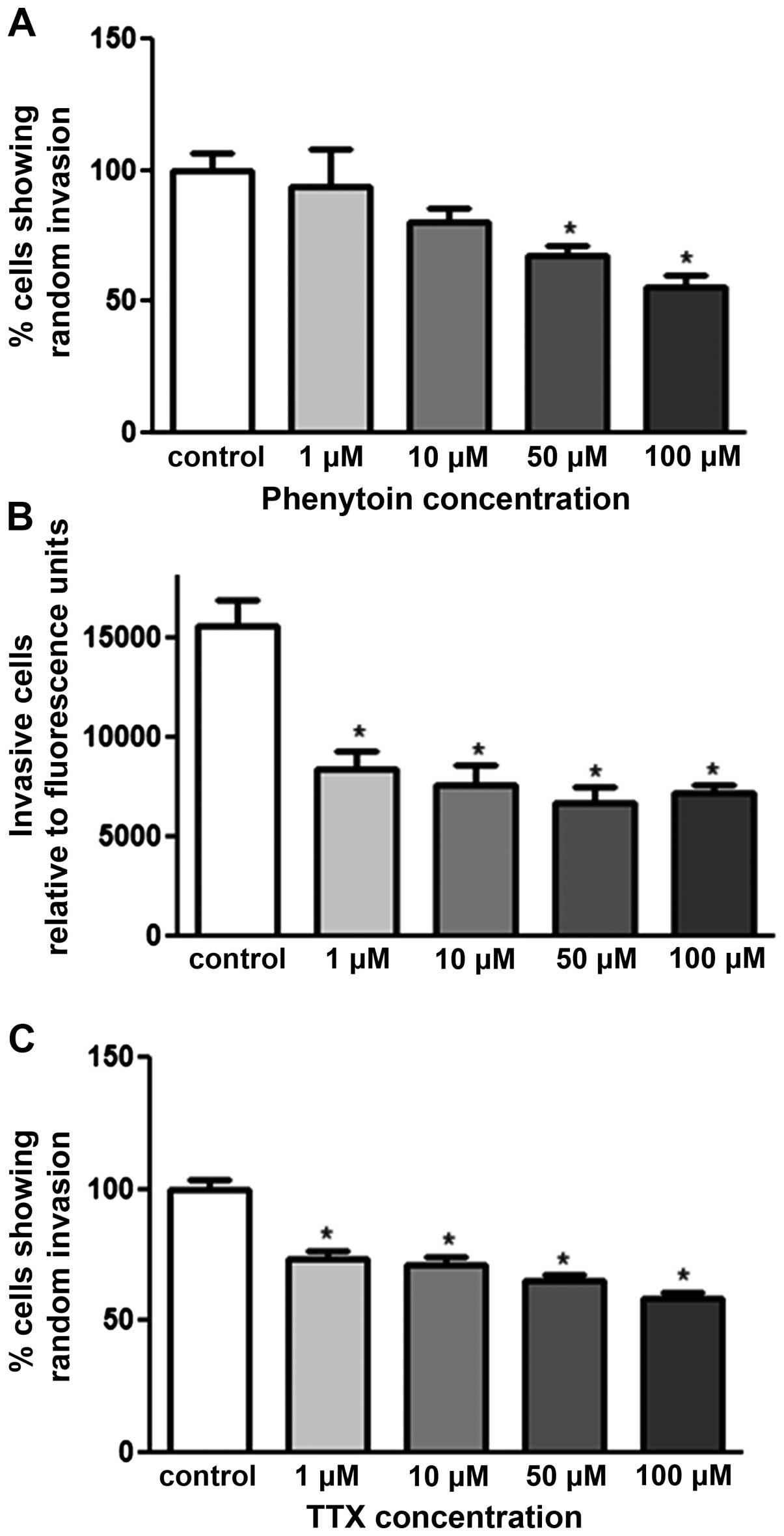
Effect of PHT and TTX on random invasion
of pII cells towards serum components
The agarose invasion assay was used to determine the
effect of VGSC inhibitors on pII invasion towards serum components.
As shown in Fig. 3A and C, both
drugs exhibited dose-dependent inhibition; PHT exerted its effect
at 50 and 100 μM with 40 and 50% inhibition respectively. TTX
exhibited its effect from a lower concentration (1 μM) with 25%
inhibition. Matrigel invasion assay was used to confirm the
anti-invasive property of PHT. Fig.
3B shows a significant inhibitory effect of PHT at all doses
used (1–100 μM) with 50% inhibition.
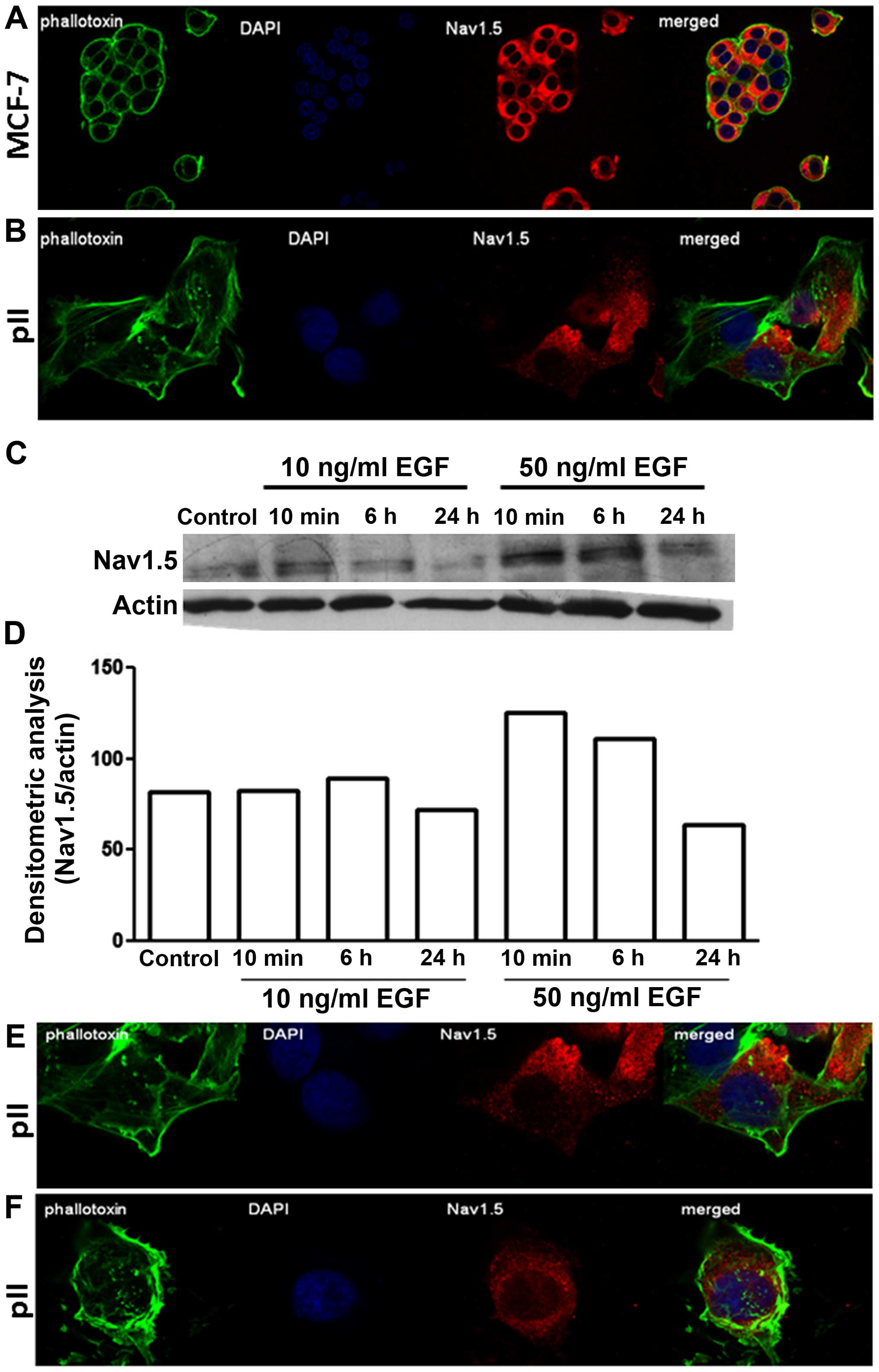
Distribution and expression profile of
VGSCs in endocrine sensitive and resistant breast cancer cell
lines
Immunofluorescence indicated a similar diffuse
cytoplasmic as well as perinuclear distribution of
Nav1.5 in both MCF-7 and pII cells (Fig. 4A and B). Measurement of total
protein by western blotting showed enhanced expression upon EGF
stimulation (at 50 ng/ml) in pII cells; the cellular distribution
of Nav1.5 remained unchanged (Fig. 4C–E).
Effect of PHT and TTX on invasion of pII
cells towards EGF
Fig. 5A and B shows
a significant increase in the invasive capacity of pII cells
towards the well containing EGF compared to vehicle (PBS, hatched
bars); this was associated with elevated ERK1/2 phosphorylation and
total MMP activity (Fig. 5C and D,
second line; and E, open circles). Both drugs (PHT and TTX) showed
similar dose-dependent inhibition of the EGF-induced invasion. As
shown in Fig. 5B and C (line 6),
the anti-invasive property of VGSCs was in part through reducing
EGF-induced ERK1/2 (but not Akt) phosphorylation as well as
reduction of EGF-induced MMP activity (Fig. 5E). In addition, by using the human
protease profiler kit, we observed that EGF stimulation
significantly enhanced the levels of cathepsin E, kallikrein-10 and
MMP-7 (Fig. 5F) relative to
controls. This effect was significantly inhibited by pre-treatment
with phenytoin.
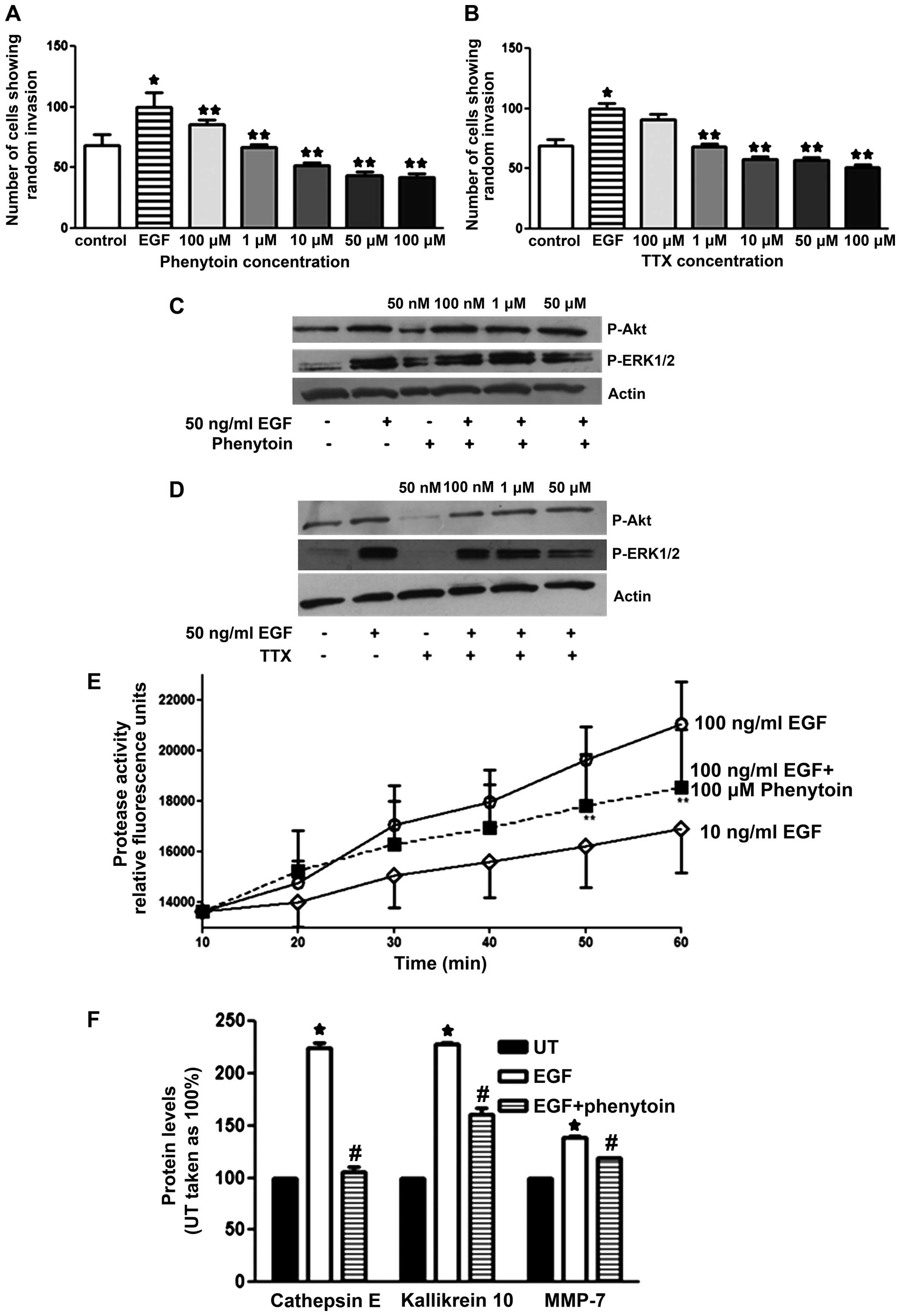 | Figure 5Effect of PHT and TTX on EGF-induced
invasion, protease levels, and MMP activity. (A and B) pII cells
were either left untreated (control, open bars), treated with EGF
(50 ng/ml, hatched bars), or pre-treated with various doses of
PHT/TTX plus EGF (solid bars). Total number of cells penetrating
into the agarose layer were manually counted as described in
Materials and methods. (C and D) pII cells were serum starved
overnight and either left untreated or stimulated with EGF in the
presence or absence of PHT/TTX. Protein lysates were
electrophoresed on SDS-PAGE and proteins transferred onto
nitrocellulose membrane and probed with antisera against P-ERK1/2,
P-Akt, or actin. (E) pII cells were seeded into 6-well plates and
grown until 90% confluent, then serum starved overnight. Cells were
then treated with EGF (10 and 100 ng/ml) with or without PHT
pre-treatment (50 μM, for 1 h). MMP activity was determined using a
fluorogenic substrate (as described in Materials and methods). (F)
Protease levels were determined in pII cells; untreated (solid
bars), treated with EGF (100 ng/ml, open bars), or treated with
phenytoin (50 μM) followed by EGF treatment (100 ng/ml, hatched
bars). Histobars represent the mean ± SEM of 3–14 independent
determinations. *Significant difference (with p≤0.05)
from control; #EGF treatment. |
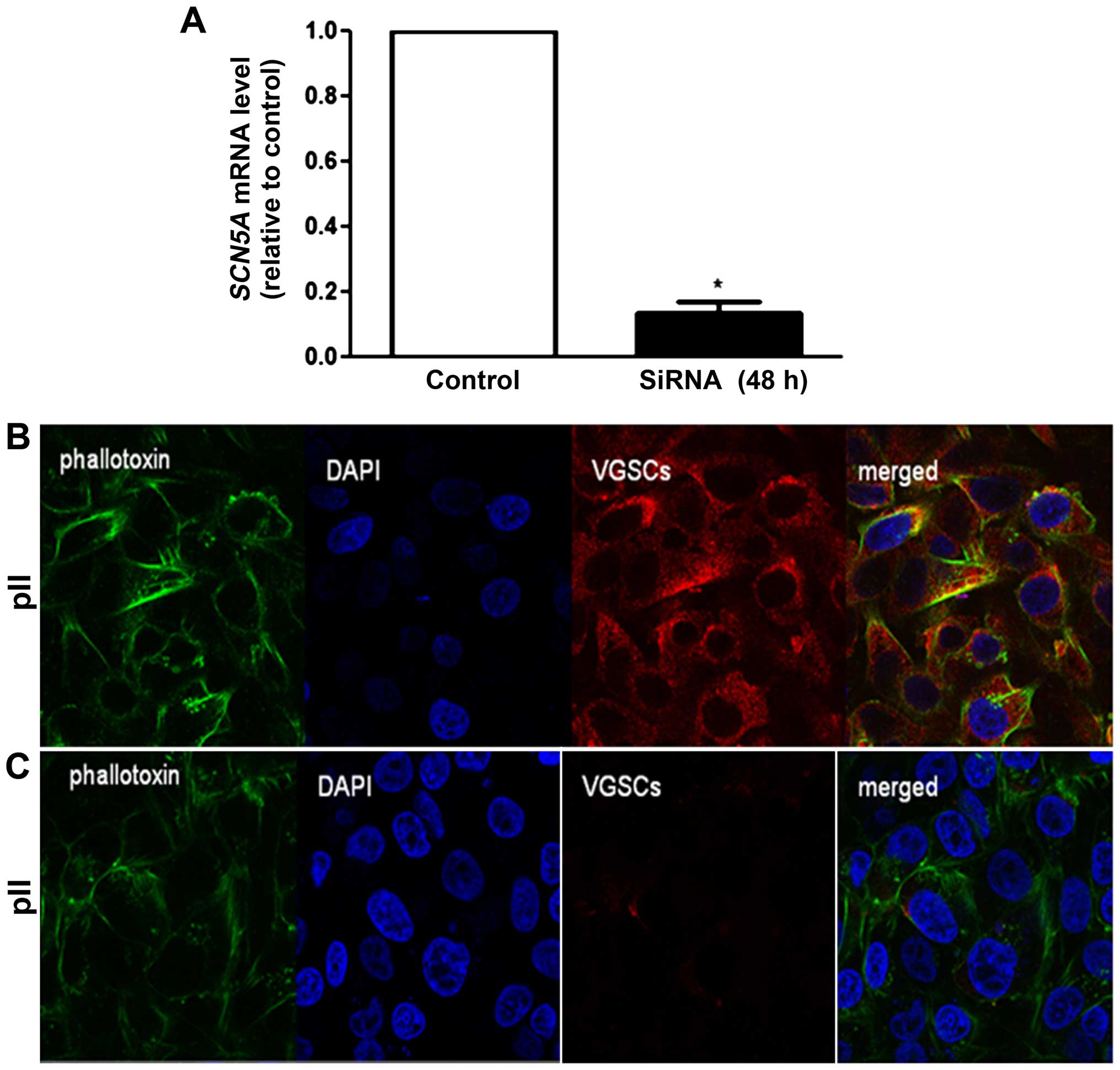
Nav1.5 knockdown by siRNA
transfection
Fig. 6A shows a
significant decrease in expression of SCN5A mRNA (80%) at 48
h post-transfection with targeting siRNA. Knockdown of
Nav1.5 protein was confirmed by immunofluorescence with
anti-VGSC antisera. Whereas there was clear cytoplasmic as well as
perinuclear staining in cells transfected with a scrambled sequence
the specific siRNA transfected cells showed almost no signal
(Fig. 6B and C). Cytoskeletal
staining with phallotoxin was the same in both.
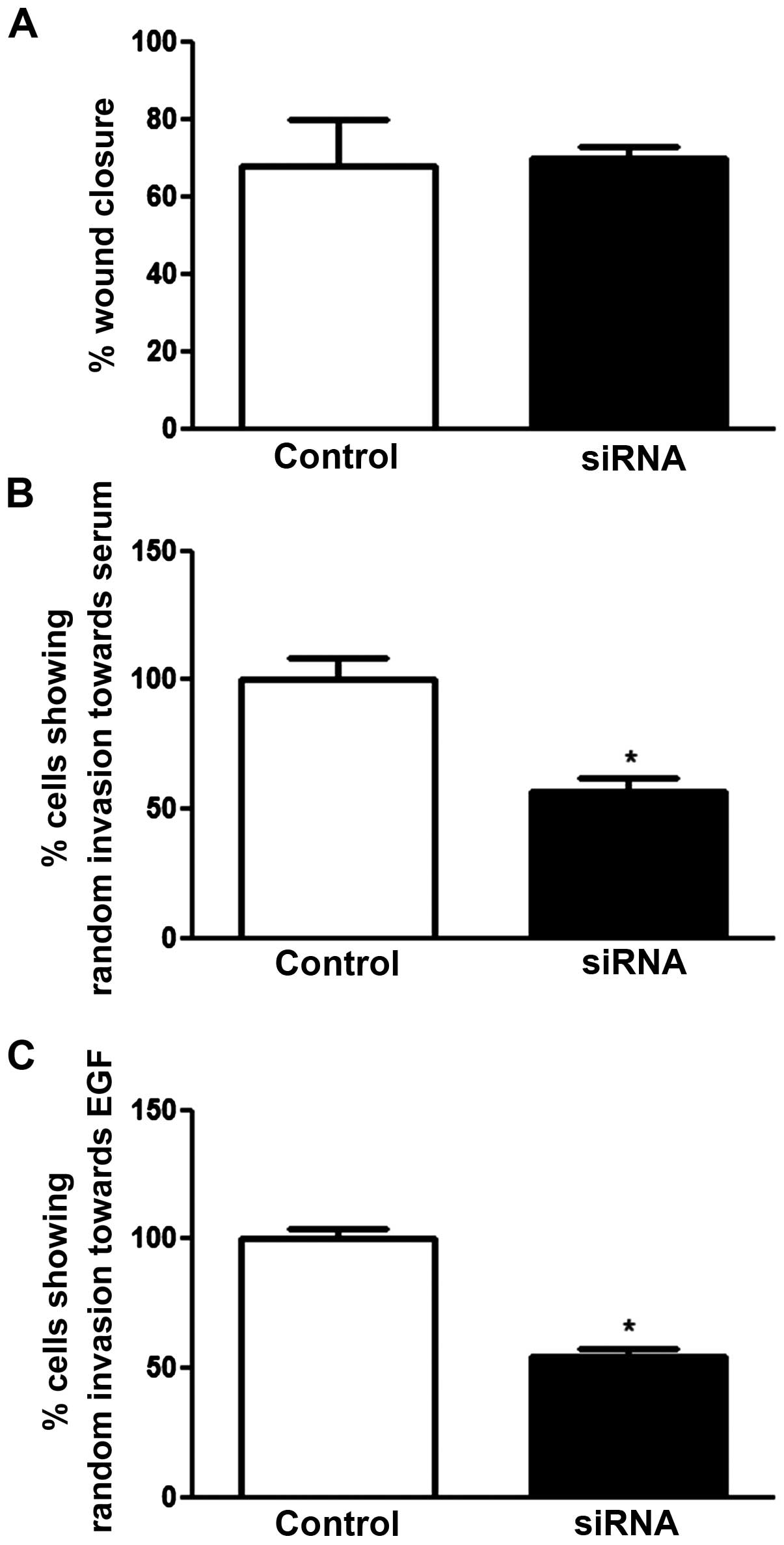
Effect of SCN5A knockdown on pII cell
motility and invasion
As shown in Fig. 7A
SCN5A mRNA knockdown did not affect pII cell motility
compared to control, but significantly inhibited invasion (50%)
towards serum components and EGF (Fig.
7B and C).
Discussion
Although VGSCs are mainly expressed in the plasma
membrane of neuronal cells (28),
these channel proteins could have roles aside from regulating
membrane potential. We showed that Nav1.5 is expressed
in the cytoplasmic and perinuclear region, as well as in the
lamellipodia, of highly invasive breast cancer cells, suggesting a
role in cell migration and invasion (3,5–7,29).
We have previously documented that the expression of SCN5A
gene (encoding Nav1.5 protein) was significantly
increased (by 4-fold) in the highly invasive ER silenced pII cells
compared to the ER+ MCF-7 cells which have little
invasive capacity (27). In view
of these findings, we investigated the expression and involvement
of VGSCs in pII cells using whole-cell patch clamp recording,
pharmacological inhibitors (PHT and TTX) and siRNA-mediated
knockdown of Nav1.5 protein. Our observations show that
VGSC currents were only detectable in pII cells, and not in MCF-7
cells, suggesting a role in modulating various functions such as
cell invasion. Upregulation of the SCN5A gene in pII cells
compared to MCF-7 might explain the absence of the VGSC current in
latter cells. In addition, cells that have undergone EMT (due to ER
loss) acquire enhanced invasive capacity and upregulate the
level/activity of various growth factor receptors, signalling and
adhesion molecules, as well as ion channels. VGSCs can promote cell
invasion, in part through modulation of EGF-induced ERK1/2
phosphorylation, protease levels, and MMP activity. We found the
same pattern of Nav1.5 expression in MCF-7 and pII
cells; this localization was not changed by EGF stimulation.
Changes in Na+ fluxes in metastatic
cancer cells have been shown to regulate intracellular pro-invasive
signaling cascades, e.g., persistent MAP kinase signaling leading
to downstream phosphorylation of ERK and other targets (30,31).
Persistent Na+ currents may contribute to invasion via
several mechanisms, including: a) allosteric regulation of the
Na+/H+ exchanger, NHE1, giving rise to
extracellular acidification (23),
b) reverse mode of the Na+/Ca2+ exchanger,
NCX, potentiating intracellular Ca2+ signaling (32), c) promotion of invadopodia
formation via src kinase activity and cortactin phosphorylation
(33), and d) regulation of
β1-mediated adhesion-dependent migration and invasion (21). These various mechanisms have been
reviewed extensively (34,35).
It has been suggested that Fyn kinase activates the
fyn-focal adhesion kinase (FAK)-ERK1/2 pathway, leading to neurite
outgrowth (36). In MDA-MB-231
breast cancer cells, fyn kinase was shown to co-localize with the
β1 subunit of VGSC, and pharmacological, as well as siRNA-mediated
fyn inhibition, resulted in inhibition of the β1-mediated process
outgrowth (which is proposed to be mediated through ERK1/2
phosphorylation) (21). In
Mat-LyLu rat prostate cancer cells, EGF treatment (for 24 h)
significantly increased VGSC current density and cell migration.
Importantly, EGF treatment in the presence of TTX (a highly
selective VGSC blocker) abolished 65% of the potentiating effect of
EGF suggesting that a significant portion of the EGF-induced
enhancement of migration occurred via VGSC activity (37). In our study, we showed that EGF
treatment significantly enhanced VGSC protein expression (Fig. 4) and siRNA-mediated knockdown of
VGSC inhibited EGF-induced invasion in pII cells (Fig. 7C), which is consistent with the
data obtained from the prostate cancer cells. We also observed a
significant inhibition of ERK1/2 phosphorylation by phenytoin and
TTX treatment (Fig. 5) consistent
with an involvement in VGSC-mediated invasion of pII cells.
Phenytoin is an anti-epileptic and class 1b
anti-arrhythmic agent which inhibits the activity of VGSCs
(38). Its binding affinity to
VGSCs increases when the channels are in the inactivated state
(39). Subtypes of VGSCs such as
Nav1.5 do not reach a complete inactivation state and
carry steady-state Na+ currents at depolarized potential
(40,41). Cancer cells possess a more
depolarized membrane potential compared to normal epithelial or
excitable cells, suggesting that some permanent Na+
current may be involved in invasion and migratory activity
(3,42). At concentrations (50 μM) that are
used in treatment of epilepsy, phenytoin significantly inhibits
both persistent and transient Na+ currents in the de
novo resistant breast cancer cells MDA-MB-231, resulting in
reduction of their invasive potential (3,43).
Furthermore, phenytoin significantly inhibits growth, invasion and
metastasis of orthotopic MDA-MB-231 breast tumors in vivo
(44). However, phenytoin had no
effect on MCF-7 cell migration or invasion (they do not express
Na+ currents), nor on cell proliferation of either
MDA-MB-231 or MCF-7 in vitro (3). TTX, also considered a highly specific
VGSC blocker, is reported to suppress metastatic behaviour in human
breast, prostate and lung cancer cells in vitro. Cell
proliferation was also not affected by TTX treatment, suggesting
involvement specifically in cell invasion (4,8,45).
We observed similar effects of PTH/TTX in pII cells as potent
anti-invasive agents (Fig. 3)
without significantly modulating either cell proliferation or
motility. A marginal decrease was seen in cell proliferation at
higher doses of TTX (50–100 μM; Fig.
2B), and a marginal decrease in cell motility with higher doses
of phenytoin treatment (50–100 μM; Fig. 1C). It should be noted that the
anti-invasive property of PHT or TTX was not due to inhibiting cell
proliferation since 24–48-h treatment (the time-point used for the
invasion assays) did not affect cell proliferation with either drug
(data not shown). Differences in effect on motility and
proliferation might be due to the lack of specificity of these
agents particularly at higher doses. Therefore, the siRNA-approach
was used to confirm these findings in a more specific way. siRNA
mediated knockdown of VGSCα isoforms has been reported to supress
breast cancer cell invasion (4,24,45),
which is in aggreement with our data in pII cells (Fig. 6B and C).
In order to metastasize, cancer cells have to
degrade the extracellular matrix (ECM) components, and VGSCs have
been suggested to play a role in this process through
Na+/H+ exchanger type I (NHE-1) activation.
Fraser et al (22) showed
that VGSCs (specifically Nav1.5) increase Na+
influx, which in turn activates the NHE-1 present in caveolae.
NHE-1 plays a role in Na+ influx regulation, leading to
extracellular acidification of the tumor microenvironment,
resulting in activation of pH-dependent extracellular matrix
degradation by cysteine cathepsins B and S, and subsequent
enhancement in cell invasion (23). Matrix metalloproteinases (MMPs) are
among the proteins involved in invasion by virtue of their ability
to degrade various ECM components including collagens, laminin,
fibronectin, vitronectin, enactin, tenascin, elastin and
proteoglycans (46). They are also
thought to play a crucial role in tumor invasion, metastasis,
migration and angiogenesis (47,48).
Pharmacological blockade of Nav1.5 channels in
MDA-MB-231 cells result in a significant decrease in MMP-9 mRNA
expression and cell invasion (49), which is also in agreement with our
finding of increased MMP activity in the presence of EGF (Fig. 4E). We showed that the most
significant inhibitory effect of phenytoin was observed on
EGF-induced cathepsin E levels (Fig.
4F). Cathepsin E has been suggested as a possible marker for
pancreatic tumors (50,51), and interestingly, a recent study
demonstrated a positive correlation between enhanced serum levels
of cathepsin E and poor clinical prognosis in breast cancer
patients. Mice overexpressing cathepsin E demonstrated enhanced
tumor growth and metastasis through induction of the EMT process
(52).
From a therapeutic viewpoint, VGSC blockade has been
reported to relieve severe cancer pain in patients receiving
chemotherapy (53–55). In fact, the newly FDA approved
drug, Riluzole, blocks VGSC activity and inhibits metabotropic
glutamine receptor, and was reported to prevent side effects
related to cancer chemotherapy (56). In addition, Riluzole reduced the
metabolic activity of tumors in patients with resectable stage III
and IV melanoma (57).
Furthermore, the use of VGSC blockers during radical prostatectomy
minimized cancer recurrence and metastasis (58,59).
Recently, it was suggested that further investigation of the
FDA-approved VGSC blockers which are already in the market (for
other diseases such as epilepsy and arrhythmia, and for inducing
local anaesthesia) should be tested for human metastatic diseases.
In conclusion, our data suggest a promising anti-metastatic role
for VGSC blockers in acquired forms of endocrine resistance in
breast cancer.
Acknowledgements
This study was supported by Kuwait University
Research Sector grant YP02/13. This study was supported in part by
grant SRUL02/13 to the Research Unit for Genomics, Proteomics and
Cellomics Studies at the HSC, Kuwait University, and by a grant
from the UK Medical Research Council (Fellowship G1000508). We
would like to acknowledge Princy Mathew for technical
assistance.
References
|
1
|
Yang LH, Tseng HS, Lin C, Chen LS, Chen
ST, Kuo SJ and Chen DR: Survival benefit of tamoxifen in estrogen
receptor-negative and progesterone receptor-positive low grade
breast cancer patients. J Breast Cancer. 15:288–295. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Al Saleh S, Sharaf LH and Luqmani YA:
Signalling pathways involved in endocrine resistance in breast
cancer and associations with epithelial to mesenchymal transition
(Review). Int J Oncol. 38:1197–1217. 2011.PubMed/NCBI
|
|
3
|
Yang M, Kozminski DJ, Wold LA, Modak R,
Calhoun JD, Isom LL and Brackenbury WJ: Therapeutic potential for
phenytoin: Targeting Na(v)1.5 sodium channels to reduce migration
and invasion in metastatic breast cancer. Breast Cancer Res Treat.
134:603–615. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Onkal R and Djamgoz MBA: Molecular
pharmacology of voltage-gated sodium channel expression in
metastatic disease: Clinical potential of neonatal Nav1.5 in breast
cancer. Eur J Pharmacol. 625:206–219. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chioni A-M, Brackenbury WJ, Calhoun JD,
Isom LL and Djamgoz MB: A novel adhesion molecule in human breast
cancer cells: Voltage-gated Na+ channel beta1 subunit.
Int J Biochem Cell Biol. 41:1216–1227. 2009. View Article : Google Scholar :
|
|
6
|
Brackenbury WJ, Djamgoz MBA and Isom LL:
An emerging role for voltage-gated Na+ channels in
cellular migration: Regulation of central nervous system
development and potentiation of invasive cancers. Neuroscientist.
14:571–583. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brackenbury WJ, Davis TH, Chen C, Slat EA,
Detrow MJ, Dickendesher TL, Ranscht B and Isom LL: Voltage-gated
Na+ channel beta1 subunit-mediated neurite outgrowth
requires Fyn kinase and contributes to postnatal CNS development in
vivo. J Neurosci. 28:3246–3256. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gillet L, Roger S, Bougnoux P, Le Guennec
J-Y and Besson P: Beneficial effects of omega-3 long-chain fatty
acids in breast cancer and cardiovascular diseases: Voltage-gated
sodium channels as a common feature? Biochimie. 93:4–6. 2011.
View Article : Google Scholar
|
|
9
|
Hernández-Plata E: Role of the
voltage-gated sodium channels in the metastatic capacity of cancer
cells. Rev Invest Clin. 64:567–575. 2012.(In Spanish).
|
|
10
|
Brackenbury WJ and Isom LL: Voltage-gated
Na channels: Potential for β subunits as therapeutic targets.
Expert Opin Ther Targets. 12:1191–1203. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Roger S, Potier M, Vandier C, Besson P and
Le Guennec J-Y: Voltage-gated sodium channels: New targets in
cancer therapy? Curr Pharm Des. 12:3681–3695. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Potier M, Joulin V, Roger S, Besson P,
Jourdan ML, Leguennec JY, Bougnoux P and Vandier C: Identification
of SK3 channel as a new mediator of breast cancer cell migration.
Mol Cancer Ther. 5:2946–2953. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Goldin AL: Resurgence of sodium channel
research. Annu Rev Physiol. 63:871–894. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Goldin AL, Barchi RL, Caldwell JH, Hofmann
F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Netter YB,
et al: Nomenclature of voltage-gated sodium channels. Neuron.
28:365–368. 2000. View Article : Google Scholar
|
|
15
|
Hernández-Plata E, Ortiz CS,
Marquina-Castillo B, Medina-Martinez I, Alfaro A, Berumen J, Rivera
M and Gomora JC: Overexpression of NaV 1.6 channels is associated
with the invasion capacity of human cervical cancer. Int J Cancer.
130:2013–2023. 2012. View Article : Google Scholar
|
|
16
|
Yildirim S, Altun S, Gumushan H, Patel A
and Djamgoz MBA: Voltage-gated sodium channel activity promotes
prostate cancer metastasis in vivo. Cancer Lett. 323:58–61. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Isom LL and Catterall WA: Na+
channel subunits and Ig domains. Nature. 383:307–308. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Diss JK, Archer SN, Hirano J, Fraser SP
and Djamgoz MB: Expression profiles of voltage-gated Na(+) channel
alpha-subunit genes in rat and human prostate cancer cell lines.
Prostate. 48:165–178. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Djamgoz MB and Onkal R: Persistent current
blockers of voltage-gated sodium channels: A clinical opportunity
for controlling metastatic disease. Recent Patents Anticancer Drug
Discov. 8:66–84. 2013. View Article : Google Scholar
|
|
20
|
Laniado ME, Lalani EN, Fraser SP, Grimes
JA, Bhangal G, Djamgoz MB and Abel PD: Expression and functional
analysis of voltage-activated Na+ channels in human
prostate cancer cell lines and their contribution to invasion in
vitro. Am J Pathol. 150:1213–1221. 1997.PubMed/NCBI
|
|
21
|
Nelson M, Millican-Slater R, Forrest LC
and Brackenbury WJ: The sodium channel β1 subunit mediates
outgrowth of neurite-like processes on breast cancer cells and
promotes tumour growth and metastasis. Int J Cancer. 135:2338–2351.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fraser SP, Diss JKJ, Chioni A-M, Mycielska
ME, Pan H, Yamaci RF, Pani F, Siwy Z, Krasowska M, Grzywna Z, et
al: Voltage-gated sodium channel expression and potentiation of
human breast cancer metastasis. Clin Cancer Res. 11:5381–5389.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Brisson L, Gillet L, Calaghan S, Besson P,
Le Guennec JY, Roger S and Gore J: Na(V)1.5 enhances breast cancer
cell invasiveness by increasing NHE1-dependent H(+) efflux in
caveolae. Oncogene. 30:2070–2076. 2011. View Article : Google Scholar
|
|
24
|
Gillet L, Roger S, Besson P, Lecaille F,
Gore J, Bougnoux P, Lalmanach G and Le Guennec JY: Voltage-gated
sodium channel Activity promotes cysteine cathepsin-dependent
invasiveness and colony growth of human cancer cells. J Biol Chem.
284:8680–8691. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Luqmani YA, Al Azmi A, Al Bader M, Abraham
G and El Zawahri M: Modification of gene expression induced by
siRNA targeting of estrogen receptor alpha in MCF7 human breast
cancer cells. Int J Oncol. 34:231–242. 2009.
|
|
26
|
Khajah MA, Al Saleh S, Mathew PM and
Luqmani YA: Differential effect of growth factors on invasion and
proliferation of endocrine-resistant breast cancer cells. PLoS One.
7:e418472012. View Article : Google Scholar
|
|
27
|
Al Saleh S, Al Mulla F and Luqmani YA:
Estrogen receptor silencing induces epithelial to mesenchymal
transition in human breast cancer cells. PLoS One. 6:e206102011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Brackenbury WJ, Calhoun JD, Chen C,
Miyazaki H, Nukina N, Oyama F, Ranscht B and Isom LL: Functional
reciprocity between Na+ channel Nav1.6 and beta1
subunits in the coordinated regulation of excitability and neurite
outgrowth. Proc Natl Acad Sci USA. 107:2283–2288. 2010. View Article : Google Scholar
|
|
29
|
Brackenbury WJ and Djamgoz MBA:
Activity-dependent regulation of voltage-gated Na+
channel expression in Mat-LyLu rat prostate cancer cell line. J
Physiol. 573:343–356. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
House CD, Vaske CJ, Schwartz AM, Obias V,
Frank B, Luu T, Sarvazyan N, Irby R, Strausberg RL, Hales TG, et
al: Voltage-gated Na+ channel SCN5A is a key regulator
of a gene transcriptional network that controls colon cancer
invasion. Cancer Res. 70:6957–6967. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
House CD, Wang BD, Ceniccola K, Williams
R, Simaan M, Olender J, Patel V, Baptista-Hon DT, Annunziata CM,
Gutkind JS, et al: Voltage-gated Na+ channel activity
increases colon cancer transcriptional activity and invasion via
persistent MAPK signaling. Sci Rep. 5:115412015. View Article : Google Scholar
|
|
32
|
Carrithers MD, Chatterjee G, Carrithers
LM, Offoha R, Iheagwara U, Rahner C, Graham M and Waxman SG:
Regulation of podosome formation in macrophages by a splice variant
of the sodium channel SCN8A. J Biol Chem. 284:8114–8126. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brisson L, Driffort V, Benoist L, Poet M,
Counillon L, Antelmi E, Rubino R, Besson P, Labbal F, Chevalier S,
et al: NaV1.5 Na(+) channels allosterically regulate the NHE-1
exchanger and promote the activity of breast cancer cell
invadopodia. J Cell Sci. 126:4835–4842. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Besson P, Driffort V, Bon É, Gradek F,
Chevalier S and Roger S: How do voltage-gated sodium channels
enhance migration and invasiveness in cancer cells? Biochim Biophys
Acta. 1848.2493–2501. 2015.
|
|
35
|
Brackenbury WJ: Voltage-gated sodium
channels and metastatic disease. Channels (Austin). 6:352–361.
2012. View Article : Google Scholar
|
|
36
|
Maness PF and Schachner M: Neural
recognition molecules of the immunoglobulin superfamily: Signaling
transducers of axon guidance and neuronal migration. Nat Neurosci.
10:19–26. 2007. View
Article : Google Scholar
|
|
37
|
Ding Y, Brackenbury WJ, Onganer PU,
Montano X, Porter LM, Bates LF and Djamgoz MB: Epidermal growth
factor upregulates motility of Mat-LyLu rat prostate cancer cells
partially via voltage-gated Na+ channel activity. J Cell
Physiol. 215:77–81. 2008. View Article : Google Scholar
|
|
38
|
Mantegazza M, Curia G, Biagini G, Ragsdale
DS and Avoli M: Voltage-gated sodium channels as therapeutic
targets in epilepsy and other neurological disorders. Lancet
Neurol. 9:413–424. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hille B: Ionic channels of excitable
membranes. Cell. 69:5791992. View Article : Google Scholar
|
|
40
|
Crill WE: Persistent sodium current in
mammalian central neurons. Annu Rev Physiol. 58:349–362. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ju YK, Saint DA and Gage PW: Hypoxia
increases persistent sodium current in rat ventricular myocytes. J
Physiol. 497:337–347. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kunzelmann K: Ion channels and cancer. J
Membr Biol. 205:159–173. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Turnbull DM, Rawlins MD, Weightman D and
Chadwick DW: ‘Therapeutic’ serum concentration of phenytoin: The
influence of seizure type. J Neurol Neurosurg Psychiatry.
47:231–234. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nelson M, Yang M, Dowle AA, Thomas JR and
Brackenbury WJ: The sodium channel-blocking antiepileptic drug
phenytoin inhibits breast tumour growth and metastasis. Mol Cancer.
14:132015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Brackenbury WJ, Chioni A-M, Diss JKJ and
Djamgoz MBA: The neonatal splice variant of Nav1.5 potentiates in
vitro invasive behaviour of MDA-MB-231 human breast cancer cells.
Breast Cancer Res Treat. 101:149–160. 2007. View Article : Google Scholar
|
|
46
|
Zucker S, Cao J and Chen WT: Critical
appraisal of the use of matrix metalloproteinase inhibitors in
cancer treatment. Oncogene. 19:6642–6650. 2000. View Article : Google Scholar
|
|
47
|
Gialeli C, Theocharis AD and Karamanos NK:
Roles of matrix metalloproteinases in cancer progression and their
pharmacological targeting. FEBS J. 278:16–27. 2011. View Article : Google Scholar
|
|
48
|
Sekton B: Matrix metalloproteinases - an
overview. Rev Bras Ter Intensiva. 23:222–227. 2011.
|
|
49
|
Gao R, Wang J, Shen Y, Lei M and Wang Z:
Functional expression of voltage-gated sodium channels Nav1.5 in
human breast cancer cell line MDA-MB-231. J Huazhong Univ Sci
Technolog Med Sci. 29:64–67. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Azuma T, Yamada M, Murakita H, Nishikawa
Y, Kohli Y, Yamamoto K and Hori H: Cathepsin E expressed in
pancreatic cancer. Adv Exp Med Biol. 362:363–366. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Azuma T, Hirai M, Ito S, Yamamoto K,
Taggart RT, Matsuba T, Yasukawa K, Uno K, Hayakumo T and Nakajima
M: Expression of cathepsin E in pancreas: A possible tumor marker
for pancreas, a preliminary report. Int J Cancer. 67:492–497. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kawakubo T, Yasukochi A, Toyama T,
Takahashi S, Okamoto K, Tsukuba T, Nakamura S, Ozaki Y, Nishigaki
K, Yamashita H, et al: Repression of cathepsin E expression
increases the risk of mammary carcinogenesis and links to poor
prognosis in breast cancer. Carcinogenesis. 35:714–726. 2014.
View Article : Google Scholar
|
|
53
|
Hagen NA, Fisher KM, Lapointe B, du Souich
P, Chary S, Moulin D, Sellers E and Ngoc AH; Canadian Tetrodotoxin
Study Group. An open-label, multi-dose efficacy and safety study of
intramuscular tetrodotoxin in patients with severe cancer-related
pain. J Pain Symptom Manage. 34:171–182. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Dib-Hajj SD, Black JA and Waxman SG:
Voltage-gated sodium channels: therapeutic targets for pain. Pain
Med. 10:1260–1269. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Clare JJ: Targeting voltage-gated sodium
channels for pain therapy. Expert Opin Investig Drugs. 19:45–62.
2010. View Article : Google Scholar
|
|
56
|
Busserolles J, Alloui A, Lazdunski M and
Eschalier A: Use of riluzole to treat or prevent the adverse
effects of antineoplastic agents. US Patent 2013/0064775 A1. Filed:
March 2, 2011; issued March 14, 2013.
|
|
57
|
Yip D, Le MN, Chan JLK, Lee JH, Mehnert
JA, Yudd A, Kempf J, Shih WJ, Chen S and Goydos JS: A phase 0 trial
of riluzole in patients with resectable stage III and IV melanoma.
Clin Cancer Res. 15:3896–3902. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Biki B, Mascha E, Moriarty DC, Fitzpatrick
JM, Sessler DI and Buggy DJ: Anesthetic technique for radical
prostatectomy surgery affects cancer recurrence: A retrospective
analysis. Anesthesiology. 109:180–187. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mao L, Lin S and Lin J: The effects of
anesthetics on tumor progression. Int J Physiol Pathophysiol
Pharmacol. 5:1–10. 2013.PubMed/NCBI
|