1. Introduction
Renal cell carcinomas (RCCs), tumors of epithelial
origin, represent the majority of adult kidney neoplasms (1,2). In
2015, 61,560 new cases were expected in USA (3.6% of all new
cancer) (3) and more than 10,380
in the UK. According to data in the Surveillance, Epidemiology, and
End Results Registry, RCC is locally advanced in 53% of cases,
regionally advanced in 20% and metastatic in 22%. Corresponding
5-year survival rates are 90, 61 and 10%, respectively (4,5). Due
to RCC chemo- and radio-resistance and the limited number of
available targeted therapies, overall response rates (ORR) and
overall survival (OS) are still unsatisfactory (6,7).
Clear cell RCC (ccRCC) represents 85% of all renal cell cancers and
arises from the proximal tubules. Both sporadic and inherited
ccRCCs are associated with mutations in the von Hippel-Lindau (VHL)
tumor suppressor gene. Mutation or inactivation (methylation) of
the VHL gene with a subsequent second somatic event, often the
deletion of the short arm of chromosome 3 (3p-), is associated with
the development of ccRCC, and results in the deregulation of
hypoxia-inducible pathways (8).
The von Hippel-Lindau tumor suppressor protein (pVHL) acts as a HIF
repressor by directly binding to hypoxia-inducible factor 1-alpha
(HIF-1α) and targeting it to the ubiquitin-dependent degradation
pathway. Mutations of VHL result in constitutive stabilization of
the HIF-1α subunit, leading to highly angiogenic malignancy
(9). This phenomenon occurs due to
overexpression of vascular endothelial growth factor (VEGF), its
receptors (VEGFR1, VEGFR2 and VEGFR3), and platelet-derived growth
factor (PDGF) receptors in response to constitutive activation of
HIF-1α (10,11).
Some early clinical observations led to the
hypothesis that RCC is a hormone-dependent tumor (12); these observations include the
significant gender difference of RCC occurrence (twice as common in
men as in women) (12,13), a correlation between the cessation
of gonadal activity and RCC development, and a reported regression
of metastatic renal cancer during administration of progestin or
androgen (occurring more often in men than in women). It was
subsequently suggested that steroid receptor signaling pathways may
become new targets for anticancer treatment. The efficacy of
progesterone-based therapy, employed after nephrectomy, is
supported by a large number of clinical observations. Primary
reports seem to confirm that hormone treatment could be considered
as a supportive treatment for metastatic RCC. Nevertheless, when
RCC tumors are not hormone-dependent, as based on receptor
expression, patients should only be treated with targeted therapy
or immunotherapy, with or without radiotherapy (12,14–16).
Multiple endocrine factors related with ccRCC development and
progression remain unidentified. The development of relevant tumor
induction models is thus instrumental to proposing strategies for
analyzing signaling pathways and verifying gene expression
data.
The role of hormone-related factors in RCC etiology
was first hypothesized over 20 years ago. Even today,
epidemiological evidence is still more profound than molecular. In
classic epidemiological studies, it was defined that, for women,
ccRCC risk was inversely related to age at first given birth
(OR=0.7; for ≥25 vs. <25 years), and that hysterectomies doubled
ccRCC risk (OR=2.3). A negative association between ccRCC and age
at menarche was also shown, suggesting a link between steroid sex
hormones and ccRCC (17).
Moreover, high parity was shown to be associated with an increased
risk of ccRCC, while oral contraceptive use was associated with a
reduced risk. Women with 5 or more births had a 2-fold increase in
ccRCC risk when compared to those with 1 or 2 births (18). Some clinical studies concluded that
due to significant gender differences, the occurrence of RCC will
be twice as common in men as in women, for whom it will vary after
the cessation of gonadal activity. These studies also showed that
the majority of cases are diagnosed in adults aged 50–70 years
(12,13,19).
At the same time clinical biochemical research revealed that serum
levels of luteinizing hormones (LH), follicle-stimulating hormones
(FSH), thyroid-stimulating hormones (TSH), luteotropic hormones
(prolactin, PRL), human chorionic gonadotropin (beta-HCG) hormones,
and parathyroid hormones (PTH) were significantly modulated in
patients with urogenital tumors including RCC. After nephrectomy,
PTH was frequently suppressed in these patients (20). Prolactin elevation was found in 45%
of ccRCC patients regardless of the stage of the disease. At the
same time, aberrant TSH and FSH were shown to be indicative of
distant metastasis in ccRCC carriers. Serum PTH was also found to
be decreased in patients with tumor dissemination. The
overexpression of human gonadotropin-releasing hormone (GnRH) and
their receptors has been demonstrated in ccRCC (21). Inhibition of somatostatin (growth
hormone-inhibiting hormone, or GHIH) release from the pituitary
gland was also shown to control the proliferation of tumor cells
through a family of G protein-coupled receptors [somatostatin
receptor (SSTR)1-SSTR5] via the autocrine and paracrine modes. The
indirect antitumor effects of SSTRs include anti-angiogenic actions
(22,23). Moreover, parathyroid
hormone-related protein (PTHrP), a cytokine-like polyprotein, was
determined to be a survival factor for human ccRCC: its expression
was found to be negatively regulated by the VHL tumor suppressor
gene at the level of messenger RNA (mRNA) stability (9), and induced phosphorylation of Akt at
S473 via activated integrin-linked kinase (ILK), which in turn
acted as either a phosphoinositide-dependent kinase (PDK2) or a
facilitator protein to phosphorylate Akt, with nuclear factor kappa
B (NF-κB) serving as the downstream Akt target (24). The examples mentioned above
underscore the possibility of alternative pathway activation in
ccRCC via hormones and illustrate the need to explore the molecular
basis of phenomena observed in clinics (25,26).
Functional cell biology analysis has also recently provided new
data. Most recently, a three-dimensional in vitro angiogenic
system consisting of microvascular endothelial cells was used to
study the influence of hormones on neovascularization and high
levels of LH were found to promote angiogenesis in ovarian cancer
models via the PI3K/AKT-mTOR pathway (27). Moreover, multiple clinical ccRCC
research papers, on disease progression and treatment response,
incorporate patient endocrine status in prognostic scores. The most
notable current results indicate that hypothyroidism can serve as a
predictive marker of therapy outcome in patients with metastatic
RCC (28). In summary, ccRCC
patients without paraneoplastic syndromes and no endocrinologic
disease have frequent abnormalities in steroid and peptide hormones
and expression of their receptors in renal cancer tumor masses,
which in consequence influence disease progression (29,30).
2. Hypothalamus hormones
Growth hormone-releasing hormone
(GHRH)
GHRH antagonists suppress the growth of ccRCC lines
xenografted into nude mice. The antitumor effects of GHRH
antagonists are exerted in part through the inhibition of the
secretion of GH from the pituitary gland and the resultant
reduction in levels of the hepatic insulin-like growth factor I
(IGF-I). The main effects of GHRH antagonists are exerted directly
on tumors, as the principal action of GHRH antagonists in
vivo appears to be the direct suppression of autocrine and/or
paracrine production and the expression of the genes encoding IGF-I
(IGF1) and IGF-II (IGF2) in tumors (31). GHRH ligands are present in cancer
cells and might function as autocrine and/or paracrine growth
factors. Pituitary-type GHRH receptors and their splice variants
are also found in tumor samples (32). The presence of the GHRH ligand has
also been demonstrated in cancer cells, suggesting that GHRH could
be a growth factor in RCC (31).
GHRH antagonists JV-1-38 and MZ-4-71 inhibit the growth of
orthotopic Caki-1 human RCC and inhibit the development of
metastases in lung and lymph nodes (31,33).
The receptors for GHRH antagonists on Caki-1 tumors are distinct
from binding sites detected in the pituitary gland (34). More recently in ACHN, A498 and
786-0 human RCC cells GHRH antagonists MIA-602, MIA-604, MIA-606
and MIA-690 inhibited the proliferation of these cells in nude
mouse xenograft models (35). More
data are still needed.
Somatostatin (growth hormone-inhibiting
hormone-SS, GHIH or SRIF)
The presence of transcripts for somatostatin
receptor (SSTR) subtypes 1, 2, 3 and 4 was proven in RCC tissues
(36) and human proximal tubular
epithelial cells (PTEC). PTECs express somatostatin, but mitogens,
epidermal growth factors (EGFs), and hydrocortisone inhibit PTEC
somatostatin secretion; however, direct stimulation by adenylate
cyclase (i.e. forskolin) and fetal bovine serum induce secretion of
somatostatin in PTEC cell cultures. These findings raise the
possibility that renal-derived somatostatin modulates tubular cell
function via autocrine and paracrine mechanisms (37). Nevertheless, phase II trials of
somatostatin analogue administration (SSA) did not result in the
control of RCC growth. Consequently, the use of SSA in advanced RCC
does not seem to be a relevant therapeutic option (38); therefore, the role of SS in ccRCC
requires further elucidation.
Corticotropin-releasing hormone
(corticotropin-releasing factor, corticoliberin CRH or CRF)
Corticotropin-releasing hormone (CRF) acts via
signaling mediated by corticotropin-releasing hormone receptors
(CRHRs). The effects of Urocortin (Ucn) are also exerted through
the activation of CRFRs and the involvement of the Ucn-CRFR system
in pathophysiological conditions, including the regulation of
angiogenesis and the inhibition of its proliferation was described
in RCC. Suppression of neovascularization through VEGF reduction
and tumor cell cycling inhibition is modulated mainly through the
activation of CRHR2 (39). The
direct activity of corticotropin-releasing hormones on ccRCC cells
was only described recently. Corticotropin-releasing
hormone-binding protein (CRHBP) gene downregulation is 33-fold on
mRNA level in RCC tissues compared to control paired normal
tissues. This CRHBP downregulation is also correlated with
aggressiveness of RCC tumors (40).
Thyrotropin-releasing hormone
(prolactin-releasing Hormone-TRH, TRF or PRH)
Thyrotropin-releasing hormone [stimulates the
release of thyrotropin (thyroid-stimulating hormone, TSH)] and
prolactin from the anterior pituitary, but may also be taken up by
kidney cells (41). Specific DNA
hypermethylation of CpG sites on TRH gene was only reported over
last years in RCC (42), and no
further clinically relevant data is available at this time.
Gonadotropin-releasing hormone
(Luteinizing hormone-releasing hormone-GnRH or LHRH)
Gonadotropin-releasing hormone (GnRH), also known as
follicle-stimulating hormone-releasing hormone (FSH-RH),
luteinizing hormone-releasing hormone (LHRH), gonadoliberin, and
luliberin mediates release of follicle-stimulating hormone (FSH)
and luteinizing hormone (LH) from the anterior pituitary but
expression of LHRH receptors was investigated in surgically removed
specimens of RCC and in human RCC cell lines (A-498, ACHN and
786-0), and positive staining (expression) was found in each of the
cases. In the tumor samples, LHRH receptor expression was found to
be very high. It was therefore hypothesized that inhibitors of LHRH
receptors (i.e. AN-201 or AEZS-108), which bind with high affinity
to LHRH receptors, can be targeted against ccRCC tumors with
overexpression of these receptors (43,44).
In Caki-1 cell line-based xenograft models, GnRH antagonists [i.e.
Cetrorelix (SB-75)] were tested and shown to effectively inhibit
the growth of RCC tumors. It was subsequently proposed that this
group of compounds should be considered in therapies for patients
with metastatic ccRCC (45). The
whole genome gene expression profile of LHRH-activated ccRCC cells
is not currently known.
Proopiomelanocortin (POMC)
It was shown that proopiomelanocortin (POMC), an
adrenocorticotropic hormone precursor, is upregulated in
VHL-mutated RCC. Regulatory mechanism in which proopiomelanocortin
(POMC) and nuclear receptor subfamily 4 group A member 1
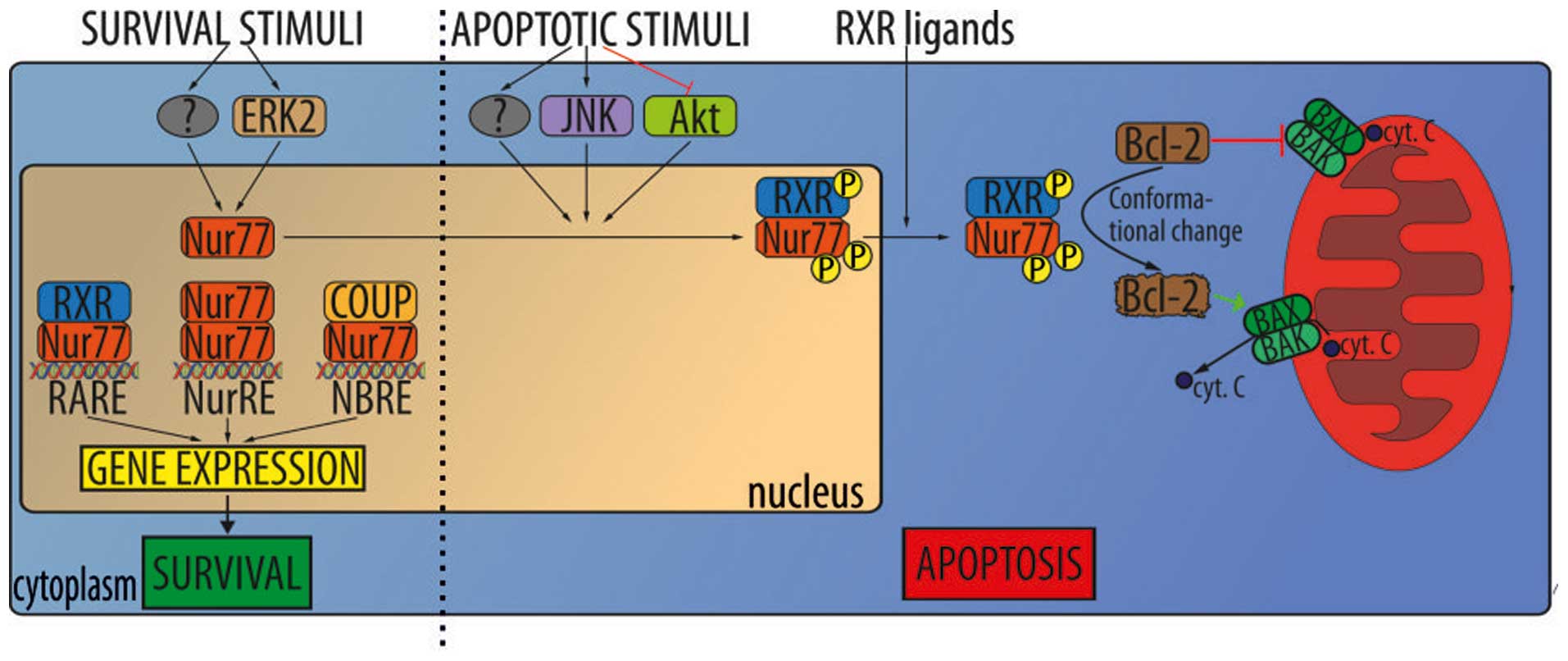
(NR4A1)/Nur77 are upregulated in VHL-mutated RCC was identified.
Nur77, a member of the orphan steroid receptor superfamily, is
believed to be activated by HIF under hypoxic conditions, and in
turn regulate the production of the peptide hormone precursor POMC
(46). Nur77 was identified as a
critical transcription factor responsible for POMC overproduction
and to be directly regulated by HIF. HIF-1α (but not HIF-2α) binds
to a HIF-responsive site in the Nur77 promoter region, activating
the expression of Nur77 during hypoxic (VHL-mutant) conditions.
Mutation or deletion of the HIF binding site in the Nur77 promoter
region significantly decreased activation of a Nur77 and its target
genes. The treatment of cells with Nur77 antisense oligonucleotides
reduces POMC transcription under hypoxic conditions. In contrast to
the normal control tissue the tumor tissues produce abnormally high
amounts of Nur77 and POMC. Taken together, these results strongly
suggest that Nur77 is both necessary and sufficient for
hypoxia-dependent transcription of POMC (Fig. 1) (46).
3. Pituitary gland hormones
Growth hormone (human growth hormone,
somatotropin-HGH, GH)
Growth hormones (GH) stimulate proliferation and
differentiation of normal human cells but have also been shown to
be involved in the development of malignant tumors by leading to
excess IGF-I production in the liver, as well as having direct
effects via GH receptors (GHR) expressed in a variety of tumors,
including RCC, colorectal and breast cancer (12,47).
Both GH and IGF-I have been shown to act as oncogenes by inducing
mitogenic and anti-apoptotic effects in a variety of tumors and
cancer-derived cell lines via endocrine and/or autocrine/paracrine
mechanisms. It should be noted that the expression of IGF-IR is
increased by the activation of oncogenes such as SV40 T antigen and
c-MYB, but decreased by the activation of tumor suppressor genes
such as p53 and WT1 (48).
Oncogenic transformation seems to be responsible for the local
expression of IGF-IR and GHR in tumor tissues including RCC. It has
been reported that acromegaly patients have an increased risk of
developing malignant tumors, although several epidemiological
studies have shown that RCC rarely co-occurs with acromegaly
(49). Several epidemiological and
experimental studies have proposed the hypothesis that elevated
GH/IGF-I levels are associated with oncogenic processes in RCC,
thyroid tumors, and colon cancer, and play a crucial role in
tumorigenesis in acromegaly and/or the growth in multiple tumors
(50). Sekizawa et al
(49) described a case in which a
56-year-old man diagnosed with acromegaly was also found to have
multiple tumors (ccRCC, colon cancer, follicular thyroid tumor and
GH-producing pituitary adenoma). This is a rare case, as the
association of acromegaly with multiple tumors other than in MEN1
has not been reported elsewhere in the literature. Nevertheless
several large-scale epidemiological studies on the co-incidence of
neoplastic diseases and acromegaly have shown coincidence with
RCC.
Adrenocorticotropic hormone (ACTH)
The biosynthesis of adrenocorticotropic hormone
(ACTH), also known as corticotropin is controlled by a multiple
transcription factors through the promotion of proopiomelanocortin
(POMC), the precursor to ACTH and other enzymes involved in the
synthesis of steroid hormones in pituitary cells. Hypoxia activates
the hypothalamic-pituitary-adrenal (HPA) axis, resulting in an
increase of ACTH and ACTH receptor expression; this suggests that
oxygen fluctuation may influence the release of cellular hormones
in the tumor niche (46).
Thyroid-stimulating hormone (TSH)
Patients with thyroid disease (TD) and abnormal TSH
levels, including on-nodular TD, solitary nodules, multinodular TD,
thyroid cancers [with either the presence or absence of
anti-thyroglobulin (TgAb)], and anti-thyroid peroxidase (TPOAb) or
anti-thyroid-stimulating hormone (TSH) receptor autoantibodies, are
at higher risk of developing kidney cancer (OR=3.40) compared to
the general population (51).
Whereas, hypothyroidism is associated with longer progression-free
survival (PFS) in sunitinib and sorafenib treatments (25,28).
The severity of vascular endothelial growth factor receptor
tyrosine kinase inhibitor (TKI) therapy-associated hypothyroidism
(TSH >10 mIU/l) is associated with improved treatment efficacy
and survival outcomes in patients with metastatic RCC (52). This year meta-analysis suggested
that development of hypothyroidism during TKI therapy is not
clearly predictive of efficacy in patients with metastatic RCC or
advantage in overall survival (OS) (53).
Follicle-stimulating hormone (FSH)
Follicle-stimulating hormones (FSHs) are released
under the influence of gonadotropin-releasing hormones (GnRH). The
FSH receptor (FSHR), which was expected to be expressed only in the
ovary and testis, was recently detected in the blood vessels of
many solid tumors, including RCC (54,55).
FSHR expression was evaluated in the endothelium of 1,336 primary
solid tumors, representing 11 tumor types, comprised mostly of
genitourinary malignancies and 64 RCC cases. The FSHR expression in
the neo-vasculature of tumors found it almost exclusively in
peripheries, in a region <1 cm inside or outside of the tumor in
70% of cases, but ~30% of samples had equal FSHR expression in
total tumor mass. FSHR expression was not detected in the blood
vessels of nonmalignant tissues (55,56).
Based on functional studies it was suggested that
FSHR may contribute to neoangiogenesis, which would make it an
interesting target for therapeutic and imaging purposes, as well as
a potentially useful prognostic and/or predictive biomarker in
genitourinary cancers. FSHR may also contribute to the development
of metastases due to its position on the luminal surface of the
endothelium. Moreover, FSHR may play a role in tumor intravasation,
allowing RCC cells to penetrate through the endothelium and into
circulation (57). In addition,
FSHR expression at the periphery of tumors (55), where the tumor interacts with the
stroma, may suggest its role in metastases. The
epithelial-to-mesenchymal transition (EMT) is believed to be
critical to the formation of metastases, while the interaction of
stromal elements with tumor cells at the tumor periphery is thought
to contribute to EMT. FSHR has also been proposed as a marker of
tumor endothelium in solid tumors (56). Expression of FSH receptor (FSHR)
expression was shown to be effective prediction markers of tumor
vasculature response to sunitinib treatment. The percentage of
FSHR-stained vessels was on average 5 times higher for patients who
responded to treatment than the control group, and almost 8 times
higher than in the non-responsive group. However, no significant
differences were detected in the total density of vessels between
these 3 groups, nor was a significant correlation found between
FSHR expression and tumor grade. Nevertheless, a far greater number
of FSHR-positive vessels were detected in patients who responded to
the treatment. The response threshold between the two groups of
patients was defined at 23% FSHR-positive vessels. Not only was a
higher density of FSHR-expressing vessels observed in the primary
tumors of patients who responded to sunitinib treatment, but the
von Willebrand factor (vWF) was detected in these vessel
(FSHR+/vWF+) (30). Plausible mechanisms that could
explain this correlation have been hypothesized. One possible
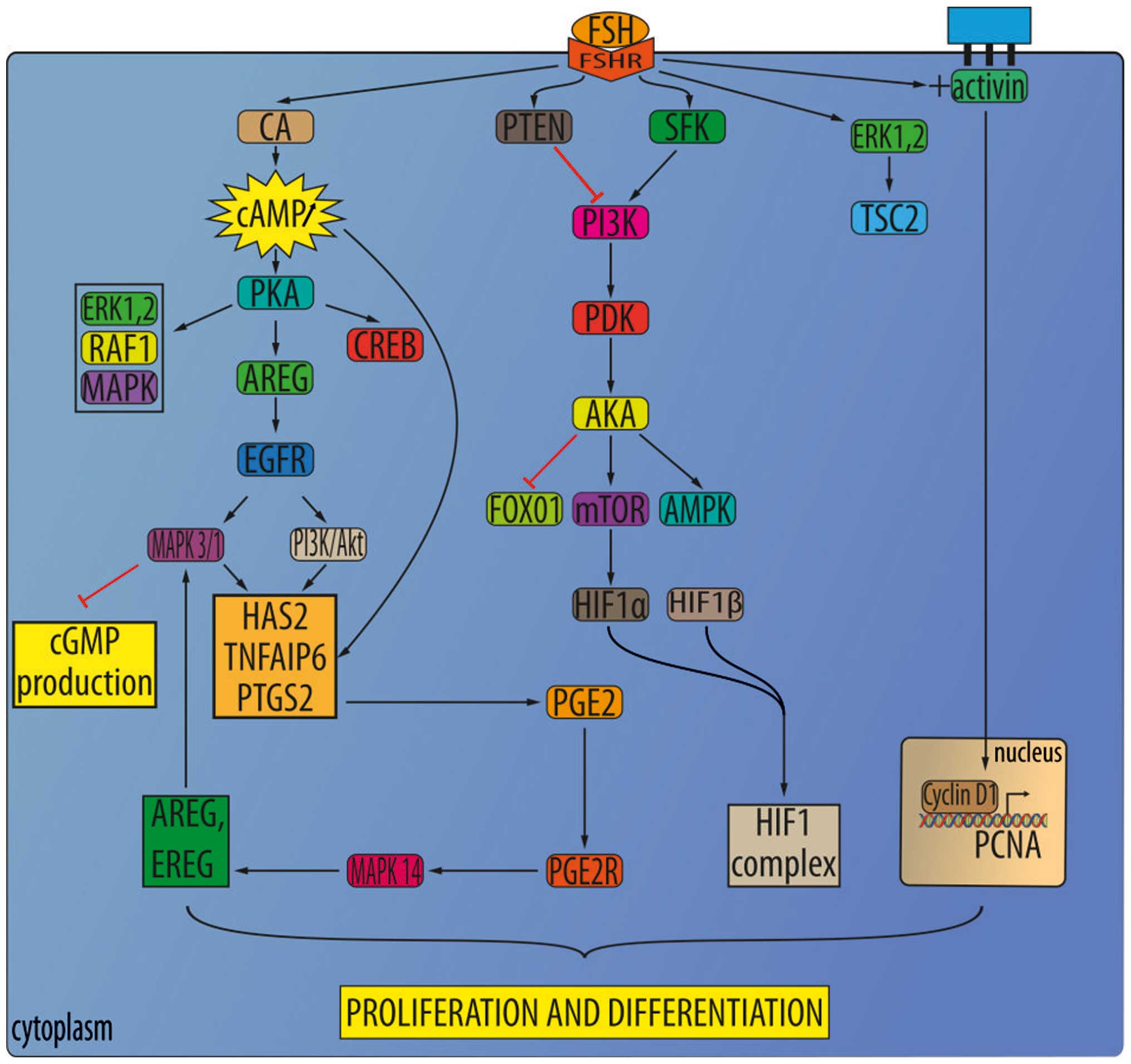
mechanism is that FSHR stimulation by FSH leads to VEGF secretion
by the ECs, which in turn stimulates VEGFR2 on the ECs as an
autocrine mechanism. In fact, it has been observed that the binding
of FSH to FSH receptors in ovarian granulosa cells induces an
increase in hypoxia-inducible factor 1α protein levels, which
consequently leads to upregulation of VEGF production (Fig. 2) (57). It was later suggested that FSHR
expression in the blood vessels of ccRCC primary tumors can be used
to predict if patients will respond to sunitinib treatment
(56). Another possible
explanation for the correlation between FSHR expression and
sunitinib efficacy is that FSHR activates one or more of the other
kinases known to be inhibited by sunitinib like c-Kit, PDGFR,
CSF-1R, FLT3 or RET (30).
Treatment
The FSH-FSHR signaling axis can be targeted with
drugs in several ways, and GnRH antagonists that decrease FSH
levels are currently available. Futhermore, the use of
FSH-neutralizing antibodies could potentially cause an even greater
reduction in FSH levels. FSHRs may be targeted by the use of
molecules with drug- or radio-immunoconjugates. In order to do so,
several classes of antagonizing compounds have been identified,
most of which are negative allosteric modulators. An example is the
thiazolidinone analogues, whose modifications in the chemical
structure of the thiazolidinone backbone allow it to divide its
generation into positive, mixed, and negative allosteric modulators
of FSHR (30). Several studies
have explored the use of FSHR allosteric modulators in vivo.
Recently, ADX61623, another negative allosteric modulator of FSHR,
was investigated. ADX61623 inhibited some elements of FSHR
signaling, such as progesterone synthesis and cAMP production, but
failed to inhibit estradiol synthesis (56). FSHR-antagonizing molecules need to
be examined further, as they may play a role in targeting FSHR in
RCC and other tumors.
Prolactin (PRL)
Prolactin (PRL) is measured by its pathological
range in patients with RCC. Even patients without paraneoplastic
syndromes experience frequent changes that are directly caused by
RCC (20), although only a single
case of hyperprolactinemia induced by RCC has thus far been
reported (58).
4. Adrenal hormones
Glucocorticoid receptors (GR)
In normal kidneys, glucocorticoid receptors (GRs)
are expressed in proximal tubules and glomeruli. In RCC, high GR
expression is a positive prognostic marker. Initial studies,
conducted in the early 1980s, and that discovered the presence of
GRs in kidney tumors, were based on ligand-binding assays (59,60).
GRs were found to be overexpressed in 66% of ccRCC cases, 26% of
pRCC cases, 14% of oncocytomas and 6% of chRCC cases. Moreover, GR
expression serves as a favorable marker and correlates with low
nuclear grade and stage. A significant correlation between GR
expression and OS in RCC patients may be hypothesized. The majority
of patients with ccRCC-expressing GRs were still alive by the end
of the follow-up, in contrast to those with expression-negative
tumors (59,61). As a result, the correlation of GR
expression with less aggressive tumor behavior was reported and the
anti-proliferative role of GR signaling in RCC was suggested.
Suppression of transcription factors, including cAMP response
element-binding protein (CREBs), nuclear factor
kappa-light-chain-enhancer of activated B cells (NF-κBs), signal
transduction activator of transcription (STATs), activator protein
1 (AP-1s), p53s, CCAAT-enhancer-binding proteins (C/EBP), and SMADs
was suggested as inhibitory mechanisms mediated by GR signaling
(59,62). In RCC, two main isoforms of GR were
analyzed. GR-α, a predominant isoform, exhibits steroid binding
activity. GR-β has a lower expression in normal kidney tissues, but
is overexpressed in inflammatory blood cells localized in the tumor
(59). In RCC, GRs have been shown
to bind not just glucocorticoids (63), but with progesterone,
diethylstilbestrol, testosterone, and aldosterone as well, albeit
with low affinity. In addition, progestin or medroxyprogesterone
easily binds to GRs. As a result of medroxyprogesterone acetate
treatment RCC tumor regression was reported (64).
Treatment
Efficacy of RU486 (Mifepristone), a glucocorticoid
receptor antagonist, on TRAIL-induced apoptosis in human RCC,
Caki-1, A498 and ACHN cell lines was measured. RU486 is known as an
anti-progesterone and anti-glucocorticoid agent. Its low dose has
no effect on apoptosis, but sensitizes Caki-1 cells to
TRAIL-induced apoptosis. As a result, RU486 enhances TRAIL-mediated
apoptosis through the downregulation of Bcl-2 and c-FLIP(L) as well
as CHOP-mediated DR5 upregulation. TRAILs bind to their receptors
(death receptors DR4 and DR5) to form death-inducing signal
complexes (DISC) by recruiting caspase-8, which in turn increases
apoptosis through the activation of caspase-3. In addition, TRAILs
activate mitochondrial apoptotic pathways: TRAILs release
cytochrome c to the cytosol via disruption of mitochondria
membrane permeability, allowing it to form apoptosomes. These
responses were also commonly observed in a variety of other cancer
cells, including SK-Hep-1 (hepatocarcinoma cells) and HT29 (colon
cancer cells) (62). In addition,
RU486 slightly inhibited XIAP expression, increased active cleaved
caspase-3 and PARP cleavage, induced cytoplasmic histone-associated
DNA fragments, and blocked chromatin in the nuclei, indicating
apoptosis. Moreover, RU486-mediated TRAIL-induced apoptosis acts
independently of GR and PR signaling, through CHOP-mediated DR5
expression and the downregulation of Bcl-2 and c-FLIP expression.
As TRAILs induce apoptosis only in tumor cells, they may be a
promising cancer treatment, and medications such as RU486 may be a
novel strategy for the recovery of TRAIL sensitivity in cancer
cells otherwise resistant to TRAIL (62). In a study conducted by Arai et
al (65), the inhibitory
effects of dexamethasone (DEX) on the growth of RCC in vivo
and in vitro were examined and suppression of NF-κB
activation was measured. DEX binds more powerfully to the
glucocorticoid receptor than cortisol. DEX has long been used to
suppress inflammatory reactions in patients with advanced cancers
by activating transcription factor NF-κB and its target genes IL-6,
IL-8 and VEGF. All the RCC cell lines tested responded to DEX
treatment, but RCC growth suppression was more remarkable in
vivo than in vitro. Caki-1 cells expressed a low level
of GR protein, and GR was translocated into the nucleus. Moreover,
after 6 weeks of treatment, mean tumor volume statistically
decreased. H&E staining showed less inflammatory cells and
necrotic tissues in DEX groups as well. Intracellular IL-6, as well
as those in the conditioned medium, was downregulated in all cell
lines following treatment. Concentrations of IL-8 in the
conditioned medium were remarkably decreased in NC65 cells, while
VEGF secretion was lowered by 30–65% in all RCC lines, with the
inhibition of the nuclear translocation of NF-κB (65). After therapy with dexamethasone,
cases of complete regression (CR) occurred, and the cases included
patients with metastases located in the brain and lungs.
Dexamethasone treatment may thus be a candidate for RCC supportive
therapy. This compound was shown to inhibit expression and
signaling mediated by NF-κB, VEGF, IL-6 and IL-8. Cortisone therapy
was also reported to be successful in some cases. Over 10 years of
complete remission (CR) was demonstrated in patients with
retroperitoneal lymph node involvement and liver metastases. This
observation supports the thesis that GR and its agonists may play
an important role in anticancer ccRCC therapy (66). Using dexamethasone in combination
with kinase inhibitors was examined (59).
Mineralocorticoid receptors (MR)
Mineralocorticoid receptors (MR) in the kidneys are
expressed in Henle's loop, distal tubules and collecting ducts. Due
to its compartment-specific expression in the kidneys, MR was
suggested as a diagnostic marker for oncocytomas and chromophobe
RCC. MR was indicated as both a highly specific and sensitive
marker of the distal nephron cells and its derived neoplasms
(67). Aside from MR, 11beta-HSD2
[11β-Hydroxysteroid dehydrogenase (HSD-11β or 11β-HSD)] was also
investigated as a RCC subtype marker. Expression of both MR and
11beta-HSD2 was detected in the distal nephrons of normal kidneys.
MR and 11beta-HSD2 were highly expressed in 90% of chromophobe RCCs
and 93% of oncocytomas, thus reflecting their histogenetic origin.
No MR staining was detected in ccRCC, since its absence corresponds
with its proximal tubule origin. Only 2.6% cases of ccRCC showed
focal positivity for 11beta-HSD2, whereas all papillary RCCs were
negative (67). In functional MR
studies, aldosterone binding was observed to be more significantly
decreased in clear cell RCCs than in normal tissues, both in the
cytosol and in the nucleus (59).
Recent studies carried out in the US and Eastern
Europe have shown that increased activity of the
renin-angiotensin-aldosterone system (RAAS) is one of the major
risk factors influencing genetic changes and is mainly observed in
cases of ccRCC. Epidemiological evidence suggests that RAAS-related
hypertension and obesity increase the risk of RCC development
(68), while epidemiological
evidence from the MRC Blood Pressure Unit in Glasgow has shown that
pharmacological suppression of the RAAS lowers the risk of
developing renal cancer in hypertensive patients as opposed to
their non-hypertensive counterparts. In experimental models, the
inhibition of angiotensin-converting enzymes restricted the growth
of human renal cancer cells in a mouse xenograft system; and in
vitro, restored the sensitivity of mouse Renca cells to the
growth-suppressing effects of TGFβ. Moreover, in several
experimental animal models of hypertension, overactivity of the
RAAS was accompanied by renal tubular hyperplasia. One potential
mechanism to explain this is the influence of the RAAS on the
expression and activity of the K-ras cellular oncogene to promote
tumor growth. King et al (68) also observed that the K-ras isoform
K-RAS4A is aldosterone-sensitive in renal cancer, and that its
overexpression contributes to the survival and increased
proliferation of RCC cells in response to RAAS activation. This was
demonstrated by adding spironolactone to RCC cultures, which
blocked mineralocorticoid receptors and led to an absolute
reduction in both cell number and K-ras expression. Thus,
mineralocorticoid activation and K-RAS4A expression were shown to
support RCC proliferation. A second study treatment involved
transfection with siRNA, which also suppressed K-ras protein
expression and decreased cell count by ~40–73% after 72 h. In
addition, it was found that K-RAS4A acts through the Raf and Akt
pathways to support the survival and growth of RCC cells. Both Raf
and Akt proteins were markedly reduced following a decrease in
K-RAS4A. According to Varela et al (69) there is evidence that K-ras may act
in tandem with the SWI/SNF/PBRM1 complex to promote the formation
of renal carcinoma. Kotelevtsev et al (70) demonstrated that the
mineralocorticoid receptor is expressed in about half of human
clear cell carcinomas. In this model, K-RAS4A expression was
induced by aldosterone treatment and its activation sustained by an
as yet unproven tyrosine kinase that may have been involved with
the epidermal growth factor or insulin-like growth factor 2
receptor; also, K-RAS4A links the phosphatidylinositol 3-kinase
pathway to ENaC activity. Thus, K-ras is also involved in cell
growth promotion via aldosterone-sensitive growth responses
(70).
Vitamin D receptors (VDR)
Vitamin D receptor (VDR) signaling regulates
multiple target genes and promotes cell differentiation,
angiogenesis, and angiogenic proliferation in multiple tissues,
including the kidneys. Long-term vitamin D serum levels were
suggested to be inversely correlated with renal cancer development
risk. VDR was shown to be overexpressed in several malignant
neoplasms, including metastatic RCC tumors, and in poorly
differentiated and sarcomatous RCCs of Fuhrman grade IV in
particular (59,71). The relationship of
1,25-Dihydroxyvitamin D3 receptors (VDR) to histological features
in RCC was further investigated (72). VDR expression was shown to be
absent in the proximal tubules. In contrast, tumors originating
from the distal nephron tested positive for VDR, including the
majority of papillary RCCs, chromophobe RCCs and oncocytomas.
Positive VDR staining could help in efforts to differentiate
between papillary RCC and clear cell RCC with papillary features
(73). VDR immunohistochemistry
results can help classify RCC tumors (73). Generally, ccRCC tests negative for
VDR. In addition, ccRCC exhibits decreased VDR mRNA levels when
compared to normal kidney tissues, and VDR staining is limited only
to the peripheral region of the tumor (73). Analysis of RCC samples exhibited
expression of 1,25-dihydroxyvitamin D3 receptors in 81% of the
tumors. Absence or loss of the receptor was associated with low
differentiated sarcomatoid tumors with a poor prognosis. On the
other hand, the expression of VDR receptors in the
receptor-positive tumors did not correlate with clinical stage or
pathological grade or RCC (72).
The expression of 1,25-(OH)2D3 receptors was
also analyzed in normal kidney tissues and primary RCC samples. In
83% of RCC cases, 1,25-(OH)2D3 receptor
expression was found, and in 65% of the cases the receptor was
overexpressed. The mean expression of the
1,25-(OH)2D3 receptor in RCCs is
significantly lower than in normal kidneys. The lower
1,25-(OH)2D3 receptor expression may be due
to lack of differentiation in the malignant-transformed renal
cells. A functional analysis of the
1,25-(OH)2D3 receptor in both normal and RCC
tissues show similarities, which would suggest that the receptor
may possess a normal cellular function in the transformed cells. A
possible correlation between 1,25-(OH)2D3
receptor expression in primary tumors and the late development of
lymph node metastases was also found (74). In an earlier study, no significant
difference between expression of VDR in ccRCC and control tissues
was found (75). Such a
discrepancy could be explained by the variable degree of
differentiation in the tumors analyzed in these two studies.
Subsequent physiological studies have revealed the
complicated role vitamin D plays in renal tissue. According to
several biochemical studies, even when exogenous vitamin D
supplementation was present, the formation of VDR-DNA complexes in
RCC was decreased. This impairment was shown to be secondary to
deregulated VDR heterodimerization with RXR (retinoid X receptor)
in tumor cells. It was shown that the expression of RXR-γ in RCC
was correlated with OS of RCC patients (59). Another study discovered that VDR
mRNA was almost undetectable in clear cell RCC as compared to
normal kidney tissue, and was accompanied by the underexpression of
CYP2R1, CYP27B1 and CYP27A1 enzymes (76). No correlation between
concentrations of VDR receptors and cancer cell DNA-ploidy was
found. The mean VDR concentration used was 8.4 fmol/mg of protein
(range, 2.8–15.9) in diploid tumors and 7.0 fmol/mg of protein
(range, 0–27.8) in DNA aneuploid tumors (11 out of 22) (74).
Treatment
Vitamin D exerts its anticancer activity by inducing
apoptosis and inhibiting cell proliferation. This effect has been
demonstrated in many cancer models, including malignant melanoma,
breast cancer, osteogenic sarcoma and acute myelogenic leukemia
(77). In one report,
proliferation of the RCC cell line, derived from a pulmonary
metastasis, was inhibited by calcitriol treatment (72). In another study, RCC tumor growth
was inhibited, pulmonary and hepatic metastates were decreased, and
the OS of mice was prolonged in relation to the dose of vitamin D
provided (78). Although vitamin
D-based supplementation therapy shows promising results, its
implementation is impeded by its toxic hypercalcemic effect
(59). Bypassing this toxicity is
possible by using alternative vitamin D derivatives. At the same
time, alkylating derivative 1,25-dihydroxyvitamin D3-3-bromoacetate
was investigated in vitro and in vivo. This study
showed that 1,25(OH)2D3-3-BE had a
significantly more potent effect on the inhibition of human RCC
cell line proliferation than an equivalent concentration of normal
vitamin D. In addition, an increase in the apoptosis rate was
observed with a reduction in cyclin A expression (79).
RAR (retinoic acid) receptors and RXR
(retinoid X) receptors
Expression of retinoic acid receptors (RARs) and
retinoid X receptors (RXRs) are cell type-specific. RAR and RXR are
expressed in proximal tubules and renal interstitial cells. RAR-β
mRNA was found to be constrictively expressed in normal kidney
cells. Furthermore, RAR and RXR expression was reported in
podocytes (74). Expression of
RXR-β was reported in proximal tubules and interstitial cells,
while RXR-α and RXR-γ were present mostly in the nuclei of proximal
tubule cells (80). In the
analysis of 49 RCC tumors, RXR-α expression was detected in 70% of
the cases, RXR-β in 47% of the cases, and RXR-γ in 85% of the cases
(80). Only RXR-γ expression was
found to be inversely correlated with the TNM stage, because
patients with RXR-γ-positive tumors were observed to have prolonged
OS (59). RAR-β probably
contributes to tumorigenesis in RCC because the deletion of its
gene on the short arm of chromosome 3 is frequently found in RCC
and other cancers. RAR-β mRNA is often not detectable in RCC cell
lines. This also suggests minimal inhibition or resistance to
13-cis-RA treatment (59).
Treatment
In clinical studies, the effect of retinoic acid on
RCC was tested in patients with multiple metastases. In a large
trial, responses to therapy with either IFN-α 2a alone or in
combination with 13-cis-retinoic acid (13-CRA) was
evaluated. This study showed that 19% of patients on combined
treatment did not show progression after 24 months.
Progression-free (PFS) and overall survival (OS) rates for patients
were significantly longer when treated with combined IFN-α 2a and
13-CRA therapy (81). In another
study, 3 different treatment arms were analyzed: i) a treatment
group given a combination of IFN-α 2a, IL-2, and fluorouracil; ii)
a treatment given a combination of IFN-α 2a, IL-2, fluorouracil,
and 13-CRA; and iii) a control group given vinblastine and IFN-α.
The results showed that group 1 and 2 had a significantly longer
progression-free period and overall survival rate, although no
significant difference in efficacy between them was demonstrated
(82). According to both of the
studies mentioned above, retinoid treatment can have a beneficial
effect, at least for a subgroup of RCC patients.
5. Sex hormones
Estrogen receptors (ER)
Two isoforms of estrogen receptors (ER) are known
and are encoded on different chromosomes: ER-α and ER-β. ER-α is
mainly expressed in reproductive organs, while ER-β is expressed in
genitourinary human tissues in the central nervous system. Both
types of estrogen receptors are also expressed in normal renal
interstitial stromal cells. In tumor samples, ERs are found in
stromal tumors, cystic nephromas and angiomyolipomas (59). The relative concentrations of these
receptors in the renal tumor is as follows: progesterone >
estrogen > androgen > glucocorticoid > mineralocorticoid
receptors (Fig. 3) (83). The affinity of the ER-β isoform to
bind with 17-β-estradiol is similar to ER-α. At the same time,
androgens and phytoestrogens are bound with greater affinity by
ER-β (84).
Clinical studies
Initial studies in estrogen and RCC covered the
procedure for experimental induction of cancers. The procedure
covers the supply of female sex hormones to male mice, or male
Syrian hamsters, as well as in female guinea pigs, after
ovariectomy, during low progesterone secretion, or before
reproductive maturity. Endocrine balancing following the resection
of ovaries has been seen to delay the occurrence of tumors in
various organs, including the kidneys. Moreover, environmental
exposure to both xeno-estrogen and estrogen is associated with
cancer development. RCC can be experimentally induced in animal
models by exposure to high levels of estrogen, which might suggest
the involvement of estrogen receptors in the etiology of renal
cancer and possibly, xeno-estrogen as well. In normal human kidney
and RCC cytosols, both estradiol and progesterone receptors have
been found, although a lower binding capacity of human renal cancer
cytosols compared to that of normal kidney cytosols was reported,
perhaps due to a nuclear translocation of the receptors in the
neoplastic tissue (12). Early
studies of ER in renal cancer showed that the expression of ERs in
RCC was highly variable (85). ERs
were detected in only 30% of the tumors and 40% of normal kidney
tissues, but in some experiments ERs were not detected in any RCC
samples. At the same time, ER expression in the interstitial cells
of the human kidney, in both adults and children, has also been
reported. ER- and PR-positive stroma, described as Müllerian-like,
was also found in normal kidneys and metaplastic kidney tissues
(86). A recent
immunohistochemical study covering 182 RCCs of different subtypes
found that ER immunoreactivity was demonstrated in 1.1% of
patients, one with ccRCC, and the other, chromophobe RCC (77). Expression of ER and PR in some
human renal tumors has led to suggestions that some of these benign
tumor types, particularly mixed epithelial-stromal tumors (MEST),
cystic nephromas, or angiomyolipomas with epithelial cysts (AMLEC),
may be related to excessive exogenous estrogens. At the same time,
the incidence of renal cancer appears to be gender-related, since
it is twice as high in men than it is in women. In addition, ER-α
genetic polymorphism in the kidneys also seems to play an important
role in the development of renal cancer (87).
Human kidneys were traditionally thought to be
unresponsive to estrogen stimulation, but renal tumors as well as
angiomyolipomas have been reported to increase in size during
pregnancy or as a secondary effect of oral contraceptive therapy.
In animal studies, tumors similar to that of human angiomyolipomas
have also been observed to grow with estrogen therapy and to
regress on tamoxifen. In hamster animal models, renal epithelial
tumors have been shown to be inducible with estrogen. Additionally,
ovarian-like stromas are reported to be quite common; but on the
other hand, they do not constitute the exclusive type of stroma in
most tumors of a specific type, and in many tumors no ovarian-like
stromas are even present. In early clinical reports, the presence
of steroid receptors in renal tissue extracted from nephrectomy
specimens was correlated with responses to progesterone-based
therapy. In this trial, tumors obtained from 23 patients were
examined. In the cytosol fraction of cancer cells, 61% were found
to contain ER and 61% were found to contain PR. Moreover, 39% of
the tumors tested positive for both ER and PR, while 17% tested
negative for both ER and PR. ER and PR were also examined in the
nuclear fraction of 3 renal cancers, and estradiol nuclear
receptors were found in 2, while progesterone nuclear receptors
were found in 3. Next, treatment response was evaluated. With few
exceptions, patients were treated with the following progestins:
medroxyprogesterone acetate (MPA) with R5020, and
17,21-dimethyl-19- norpregna-4,9-diene-3,20-dione, after
nephrectomy. The best responders to progestin treatment were those
patients with hormone-dependent tumors (ER+
PR− or vice versa). These patients did not develop
metastases for up to 22 months following nephrectomy. Other
patients, also treatment responders, also achieved disease
stabilization (SD) with prolonged OS (12). In another study that included
primarily perimenopausal female patients (and 1 male patient with a
history of diethylstilbestrol treatment after prostatic
adenocarcinoma), ER expression in RCC was observed. The excessive
growth of RCC in these patients was suggested to be stimulated by
therapeutic hormones, overproduction of estrogen, or perimenopausal
hormonal abnormalities (88).
Functional cell biology
A novel role for estrogen-induced cathepsin D in
hamster kidneys during tumorigenesis that involves renal tubular
damage followed by cell proliferation may contribute to renal tumor
formation. Primary estrogen-induced RCCs and their metastases
showed significantly increased levels of all 3 cathepsin D
isoforms. A concomitant increase of cathepsin D, along with
estrogen receptor proteins, indicates that cathepsin D gene
expression is under the control of estrogen and is possibly
mediated via induced renal estrogen receptors. Cathepsin D is
considered to be an early estrogen-response gene. Oncogenes such as
c-Myc, c-Fos and c-Jun (and all early estrogen-response genes) are
overexpressed in the kidneys after only four months of estrogen
treatment. Either tamoxifen or dihydrotestosterone (DHT) prevented
the rise in cathepsin D and estrogen receptor content observed
after estrogen treatment alone (89). Numerous in vitro studies
have suggested the possibility that potentially reactive
intermediates of estrogen may be the causative factors contributing
to renal cell injury during chronic and prolonged exposure to
estrogen (18,90). Estrogen-mediated cytotoxicity were
confirmed in various tissues including kidney following chronic
estrogen treatment (27).
ER-α
Single nucleotide polymorphisms of the ER-α gene in
RCC samples were investigated. Six different polymorphic loci of
this gene were analyzed in 113 RCCs to determine their frequency.
The results showed that the distribution of genotypes of codon 10
varied between RCC patients and healthy controls, and the relative
risk of this genotype was calculated as HR=2.51. Analysis of DNA
from pairs of cancerous and normal tissues detected genotypic
changes in 9% of cancer samples on (and exclusively) exon 1 (codons
10 and 87) of ER-α. This leads to the conclusion that codon 10
polymorphism on exon 1 of ER-α may be involved in RCC development.
No differences were observed between men and women in the
distribution of codon 10 genotypes in the control group. No
association was observed between gender, age, or stage of renal
cancer with polymorphisms of other genes such as p53 (91,92).
Estrogen receptor-α (ER-α) was recently found to be a novel
proteasomal degradation target of the pVHL E3 ligase.
Overexpression of VHL suppresses ER-α expression in RCC, whereas
downregulation of pVHL can increase ER-α expression (8). Overexpression of ER-α may increase
HIF-1α transcription factor activity. In VHL-deficient cells, the
expression of ER-α and HIF-1α is retained, and blocking ER-α using
its inhibitor could suppress the proliferation of VHL-deficient
cells as effectively as hypoxia-induced growth suppression. The
experiment also showed the anti-proliferative effect of faslodex
(ER-α inhibitor) in VHL-deficient cells by inducing p53 expression
(8). It was also shown that after
binding to ER-α, the estrogen complex promotes the transcription of
growth-related factors that enhance gene expression and mitosis and
promotes proliferation, leading to cancer development and tumor
progression (93).
ER-β
Previous studies indicated that ER-β has
anti-proliferative and apoptosis-inducing functions (69). In RCC ER-β was suggested as tumor
suppressor. ER-β is highly expressed in RCC cell lines, and
estrogen-activated ER-β reduced growth hormone downstream signaling
activation of the AKT, ERK, NF-κB, MMP9 and JAK signaling pathways
but also increased apoptotic cascade activation (caspase-3, -8, -9,
BID activation and reduced Bcl-2 and survivin expression). ER-β
signaling increases RCC cell migration via induction of the
VEGFa/HIF2α pathway. Moreover, in RCC tumors, infiltrating
neutrophils modulate the expression of ER-β and in turn promote RCC
migration (94). ER-β has higher
expression in normal renal tissue than in tumorous tissues. In
contrast, as no ER-α expression was observed in these cell lines,
only ER-β was activated through estrogen stimulation. Estrogen
treatment significantly decreased the proliferation, migration,
invasion, and increased apoptosis of 786-O (high endogenous ER-β),
and ER-β siRNA-induced silencing attenuated the estrogen-induced
effects. Ectopic expression of ER-β in A498 (with low endogenous
ER-β) increased sensitivity to estrogen. Therefore, estrogen is
believed to activate the ER-β suppressive function, resulting in
the elimination of cancer cells (93) which leads to different RCC
incidence rates between males and females. It also implies that
ER-β may be a useful prognostic marker for RCC progression and a
novel developmental direction for improved RCC treatment (95).
Treatment
Chronic treatment with diethylstilbestrol (DES) and
polydiethylstilbestrol phosphate produced renal tumors in male
hamsters. The presence of renal tumors results in increased
activity of hepatic glucuronyl transferase that diminishes DES in
chronically treated hamsters. The antiestrogen nafoxidine,
administered along with DES, completely inhibits tumor formation
(96). Ten patients with advanced
RCC were given combined chemoendocrine treatment with tegafur and
tamoxifen. One out of 2 patients with ER-positive tumors and 3 out
of 4 patients with ER-negative tumors responded favorably to this
treatment (97).
Androgen receptors (AR)
In normal kidneys, androgen receptors (AR) are
constitutively expressed in the proximal and distal tubules and
localized in cell nuclei. They are also focally expressed in some
Bowman's capsule cells. Expression of AR is higher in adjacent
normal kidneys than in RCC tissues. AR expression in RCC is
negatively correlated with pT stage and Fuhrman's grade (97). In RCC tumors, ARs are detectable in
clear cells, papillaries and chromophobe RCCs. No difference of AR
immunoreactivity was detected between histological subtypes
(77). Upregulated expression of
ARs was shown as a favorable marker in RCC (77,98).
In another study by Brown et al (99), which included primary clear cell
RCCs and their metastases, AR immunoreactivity was mainly present
in primary tumors, but not in their respective metastases. AR
expression was higher in adjacent normal kidneys (90.9%) than in
RCC tissues or control group human ccRCC cell lines. Specifically,
there were 40.7% AR-positive cases in pT1 compared with 8.0% in
pT3, and 50.0% of grade I cases were found to be AR-positive
compared with 12.9% in grade III. AR expression was more abundant
in primary RCC tissues (12.5%) than in their respective metastases
(0%). There was no significant difference found in AR-positive
rates between male and female RCC patients from the same subgroups
who had the same pT stages or Fuhrman's grades. Immunohistochemical
analysis of 182 RCC tumors for ER, PR, and AR expression in
relation to associations with histological subtype, pT stage,
grading, gender and impact on disease-free survival was conducted.
AR expression was found in 27 of 182 tumors (14.8%), 24 males and 3
females. AR expression was significantly associated with a lower
stage and grade, moderate, or high differentiation of tumor cells.
Outcome expectancy decreased with de-differentiation and tumor
growth. AR-positive RCCs showed a significantly better prognosis
(77). Another study discovered AR
to be inversely correlated with TNM stage pT1 tumors being
AR-positive for 27% of cases, in contrast to pT3 tumors being 4%
positive. Additionally, the presence of AR was inversely correlated
with nuclear grade. Thus, patients with AR-positive tumors were
observed to have a longer progression-free condition and overall
survival rate (77). Recently,
high AR expression was associated with favorable prognostic
factors, such as low pT stage and low histologic Fuhrman's grade
among the sample of 120 primary RCCs (100,101). In functional studies androgen
receptors (AR) were shown to induce HIF2α/VEGF signals that
potentially drive RCC progression. Anti-AR targeting inhibits RCC
cell migration and invasion (102). Normal kidney cells that were
transformed into cancerous versions had decreased AR expression
rates or more localized cell nuclei. Observations of generally high
levels of AR in hamster renal tumors are consistent with the
finding that the growth rate of transplanted primary renal tumors
is stimulated by testosterone propionate (83). Dihydrotestosterone-specific
receptors were present in all RCC samples examined (20 of 20) and
in 13 of 14 normal renal parenchyma samples. Testosterone receptors
were found in a smaller number of cases. Moreover, significantly
higher levels of the dihydrotestosterone receptor were found in
high-stage compared to low-stage tumors (103). In summary, the utility of AR in
RCC as a prognostic, diagnostic or therapeutic factor is uncertain
and requires further investigation and observation.
Treatment
Hormonal manipulation in patients with RCC seems to
increase survival in patients with these receptors. The survival
rate of patients with 1 or more receptors was significantly higher
than that of patients with no receptors (104). Ahmed et al (105) investigated the efficacy of
flutamide (an anti-androgenic drug) treatment in Phase II
disease-oriented drug trials on patients with advanced
bidimensional RCC. Of 25 treated cases, 1 experienced partial
cancer remission and 2 were deemed progression-free. Flutamide
showed no benefits in patients with disseminated RCC. Sixty-two
specimens were evaluated for steroid binding sites: 33 of 62
specimens showed no hormone-binding sites, and only 12 cases
exhibited androgen binding.
Progesterone receptors (PR)
In normal human kidneys, 30% of investigated tissue
samples were positive for progesterone receptors (PRs) in the
mesangial cells of glomeruli, in interstitial stromal cells and in
several tubules (86). Two
predominant isoforms of PR were found, PR-α and PR-β, both of which
are derived from 1 gene due to alternative promoter usage. Their
DNA binding and steroid hormone activities are similar, although
higher transcriptional activating potential was observed in the
case of PR-β (59). The PR was
found in 40% and 30% of normal and carcinomatous kidney tissue,
respectively (105). PR
expression was decreased in 10% of tumors and increased in only 1%
of patients, one with ccRCC and one with pRCC (77). Less frequent than ER, PR was found
in stromal cells of benign renal carcinomas: angiomyolipomas,
cystic nephromas, oncocytomas, mixed epithelial and stromal tumors,
and also in chromophobe RCC (86).
Expression of PR in tumor stroma was also reported in benign renal
tumors as well as in normal kidneys and metaplastic nodules
(86). In general, PR appears to
be a highly specific marker for chromophobe RCC. It is also a
highly specific and sensitive marker for oncocytomas. In
particular, PR immunoreactivity is more abundant in oncocytomas
than in chromophobe cancer, which can be used to distinguish
between these two tumor types. Moreover, PR expression is not
detectable in other subtypes of RCC tumors, such as pRCC or ccRCC
with eosinophilic cytoplasm (59).
Treatment
Estradiol receptor (ER) and progesterone receptor
(PR) expression was evaluated in 27 RCCs in an attempt to predict
the response to progestational therapy. Patients whose tumors were
positive either for ER or PR (or both) had favorable outcomes from
progestational therapy. Three patients with ER-PR-renal cancer
showed negative results in follow-up therapy (12). In the study of McDonald et
al (13), PR was measured in
eight RCCs compared to nine normal renal tissues and one melanoma
tissue sample. PR was identified in all of the samples, with the
exception of one RCC. Three patients, all of whom had
receptor-positive tumors, were treated with medroxyprogesterone
acetate for metastatic disease. In one of these patients, an
objective response to treatment was achieved. Nakano et al
(104) also demonstrated that
hormonal manipulation in patients with one, or more receptors,
results in a significantly higher survival rate.
6. Summary and conclusions
In the past several decades, clinical observations
and molecular studies have led to the hypothesis that RCC is a
hormone-dependent tumor. Steroid receptors are transcription
factors that control cell differentiation, proliferation and death
(12). Active hormone receptors
are found to be abnormally expressed in RCC cells, while abnormal
endocrine stimulation is thought to influence cell proliferation,
migration and angiogenesis. The expression of steroid receptors
varies between normal kidney tissues and RCC tumors (59). To date, the study of these
receptors in RCC has been confined to estrogen (ER) and
progesterone receptors (PR), but the employment of novel molecular
biology and cell biology techniques have supplemented the data on
steroid hormone RCC dependence. More recently, immunocytochemistry,
tissue and protein microarray platforms, mass spectrometry,
quantitative reverse real-time PCR, whole genome cDNA analysis, and
DNA sequencing have served as functional studies of steroid hormone
receptors in renal cancers (68,106). The molecular role of each hormone
in RCC pathophysiology is currently still being elucidated, in
order to provide a precise model of hormonal interactions with
oncogenesis. RCC patients without any paraneoplastic syndromes have
experienced frequent changes in peptide hormone balance, which is
either directly or indirectly caused by renal cancer tumor masses
and which may in turn influence disease biology. On the basis of
immunohistochemistry staining, specific hormones can be used as
potential biomarkers of progression of oncogenesis, or to aid in
the identification of tumor types. Yet, another targeted approach
would be to use hormonal receptors for treatment, so as to inhibit
hormonal activity with chemical inhibitors. The employment of
techniques such as protein and tissue microarray technology, whole
genome arrays, mass spectrometry, DNA sequencing, and cell cultures
has helped to reveal the expression and role of steroid receptors
and their signaling pathways.
Starting with hypothalamus hormones, the presence
of the GHRH ligand has been demonstrated in cancer cells,
suggesting that GHRH could be a growth factor. In turn, GHRH
antagonists exhibit antitumor effects by suppressing the growth of
ccRCC lines xenografted into nude mice and inhibiting the growth of
orthotopic Caki-1 human RCC, as well as the development of
metastases in lung and lymph nodes. The main action of GHRH
antagonists in vivo appears to be the direct suppression,
through specific binding sites, of autocrine and/or paracrine
production in the pituitary gland and the reduction of expression
of the genes encoding IGF-I (IGF1) and IGF-II (IGF2) in tumors. The
overexpression of GnRH and its receptor has been found in ccRCC. As
LHRH receptor expression was found to be very high in RCCs, its
inhibitors can be targeted to ccRCC tumors expressing these
receptors for therapy. GnRH antagonists effectively inhibited the
growth of tumors in the Caki-1 cell line xenografts of nude mice.
As a result, this group of compounds was proposed as a therapy for
patients with metastatic or recurrent ccRCC. The role of
somatostatin is still unproven and requires further elucidation. On
the one hand, in Phase II trials somatostatin analogue
administration did not result in the control of RCC growth.
However, the findings did raise the possibility of using
renal-derived somatostatin to modulate tubular cell function
through the family of G protein-coupled receptors via autocrine and
paracrine modes. It was proposed that the indirect antitumor
effects of somatostatin receptors are anti-angiogenic actions. CRH
and Ucn were reported to suppress neovascularization through the
reduction of VEGF and the inhibition of tumor cell cycling. POMC
was found to be constitutively upregulated in VHL-mutated renal
cell carcinoma via Nur77: a critical transcription factor
responsible for POMC overproduction that is directly regulated by
HIF-1α during hypoxic conditions.
Biochemical analyses revealed that serum levels of
pituitary gland hormones are also significantly modulated in
patients with urogenital tumors. Elevated GH and IGF-I levels are
associated with oncogenic transformation in a variety of tumors
affecting the general population, and induce mitogenic and
anti-apoptotic effects; for instance, in RCC, thyroid tumors, and
colon cancer, due to local expression of IGF-IR stimulated by
excess production in the liver and direct GHR effects in the tumor
tissues. These mechanisms can be easily observed in acromegaly
patients, who can have multiple cancers simultaneously. ACTH and
ACTH receptors are stimulated by the HPA axis in hypoxia, as their
production is controlled by the main transcription factor Nur77,
the same as in POMC production. Retinoic acid was shown to inhibit
Nur77 in the prevention of Cushing's syndrome. TSH and PRL are
measured by pathological range in patients with RCC, and are
sensitive to indicating distant metastasis in ccRCC carriers.
Moreover, hypothyroidism is associated with longer PFS in sunitinib
and sorafenib treatments. Prolactin elevation was found in 45% of
ccRCC patients, and its level was unrelated to the stage of the
disease. Serum PTH was found to be decreased in patients with tumor
dissemination. The FSHR was identified in the tumor endothelium of
many genitourinary malignancies, including RCC, in which its
expression was mostly observed as being equally located throughout
the tumor's neovasculature. According to one hypothesis, FSH
signaling could induce VEGF in tumor endothelium, which could
contribute to the development of metastatic disease. On the other
hand, it makes FSHR an appealing target for therapeutic and imaging
purposes and as a prognostic biomarker in genitourinary cancers.
FSHR antagonists, such as thiazolidinone analogues, neutralizing
antibodies, and negative allosteric modulators should be further
examined in clinical trials. GRs are abundantly expressed in ccRCC
tumors. Their expression was correlated with a low nuclear grade
and tumor stage. High GR expression is considered to be a marker of
less aggressive RCC tumors. Prolonged OS was reported in RCC
patients with GR-positive tumors. GR signaling in RCC cells likely
results in the suppression of other transcription factors induced
by signaling in cancer cells. MR appears to be a specific and
sensitive marker of the distal nephron and its derived carcinomas:
chromophobe RCCs and oncocytomas. In the case of ccRCC, MR staining
was rarely detected. Therefore, MR serves as a marker for the
expression of subtypes of the major types of renal cell neoplasms.
In addition, recent studies have also shown that increased activity
of the renin-angiotensin-aldosterone system (RAAS), mostly due to
hypertension and obesity, is one of the major risk factors
influencing genetic changes, and is mostly observed in clear cell
forms of RCC. Inhibition of angiotensin-converting enzymes
restricts the growth of human renal cancer cells in a mouse
xenograft system, and in several experimental animal models of
hypertension, overactivity of the RAAS is accompanied by renal
tubular hyperplasia. A potential mechanism for this is the
influence of isoforms of the K-RAS oncogene, whose overexpression
contributes to the survival and increased proliferation of renal
cancer cells in response to activation of the RAAS. The expression
of VDR is inversely correlated with RCC development risk. The
downregulation or loss of receptor expression was only reported in
poorly differentiated sarcomatoid tumors with a poor prognosis.
However, the amount of the receptor in the receptor-positive tumors
did not relate to the other clinical and pathological features of
the patients. Vitamin D, depending on dosage, inhibited cancer
growth, prolonged overall survival in mice, and reduced hepatic and
pulmonary metastates. Because of the vitamin D hypercalcemic toxic
effect, alternative vitamin D-like molecules have been explored and
have shown promising results; these include alkylating derivatives
of 1,25(OH)2D3. RAR and RXR are normally
expressed in proximal tubules and interstitial cells. RAR-β is
involved in solid tumorigenesis, as it is associated with the
deletion of the short arm of chromosome 3, where it has been
mapped. RAR-β mRNA was not detected in renal cancer cell lines,
suggesting either resistance to or minimal inhibition with
13-cis-RA treatment. RXR-γ was found to be a favorable
marker in RCC, with an inverse correlation with clinical and
pathological stages. Patients with RXR-γ-positive tumors were
observed to have prolonged OS. Moreover, retinoid treatment with
13-cis-RA resulted in prolonged PFS and OS in RCC
patients.
Both ER and PR have been found in cytosols of human
RCC, but with a lower binding capacity of cancer cytosols compared
to that of normal kidneys, likely due to a nuclear translocation of
the receptors in the neoplastic tissue. In clinical studies,
positive responses to progestin treatment were demonstrated in
patients with hormone-dependent tumors (ER+
PR− or vice versa), and in those who either did not
develop metastases within 18 to 22 months of nephrectomy or had at
least achieved stabilization of the disease and a longer period of
survival. The question of the influence of exogenous estrogens is
still unclear. First, the expression of ER and PR in some benign
human renal tumors has led to suggestions that they may be related
to excessive exogenous estrogens, as human kidneys are
traditionally thought to be unresponsive to estrogen. Furthermore,
angiomyolipomas have been reported to increase in size during
pregnancy or secondary oral contraceptive therapy. RCC can be
experimentally induced by exposure to estrogens in animal models.
Moreover, renal cancer incidence seems to be gender-related, with
an incidence that is 2 times higher in men than in women. The data
strongly suggest an environmental, exogenous influence of estrogens
in RCC etiology, but more research is still needed. The frequency
of ER expression in human RCCs was highly variable in different RCC
studies. Many reports provide evidence that ER is most often not
expressed in RCCs. Moreover, cathepsin D was found to be
estrogen-induced in hamster kidneys, leading to tumorigenesis by
the mediation of renal tubular damage following reparation of cell
proliferation that contributes to RCC formation. In primary
estrogen-induced renal tumors and their metastases, significantly
elevated levels of all 3 cathepsin D isoforms were detected. ER-α
was found to be a novel proteasomal degradation target of the pVHL
E3 ligase, thus downregulation of pVHL (in VHL-deficient cells) can
increase ER-α expression, which in turn can increase the
transcription factor activity of HIF-1α by binding to it and then
promoting tumor progression. Using ER-α inhibitors (i.e. faslodex)
could suppress the proliferation of VHL-deficient cells, as could
inducing p53 expression. ER-β might also play a tumor suppressive,
anti-proliferative role. Estrogen-activated ER-β reduced growth
hormone downstream signaling pathways and also increased apoptotic
cascade activation. Estrogen treatment significantly decreased the
proliferation, migration, invasion, and increased apoptosis of cell
lines with high endogenous ER-β. Therefore, estrogen is believed to
activate ER-β's suppressive function, resulting in the elimination
of cancer cells; this could explain different RCC incidence rates
between males and females. It is implied that ER-β may be a useful
prognostic marker for RCC progression and a novel direction for RCC
treatment. AR expression was found to be higher in adjacent normal
kidneys than in RCC tissues, and was slightly higher in primary RCC
tissues than in their respective metastases (no expression). Normal
kidney cells that were transformed into cancerous versions had
decreased AR expression rates or more localized cell nuclei. They
were also negatively associated with pT stage and Fuhrman's grade,
although results did not show any significant difference between
male and female RCC patients in the subgroups which had the same pT
stage or Fuhrman's grade. In contrast, androgen receptor-induced
HIF2α/VEGF signals that drive RCC progression and AR targeting
inhibited RCC cell migration and invasion. Additionally,
significantly higher levels of the dihydrotestosterone receptor
were found in high-stage compared to low-stage tumors. PR
immunoreactivity serves as a highly specific and sensitive
diagnostic marker for chromophobe RCC and oncocytomas, which could
be used for distinguishing between these 2 subtypes. PRs are also
found in clear cell RCCs, but to a lesser extent. The increased
expression of PRs is a favorable prognostic marker in RCC.
Prolonged estrogen treatment augments the levels of specific
progesterone binding. PRs were found, although less frequently than
ER, in mostly ovarian-like stromal cells of benign renal neoplasms.
Patients whose tumors were positive either for ER or PR (or both)
had favorable outcomes from progestational therapy. The
above-mentioned examples underscore the possibility of alternative
pathway activation in ccRCC via hormones, and demonstrate the need
to explore the molecular basis of such phenomena as they are
observed in clinics.
Acknowledgements
The present review was supported by the National
Science Centre (NCN) grant no. UMO-2012/05/D/NZ5/01844 and WIM
intramural grant nr 1/8863 (355).
References
|
1
|
Czarnecka AM, Szczylik C and Rini B: The
use of sunitinib in renal cell carcinoma: Where are we now? Expert
Rev Anticancer Ther. 14:983–999. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Motzer RJ, Bander NH and Nanus DM:
Renal-cell carcinoma. N Engl J Med. 335:865–875. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wood LS: Renal cell carcinoma: Screening,
diagnosis, and prognosis. Clin J Oncol Nurs. 13(Suppl): 3–7. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
DeSantis CE, Lin CC, Mariotto AB, Siegel
RL, Stein KD, Kramer JL, Alteri R, Robbins AS and Jemal A: Cancer
treatment and survivorship statistics, 2014. CA Cancer J Clin.
64:252–271. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Molina AM, Lin X, Korytowsky B, Matczak E,
Lechuga MJ, Wiltshire R and Motzer RJ: Sunitinib objective response
in metastatic renal cell carcinoma: Analysis of 1059 patients
treated on clinical trials. Eur J Cancer. 50:351–358. 2014.
View Article : Google Scholar
|
|
7
|
Gore ME, Szczylik C, Porta C, Bracarda S,
Bjarnason GA, Oudard S, Lee SH, Haanen J, Castellano D, Vrdoljak E,
et al: Final results from the large sunitinib global
expanded-access trial in metastatic renal cell carcinoma. Br J
Cancer. 113:12–19. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jung YS, Lee SJ, Yoon MH, Ha NC and Park
BJ: Estrogen receptor α is a novel target of the Von Hippel-Lindau
protein and is responsible for the proliferation of VHL-deficient
cells under hypoxic conditions. Cell Cycle. 11:4462–4473. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Danilin S, Sourbier C, Thomas L, Rothhut
S, Lindner V, Helwig JJ, Jacqmin D, Lang H and Massfelder T: von
Hippel-Lindau tumor suppressor gene-dependent mRNA stabilization of
the survival factor parathyroid hormone-related protein in human
renal cell carcinoma by the RNA-binding protein HuR.
Carcinogenesis. 30:387–396. 2009. View Article : Google Scholar
|
|
10
|
Buczek M, Escudier B, Bartnik E, Szczylik
C and Czarnecka A: Resistance to tyrosine kinase inhibitors in
clear cell renal cell carcinoma: From the patient's bed to
molecular mechanisms. Biochim Biophys Acta. 1845:31–41. 2014.
|
|
11
|
Kornakiewicz A, Solarek W, Bielecka ZF,
Lian F, Szczylik C and Czarnecka AM: Mammalian target of rapamycin
inhibitors resistance mechanisms in clear cell renal cell
carcinoma. Curr Signal Transduct Ther. 8:210–218. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Concolino G, Marocchi A, Conti C, Tenaglia
R, Di Silverio F and Bracci U: Human renal cell carcinoma as a
hormone-dependent tumor. Cancer Res. 38:4340–4344. 1978.PubMed/NCBI
|
|
13
|
McDonald MW, Diokno AC, Seski JC and Menon
KM: Measurement of progesterone receptor in human renal cell
carcinoma and normal renal tissue. J Surg Oncol. 22:164–166. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Concolino G, Di Silverio F, Marocchi A and
Bracci U: Renal cancer steroid receptors: Biochemical basis for
endocrine therapy. Eur Urol. 5:90–93. 1979.PubMed/NCBI
|
|
15
|
Concolino G, Di Silverio F, Marocchi A and
Bracci U: Renal cancer steroid receptors: Biochemical basis for
endocrine therapy. Eur Urol. 5:319–322. 1979.PubMed/NCBI
|
|
16
|
Jang JH, Min KJ, Kim S, Park JW and Kwon
TK: RU486 induces pro-apoptotic endoplasmic reticulum stress
through the induction of CHOP expression by enhancing C/EBPdelta
expression in human renal carcinoma Caki cells. J Cell Biochem.
117:361–369. 2015. View Article : Google Scholar
|
|
17
|
Zucchetto A, Talamini R, Dal Maso L, Negri
E, Polesel J, Ramazzotti V, Montella M, Canzonieri V, Serraino D,
La Vecchia C, et al: Reproductive, menstrual, and other
hormone-related factors and risk of renal cell cancer. Int J
Cancer. 123:2213–2216. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kabat GC, Silvera SA, Miller AB and Rohan
TE: A cohort study of reproductive and hormonal factors and renal
cell cancer risk in women. Br J Cancer. 96:845–849. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Qu Y, Chen H, Gu W, Gu C, Zhang H, Xu J,
Zhu Y and D: Age-dependent association between sex and renal cell
carcinoma mortality: A population-based analysis. Sci Rep.
5:91602015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dunzendorfer U, Drahovsky D and
Schmidt-Gayk H: Peptide hormones LH, FSH, TSH, prolactin, beta-HCG
and PTH in patients with urogenital tumors. Onkologie. 4:188–192.
1981.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aguilar-Rojas A and Huerta-Reyes M: Human
gonadotropin-releasing hormone receptor-activated cellular
functions and signaling pathways in extra-pituitary tissues and
cancer cells (Review). Oncol Rep. 22:981–990. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guillermet-Guibert J, Lahlou H, Cordelier
P, Bousquet C, Pyronnet S and Susini C: Physiology of somatostatin
receptors. J Endocrinol Invest. 28(Suppl Int): 5–9. 2005.
|
|
23
|
Lu HT, Salamon H and Horuk R: The biology
and physiology of somatostatin receptors. Expert Opin Ther Targets.
5:613–623. 2001. View Article : Google Scholar
|
|
24
|
Agouni A, Sourbier C, Danilin S, Rothhut
S, Lindner V, Jacqmin D, Helwig JJ, Lang H and Massfelder T:
Parathyroid hormone-related protein induces cell survival in human
renal cell carcinoma through the PI3K Akt pathway: Evidence for a
critical role for integrin-linked kinase and nuclear factor kappa
B. Carcinogenesis. 28:1893–1901. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Riesenbeck LM, Bierer S, Hoffmeister I,
Köpke T, Papavassilis P, Hertle L, Thielen B and Herrmann E:
Hypothyroidism correlates with a better prognosis in metastatic
renal cancer patients treated with sorafenib or sunitinib. World J
Urol. 29:807–813. 2011. View Article : Google Scholar
|
|
26
|
Baldazzi V, Tassi R, Lapini A, Santomaggio
C, Carini M and Mazzanti R: The impact of sunitinib-induced
hypothyroidism on progression-free survival of metastatic renal
cancer patients: A prospective single-center study. Urol Oncol.
30:704–710. 2012. View Article : Google Scholar
|
|
27
|
Liao H, Zhou Q, Gu Y, Duan T and Feng Y:
Luteinizing hormone facilitates angiogenesis in ovarian epithelial
tumor cells and metformin inhibits the effect through the mTOR
signaling pathway. Oncol Rep. 27:1873–1878. 2012.PubMed/NCBI
|
|
28
|
Schmidinger M, Vogl UM, Bojic M, Lamm W,
Heinzl H, Haitel A, Clodi M, Kramer G and Zielinski CC:
Hypothyroidism in patients with renal cell carcinoma: Blessing or
curse? Cancer. 117:534–544. 2011. View Article : Google Scholar
|
|
29
|
Dunzendorfer U, Drahovsky D, Schmidt-Gayk
H and Zahradnik HP: Clinical significance of peptide hormones LH,
FSH, TSH, prolactin, HCG, parathormone, calcitonin and
prostaglandin F2 alpha in kidney neoplasms. Z Urol Nephrol.
74:13–19. 1981.(In German). PubMed/NCBI
|
|
30
|
Siraj MA, Pichon C, Radu A and Ghinea N:
Endothelial follicle stimulating hormone receptor in primary kidney
cancer correlates with subsequent response to sunitinib. J Cell Mol
Med. 16:2010–2016. 2012. View Article : Google Scholar
|
|
31
|
Schally AV and Varga JL: Antagonistic
analogs of growth Hormone-releasing hormone: New potential
antitumor agents. Trends Endocrinol Metab. 10:383–391. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schally AV, Varga JL and Engel JB:
Antagonists of growth-hormone-releasing hormone: An emerging new
therapy for cancer. Nat Clin Pract Endocrinol Metab. 4:33–43. 2008.
View Article : Google Scholar
|
|
33
|
Jungwirth A, Schally AV, Pinski J, Groot
K, Armatis P and Halmos G: Growth hormone-releasing hormone
antagonist MZ-4–71 inhibits in vivo proliferation of Caki-I renal
adenocarcinoma. Proc Natl Acad Sci USA. 94:5810–5813. 1997.
View Article : Google Scholar
|
|
34
|
Halmos G, Schally AV, Varga JL, Plonowski
A, Rekasi Z and Czompoly T: Human renal cell carcinoma expresses
distinct binding sites for growth hormone-releasing hormone. Proc
Natl Acad Sci USA. 97:10555–10560. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rick F, Szalontay L, Abi-Chaker A, Block
NL, Halmos G and Schally AV: Effect of novel growth
hormone-releasing hormone antagonists on growth of experimental
renal cell carcinomas. J Clin Oncol. 31:abstr 469. 2013.
|
|
36
|
Vikić-Topić S, Raisch KP, Kvols LK and
Vuk-Pavlović S: Expression of somatostatin receptor subtypes in
breast carcinoma, carcinoid tumor, and renal cell carcinoma. J Clin
Endocrinol Metab. 80:2974–2979. 1995.
|
|
37
|
Turman MA and Apple CA: Human proximal
tubular epithelial cells express somatostatin: Regulation by growth
factors and cAMP. Am J Physiol. 274:F1095–F1101. 1998.PubMed/NCBI
|
|
38
|
Freudenberg LS, Gauler T, Görges R, Bauer
S, Stergar H, Antoch G, Bockisch A and Schütte J: Somatostatin
receptor scintigraphy in advanced renal cell carcinoma. Results of
a phase II-trial of somatostatine analogue therapy in patients with
advanced RCC. Nuklearmedizin. 47:127–131. 2008.PubMed/NCBI
|
|
39
|
Tezval H, Jurk S, Atschekzei F, Becker JU,
Jahn O, Serth J and Kuczyk MA: Urocortin and
corticotropin-releasing factor receptor 2 in human renal cell
carcinoma: Disruption of an endogenous inhibitor of angiogenesis
and proliferation. World J Urol. 27:825–830. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tezval H, Atschekzei F, Peters I, Waalkes
S, Hennenlotter J, Stenzl A, Becker JU, Merseburger AS, Kuczyk MA
and Serth J: Reduced mRNA expression level of
corticotropin-releasing hormone-binding protein is associated with
aggressive human kidney cancer. BMC Cancer. 13:1992013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Thwaites DT, Hirst BH and Simmons NL:
Passive transepithelial absorption of thyrotropin-releasing hormone
(TRH) via a para-cellular route in cultured intestinal and renal
epithelial cell lines. Pharm Res. 10:674–681. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Arai E, Chiku S, Mori T, Gotoh M, Nakagawa
T, Fujimoto H and Kanai Y: Single-CpG-resolution methylome analysis
identifies clinicopathologically aggressive CpG island methylator
phenotype clear cell renal cell carcinomas. Carcinogenesis.
33:1487–1493. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Keller G, Schally AV, Gaiser T, Nagy A,
Baker B, Halmos G and Engel JB: Receptors for luteinizing hormone
releasing hormone expressed on human renal cell carcinomas can be
used for targeted chemotherapy with cytotoxic luteinizing hormone
releasing hormone analogues. Clin Cancer Res. 11:5549–5557. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Engel J, Emons G, Pinski J and Schally AV:
AEZS-108: A targeted cytotoxic analog of LHRH for the treatment of
cancers positive for LHRH receptors. Expert Opin Investig Drugs.
21:891–899. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jungwirth A, Schally AV, Halmos G, Groot
K, Szepeshazi K, Pinski J and Armatis P: Inhibition of the growth
of Caki-I human renal adenocarcinoma in vivo by luteinizing
hormone-releasing hormone antagonist Cetrorelix, somatostatin
analog RC-160, and bombesin antagonist RC-3940-II. Cancer.
82:909–917. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Choi JW, Park SC, Kang GH, Liu JO and Youn
HD: Nur77 activated by hypoxia-inducible factor-1alpha overproduces
proopiomelanocortin in von Hippel-Lindau-mutated renal cell
carcinoma. Cancer Res. 64:35–39. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Waters MJ: The growth hormone receptor.
Growth Horm IGF Res. Jun 7–2015. View Article : Google Scholar : (Epub ahead of
print). PubMed/NCBI
|
|
48
|
Gan Y, Buckels A, Liu Y, Zhang Y, Paterson
AJ, Jiang J, Zinn KR and Frank SJ: Human GH receptor-IGF-1 receptor
interaction: Implications for GH signaling. Mol Endocrinol.
28:1841–1854. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sekizawa N, Hayakawa E, Tsuchiya K,
Yoshimoto T, Akashi T, Fujii T, Yamada S and Hirata Y: Acromegaly
associated with multiple tumors. Intern Med. 48:1273–1278. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Asai K, Shimoyama S, Sanno N, Kaminishi M
and Oohara T: A rare case of gastric cancer in an acromegalic
patient. J Gastroenterol. 32:528–532. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Prinzi N, Sorrenti S, Baldini E, De Vito
C, Tuccilli C, Catania A, Coccaro C, Bianchini M, Nesca A, Grani G,
et al: Association of thyroid diseases with primary extra-thyroidal
malignancies in women: Results of a cross-sectional study of 6,386
patients. PLoS One. 10:e01229582015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bailey EB, Tantravahi SK, Poole A, Agarwal
AM, Straubhar AM, Batten JA, Patel SB, Wells CE, Stenehjem DD and
Agarwal N: Correlation of degree of hypothyroidism with survival
outcomes in patients with metastatic renal cell carcinoma receiving
vascular endothelial growth factor receptor tyrosine kinase
inhibitors. Clin Genitourin Cancer. 13:e131–e137. 2015. View Article : Google Scholar
|
|
53
|
Nearchou A, Valachis A, Lind P, Akre O and
Sandström P: Acquired hypothyroidism as a predictive marker of
outcome in patients with metastatic rmenal cell carcinoma treated
with tyrosine kinase inhibitors: A literature-based meta-analysis.
Clin Genitourin Cancer. 13:280–286. 2015. View Article : Google Scholar
|
|
54
|
Siraj A, Desestret V, Antoine M, Fromont
G, Huerre M, Sanson M, Camparo P, Pichon C, Planeix F, Gonin J, et
al: Expression of follicle-stimulating hormone receptor by the
vascular endothelium in tumor metastases. BMC Cancer. 13:2462013.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Radu A, Pichon C, Camparo P, Antoine M,
Allory Y, Couvelard A, Fromont G, Hai MT and Ghinea N: Expression
of follicle-stimulating hormone receptor in tumor blood vessels. N
Engl J Med. 363:1621–1630. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gartrell BA, Tsao CK and Galsky MD: The
follicle-stimulating hormone receptor: A novel target in
genitourinary malignancies. Urol Oncol. 31:1403–1407. 2013.
View Article : Google Scholar
|
|
57
|
Alam H, Weck J, Maizels E, Park Y, Lee EJ,
Ashcroft M and Hunzicker-Dunn M: Role of the
phosphatidylinositol-3-kinase and extracellular regulated kinase
pathways in the induction of hypoxia-inducible factor (HIF)-1
activity and the HIF-1 target vascular endothelial growth factor in
ovarian granulosa cells in response to follicle-stimulating
hormone. Endocrinology. 150:915–928. 2009. View Article : Google Scholar :
|
|
58
|
Stanisic TH and Donovan J: Prolactin
secreting renal cell carcinoma. J Urol. 136:85–86. 1986.PubMed/NCBI
|
|
59
|
Yakirevich E, Matoso A, Morris D and
Resnick M: Steroid receptors in renal cell carcinoma. Emerging
Research and Treatments in Renal Cell Carcinoma. Amato RJ: InTech;
Rijeka, Croatia: 2012, View
Article : Google Scholar
|
|
60
|
Iwai A, Fujii Y, Kawakami S, Takazawa R,
Kageyama Y, Yoshida MA and Kihara K: Down-regulation of vascular
endothelial growth factor in renal cell carcinoma cells by
glucocorticoids. Mol Cell Endocrinol. 226:11–17. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yakirevich E, Matoso A, Sabo E, Wang LJ,
Tavares R, Meitner P, Morris DJ, Pareek G, Delellis RA and Resnick
MB: Expression of the glucocorticoid receptor in renal cell
neoplasms: An immunohistochemical and quantitative reverse
transcriptase polymerase chain reaction study. Hum Pathol.
42:1684–1692. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Min KJ, Jang JH, Lee JT, Choi KS and Kwon
TK: Glucocorticoid receptor antagonist sensitizes TRAIL-induced
apoptosis in renal carcinoma cells through up-regulation of DR5 and
down-regulation of c-FLIP(L) and Bcl-2. J Mol Med Berl. 90:309–319.
2012. View Article : Google Scholar
|
|
63
|
Bojar H, Maar K and Staib W: The endocrine
background of human renal cell carcinoma. III Role of inhibitors of
R 5020 binding in tumour cytosol. Urol Int. 34:321–329. 1979.
View Article : Google Scholar
|
|
64
|
Bojar H, Maar K and Staib W: The endocrine
background of human renal cell carcinoma. IV Glucocorticoid
receptors as possible mediators of progestogen action. Urol Int.
34:330–338. 1979. View Article : Google Scholar
|
|
65
|
Arai Y, Nonomura N, Nakai Y, Nishimura K,
Oka D, Shiba M, Nakayama M, Takayama H, Mizutani Y, Miki T, et al:
The growth-inhibitory effects of dexamethasone on renal cell
carcinoma in vivo and in vitro. Cancer Invest. 26:35–40. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Christophersen AO, Lie AK and Fosså SD:
Unexpected 10 years complete remission after cortisone mono-therapy
in metastatic renal cell carcinoma. Acta Oncol. 45:226–228. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yakirevich E, Morris DJ, Tavares R,
Meitner PA, Lechpammer M, Noble L, de Rodriguez AF, Gomez-Sanchez
CE, Wang LJ, Sabo E, et al: Mineralocorticoid receptor and
11beta-hydroxysteroid dehydrogenase type II expression in renal
cell neoplasms: A tissue microarray and quantitative RT-PCR study.
Am J Surg Pathol. 32:874–883. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
King S, Bray S, Galbraith S, Christie L
and Fleming S: Evidence for aldosterone-dependent growth of renal
cell carcinoma. Int J Exp Pathol. 95:244–250. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Varela I, Tarpey P, Raine K, Huang D, Ong
CK, Stephens P, Davies H, Jones D, Lin ML, Teague J, et al: Exome
sequencing identifies frequent mutation of the SWI/SNF complex gene
PBRM1 in renal carcinoma. Nature. 469:539–542. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kotelevtsev Y, Brown RW, Fleming S, Kenyon
C, Edwards CR, Seckl JR and Mullins JJ: Hypertension in mice
lacking 11beta-hydroxysteroid dehydrogenase type 2. J Clin Invest.
103:683–689. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Khan MI, Bielecka ZF, Najm MZ, Bartnik E,
Czarnecki JS, Czarnecka AM and Szczylik C: Vitamin D receptor gene
polymorphisms in breast and renal cancer: Current state and future
approaches (Review). Int J Oncol. 44:349–363. 2014.
|
|
72
|
Nagakura K, Hayakawa M, Hata M and
Nakamura H: 1,25-Dihydroxyvitamin D3 receptors and their
relationship to histological features in renal cell carcinoma. J
Urol. 137:1300–1303. 1987.PubMed/NCBI
|
|
73
|
Liu W, Tretiakova M, Kong J, Turkyilmaz M,
Li YC and Krausz T: Expression of vitamin D3 receptor in kidney
tumors. Hum Pathol. 37:1268–1278. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Trydal T, Bakke A, Aksnes L and Aarskog D:
1,25-Dihydro−xyvitamin D3 receptor measurement in
primary renal cell carcinomas and autologous normal kidney tissue.
Cancer Res. 48:2458–2461. 1988.PubMed/NCBI
|
|
75
|
Madej A, Puzianowska-Kuznicka M, Tanski Z,
Nauman J and Nauman A: Vitamin D receptor binding to DNA is altered
without the change in its expression in human renal clear cell
cancer. Nephron Exp Nephrol. 93:e150–e157. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Blomberg Jensen M, Andersen CB, Nielsen
JE, Bagi P, Jørgensen A, Juul A and Leffers H: Expression of the
vitamin D receptor, 25-hydroxylases, 1alpha-hydroxylase and
24-hydroxylase in the human kidney and renal clear cell cancer. J
Steroid Biochem Mol Biol. 121:376–382. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Langner C, Ratschek M, Rehak P, Schips L
and Zigeuner R: Steroid hormone receptor expression in renal cell
carcinoma: An immunohistochemical analysis of 182 tumors. J Urol.
171:611–614. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Fujioka T, Hasegawa M, Ishikura K,
Matsushita Y, Sato M and Tanji S: Inhibition of tumor growth and
angiogenesis by vitamin D3 agents in murine renal cell carcinoma. J
Urol. 160:247–251. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Lambert JR, Eddy VJ, Young CD, Persons KS,
Sarkar S, Kelly JA, Genova E, Lucia MS, Faller DV and Ray R: A
vitamin D receptor-alkylating derivative of 1α,25-dihydroxyvitamin
D3 inhibits growth of human kidney cancer cells and suppresses
tumor growth. Cancer Prev Res (Phila). 3:1596–1607. 2010.
View Article : Google Scholar
|
|
80
|
Obara W, Konda R, Akasaka S, Nakamura S,
Sugawara A and Fujioka T: Prognostic significance of vitamin D
receptor and retinoid X receptor expression in renal cell
carcinoma. J Urol. 178:1497–1503. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Motzer RJ, Murphy BA, Bacik J, Schwartz
LH, Nanus DM, Mariani T, Loehrer P, Wilding G, Fairclough DL, Cella
D, et al: Phase III trial of interferon alfa-2a with or without
13-cis-retinoic acid for patients with advanced renal cell
carcinoma. J Clin Oncol. 18:2972–2980. 2000.PubMed/NCBI
|
|
82
|
Atzpodien J, Kirchner H, Jonas U, Bergmann
L, Schott H, Heynemann H, Fornara P, Loening SA, Roigas J, Müller
SC, et al; Prospectively Randomized Trial of the German Cooperative
Renal Carcinoma Chemoimmunotherapy Group (DGCIN). Interleukin-2-
and interferon alfa-2a-based immunochemotherapy in advanced renal
cell carcinoma: A Prospectively Randomized Trial of the German
Cooperative Renal Carcinoma Chemoimmunotherapy Group (DGCIN). J
Clin Oncol. 22:1188–1194. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Li JJ, Li SA and Cuthbertson TL: Nuclear
retention of all steroid hormone receptor classes in the hamster
renal carcinoma. Cancer Res. 39:2647–2651. 1979.PubMed/NCBI
|
|
84
|
Vrtačnik P, Ostanek B, Mencej-Bedrač S and
Marc J: The many faces of estrogen signaling. Biochem Med Zagreb.
24:329–342. 2014. View Article : Google Scholar
|
|
85
|
Hemstreet GP III, Wittliff JL, Sarrif AM,
Hall ML III, McRae LJ and Durant JR: Comparison of steroid receptor
levels in renal-cell carcinoma and autologous normal kidney. Int J
Cancer. 26:769–775. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Tickoo SK, Gopalan A, Tu JJ, Harik LR,
Al-Ahmadie HA, Fine SW, Olgac S and Reuter VE: Estrogen and
progesterone-receptor-positive stroma as a non-tumorous
proliferation in kidneys: A possible metaplastic response to
obstruction. Mod Pathol. 21:60–65. 2008. View Article : Google Scholar
|
|
87
|
Fucic A, Gamulin M, Ferencic Z, Katic J,
Krayer von Krauss M, Bartonova A and Merlo DF: Environmental
exposure to xenoestrogens and oestrogen related cancers:
Reproductive system, breast, lung, kidney, pancreas, and brain.
Environ Health. 11(Suppl 1): S82012. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Adsay NV, Eble JN, Srigley JR, Jones EC
and Grignon DJ: Mixed epithelial and stromal tumor of the kidney.
Am J Surg Pathol. 24:958–970. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Li SA, Liao DZ, Yazlovitskaya EM, Pantazis
CG and Li JJ: Induction of cathepsin D protein during estrogen
carcinogenesis: Possible role in estrogen-mediated kidney tubular
cell damage. Carcinogenesis. 18:1375–1380. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Jakse G and Müller-Holzner E: Hormone
receptors in renal cancer: An overview. Semin Surg Oncol.
4:161–164. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Tanaka Y, Sasaki M, Kaneuchi M, Fujimoto S
and Dahiya R: Estrogen receptor alpha polymorphisms and renal cell
carcinoma - a possible risk. Mol Cell Endocrinol. 202:109–116.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Tanaka Y, Sasaki M, Kaneuchi M, Fujimoto S
and Dahiya R: Single nucleotide polymorphisms of estrogen receptor
alpha in human renal cell carcinoma. Biochem Biophys Res Commun.
296:1200–1206. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Yu CP, Ho JY, Huang YT, Cha TL, Sun GH, Yu
DS, Chang FW, Chen SP and Hsu RJ: Estrogen inhibits renal cell
carcinoma cell progression through estrogen receptor-β activation.
PLoS One. 8:e566672013. View Article : Google Scholar
|
|
94
|
Song W, Yeh CR, He D, Wang Y, Xie H, Pang
ST, Chang LS, Li L and Yeh S: Infiltrating neutrophils promote
renal cell carcinoma progression via VEGFa/HIF2α and estrogen
receptor β signals. Oncotarget. 6:19290–19304. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Ivantsov AO, Imianitov EN, Moiseenko VM,
Matsko DE and Artem'eva AS: Expression of Ki-67, p53, bcl-2,
estrogen receptors alpha in patients with clear cell renal
carcinoma and epidermal growth factor receptor mutation. Arkh
Patol. 73:6–7. 2011.(In Russian). PubMed/NCBI
|
|
96
|
Antonio P, Gabaldón M, Lacomba T and Juan
A: Effect of the antiestrogen nafoxidine on the occurrence of
estrogen-dependent renal tumors in hamster. Horm Metab Res.
6:522–524. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Wada T, Nishiyama K, Maeda M, Hara S,
Tanaka N, Yasutomi M and Kurita T: Combined chemoendocrine
treatment with tegafur and tamoxifen for advanced renal cell
carcinoma. Anticancer Res. 15:1581–1584. 1995.PubMed/NCBI
|
|
98
|
Kimura N, Mizokami A, Oonuma T, Sasano H
and Nagura H: Immunocytochemical localization of androgen receptor
with polyclonal antibody in paraffin-embedded human tissues. J
Histochem Cytochem. 41:671–678. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Brown DF, Dababo MA, Hladik CL, Eagan KP,
White CL III and Rushing EJ: Hormone receptor immunoreactivity in
heman-gioblastomas and clear cell renal cell carcinomas. Mod
Pathol. 11:55–59. 1998.PubMed/NCBI
|
|
100
|
Putz J, Wirth MP, Froehner M and Jahn S:
Re: Zhu et al: The expression and evaluation of androgen receptor
in human renal cell carcinoma (Urology 2014;83:510.e19–24).
Urology. 84:734–735. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Zhu G, Liang L, Li L, Dang Q, Song W, Yeh
S, He D and Chang C: The expression and evaluation of androgen
receptor in human renal cell carcinoma. Urology. 83:510.e519–524.
2014. View Article : Google Scholar
|
|
102
|
He D, Li L, Zhu G, Liang L, Guan Z, Chang
L, Chen Y, Yeh S and Chang C: ASC-J9 suppresses renal cell
carcinoma progression by targeting an androgen receptor-dependent
HIF2α/VEGF signaling pathway. Cancer Res. 74:4420–4430. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Noronha RF and Rao BR: Increased
dihydrotestosterone receptor levels in high-stage renal
adenocarcinoma. Cancer. 56:134–137. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Nakano E, Tada Y, Fujioka H, Matsuda M,
Osafune M, Kotake T, Sato B, Takaha M and Sonoda T: Hormone
receptor in renal cell carcinoma and correlation with clinical
response to endocrine therapy. J Urol. 132:240–245. 1984.PubMed/NCBI
|
|
105
|
Ahmed T, Benedetto P, Yagoda A, Watson RC,
Scher HI, Herr HW, Sogani PC, Whitmore WF and Pertschuk L:
Estrogen, progesterone, and androgen-binding sites in renal cell
carcinoma. Observations obtained in Phase II trial of flutamide.
Cancer. 54:477–481. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Liu Z and Lu Y, He Z, Chen L and Lu Y:
Expression analysis of the estrogen receptor target genes in renal
cell carcinoma. Mol Med Rep. 11:75–82. 2015.
|