Introduction
As a malignant disease of bone marrow and blood,
leukemia is the most common form of cancer in children. Pediatric
acute myeloid leukemia (AML) represents 15–20% of all pediatric
acute leukemias. The clinical outcomes of pediatric AML have
improved significantly over the past few decades so that the
long-term survival rate stands at ~70%. Improvement is due to
intensification of chemotherapeutic regimens, optimal risk-group
stratification, timely salvage for relapse and enhanced supportive
cares (1–3). Despite intensive treatment, ~30% of
pediatric patients relapsed with poor outcomes. In addition, 30–40%
of patients survived in the largest and most recent series
(4,5). Chemoresistance of leukemia cells
constitutes a great challenge for successful therapeutics.
Autophagy has been shown to played important roles in conferring
resistance to chemotherapy, radiation therapy and immunotherapy in
cancer cells (6,7). However, exact molecular mechanisms by
which autophagy induces drug resistance in cancer cells,
specifically leukemia cells, have been poorly defined.
The Wiskott-Aldrich syndrome (WAS) protein (WASP)
and WASP family verprolin-homologous protein (WAVE) family is
composed of 5 members, i.e. WASP, N-WASP, WAVE1, WAVE2 and WAVE3.
They serve as key links between GTPases and actin cytoskeleton
(8). Essential for cell
morphologic changes, motility and apoptosis, cytoskeleton
reorganization is also an important regulator of autophagy
(9–11). Actin microfilaments and
microtubules are vital for initial formation of autophagosomes in
starved cells (11). Moreover,
cytoskeleton regulatory proteins such as dynein, myosin and kinesin
play important regulatory roles during autophagy (12–16).
Similar to other intracellular trafficking events, autophagosomal
movement employs microtubule-dependent machinery in mammalian cells
(17,18). Moreover, these observations have
provided evidence for the relevance of cytoskeleton during
autophagy.
WAVE1 (also known as Scar1), a suppressor of cyclic
AMP receptor 1, was first identified as a regulator of actin
cytoskeleton through interactions with Arp2/3 downstream of Rac
(19,20). WAVE1 is expressed most abundantly
in murine brain tissue and leukemia cells and at extremely low
levels in other tissues including heart, liver, lung, kidney,
pancreas and peripheral blood (10,21).
In addition to its roles in the nervous system development and
mammalian fertilization, WAVE1 also functions as an important
molecule for regulating tumor development, tumor invasion and
metastasis (22–24). Our previous studies have shown that
WAVE1 was involved in multidrug resistance and oxidative stress of
human leukemia cells and functioned as a negative regulator of
apoptosis (10,25,26).
As autophagy and apoptosis are currently regarded as different
aspects of the same cell death continuum, their regulations become
intimately connected. In addition, the same regulators could
control both apoptosis and autophagy (27). In light of the fact that
cytoskeleton reorganization is also an important regulator of
autophagy, the role of WAVE1 in leukemia cell apoptosis is expected
to shed some light on its role in autophagy.
It was shown in the present study that WAVE1 was
overexpressed in both human hematological cancer cell lines and
primary BMMCs from patients with pediatric leukemia. WAVE1
expression was positively correlated with clinical status in
pediatric leukemia. Suppression of WAVE1 expression blocked
drug-induced autophagic reactions and increased the sensitivity of
leukemia cells to anti-neoplastic agents. Furthermore, our data
suggested a regulatory role for WAVE1 during autophagy through a
formation of Beclin1-PI3K-III complex and a disassociation of
Bcl-2-Beclin1 complex. Thus, WAVE1 is a potential drug target of
therapeutic interventions for leukemia.
Materials and methods
Reagents and antibodies
The antibodies to WAVE1 and p62 were purchased from
Santa Cruz Biotechnology (Santa Cruz, CA, USA); antibodies to LC3,
actin and tubulin from Sigma Inc. (St. Louis, MO, USA); antibody to
mitochondria HSP70 (mHSP70) from Abcam (Cambridge, MA, USA);
antibodies to class III phosphoinositide 3-kinase (PI3K-III),
p-4EBP1, Beclin1 and Bcl-2 from Cell Signaling Technology (Danvers,
MA, USA); 3-methyladenine (3-MA), vincristine (VCR), cytosine
arabinoside (Ara-C), adriamycin (ADM), E64D and pepstatin from
Sigma.
Cell culture
HL-60 acute promyelocytic leukemia cells, K562
chronic myelogenous leukemia cells, THP-1 acute monocytic leukemia
cells, adriamycin-resistant HL-60/ADR cells, A549 human lung cancer
cells, human umbilical vein endothelial cells, human lung A549
cancer cells and CNE2 nasopharyngeal carcinoma cells (Xiangya
School of Medicine Type Culture Collection, Xiangya, China) were
cultured in RPMI-1640 or DMEM medium with 10% heat-inactivated
fetal bovine serum (FBS) in 5% CO2 and 95% ambient
air.
Gene transfection and RNAi
WAVE1 small hairpin RNA (shRNA) lentiviral knockdown
(GeneCopoeia, Guangzhou, China) or shRNA non-target control (NTC)
were packaged with HIV-based packaging mix (GeneCopoeia) for
infecting HL-60 cells to establish cells constitutively repressing
WAVE1. Stable clones were selected by puromycin. PI3K-III shRNA
from Sigma were constructed with FuGENE HD transfection reagent
(Roche Applied Science, Stockholm, Sweden) according to the
manufacturer's instructions.
Cell viability assay
After drug dosing, cell viability was evaluated by
Cell Counting kit-8 (CCK-8) (Dojindo Molecular Technologies, Tokyo,
Japan) according to the manufacturer's instructions.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was isolated by TRIzol reagent
(Invitrogen, Carlsbad, CA, USA) according to the manufacturer's
instructions. RNA concentration and purity were measured with a
spectrophotometer at A260 and A260/280, respectively. RNA was
reverse-transcribed into cDNA using a Primescript™ RT reagent kit
(Invitrogen) according to the manufacturer's instructions. The
sequences of primers used were as follows: for β-actin: forward,
5′-TCCTTCCTGGGCATGGAGTC-3′ and reverse,
5′-GTAACGCAACTAAGTCATAGTC-3′. For WAVE1: forward,
5′-TCTGGGCTACATCCAACTCC-3′ and reverse, 5′-CCTGTTCACGCTGCTCTTCT-3′.
β-actin was used as an internal control for evaluating the relative
expressions of WAVE1. The conditions of polymerase chain reaction
(PCR) for WAVE1 were as follows: denaturation at 94°C for 2 min,
followed by 30 cycles of 94°C for 30 sec, 56°C for 30 sec (β-actin,
50°C for 30 sec), 72°C for 30 sec and ultimately by a 5-min
elongation at 72°C. The PCR products were analyzed with 1.0%
agarose gel electrophoresis, ethidium bromide-stained, photographed
and scanned by Band Leader software for grey scale
semi-quantitative analysis.
Western blot analysis
After rinsing with phosphate-buffer solution (PBS),
the cells were collected, resuspended in lysis buffer (Beyotime
Institute of Biotechnology, Beijing, China) and maintained on ice
for 15 min. Cell extracts were cleared by microcentrifugation at
14,000 × g for 30 min at 4°C. Whole cell lysate was separated by 8%
(10%/12%) sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) and electrophoretically transferred onto polyvinylidene
difluoride (PVDF) blotting membrane (Beyotime Institute of
Biotechnology). After blocking with 5% non-fat dry milk in TBST (50
mM Tris pH 7.5, 100 mM NaCl, 0.15% Tween-20), the membranes were
incubated with diluted primary antibodies for 12 h at 4°C and
washed thrice with TBST for 10 min. After incubating for 12 h at
4°C with various secondary antibodies, detection was made with
enhanced chemiluminescence (ECL) reagents (Pierce, Rockford, IL,
USA) after rinsing thrice with TBST for 10 min. The membranes were
exposed to X-ray film and the expressions of targeted proteins
quantified by detecting specific bands. In addition, BandScan 5.0
system was used for quantifying and analyzing each specific
blotting band (16).
Immunoprecipitation
Cells were lysed at 4°C in ice-cold lysis buffer (50
mM Tris-HCl, pH 7.4, containing 150 mM NaCl, 1% NP-40, 0.5%
nadeoxycholate, 0.1% SDS, protease inhibitor cocktail) and cell
lysates centrifugated at 12 000 × g for 10 min. The concentrations
of proteins in supernatant were determined by bicinchoninic acid
(BCA) assay. Prior to immunoprecipitation, the samples containing
equal amounts of proteins were pre-cleared with protein A or
protein G agarose/sepharose (Santa Cruz Biotechnology, Santa Cruz,
CA, USA) (4°C, 3 h) and subsequently incubated with various
irrelevant IgG or specific antibodies (5 mg/ml) in the presence of
protein A or G agarose/sepharose beads for 2 h or overnight at 4°C
with gentle vortexing. After incubation, agarose/sepharose beads
were rinsed thoroughly with PBS, and the proteins were eluted by
boiling in 2X SDS sample buffer prior to SDS-PAGE.
Apoptosis assays
Cellular apoptosis was assessed by FITC Annexin V
apoptosis detection kit [Annexin V-FITC, propidium iodide (PI)
solution and Annexin V binding buffer]. This assay involved
staining cells with Annexin V-FITC (a phospholipid-binding protein
binding to disrupted cell membranes) in combination with PI (a
vital dye binding to DNA penetrating into apoptotic cells). Flow
cytometry (FACS) was performed for determining the percentage of
apoptotic cells (Annexin V+/PI).
Electron microscopy
After collection, NB4 cells were fixed in 2.5%
glutaraldelhyde for at least 3 h and subsequently treated with 2%
paraformaldehyde at room temperature for 60 min, 0.1%
glutaraldehyde in 0.1 M sodium cacodylate for 2 h and post-fixation
with 1% osmium tetroxide (OsO4) for 1.5 h. After the
second rinsing, the samples were dehydrated with graded acetone and
ultimately embedded in Quetol 812. Ultrathin sections were observed
under Hitachi H7500 electron microscope (Hitachi, Tokyo,
Japan).
Caspase activity assay
Caspase-3 activity was assayed by caspase-3
colorimetric assay kit (Calbiochem, Berlin, Germany) according to
the manufacturer's instructions.
Statistical analysis
The quantitative data were presented as means ±
standard deviation. In addition, analysis was performed with
specified statistical methods using GraphPad Prism (version 5.04).
For calculating P-value, two-tailed parameters with a confidence
interval of 95% were used. A P-value <0.05 was considered to
indicate a statistically significant result.
Results
Correlation between WAVE1 expression and
clinical status in pediatric AML
A total of 30 patients aged 1–13 years with newly
diagnosed AML at our department were enrolled from January 2008 to
December 2014. There were 17 males and 13 females (Table I). The median age was 6 years
(range, 1–13 years). According to the French-American-British (FAB)
classification scheme, the types were M0 (n=1), M1 (n=1), M2
(n=16), M4 (n=7), M5 (n=4) and M7 (n=1). At the time of diagnosis,
the median count of white blood cells (WBC) was
14.9×109/l (range, 0.8–102.7×109/l), median
hemoglobin level 66.5 g/l (range, 31–241 g/l) and median platelet
count 31.2×109/l (range, 2–104.6×109/l). The
median relative level of WAVE1 mRNA expression was 0.4635 (range,
0.0–0.781), and 22 (73.3%) achieved a complete remission (CR) with
1 or 2 induction chemotherapies. The survival outcomes revealed
that 16/30 patients (48.4%) achieved CR after 6-month chemotherapy,
and 10 of them (33.3%) died. Relapsing and refractory leukemias
were the commonest cause of mortality (n=8), followed by infection
(n=1) and hemorrhage (n=1). All of them received anthracycline and
cytarabine-based chemotherapeutic regimens, and 4 patients received
hematopoietic stem cell transplantation (HSCT) after induction
therapy.
 | Table IClinical characteristics of 30
pediatric AML patients. |
Table I
Clinical characteristics of 30
pediatric AML patients.
| Subtype | No. | Age (years) | Gender | Initial leukocyte
count (×109/l) | BM evaluation after
1/2 induction chemotherapies | Relative WAVE1 mRNA
expression | Survival
outcome |
|---|
| M2 | 1 | 6 | Male | 6.5 | CR | 0.335 | CR |
| M2 | 2 | 8 | Female | 63.2 | CR | 0.589 | HSCT |
| M4 | 3 | 2 | Male | 7.8 | IR (BM blasts,
22.8%) | 0.321 | Deceased
(hemorrhage) |
| M2 | 4 | 5 | Female | 24.7 | CR | 0.582 | Deceased
(infection) |
| M1 | 5 | 1 | Male | 3.6 | CR | 0.378 | CR |
| M2 | 6 | 3 | Male | 1.4 | CR | 0 | CR |
| M2 | 7 | 11 | Male | 18.5 | IR (BM blasts,
10.4%) | 0.434 | CR |
| M5 | 8 | 6 | Male | 26.3 | IR (BM blasts,
8.3%) | 0.391 | CR |
| M2 | 9 | 10 | Female | 2.1 | CR | 0.361 | CR |
| M4 | 10 | 13 | Female | 46.7 | CR | 0.528 | Deceased
(relapsing) |
| M4 | 11 | 8 | Male | 34.6 | CR | 0.551 | HSCT |
| M4 | 12 | 3 | Male | 10.2 | CR | 0.580 | HSCT |
| M5 | 13 | 5 | Female | 19.8 | CR | 0.628 | Deceased
(refractory) |
| M2 | 14 | 7 | Female | 102.7 | IR (BM blasts,
11%) | 0.684 | Deceased
(relapsing) |
| M2 | 15 | 6 | Female | 73.1 | CR | 0.617 | Deceased
(refractory) |
| M2 | 16 | 13 | Male | 90.5 | IR (BM blasts,
37%) | 0.699 | Deceased
(refractory) |
| M2 | 17 | 9 | Male | 4.7 | CR | 0 | CR |
| M5 | 18 | 3 | Male | 88.4 | CR | 0.781 | Deceased
(relapsing) |
| M5 | 19 | 5 | Male | 41.3 | CR | 0.508 | CR |
| M4 | 20 | 2 | Female | 9.7 | CR | 0.702 | Deceased
(refractory) |
| M2 | 21 | 7 | Male | 0.8 | CR | 0.257 | CR |
| M2 | 22 | 4 | Male | 47.6 | IR (BM blasts,
15%) | 0.463 | CR |
| M0 | 23 | 7 | Female | 55.8 | CR | 0.523 | HSCT |
| M2 | 24 | 11 | Female | 3.7 | CR | 0.295 | CR |
| M2 | 25 | 5 | Female | 2.5 | CR | 0.357 | CR |
| M4 | 26 | 2 | Female | 11.3 | CR | 0.346 | CR |
| M7 | 27 | 12 | Male | 57.6 | IR (BM blasts,
75%) | 0.663 | Deceased
(relapsing) |
| M2 | 28 | 10 | Male | 2.5 | CR | 0.362 | CR |
| M2 | 29 | 4 | Female | 1.0 | IR (BM blasts,
26%) | 0 | CR |
| M4 | 30 | 8 | Male | 3.9 | CR | 0.284 | CR |
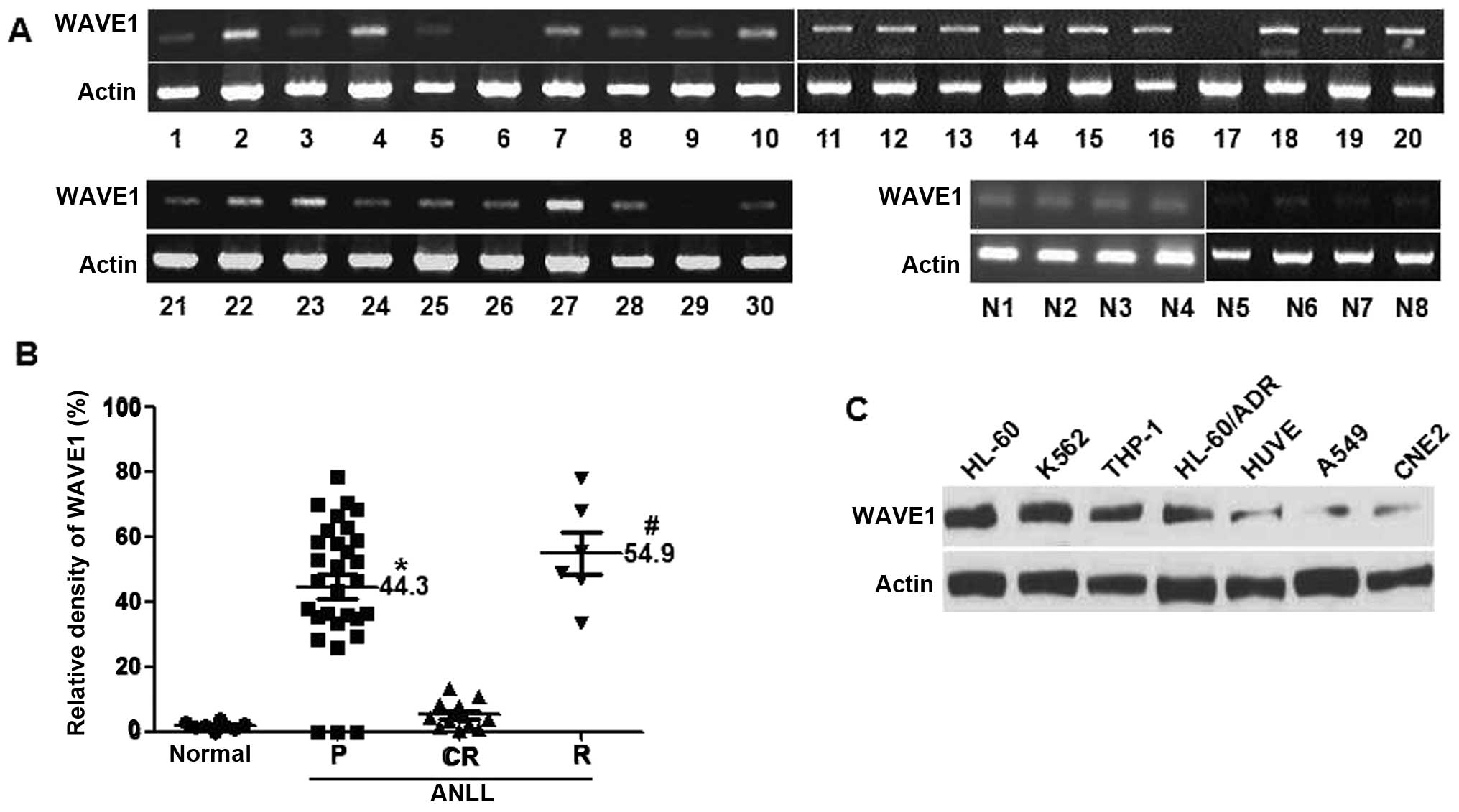
WAVE1 expression patterns were detected by RT-PCR in
30 newly-diagnosed AML patients and 8 normal healthy subjects
(Fig. 1A). The relative WAVE1 mRNA
levels were summarized in Table I.
There was a general trend for a higher incidence of relapsing or
refractory leukemia in WAVE1 high-expression patients. Also a poor
prognosis was found in patients with leucocytosis at diagnosis
(Table II). Moreover, the
relative WAVE1 mRNA expression levels were examined for 30
pediatric AML patients at varying clinical status. Higher levels of
WAVE1 expression were found in bone marrow mononuclear cells
(BMMCs) from patients with primary (n=30) and relapsing (n=6)
leukemias (Fig. 1B). By contrast,
WAVE1 was not detectable in BMMCs from CR patients (n=12) or normal
healthy subjects (n=8) (Fig. 1B).
Thus WAVE1 was well-correlated with the clinical status of
pediatric AML.
| Table IIResults of various patient variables
during BM evaluation. |
Table II
Results of various patient variables
during BM evaluation.
| CR group
(n=16) |
Relapsing/refractory group (n=8) | P-valuea |
|---|
| WAVE1 | 0.298
(1–0.508)b | 0.662
(0.530–0.780) | 0.00 |
| WBCs
(×109/l) at diagnosis | 11.1
(0.8–47.6) | 61.1
(9.7–102.7) | 0.00 |
| Hb (g/l) at
diagnosis | 70.1 (31–105) | 84 (58–169) | 0.276 |
| Platelet
(×109/l) at diagnosis | 48.5 (2–149) | 49.3 (5–106) | 0.793 |
| Age at diagnosis
(years) | 6.3 (1–11) | 7.6 (2–13) | 0.438 |
Furthermore, the expression levels of WAVE1 were
determined by western blot analysis in 4 leukemia cell lines of
HL-60, K562, HL-60/ADR and THP-1, their levels were all upregulated
(Fig. 1C). In contrast, the
constitutive expression levels of WAVE1 were noticeably lower in
non-hematological cancer cell lines, including human lung A549
cancer cells, human umbilical vein endothelial cells and CNE2
nasopharyngeal carcinoma cells. Thus, a different role of WAVE1 is
implicated for leukemic tumorigenesis.
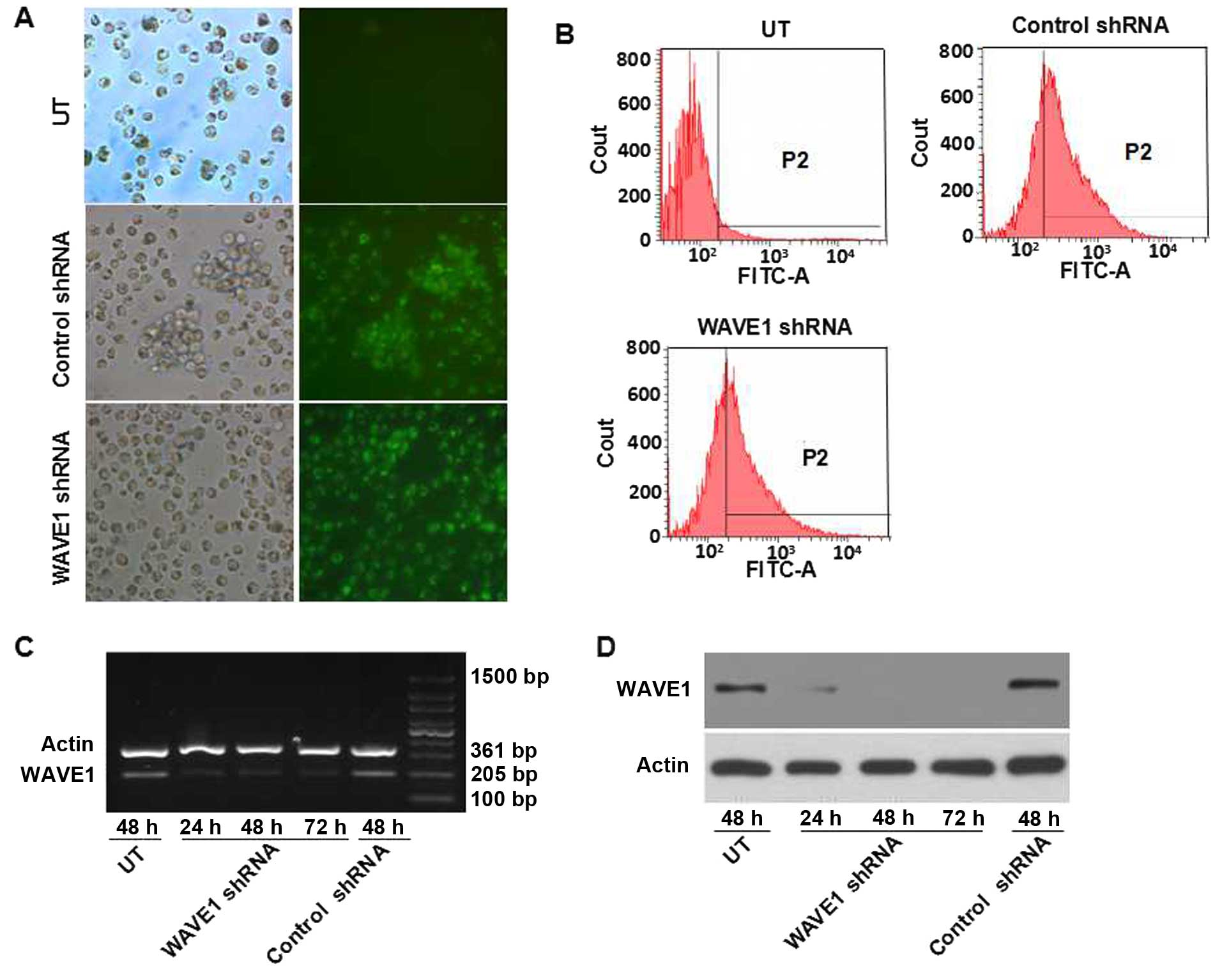
Targeting shRNA-mediated silencing of
WAVE1 expression
A specific shRNA against WAVE1 was transferred into
HL-60 for knocking down the expression of WAVE1. The cellular
transfection efficiency was determined by visualizing eGFP-positive
cells under microscope and confirmed by flow cytometry (Fig. 2A and B). Moreover, RT-PCR and
western blot analysis were performed for detecting the expression
of WAVE1. Transfection of WAVE1 shRNA resulted in a marked
downregulation of WAVE1. In contrast, transfection of blank vector
did not interfere with WAVE1 expression at the level of either mRNA
or protein (Fig. 2C and D).
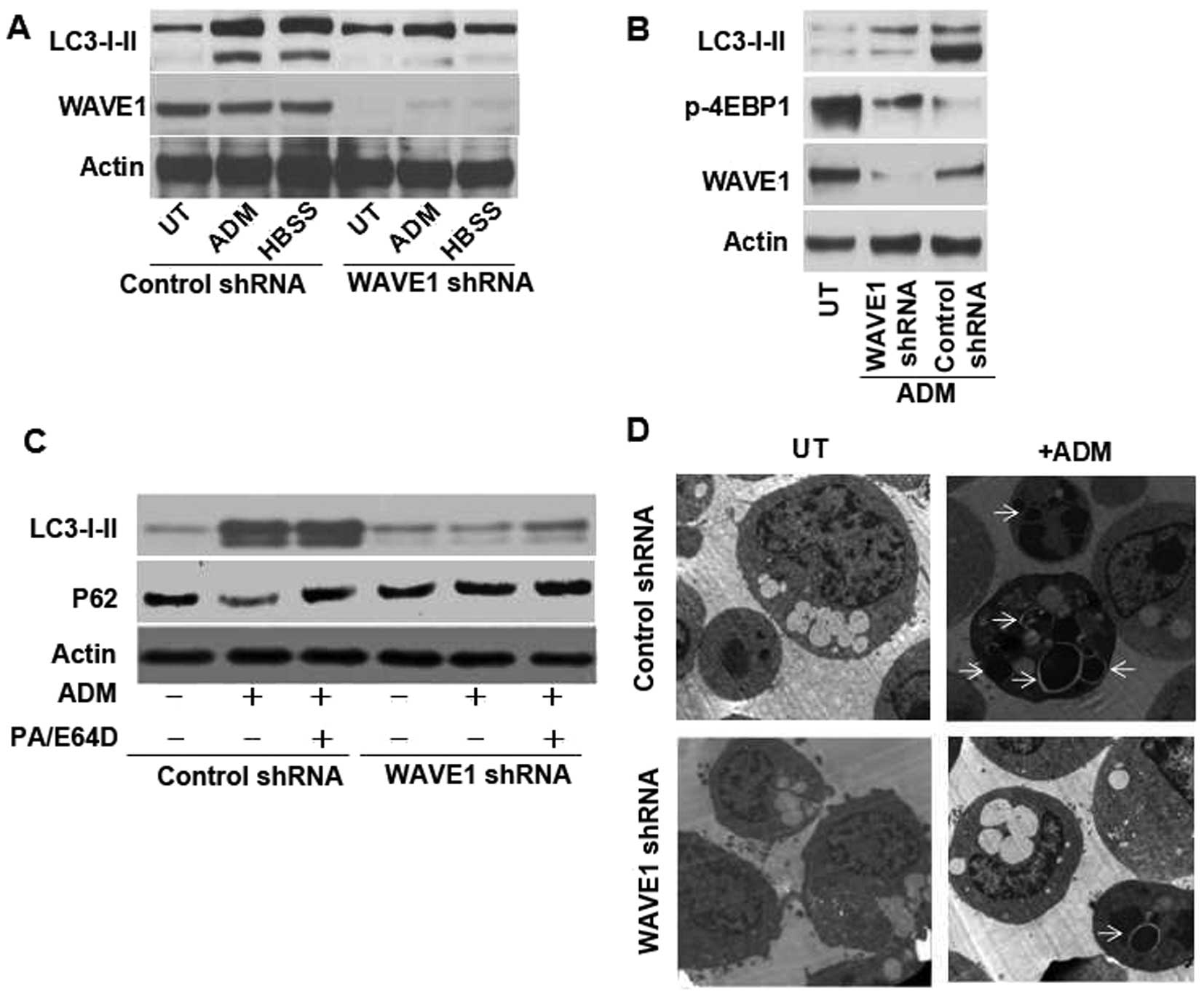
Knockdown of WAVE1 expression inhibits
the initiation of autophagy
As a dynamic process for degrading such cytosolic
components as dysfunctional organelles and proteins, autophagy
offered a pathway of generating metabolic substrates (28,29).
A host of existing cancer therapeutics, including DNA-damaging
chemotherapy, radiation therapy and molecular targeted therapies,
could induce autophagy in cell culture and animal models (6,30).
To investigate whether WAVE1 is a direct activator of autophagy,
immunoblot was employed for determining microtubule-associated
protein light chain 3 (LC3) and its conversion products (LC3-I to
LC3-II) (31). With a depletion of
WAVE1 expression, the classical autophagic stimuli, such as
starvation (Hank's balanced salt solution, HBSS), decreased the
expression of LC3-II versus control groups (Fig. 3A). Similar to HBSS, ADM at a
therapeutic dose of 1 μg/ml also decreased LC3-II expression
(Fig. 3A), suggesting targeted
deletion of WAVE1 inhibited ADM-induced autophagy.
The inhibition of mammalian target of rapamycin
(mTOR) signaling pathway has been considered as an important step
in the initiation of autophagy (6,29,32),
and mTOR activity was further determined through monitoring 4EBP1
(a mTOR substrate) phosphorylation (33). The levels of phosphor-4EBP1
(p-4EBP1) decreased in transfection vector control while a
depletion of WAVE1 expression partially rescued p-4EBP1 expression
compared with control group (Fig.
3B). An important method for detecting autophagic flux is
measuring enhanced degradation of p62 (sequestosome-1), a
long-lived scaffolding protein involved in the transport of
ubiquitinated protein for proteasomal digestion (34). p62 has LC3 binding domains
targeting this protein for incorporation into autophagosome. Thus,
it served as a selective substrate of autophagy. Knockdown of WAVE1
decreased the level of LC3-II, yet, the level of p62 increased
compared with control group. It suggested that p62 degradation was
dependent on WAVE1-induced autophagy (Fig. 3C). Moreover, LC3 accumulation and
p62 expression were exaggerated after treatment with lysosomal
protease inhibitor E64d and pepstatin A vs. WAVE1 shRNA group
(Fig. 3C). Thus, the resulting
elevation of LC3-II was not due to a decreased degradation of
lipidated LC3, but rather to a higher autophagic flux.
The most reliable and conventional technique for
visualizing autophagic vacuolization is transmission electron
microscopy capable of revealing the presence of multiple
autophagosome-like vacuoles with double-membrane structures
(31). Ultrastructural analysis
revealed that HL-60 cells exhibited fewer autophagosomes after
WAVE1 RNAi treatment vs. control group (Fig. 3D). Altogether, these data
demonstrate that WAVE1 is required for initiating autophagy in
leukemia cells.
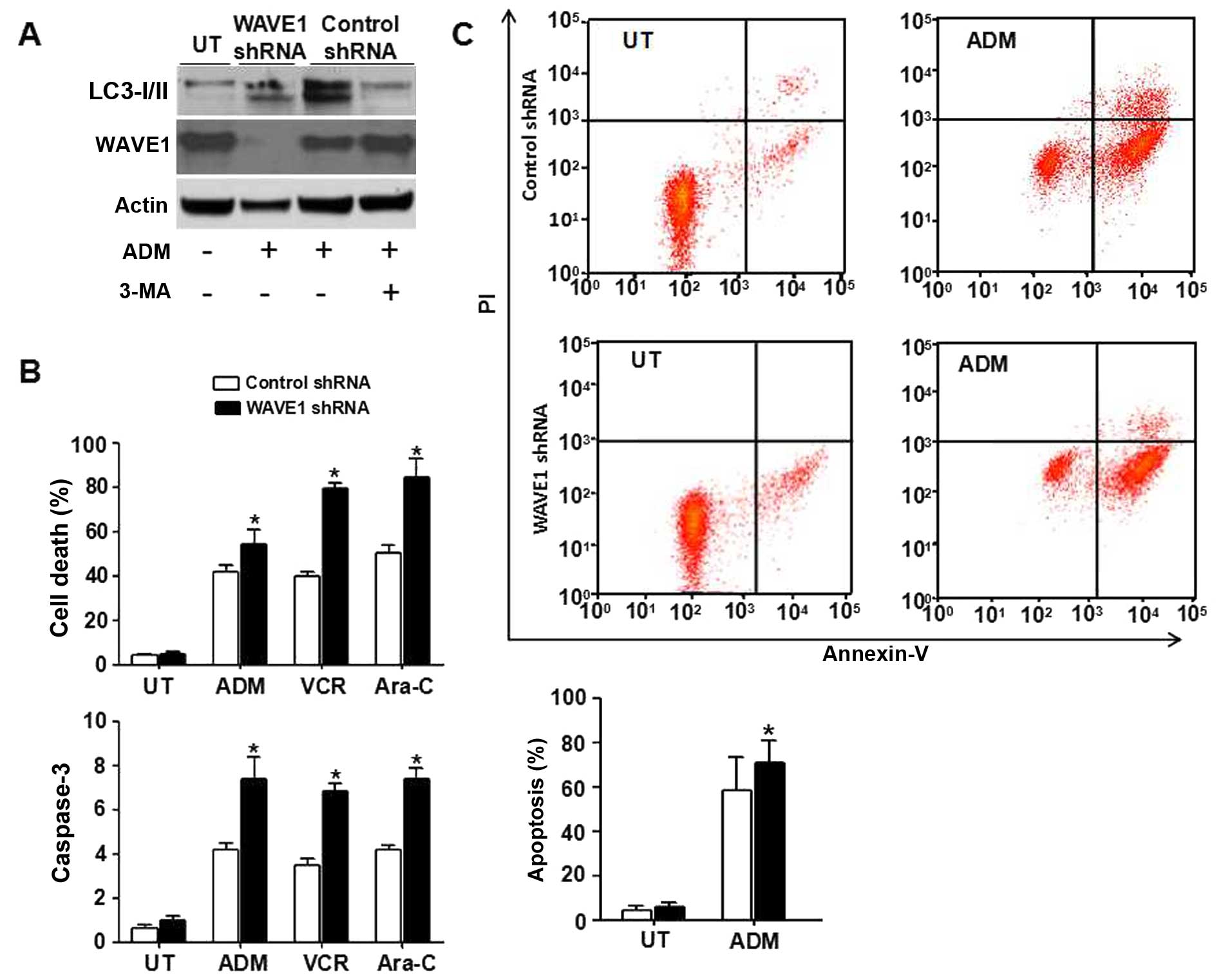
Knockdown of WAVE1 expression increases
chemotherapy sensitivity
Chemoresistance has become a major obstacle for
successful therapeutics of leukemia. A growing body of evidence has
suggested that autophagy is an important resistance mechanism for
chemotherapy in hematological malignancies (35–37).
To characterize the role of WAVE1-mediated autophagy in
chemosensitivity of leukemia cells, HL-60 cells were treated with
several common chemotherapeutic drugs such as ADM (1 μg/ml), VCR (1
μg/ml) and Ara-C (0.2 μM) (32).
After transfection with WAVE1 shRNA or control shRNA, autophagy was
suppressed by 3-MA, a PI3K inhibitor. The expression of LC3-II
significantly decreased in control group. Similar to using 3-MA, a
depletion of WAVE1 expression also decreased LC3-II expression,
suggesting that WAVE1 was required for ADM-induced autophagy
(Fig. 4A). Moreover, a knockdown
of WAVE1 expression in HL-60 cells rendered it significantly more
sensitive to ADM, VCR and Ara-C-induced apoptosis associated with
high levels of caspase-3 activities (Fig. 4B). Furthermore, a depletion of
WAVE1 expression increased early apoptosis with ADM treatment vs.
control group (Fig. 4C),
supporting a potential prosurvival role for WAVE1-induced autophagy
in leukemia cells under chemotherapy.
WAVE1 regulates autophagy in
Beclin1/Bcl-2 and Beclin1/PI3K-III-dependent pathway
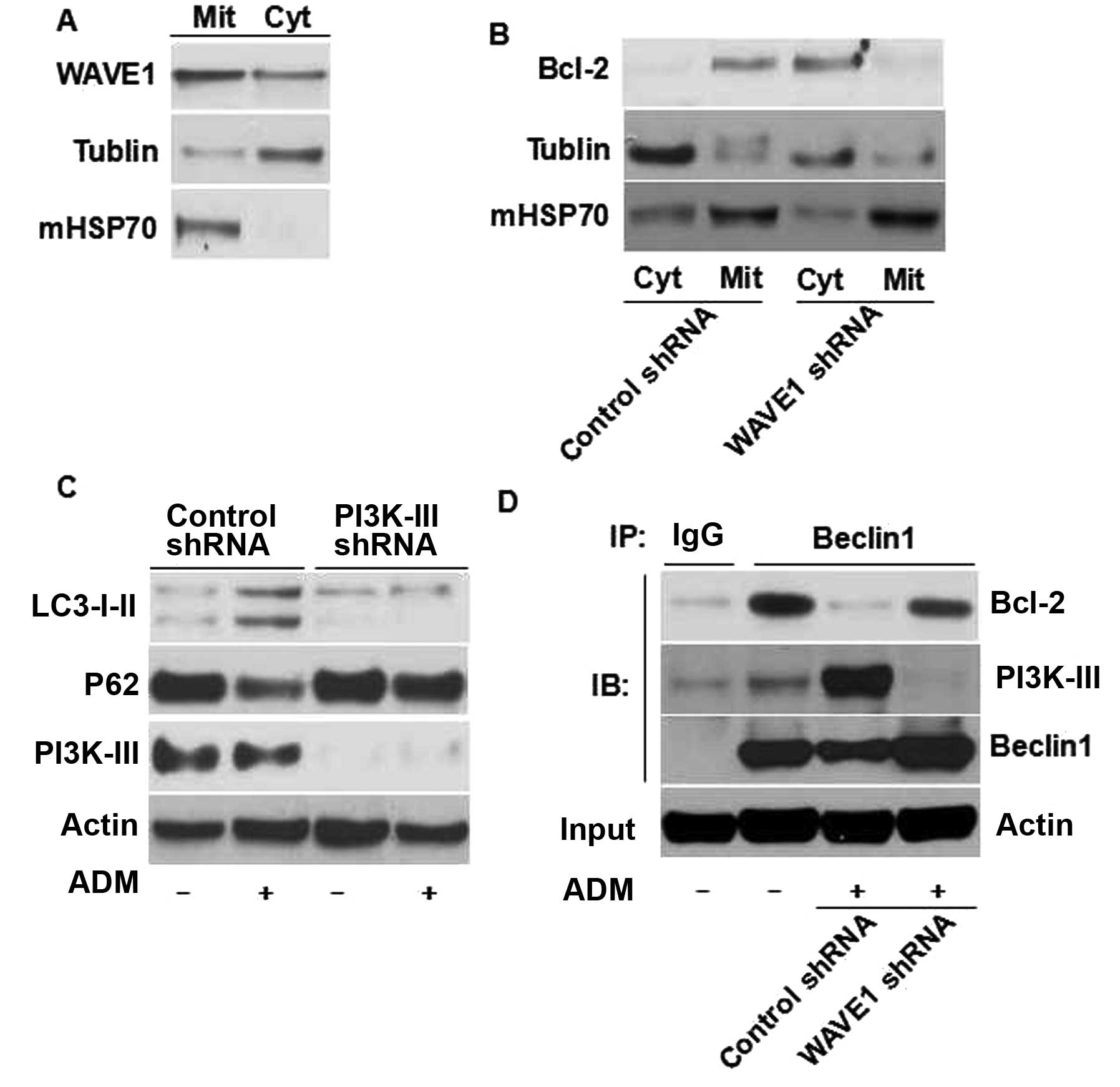
WAVE1 is expressed abundantly in mitochondria of
neuronal cells (37). Similar to
neuronal cells, our previous study showed that WAVE1 was localized
to mitochondria and cytoplasm in leukemia cells (10). In the present study the expression
of WAVE1 was confirmed by western blot analysis in both
mitochondria and cytoplasm (Fig.
5A). Its mitochondrial localization provided a basis for its
molecular interaction with different mitochondrial proteins. Bcl-2,
an integral membrane protein on endoplasmic reticulum (ER) and
mitochondria, is an important inhibitor of apoptosis. Yet, its
mechanism of localization is not fully understood (38). WAVE1 knockdown decreased the
mitochondrial levels of (Mit)-Bcl-2, but increased the cytoplasmic
levels of Bcl-2 (Fig. 5B). Thus, a
specific role for WAVE1 was evident in the regulation of Bcl-2
intracellular localization in leukemia cells.
An important molecular event in autophagic vesicle
nucleation is the disassociation of Bcl-2-Beclin1 complex and the
formation of Beclin1-PI3K-III complex (31,40).
The disassociation of Bcl-2-Beclin1 complex sustained autophagy
(39). In the present study
knockdown of WAVE1 blocked the disassociation of Beclin1-Bcl-2
during enhanced autophagy (Fig.
5D). As PI3K-III is required for autophagy initiation (40), the role of PI3K-III was examined in
the regulation of WAVE1-mediated autophagy using a target-specific
shRNA against PI3K-III expression. Transfection of PI3K-III-shRNA
led to a significant decrease in PI3K-III protein and significantly
inhibited WAVE1-mediated autophagy (Fig. 5C), suggesting that PI3K-III is
required for WAVE1-mediated autophagy. Depletion of WAVE1
expression blocked the interaction between Beclin1 and PI3K-III
(Fig. 5D), suggesting that WAVE1
promotes vesicle nucleation. The overall data indicate that
WAVE1-mediated autophagy may occur through Beclin1/Bcl-2 and
Beclin1/PI3K-III-dependent pathway in HL-60 cells.
Discussion
A novel function of WAVE1 is to modulate
chemotherapy sensitivity by inducing autophagy in leukemia cells.
WAVE1 expression was positively correlated with clinical status in
pediatric AML. WAVE1 expression was significantly higher in active
phase (such as in primary and relapsing phase) and returned to
normal in CR. It was also expressed abundantly in various leukemia
cell lines, yet scantly in non-hematological cell lines. Thus, a
potentially contributory role of WAVE1 was implicated for the
pathogenesis of AML.
Currently chemoresistance is a major obstacle to
successful therapeutics of leukemia. Multiple mechanisms of drug
resistance are well-recognized, such as drug export transporters
(e.g. permeability-glycoprotein), more effective DNA repair,
altered pharmacokinetics, resistant hematopoietic stem cells and
resistance to apoptosis (30,32,41).
Although these mechanisms of drug resistance have been proposed,
exact mechanisms remain to be established. Detailed mechanistic
understandings of leukemia might help to predict and overcome drug
resistance through enhancing chemotherapy and ultimately improving
the patient outcomes. WAVE1 has been found to be involved in
multidrug resistance and oxidative stress of human leukemia cells,
however, its pathogenic role in leukemia was poorly elucidated
(26,42). WAVE1 enhanced leukemia cell
chemoresistance potentially through the regulation of
P-glycoprotein expression (25).
Moreover, WAVE1 was associated with mitochondrial Bcl-2, and its
depletion caused a mitochondrial release of Bcl-2 and
phosphorylation of ASK1/JNK and Bcl-2 in leukemia cells (10). In the present study, it was
demonstrated that a knockdown of WAVE1 expression could sensitize
leukemia cells to chemotherapeutic drugs and apoptosis. While WAVE1
is essential for leukemic apoptosis, its role in the regulation of
leukemia autophagy for drug resistance has been poorly defined. In
general, autophagy is a ‘programmed cell survival’ mechanism
because cells employ autophagy for preventing an accumulation of
damaged or unnecessary components, but also functions to facilitate
the recycling of these components for sustaining homeostasis.
Autophagy and apoptosis are currently regarded as different aspects
of the same cell death continuum, their regulations are intimately
connected, and the same regulators could sometimes control both
apoptosis and autophagy (43). The
role of WAVE1 in cell apoptosis is expected to shed some light also
on its role in autophagy.
Numerous studies have demonstrated that autophagy
combats various types of adverse stresses and maintains the
survival of tumor cells. As a programmed cell survival mechanism
responding to cytotoxic chemotherapy or irradiation, it is
responsible for drug resistance (7). For example, a loss of
autophagy-related genes resulted in the sensitization of resistant
carcinoma cells to radiation (44). Furthermore, combined application of
chloroquine (an autophagy inhibitor) and imatinib significantly
increased the death rate of chronic myeloid leukemia cells, even in
imatinib-resistant cases (35).
Even though autophagy has been functionally connected with the
chemoresistance of tumor cells, little is known about its impact on
chemoresistance and its connection with WAVE1 in leukemia cells.
The present study demonstrated that WAVE1 is a direct regulator of
autophagy in leukemia cells thereby controlling LC3 conversion
(LC3-I to LC3-II), a degradation of p62 and a maturation of
autophagosomes. Moreover, a depletion of WAVE1 expression inhibited
autophagy, promoted sensitivity of leukemia cells to
chemotherapeutic drugs and enhanced apoptosis. There was a dominant
role of WAVE1 in regulating autophagy, and chemoresistance of
leukemia cells has been highly correlated with autophagy.
The process of mammalian autophagy is divided into
several steps of initiation, nucleation, elongation, closure,
maturation and degradation or extrusion (45). As a key regulator of autophagy,
Beclin1 is a core component of class III PI3K/Vps34 complex
required for autophagosomal formation and maturation (46), and it interacts with autophagy
regulators, organelle membrane anchor proteins, Bcl-2 and Bcl-xL.
The disassociation of Bcl-2-Beclin1 complex plays an important role
in autophagic vesicle nucleation (39,46).
As an integral membrane protein on ER and within mitochondria,
Bcl-2 is an important inhibitor of apoptosis. WAVE1 might function
as an anchoring protein for mitochondrial Bcl-2 so as to affect the
mitochondrial release and phosphorylation of Bcl-2 (10). WAVE1 was localized to mitochondria
of leukemia cells and a knockdown of WAVE1 expression induced the
translocation of Bcl-2 from mitochondria into the cytoplasm.
Moreover, WAVE1 potentially confers its pro-autophagic activities
by controlling Beclin1-Bcl-2 complex formation through disrupting
the interactions between Beclin1 and Bcl-2. Thus, the
disassociation of Beclin1-Bcl-2 complex is an important mechanism
of WAVE1-regulated autophagy.
PI3K (PI3-kinase/PI3K) family is divided into three
classes of I, II and III. PI3K-III activity was essential for
autophagy whereas PI3K-I activity had some inhibitory effects on
autophagy (31). PI3K-III, a
mammalian homolog of Vps34, was first identified in yeast. It has
been implicated in such a wide range of cellular phenomena as
autophagy, phagocytosis and post-endocytic receptor sorting
(40,47,48).
In yeast models, Vps34/class III PI3K formed macromolecular
complexes with autophagic protein Apg6/Beclin1 (46). It was found that genetic inhibition
of PI3K-III by specific shRNA suppressed ADM-induced autophagy,
suggesting that PI3K-III activity is required for ADM-induced
autophagy. Furthermore, a depletion of WAVE1 expression suppressed
the interaction between Beclin1 and PI3K-III. There was a
dominating role of WAVE1 in regulating Beclin1-PI3K-III complex
formation of autophagy.
In summary, overexpression of WAVE1 is an
unfavorable prognostic factor in pediatric AML and leukemia cells.
As a positive regulator of autophagy, it can enhance the
chemoresistance of leukemia cells and regulate the formative
autophagosomal interaction of Beclin1-Bcl-2 and Beclin1-PI3K-III
complexes. The above findings further confirm its roles in
autophagy and chemoresistance of leukemia cells.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81400138,
31171328 and 81270616) and the Guang Dong Provincial Medical
Scientific Research Foundation (grant no. A2013585).
References
|
1
|
Abrahamsson J, Forestier E, Heldrup J,
Jahnukainen K, Jónsson OG, Lausen B, Palle J, Zeller B and Hasle H:
Response-guided induction therapy in pediatric acute myeloid
leukemia with excellent remission rate. J Clin Oncol. 29:310–315.
2011. View Article : Google Scholar
|
|
2
|
Felice MS, Rossi JG, Alonso CN, Gallego
MS, Eberle SE, Alfaro EM, Guitter MR, Bernasconi AR, Rubio PL,
Coccé MC, et al: Experience with four consecutive BFM-based
protocols for treatment of childhood with non-promyelocytic acute
yeloblastic leukemia in Argentina. Leuk Lymphoma. 6:1–10.
2016.(Epub ahead of print). View Article : Google Scholar
|
|
3
|
Gibson BE, Wheatley K, Hann IM, Stevens
RF, Webb D, Hills RK, De Graaf SS and Harrison CJ: Treatment
strategy and long-term results in paediatric patients treated in
consecutive UK AML trials. Leukemia. 19:2130–2138. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sander A, Zimmermann M, Dworzak M,
Fleischhack G, von Neuhoff C, Reinhardt D, Kaspers GJ and Creutzig
U: Consequent and intensified relapse therapy improved survival in
pediatric AML: Results of relapse treatment in 379 patients of
three consecutive AML-BFM trials. Leukemia. 24:1422–1428. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kaspers GJL, Zimmermann M, Reinhardt D,
Gibson BES, Tamminga RYJ, Aleinikova O, Armendariz H, Dworzak M, Ha
SY, Hasle H, et al: Improved outcome in pediatric relapsed acute
myeloid leukemia: Results of a randomized trial on liposomal
daunorubicin by the International BFM Study Group. J Clin Oncol.
31:599–607. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
White E and DiPaola RS: The double-edged
sword of autophagy modulation in cancer. Clin Cancer Res.
15:5308–5316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shimizu S: Development of anti-cancer
drugs mediated by apoptosis and autophagy. Nihon Rinsho.
73:1302–1307. 2015.(In Japanese). PubMed/NCBI
|
|
8
|
Takenawa T and Suetsugu S: The WASP-WAVE
protein network: Connecting the membrane to the cytoskeleton. Nat
Rev Mol Cell Biol. 8:37–48. 2007. View
Article : Google Scholar
|
|
9
|
Gourlay CW, Carpp LN, Timpson P, Winder SJ
and Ayscough KR: A role for the actin cytoskeleton in cell death
and aging in yeast. J Cell Biol. 164:803–809. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kang R, Tang D, Yu Y, Wang Z, Hu T, Wang H
and Cao L: WAVE1 regulates Bcl-2 localization and phosphorylation
in leukemia cells. Leukemia. 24:177–186. 2010. View Article : Google Scholar
|
|
11
|
Monastyrska I, Rieter E, Klionsky DJ and
Reggiori F: Multiple roles of the cytoskeleton in autophagy. Biol
Rev Camb Philos Soc. 84:431–448. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Di Bartolomeo S, Corazzari M, Nazio F,
Oliverio S, Lisi G, Antonioli M, Pagliarini V, Matteoni S, Fuoco C,
Giunta L, et al: The dynamic interaction of AMBRA1 with the dynein
motor complex regulates mammalian autophagy. J Cell Biol.
191:155–168. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Batlevi Y, Martin DN, Pandey UB, Simon CR,
Powers CM, Taylor JP and Baehrecke EH: Dynein light chain 1 is
required for autophagy, protein clearance, and cell death in
Drosophila. Proc Natl Acad Sci USA. 107:742–747. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wrighton KH: Autophagy: Myosin II moves in
on autophagosomes. Nat Rev Mol Cell Biol. 12:772011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cardoso CM, Groth-Pedersen L, Høyer-Hansen
M, Kirkegaard T, Corcelle E, Andersen JS, Jäättelä M and Nylandsted
J: Depletion of kinesin 5B affects lysosomal distribution and
stability and induces peri-nuclear accumulation of autophagosomes
in cancer cells. PLoS One. 4:e44242009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tang HW, Wang YB, Wang SL, Wu MH, Lin SY
and Chen GC: Atg1-mediated myosin II activation regulates
autophagosome formation during starvation-induced autophagy. EMBO
J. 30:636–651. 2011. View Article : Google Scholar :
|
|
17
|
Jahreiss L, Menzies FM and Rubinsztein DC:
The itinerary of autophagosomes: From peripheral formation to
kiss-and-run fusion with lysosomes. Traffic. 9:574–587. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fass E, Shvets E, Degani I, Hirschberg K
and Elazar Z: Microtubules support production of starvation-induced
autophagosomes but not their targeting and fusion with lysosomes. J
Biol Chem. 281:36303–36316. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Miki H, Suetsugu S and Takenawa T: WAVE, a
novel WASP-family protein involved in actin reorganization induced
by Rac. EMBO J. 17:6932–6941. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Machesky LM and Insall RH: Scar1 and the
related Wiskott-Aldrich syndrome protein, WASP, regulate the actin
cytoskeleton through the Arp2/3 complex. Curr Biol. 8:1347–1356.
1998. View Article : Google Scholar
|
|
21
|
Suetsugu S, Miki H and Takenawa T:
Identification of two human WAVE/SCAR homologues as general actin
regulatory molecules which associate with the Arp2/3 complex.
Biochem Biophys Res Commun. 260:296–302. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dahl JP, Wang-Dunlop J, Gonzales C, Goad
ME, Mark RJ and Kwak SP: Characterization of the WAVE1 knock-out
mouse: Implications for CNS development. J Neurosci. 23:3343–3352.
2003.PubMed/NCBI
|
|
23
|
Rawe VY, Payne C, Navara C and Schatten G:
WAVE1 intra-nuclear trafficking is essential for genomic and
cytoskeletal dynamics during fertilization: Cell-cycle-dependent
shuttling between M-phase and interphase nuclei. Dev Biol.
276:253–267. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yamaguchi H and Condeelis J: Regulation of
the actin cytoskeleton in cancer cell migration and invasion.
Biochim Biophys Acta. 1773:642–652. 2007. View Article : Google Scholar
|
|
25
|
Yang MH, Zhao MY, Wang Z, Kang R, He YL,
Yin XC, Liu LY, Yang LC, Zhan CX, Wu XS, et al: WAVE1 regulates
P-glycoprotein expression via Ezrin in leukemia cells. Leuk
Lymphoma. 52:298–309. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kang R, Cao LZ, Yu Y, Hu T, Wang Z, Xu WQ
and Xie M: Role of WAVE1 in drug resistance of K562/A02 leukemia
cells. Zhonghua Xue Ye Xue Za Zhi. 28:379–382. 2007.(In Chinese).
PubMed/NCBI
|
|
27
|
Thorburn A: Apoptosis and Autophagy:
regulatory connections between two supposedly different processes.
Apoptosis. 13:1–9. 2008. View Article : Google Scholar :
|
|
28
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Levine B: Cell biology: Autophagy and
cancer. Nature. 446:745–747. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu
Y, Xie M, Yin X, Livesey KM, Loze MT, et al: DAMP-mediated
autophagy contributes to drug resistance. Autophagy. 7:112–114.
2011. View Article : Google Scholar :
|
|
31
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang L, Yu Y, Kang R, Yang M, Xie M, Wang
Z, Tang D, Zhao M, Liu L, Zhang H, et al: Up-regulated autophagy by
endogenous high mobility group box-1 promotes chemoresistance in
leukemia cells. Leuk Lymphoma. 53:315–322. 2012. View Article : Google Scholar
|
|
33
|
Klionsky DJ, Abeliovich H, Agostinis P,
Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA,
Ballabio A, et al: Guidelines for the use and interpretation of
assays for monitoring autophagy in higher eukaryotes. Autophagy.
4:151–175. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bellodi C, Lidonnici MR, Hamilton A,
Helgason GV, Soliera AR, Ronchetti M, Galavotti S, Young KW, Selmi
T, Yacobi R, et al: Targeting autophagy potentiates tyrosine kinase
inhibitor-induced cell death in Philadelphia chromosome-positive
cells, including primary CML stem cells. J Clin Invest.
119:1109–1123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sehgal AR, Konig H, Johnson DE, Tang D,
Amaravadi RK, Boyiadzis M and Lotze MT: You eat what you are:
Autophagy inhibition as a therapeutic strategy in leukemia.
Leukemia. 29:517–525. 2015. View Article : Google Scholar
|
|
37
|
Sung JY, Engmann O, Teylan MA, Nairn AC,
Greengard P and Kim Y: WAVE1 controls neuronal activity-induced
mitochondrial distribution in dendritic spines. Proc Natl Acad Sci
USA. 105:3112–3116. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hockenbery D, Nuñez G, Milliman C,
Schreiber RD and Korsmeyer SJ: Bcl-2 is an inner mitochondrial
membrane protein that blocks programmed cell death. Nature.
348:334–336. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Petiot A, Ogier-Denis E, Blommaart EFC,
Meijer AJ and Codogno P: Distinct classes of phosphatidylinositol
3′-kinases are involved in signaling pathways that control
macroautophagy in HT-29 cells. J Biol Chem. 275:992–998. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sakamoto KM, Grant S, Saleiro D, Crispino
JD, Hijiya N, Giles F, Platanias L and Eklund EA: Targeting novel
signaling pathways for resistant acute myeloid leukemia. Mol Genet
Metab. 114:397–402. 2015. View Article : Google Scholar :
|
|
42
|
He YL, Cao LZ, Yang J, Yang MH, Xu WQ, Xie
M and Shi Z: Expression of WAVE1 and p22phox in children with acute
lymphocytic leukemia and the relationship of WAVE1 with oxidative
stress. Zhongguo Dang Dai Er Ke Za Zhi. 11:88–92. 2009.(In
Chinese). PubMed/NCBI
|
|
43
|
Abraham MC and Shaham S: Death without
caspases, caspases without death. Trends Cell Biol. 14:184–193.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Apel A, Herr I, Schwarz H, Rodemann HP and
Mayer A: Blocked autophagy sensitizes resistant carcinoma cells to
radiation therapy. Cancer Res. 68:1485–1494. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yang Z and Klionsky DJ: Eaten alive: A
history of macroautophagy. Nat Cell Biol. 12:814–822. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sinha S and Levine B: The autophagy
effector Beclin 1: A novel BH3-only protein. Oncogene. 27(Suppl 1):
S137–S148. 2008. View Article : Google Scholar
|
|
47
|
Fratti RA, Backer JM, Gruenberg J, Corvera
S and Deretic V: Role of phosphatidylinositol 3-kinase and Rab5
effectors in phagosomal biogenesis and mycobacterial phagosome
maturation arrest. J Cell Biol. 154:631–644. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Siddhanta U, McIlroy J, Shah A, Zhang Y
and Backer JM: Distinct roles for the p110alpha and hVPS34
phosphatidylinositol 3′-kinases in vesicular trafficking,
regulation of the actin cytoskeleton, and mitogenesis. J Cell Biol.
143:1647–1659. 1998. View Article : Google Scholar : PubMed/NCBI
|