Introduction
Hypoxia as a consequence of low oxygenation caused
by impaired and aberrant vascularization is a common feature of
many malignant tumors. Hypoxia leads to reduced apoptosis,
increased proliferation and angiogenesis predominantly via altered
gene expression. The main factor mediating this oxygen sensitive
response is the hypoxia-inducible factor-1 (HIF-1) that consists of
a constitutive β subunit and an oxygen-sensitive α subunit that is
regulated through O2-dependent degradation by prolyl
hydroxylation. Oxygenation of cells results in the binding of the
von Hippel-Lindau tumor-suppressor protein specifically to
hydroxylated HIF-1α which ensures its ubiquitylation and rapid
proteasomal degradation (1).
During hypoxia these processes are inhibited, HIF-1α accumulates,
dimerizes with HIF-1β to generate the functional HIF-1 that
regulates many genes responsible for adaptive responses, which are
important for cell survival at low oxygen levels (2).
One of the proteins, which support adaptation of
tumor cells to hypoxia is carbonic anhydrase IX (CA IX). CA IX is a
tumor-associated, membrane located metalloenzyme catalyzing the
reversible conversion of carbon dioxide to bicarbonate ion and
proton (reviewed in ref. 3). Its
activity, dependent on the phosphorylation of Thr443 residue by
protein kinase A contributes to intracellular pH maintenance
(4), supporting cancer cell
survival in the conditions of hypoxia and related acidosis and
promoting their migration and invasion (5). Moreover, through its unique
extracellular proteoglycan domain it is involved in cell adhesion
and spreading (6). Transcription
of CA9 gene is primarily regulated by the hypoxia-inducible
HIF-1 transcription factor that binds to the hypoxia response
element (HRE) located next to the transcription initiation site
(7) and CA IX is considered as a
marker of tumor hypoxia. CA IX is expressed in a broad range of
tumors where its strong expression often associates with worse
prognosis. Due to its tumor-related expression and its role in
pro-survival and pro-metastatic processes of tumor cells CA IX
represents a promising target for antitumor therapy (8).
Recently, it was described that hypoxia also occurs
as a result of the inflammation when HIF-1α can be regulated
independently of the vascularization level induced by growth
factors, pro-inflammatory cytokines, reactive oxygen and nitrogen
species or mitochondrial stress (9). Moreover, hypoxia can actively
participate in the development of the inflammatory microenvironment
through the promotion of many pro-inflammatory genes (10) governed by nuclear factor-kappaB
transcription factor (NF-κB) (11,12).
Mammalian NF-κB family consists of: NF-κB1 (p50/p105), NF-κB2
(p52/p100), RelA (p65), RelB and c-Rel. The active NF-κB is a
heterodimer typically consisting of NF-κB1 and RelA subunits. In
unstimulated cells, a latent protein complex is sequestered in the
cytoplasm by the associated inhibitor IκB. The activation of the
NF-κB is initiated by the phosphorylation of IκB proteins mediated
via the signal-induced activation of IκB kinase (IKK). This leads
to the ubiquitylation and degradation of IκB in the proteasome
which results in a release of the NF-κB complex and its subsequent
relocation to the nucleus (13,14).
Transcriptional targets of NF-κB transcription factor include
mostly pro-inflammatory genes encoding cytokines, chemokines,
adhesion molecules as well as angiogenic factors and key enzymes
involved in prostaglandin synthase or nitric oxide synthase
pathways. In this way, NF-κB directly contributes to the
development of inflammation. A number of studies provided evidence
on aberrant regulation of NF-κB in many cancers where it actively
participates in tumor initiation and progression (15).
Immunosuppressive properties and their potent
ability to reduce inflammation make synthetic glucocorticoids (GCs)
the most prescribed drugs used in the treatment of different
disorders, such as asthma, arthritis or dermatitis. GCs are also
used to treat patients suffering from a wide range of hematological
and non-hematological cancers either because of their inhibiting
effects on cell cycle progression and apoptosis promotion or for
their beneficial properties, e.g. decreasing oedema, pain, nausea
and reducing toxicity of the standard chemotherapy regimens in
healthy tissues (16,17). The effect of GCs is executed
through glucocorticoid receptors (GRs) which belong to the nuclear
receptor family of ligand-dependent transcription factors (18).
Among synthetic GCs a potent and widely used one is
dexamethasone (DEX). In multiple cases dexamethasone was shown to
affect angiogenesis. This action is achieved by dexamethasone
regulation of mRNA and protein levels of VEGF, which belongs to
HIF-1 targets. Decrease of VEGF levels by DEX was reported in brain
astrocytes and pericytes (19),
hyalocytes (20) and also in
several cell lines derived from tumors, such as renal carcinoma
(21), rat glioma (22), prostate tumor (23) and meningiomas (24), where it was linked with HIF-1
involvement. A substantial reduction of HIF-1 and VEGF expression
also occurred in hypoxic mice treated with dexamethasone (25). Microarray analysis of renal
proximal tubular epithelial cells showed that hypoxia
transcriptionally upregulated glucocorticoid receptors, and this
effect was confirmed at the protein level (26). These results indicate a possible
cross-talk between glucocorticoid and hypoxia-dependent signaling
pathways.
In the present study, we have investigated the
interplay between GCs and hypoxia-mediated signaling and a possible
involvement of dexamethasone, a potent synthetic GC commonly used
in various clinical treatments, in the regulation of
tumor-associated carbonic anhydrase IX level. We have confirmed
that DEX reduced HIF-1α expression and activity, showed that it
decreased CA IX expression in 2D and 3D cellular models and
proposed a mechanism of CA9 transcriptional regulation by
glucocorticoids. The obtained results linking CA9 expression
with dexamethasone bring new insight into a clinically relevant
situation which can occur during dexamethasone treatment of
patients with solid tumors.
Materials and methods
Cell culture and spheroids
preparation
RKO colorectal carcinoma and MCF-7 breast cancer
cell lines were cultured under standard conditions in Dulbecco's
modified Eagle's medium (DMEM) supplemented with 10% fetal calf
serum (FCS; Biowhittaker, Lancaster, MA, USA) and gentamicin
(Sandoz) in humidified air containing 5% CO2 at 37°C
(normoxia). Cells were obtained from the ATCC and regularly tested
for mycoplasma contamination. Before experiments, cells were
counted, seeded in culture dishes and incubated in normoxic
conditions for 24 h. The following day, culture medium was replaced
with the fresh one containing 10 or 100 μM dexamethasone
(Sigma-Aldrich, dissolved in ethanol) or ethanol only (in
appropriate concentrations, as control samples) and moved to the
anaerobic workstation (Ruskinn Technology Ltd., Bridgend, UK) with
hypoxic conditions (2% O2, 2% H2, 5%
CO2, 91% N2) for additional 24 h.
MCF-7 spheroids were pre-formed from 600 cells/20 μl
of culture medium in drops hanging on the lid of tissue culture
dish for 7 days at 37°C. The resulting cell aggregates were
transferred to Petri dish with a non-adherent surface and
cultivated in suspension for additional 14 days. During this period
dexamethasone (treated groups) or ethanol (control groups) were
added every fourth day in fresh medium. At the end of the
experiment, images were taken and spheroids size of at least 20
aggregates from each group was analyzed using AxioVision software.
Finally, spheroids were collected by centrifugation and used for
RNA or protein isolation.
Real-time cell proliferation assays
To assess the cell growth of cells treated with
dexamethasone, a real-time characterization was performed using the
xCELLigence system (ACEA Biosciences Inc., San Diego, CA, USA). The
xCELLigence® system was placed in a hypoxic cabinet with
the O2 controller at 2% O2 (Coy Laboratory
Products Inc., Grass Lake, MI, USA), 5% CO2 and 37°C.
For the proliferation assay, 10×103 RKO colorectal
cancer cells/well were seeded into an E-plate 16 (ACEA Biosciences)
in DMEM medium with dexamethasone or ethanol as a control. The
plates were placed into the xCEL-Ligence system performing a
real-time, impedance-based, label-free monitoring of cell
proliferation. Data were collected every 15 min and are presented
as a dimensionless parameter called the cell index. Cell index
reflects an increase in the area covered by cells, corresponding to
increasing cell number during proliferation.
Transient transfections and promoter
analysis
The cells were plated onto 35-mm Petri dishes to
reach ~70% monolayer density on the following day. For luciferase
reporter assay, transfection was performed with 2 μg of the pGL3
luciferase vector containing CA9 promoter (−1500/+37,
−174/+37 or −174/+37_HRE-mut) or luciferase vector containing
hypoxia-response elements (HRE-luc) and 100 ng pRL-TK
Renilla vector using Attractene transfection reagent
(Qiagen). Human promoter constructs were generated by an insertion
of PCR-amplified −1500/+37 and −174/+37 CA9 genomic
fragments upstream of the firefly luciferase gene in pGL3-Basic
vector (Promega) (27). The
pGL3-174/+37_HRE-mut construct with mutated HRE upstream of the
firefly luciferase gene was created from the original pBMN5HREmut
plasmid (28). shRNA targeting
human HIF-1α cloned in pSUPER plasmid was kindly provided by Dr
Nicholas Denko, (Ohio State University Wexner Medical Center,
Columbus, OH, USA). Luciferase vector containing a trimer of
hypoxia-response elements (HRE-luc) of the lactate dehydrogenase A
gene was kindly provided by Dr Kaye Williams (University of
Manchester, Manchester, UK). Transient silencing of HIF-1α and
NF-κB was performed using specific si/shRNA: 2 μg of plasmid vector
pSuper_shHIF-1α vs. pSuper_shCtrl containing non-targeting
sequence, 50 nM siNF-κB (Thermo Fisher Scientific) vs.
non-targeting control (Thermo Fisher Scientific). The following
day, the transfected cells were plated according to the type of
further experiment and cultured in hypoxia for additional 24 h.
Reporter gene expression was assessed using the Dual-luciferase
reporter assay system (Promega), and the luciferase activity was
normalized against the Renilla activity.
Quantitative PCR
Total RNA was isolated using TRIzol solution
(Sigma-Aldrich) and reverse transcription of 3 μg RNA for each
sample was performed with the High-Capacity cDNA Reverse
Transcription kit (Applied Biosystems). Quantitative PCR was
carried out using Maxima SYBER-Green PCR Master Mix (Thermo Fisher
Scientific). Sample Ct values were normalized to actin. Relative
expression was calculated using the ΔΔCt method. All amplifications
were performed in triplicate. Oligonucleotides used for real-time
qPCR were as follows: Actin sense 5′-CCAACCGCGAGAAGATGACC-3′
and actin antisense 5′-GATCTTCATGAGGTAGTCAGT-3′; VEGF
sense 5′-CTTGCTGCTCTACCTCCACCAT-3′ and VEGF antisense
5′-CACACAGGATGGCTTGAAGATG-3′, Glut1 sense
5′-CTCCTTTCTCCAGCCAGCAATG-3′ and Glut1 antisense
5′-CCAGCAGAACGGGTGGCCATAG-3′; LDHa sense
5′-TGGCAGCCTTTTCCTTAGAA-3′ and LDHa antisense
5′-ACTTGCAGTTCGGGCTGTAT-3′; HIF-1α sense
5′-GCTTGGTGCTGATTTGTGAACC-3′ and HIF-1α antisense
5′-GCATCCTGTACTGTCCTGTGGTG-3′; CA9 sense
5′-CCGAGCGACGCAGCCTTTGA-3′ and CA9 anti-sense
5′-GGCTCCAGTCTCGGCTACCT-3′.
Western blotting
Protein extracts were prepared using lysis buffer
(1% Triton X-100; 50 mM Tris pH 7.5; 150 mM NaCl; 0.5% Nonidet
P-40) containing protease (Roche) and phosphatase inhibitor
cocktail (Sigma-Aldrich), disrupted by sonication and cleared by
centrifugation. Concentrations were quantified using the BCA
protein assay kit (Pierce). A total of 100 μg of proteins/lane were
resolved in 8% SDS-PAGE, transferred to a PVDF membrane
(Macherey-Nagel, Düren, Germany) and visualized using an enhanced
chemiluminescence kit (GE Healthcare Life Sciences). Protein bands
were quantified in ImageJ software, all results were normalized to
actin. Antibodies used for specific proteins were as follows:
HIF-1α (dilution 1:250, 610959; BD Transduction Laboratories, San
Jose, CA, USA), CA IX (in-house generated M75, dilution 1:3), actin
(dilution 1:1,000, sc1615; Santa Cruz Biotechnology, Santa Cruz,
CA, USA), NF-κB1-p105/p50 (dilution 1:1,000, 13586; Cell Signaling
Technology, Danvers, MA, USA), appropriate secondary antibodies
conjugated with horseradish peroxidase were purchased from
Dako.
Proteome profiler array
For analyzing the expression profile of cell
stress-related proteins in MCF-7 spheroids treated with
dexamethasone, we performed Human Cell Stress Proteome Profiler
Array kit (R&D Systems, Minneapolis, MN, USA). All steps were
carried out according to the manufacturer's instructions.
Cignal finder reporter array
For analyzing the activity of cancer-related
transduction pathways, we performed Cignal finder reporter array
(SABiosciences) according to the instructions of the manufacturer.
Dual-luciferase results were calculated for each transfectant and
analyzed by the data analysis software (SABiosciences). Changes in
the activity of each signaling pathway were determined by comparing
the normalized luciferase activities of the reporter in treated vs.
untreated transfected cells or hypoxic vs. normoxic cells.
Bioinformatics
In silico analysis of the CA9 promoter
was performed using MatInspector program (https://www.genomatix.de) (29,30).
Promoter sequence (663 bp) was extracted directly from ElDorado
genome database after gene submission. Accurate positions of
predicted binding elements were calculated according to the
transcription start site.
Statistical analysis
Results were analyzed by two-tailed unpaired t-test
(Student's t-test), and P<0.05 was considered significant.
P<0.05 is denoted as *, P<0.01 as **
and P<0.001 as ***.
Results
Dexamethasone reduces protein level of
HIF-1α as well as its activity and affects transcription of HIF-1
target genes
We investigated the effect of dexamethasone on the
regulation of hypoxia-induced genes in colorectal carcinoma RKO
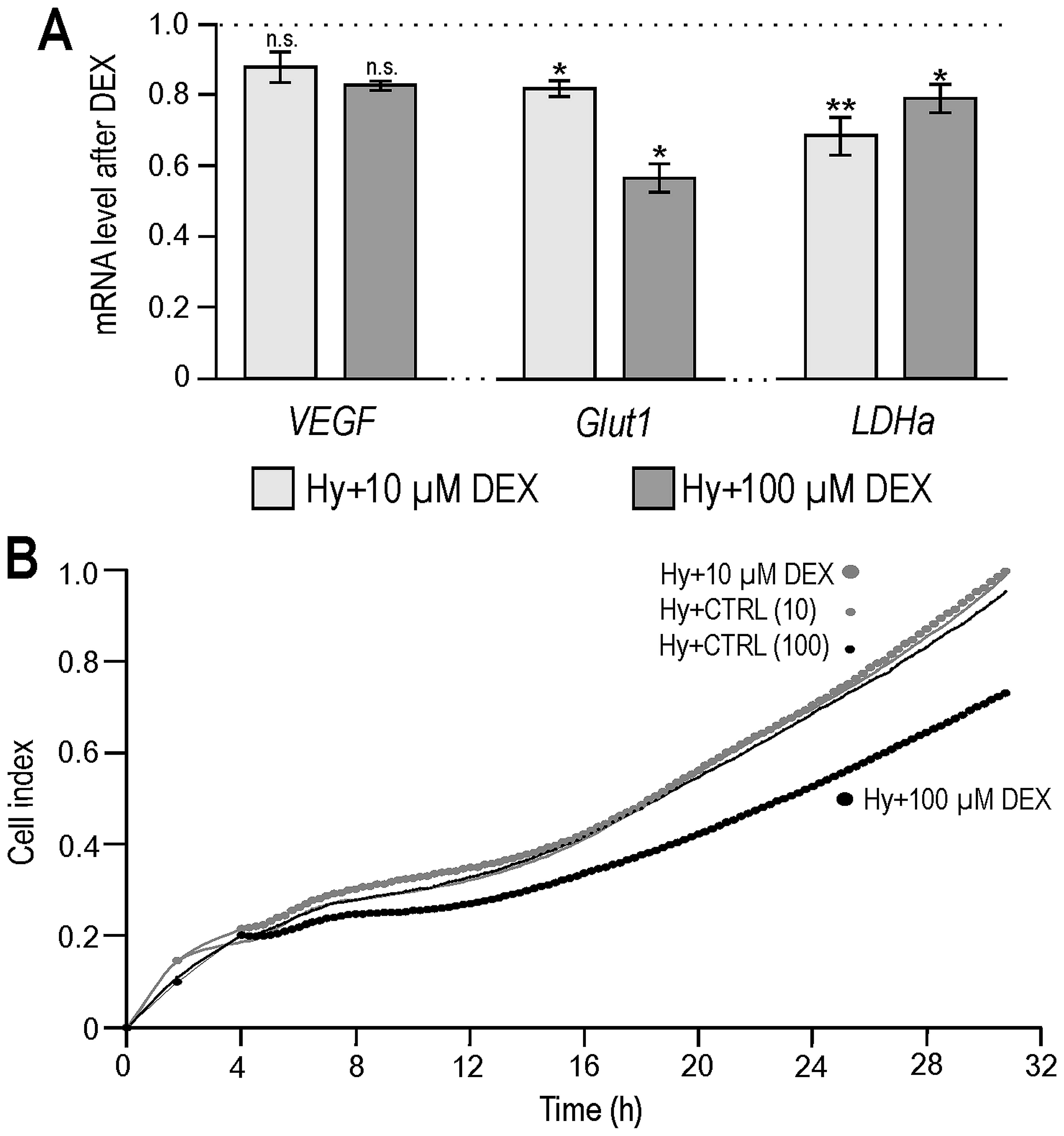
cells cultured for 24 h under 2% hypoxia. As shown in Fig. 1A, both doses of dexamethasone (10
and 100 μM) reduced the expression of several HIF-1 targets at mRNA
level, such as VEGF, Glut1 and LDHa in our colorectal carcinoma
model.
We then performed a real-time monitoring of the
proliferation of hypoxic RKO cells under dexamethasone treatment
(appropriate ethanol concentrations were used as negative controls)
using an impedance-based xCELLigence platform. As our results show
(Fig. 1B) DEX (10 μM) did not
affect the growth of hypoxic RKO cells. The higher dose of
dexamethasone (100 μM) slightly decreased the growth rate of RKO
cells which is in agreement with a known dose-dependent bimodal
effect of glucocorticoids on cell proliferation (31).
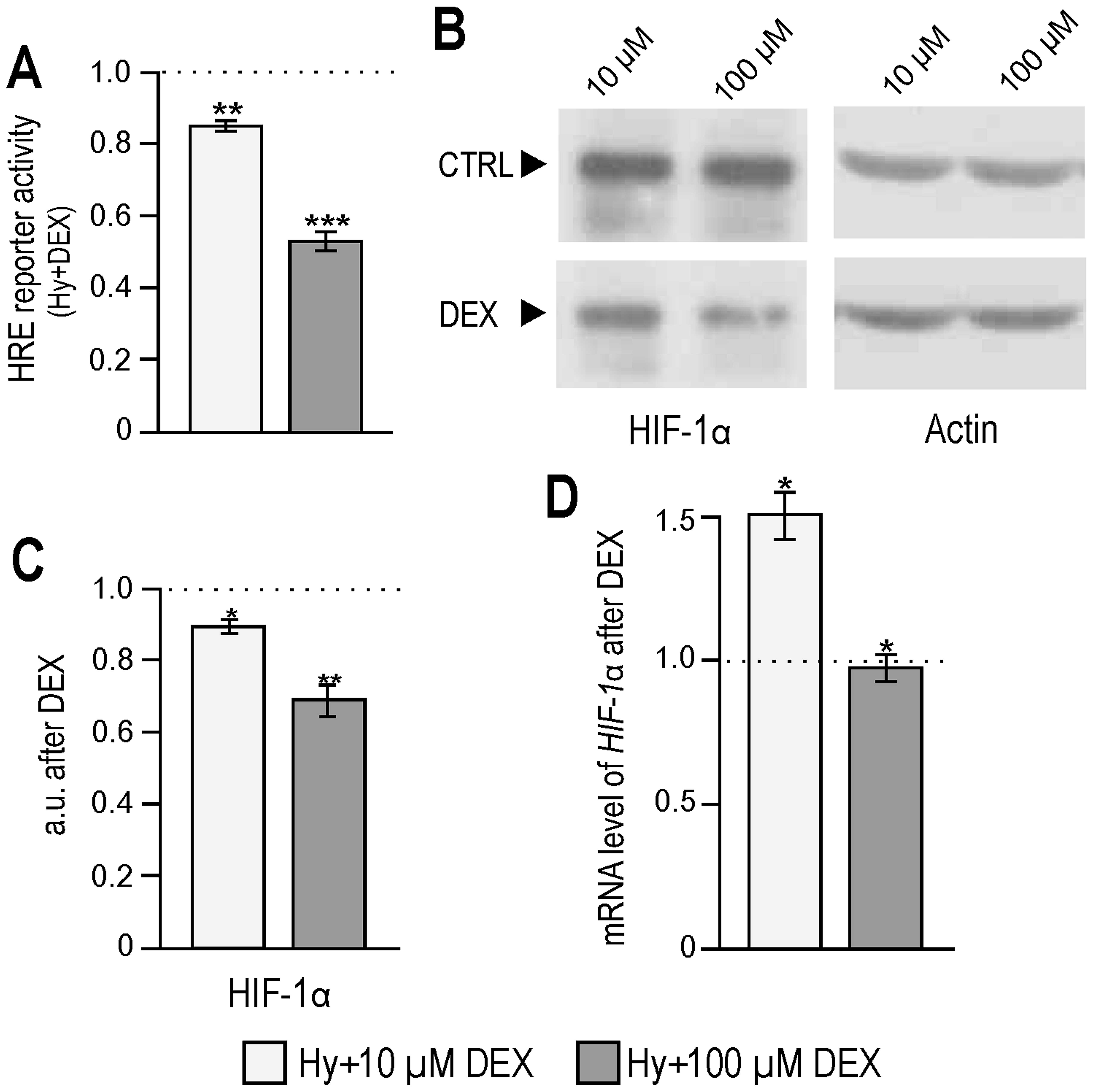
As HIF-1 is a major regulator of hypoxic responses
at the transcriptional level, we investigated if dexamethasone
influenced its transcriptional activity. Using luciferase reporter
containing HRE, we detected a significant dose-dependent decrease
in the activity of HIF-1 transcription factor with an increasing
concentration of dexamethasone (Fig.
2A). At the same time, the levels of the HIF-1α protein in the
hypoxic RKO cells subjected to dexamethasone treatment were reduced
when compared to their controls (Fig.
2B and C). Quantitative PCR analysis revealed 1.5-fold
elevation of the HIF-1α mRNA level when hypoxic RKO cells were
treated with 10 μM dexamethasone, whereas no change was detected
after 100 μM concentration (Fig.
2D) suggesting that DEX interferes with the translation and/or
degradation pathways of HIF-1α.
Dexamethasone decreases expression of CA
IX in hypoxic cancer cell monolayer and in spheroids
Tumor-associated carbonic anhydrase IX is one of the
best hypoxia-responsive targets which is often used as a marker of
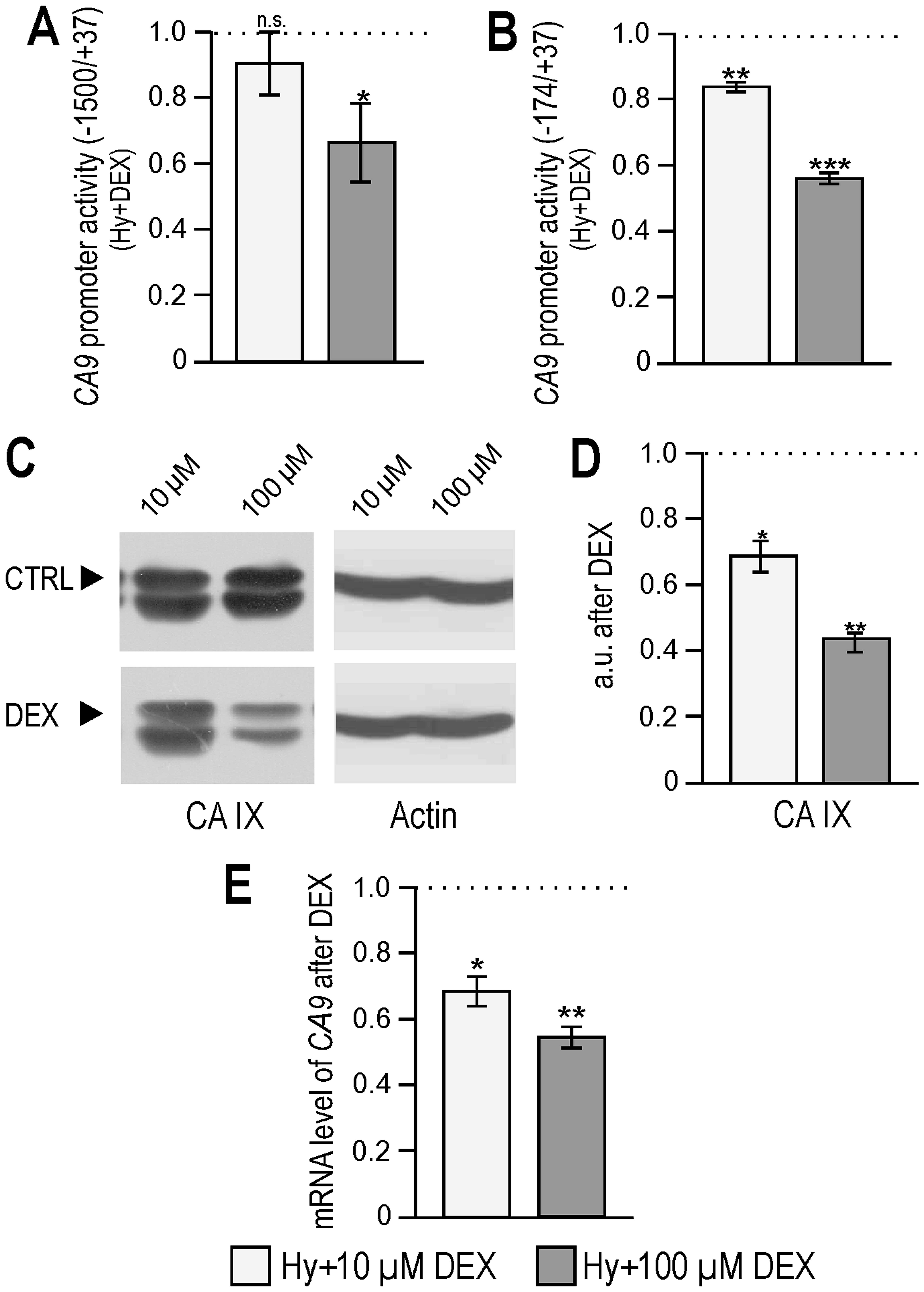
tumor hypoxia in clinical samples. Here, we show that dexamethasone
reduced the activity of the CA9 promoter in RKO cells
cultured in hypoxia in a dose-dependent manner (Fig. 3A and B). Reduced activity was
accompanied by a significantly decreased expression of CA IX at
both protein and mRNA levels (Fig.
3C–E). Measurement of extracellular pH of hypoxic RKO
monolayers showed a significantly reduced acidification after 24 h
of 100 μM DEX treatment (ΔpH=0.047 compared to control) which is in
agreement with the reduced CA IX expression described above. As
HIF-1 is the main regulator of CA9 transcription this result
is in agreement with the reduced HIF-1α protein and its lower
transcriptional activity after dexamethasone treatment. Testing of
dexamethasone treatment on another tumor cell line (MCF-7
monolayer) yielded similar results as on RKO cells, decreased
transcriptional activity of HIF-1 and reduced activity of the
CA9 promoter (Fig. 4A and
B).
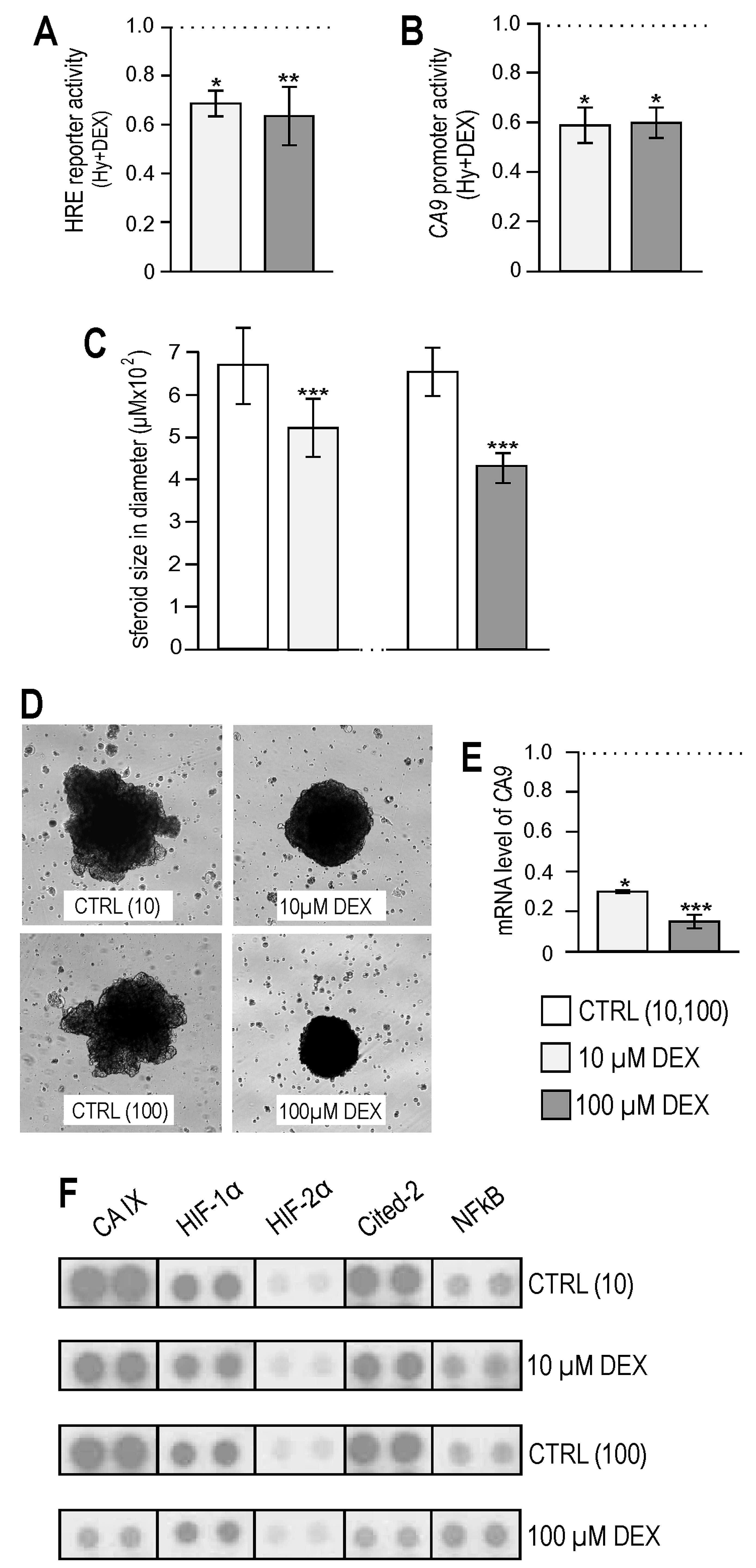
Several studies showed reduced primary tumor growth
and proliferation after knocking down the CA IX expression
(32–34). Well described was also an
inhibitory effect of dexamethasone on tumor volume e.g. in in
vivo models of the brain or prostate cancer (23,35).
As possible effects of dexamethasone on CA IX which promotes tumor
cell survival could be important, especially during DEX treatment
of cancer patients, we tested the effect of dexamethasone in the
physiologically more relevant 3D environment. We used the
established MCF-7 breast carcinoma 3D model of the spheroid
formation in hanging drops. Spheroids were treated every 4th day
during a two-week period. At the end of the experiment, spheroids
exposed to dexamethasone had a significantly smaller diameter than
controls, by almost 20% in 10 μM group and 35% in 100 μM group
(Fig. 4C) and their morphology was
also altered (Fig. 4D). Analysis
of spheroids by quantitative PCR and proteome profiler array showed
that an increasing amount of dexamethasone considerably lowered
transcription (Fig. 4E) and
protein level of CA IX (Fig. 4F)
also in this breast cancer 3D model. Our findings indicate that
dexamethasone-mediated effect of spheroid growth inhibition is at
least partially associated with its effect on CA IX levels. In
addition, reduction of HIF-1α protein was confirmed in MCF-7
spheroids. Notably, the protein level of hypoxia-inducible
factor-2α, which is also capable of activating transcription via
binding of HRE and is regulated in the same way as HIF-1α (36,37),
was unchanged (Fig. 4F).
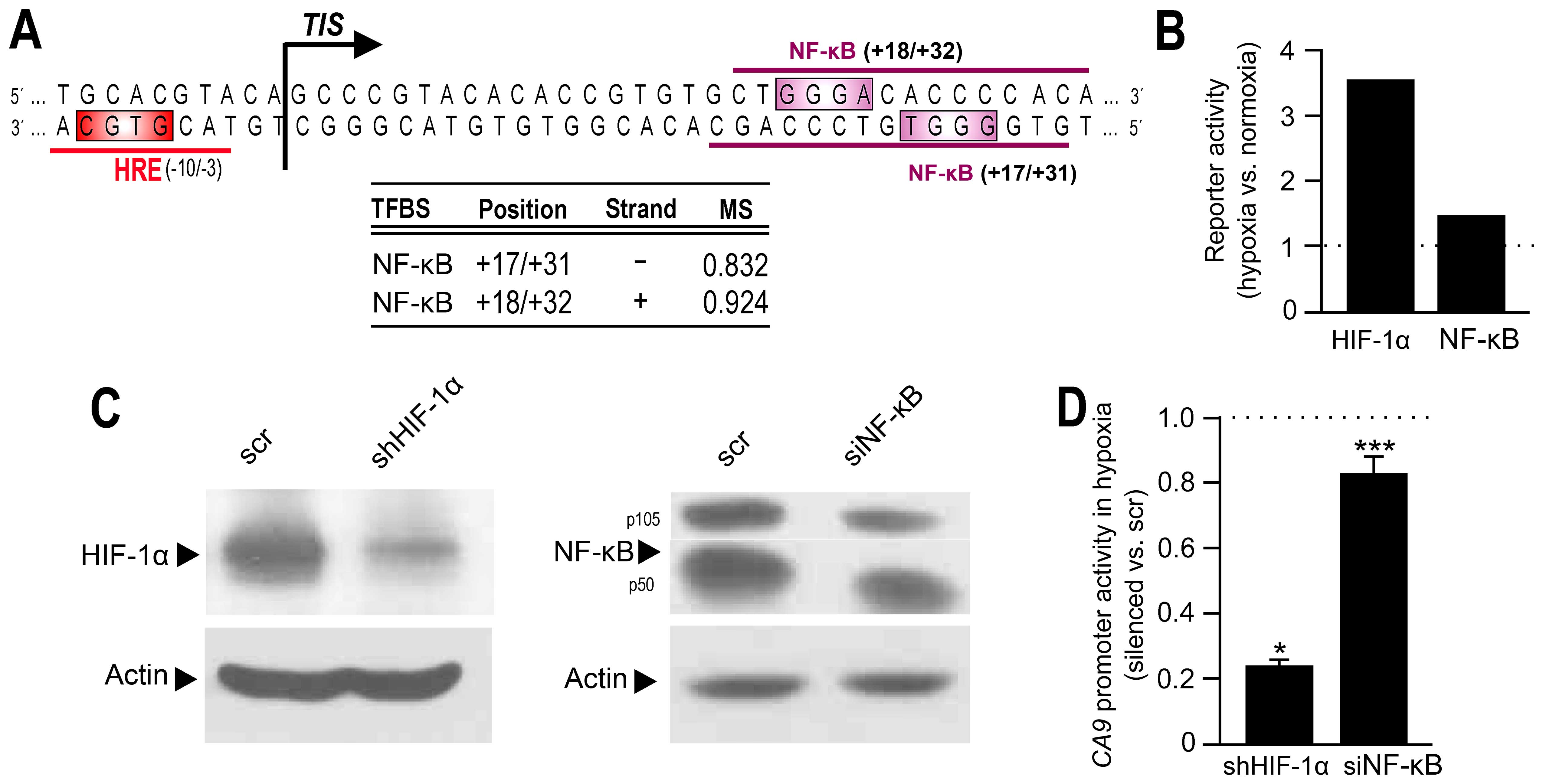
In silico analysis predicts potential
NF-κB binding sites in the promoter of CA9 gene
As mentioned above HIF-1 transcription factor is the
major transcriptional regulator of CA IX during tumor hypoxia.
HIF-1 binds to an HRE sequence localized immediately before the
transcription initiation site (7)
and represents a critical component of the core promoter of the
CA9 gene. Luciferase assay (Fig. 3A and B) showed that dexamethasone
acts equally on the activity of both CA9 promoter
constructs: longer (−1500/+37) and shorter (−174/+37) one. These
data lead to the conclusion that the reduction of CA9
transcription by dexamethasone could be mediated mainly by a
lowered binding activity of HIF-1α to the core promoter region of
CA9.
However, CA9 promoter contains additional
cis-elements that may have modulatory effects on CA9
transcription (28). Detailed
in silico analysis of the CA9 regulatory region
revealed several transcription factor binding sites in the close
proximity to the HRE, the most interesting of them were those for
NF-κB (Fig. 5A). NF-κB has been
shown as hypoxia-responsive (26,38,39).
Therefore, we examined their possible participation in the basal
hypoxic induction of CA IX. First, we tested NF-κB transcriptional
activity during hypoxia, in the absence of dexamethasone treatment
(Fig. 5B). Results indicated that
also in colorectal RKO cells hypoxia could trigger the
transactivation of NF-κB. Next, we analyzed the activity of the
CA9 promoter in hypoxic nontreated cells after transiently
silencing the NF-κB expression (Fig.
5C and D). Interestingly, NF-κB suppression led to a
downregulation of the CA9 promoter activity by almost 20%
under hypoxic conditions (in the absence of DEX).
Contribution of NF-κB to
dexamethasone-mediated reduction of CA IX expression
As synthetic glucocorticoids are often used during
chemotherapy of various tumor patients, we examined the effects of
dexamethasone on cancer-related signaling pathways in in
vitro colorectal carcinoma model. Cignal finder reporter array
(Fig. 6A) showed that
dexamethasone treatment of hypoxic RKO cells caused an additional
activation of NF-κB signaling in hypoxia in a
concentration-dependent manner, which is in agreement with higher
protein level of NF-κB detected in DEX-treated MCF-7 spheroids
(Fig. 4F). Besides gene
transactivation, glucocorticoids can also act as repressors. This
inhibitory effect of GCs can result from the interaction between
ligand-activated GR and another transcription factor e.g. AP-1,
thus, preventing them from binding to their specific site.
Dexamethasone decreased the transactivation ability of AP-1
(Fig. 6A), as well as other
proteins involved in GC mediated downregulation (SMADs and
STATs).
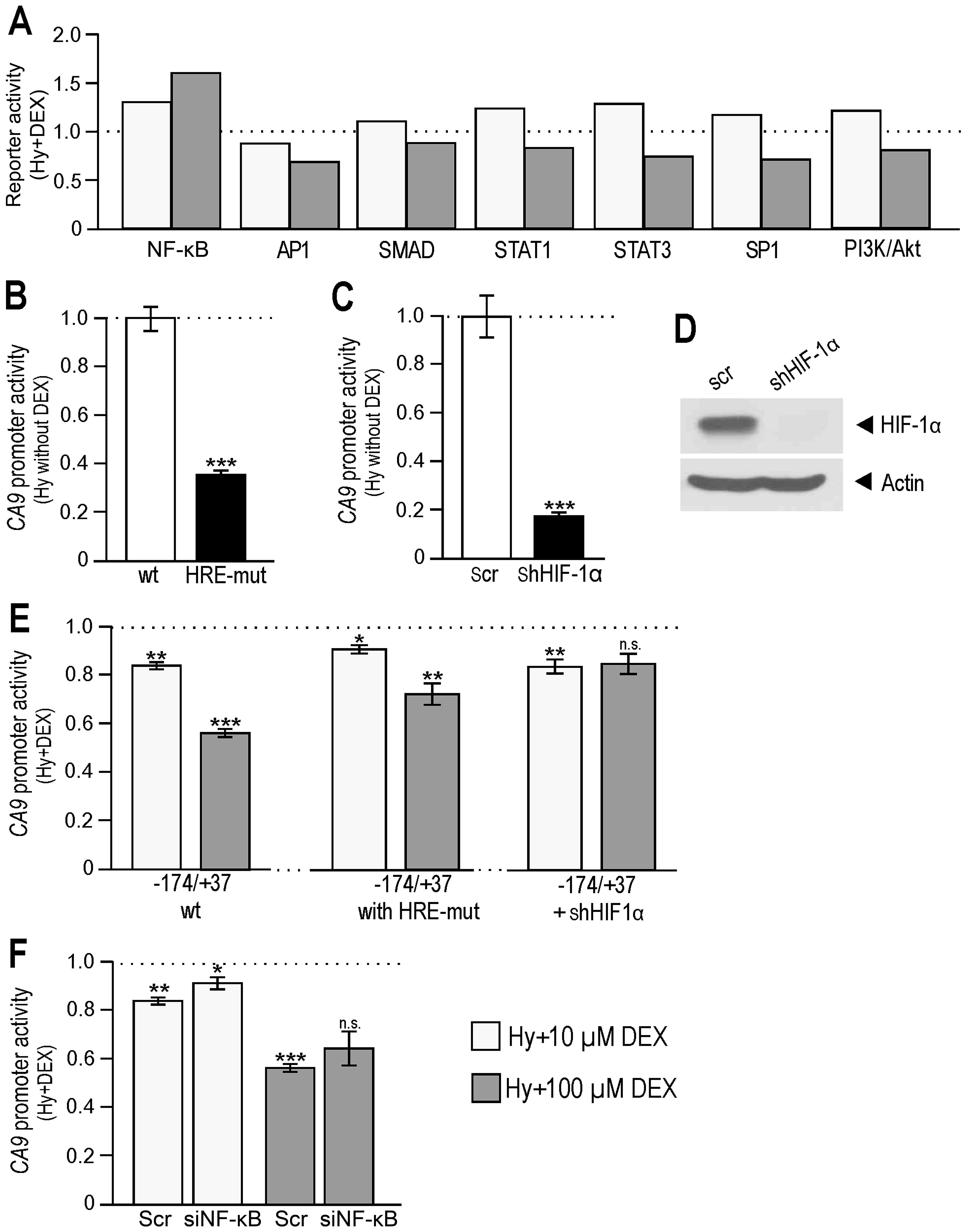 | Figure 6Participation of NF-κB in the
regulation of CA9 gene (A) Molecular response of RKO cells
to hypoxia and DEX. Cell-based dual luciferase reporter array of
hypoxic RKO cells (control, ethanol 10 and 100) vs. hypoxic RKO
cells treated with DEX (10 and 100 μM) revealed alterations of
signal transduction pathways leading to changes in transactivation
activities of NF-κB and other signaling pathways, such as SP1,
STAT3, SMAD and PI3K/Akt. (B) Graph gives the activity of
pGL3-CA9 (−174/+37) promoter construct with mutated HRE vs.
wild-type construct (set to 1) transfected into RKO cells and
cultured in hypoxia (without DEX). The results of HRE-mut samples
were compared to wild-type counterparts (***P<0.001
significant difference). (C) Graph shows the activity of the
wild-type pGL3-CA9 (−174/+37) promoter construct in RKO
stable transfectants without HIF-1α expression vs. scrambled
control (set to 1) cultured in hypoxia (wihout DEX). The results of
silenced samples were compared to scrambled counterparts
(***P<0.001 significant difference). (D) Western blot
analysis of hypoxic RKO cells stable-transfected with shHIF-1α. (E)
Left part represents the analysis of the pGL3-CA9
(−174/+37_wt) in hypoxic RKO cells treated with DEX. Middle part
represents the analysis of the pGL3-CA9 with mutated HRE
sequence (−174/+37_HRE-mut) in hypoxic RKO cells treated with DEX.
Right part gives the analysis of the pGL3-CA9 (−174/+37_wt)
in hypoxic RKO cells with HIF-1α stable knock-down, all after DEX
treatment. The data show that DEX reduced CA9 promoter
activity also in the conditions when HIF-1α was unable to bind to
its HRE. The results in E were related to their control, untreated,
samples (containing ethanol) which were set to 1. T-tests were
performed among DEX-treated samples (−174/+37 wt, −174/+37 with
HRE-mut, −174/+37 +shHIF-1α) and their appropriate untreated
controls. P<0.05 was considered significant.
*P<0.05, **P<0.01 and
***P<0.001. (F) Analysis of the CA9 promoter
activity in hypoxic RKO cells with suppressed expression of NF-κB
after DEX treatment. RKO cells were co-transfected with
pGL3-CA9 (−174/+37), pRL-TK plasmids and siNF-κB, treated
with DEX, incubated in hypoxia and finally analyzed by
Dual-luciferase assay. The data show that transient silencing of
NF-κB caused a smaller reduction of CA9 promoter activity
indicating that NF-κB contributes to CA9 downregulation in
the presence of DEX. Differences of DEX treated samples with
silenced NF-κB in F were evaluated against their corresponding
DEX-treated scrambled controls. P<0.05 was considered
significant. *P<0.05, **P<0.01 and
***P<0.001. |
As a next step we wanted to investigate whether
different mechanisms beyond diminished HIF-1α function could
participate in the observed dexamethasone-triggered down-regulation
of CA IX. Therefore, we created the situation when HIF-1α was
knocked-down or unable to bind to the CA9 promoter. This was
achieved by transfection of −174/+37 construct containing a
mutation in the corresponding HRE (Fig. 6B) or by the transfection of
−174/+37 wild-type construct into cells in which HIF-1α was
completely knocked-down by a stable transfection of pSuper_shHIF-1α
(Fig. 6C and D). Our results
showed (middle and right graph in Fig.
6E) that dexamethasone was capable of reducing the activity of
CA9 promoter in both conditions when compared to control
samples of HRE-mutant and silenced cells not treated by DEX. These
findings confirm that an additional repressive mechanism (mediated
by other trasncription factors than HIF-1α) was involved in CA IX
regulation during the treatment. Moreover, simultaneous suppression
of NF-κB led to an increase in CA9 promoter activity when
compared to the scrambled dexamethasone group (Fig. 6F). Our data indicate that NF-κB
contributes to dexamethasone-mediated modulation of the hypoxic
expression of CA IX.
Discussion
Natural glucocorticoids are steroid hormones
synthesized and secreted by the adrenal cortex. In man, they are
involved in the regulation of a variety of physiologic functions
such as growth, development, metabolism, immune response and in the
maintenance of the basal and stress-related homeostasis. One of the
main effects of glucocorticoids (GCs) is their ability to reduce
inflammatory responses. Therefore, synthetic GCs belong to the most
used agents in the treatment of different diseases which are
typically accompanied by inflammation, including cancer.
Dexamethasone (DEX) is a frequently prescribed synthetic
glucocorticoid often used in cancer treatment in a number of
different ways. Its beneficial properties include mainly prevention
of allergic reactions, nausea and vomiting caused by certain
chemotherapy drugs, and reduction of swelling, especially for
tumors in the brain, spinal cord or bones. Several studies reported
a decreased tumor volume as the effect of dexamethasone treatment
(23,35,40).
DEX was also shown to affect angiogenesis in vitro as well
as in vivo cancer models by means of reduced VEGF expression
(21–23). Recently, the involvement of HIF-1
in glucocorticoid-mediated downregulation of VEGF was shown
(24).
In the present study, we also showed DEX-mediated
downregulation of hypoxia-induced VEGF as well as other
hypoxia-induced targets, including CA IX, Glut1 and LDHa. In
accordance with the known data, DEX caused a decrease in HIF-1
transcriptional activity and reduced protein levels of HIF-1α
oxygen-sensitive subunit in both 2D colorectal carcinoma and 3D
breast cancer models. However, no down-regulation of HIF-1α was
detected at the transcriptional level after DEX treatment of RKO
cell monolayer, 10 μM concentration even caused an elevation of
HIF-1α mRNA, indicating post-transcriptional action of DEX on
HIF-1α. A wide spectrum of modulators regulates transcription of
the gene encoding HIF-1α. Although hypoxia regulates HIF-1α mainly
at the post-translational level, several hypoxia-responsive factors
initiate its transcription. Among them NF-κB contributes to its
upregulation through a binding element located at −197/−188 bp
upstream from the transcription initiation site (TIS) of the HIF-1α
gene (41). Another
hypoxia-increased protein SP1 was defined as important for
maintaining HIF-1α transcription. Multiple SP1 binding sites have
been described at −85/−65 bp region from the TIS (42) and their deletion resulted in a
decrease in HIF-1α promoter activity (43). Furthermore, JAK/STAT signaling
pathway, which is implicated in mediating the inflammatory
responses, was linked to the expression of HIF-1α. Particularly
STAT3 was shown to bind to a DNA-binding motif placed at −363/−355
bp (44). In this study, we showed
that DEX changed cellular signaling during hypoxia and that these
effects varied depending on DEX amount. In line with known
transcriptional regulation of HIF-1α gene, the activation of NF-κB,
SP1 and STAT3 signaling caused by 10 μM DEX (Fig. 6A) could participate in the observed
increase of HIF-1α at transcriptional level.
Since we did not detect any downregulation in the
HIF-1α transcription, we propose that DEX probably affects its
degradation and/or translation. Besides the negative regulation of
HIF-1α by proteasomal degradation, a number of other mechanisms
contribute to the reduction of HIF-1α protein level or activity.
Among them Cited2, activated in hypoxia, was found to downregulate
HIF-1-mediated trans-activation (45). However, our analysis of MCF-7
spheroids showed DEX-induced reduction of Cited2 at the protein
level (Fig. 4D). Stabilization of
HIF-1α can also be modulated by other signaling pathways including
PI3K/Akt kinase cascade, which is known to operate via positive as
well as negative regulation, depending on downstream effectors.
Downstream target of Akt implicated in the positive regulation of
HIF-1α is the mammalian target of rapamycin, mTOR, which likely
upregulates HIF-1α through a translation-dependent pathway
(46,47). DEX was shown to repress protein
synthesis through inhibition of mTOR signaling by reducing
phosphorylation of the downstream targets S6K1 and 4E-BP1 (48). Heat shock protein expression also
contributes to the stabilization of HIF-1α (49,50).
In hypoxia, Hsp90 binding protects HIF-1α from degradation by
non-VHL mediated ubiquitination (50) and its inhibition results in
decreased HIF-1α accumulation and activation. Maheshwari et
al (51) detected significant
reduction of Hsp90 in HD150Q cells and HD mice cortex after DEX
treatment. Recently, glucocorticoid-induced leucine zipper (GILZ)
was shown to suppress the expression of HIF-1α at the protein level
via the proteasomal pathway (52).
It has also been demonstrated that DEX directly reduced
phosphorylation of the serine/threonine kinase Akt (53) which, in turn, can lead to the
activation of downstream target GSK3β, thus, preventing further
HIF1α accumulation (54,55). All these data support our
conclusions denoting a key role of the synthetic glucocorticoid
dexamethasone in HIF-1α destabilization and subsequent reduction of
HIF-1 transcriptional activation.
HIF-1 activates transcription of many genes
mediating adaptive responses to hypoxia. Among them the gene
encoding carbonic anhydrase IX is one of the most strongly
activated. Within the regulatory region of CA9 gene a
hypoxia-responsive element (HRE) is localized at −10/−3 bp next to
the transcription initiation site (7). In line with DEX-mediated reduction of
HIF-1α expression and activity it is not surprising that DEX
reduced the CA9 promoter activation, transcription and CA IX
protein level in both RKO and MCF-7 cancer models. However, our
data indicate also the existence of an additional mechanism
contributing to the decreased CA IX expression after DEX treatment
besides impaired HIF-1 function (Fig.
6E).
At the cellular level, the mechanism of GCs actions
is mediated through glucocorticoid receptors (GRs) which function
as ligand-dependent transcription factors. Basic genomic regulation
involves a direct receptor binding to DNA on its specific sequences
termed glucocorticoid response elements (GREs) providing gene
transactivation. However, regulation mediated by GRs can lead also
to the transrepression depending on a promoter context and GRE
sequence present (56). Direct
negative regulation by GCs occurs via the interaction of GR with a
negative GRE site the actual sequence of which is poorly defined.
In silico analysis of the regulatory region of CA9
gene revealed two potential GREs (−8/+10 and +3/+21), both
localized in a close proximity to the TIS (data not shown). A
precise regulatory action of these GREs remains undetermined and
requires further study. Alternatively, ligand-activated GR can
mediate a transrepression independently of GRE by means of a
protein-protein interaction with another transcription factor, thus
preventing them from binding to their specific site. Most of
anti-inflammatory effects of GCs are mediated through this pathway,
especially via GR cross-talk with pro-inflammatory NF-κB or AP-1.
AP-1-responsive element was described in CA9 promoter up-stream of
HRE by Kaluz et al (57).
This element functionally contributes to CA9 transcription
and, therefore, DEX could repress it via GR and AP-1 interaction.
This conclusion is also supported by decreased transactivation
ability of AP-1 after DEX treatment as determined by Cignal
reporter array (Fig. 6A).
Moreover, we also identified potential NF-κB-binding sites
(Fig. 5A) and suggested their
involvement in DEX-mediated inhibition of CA9 transcription
as a knock-down of NF-κB resulted in a weaker effect of DEX on
CA9 promoter activity (Fig.
6F). However, DEX treatment increased protein level and
subsequent transcriptional activity of NF-κB in the Cignal array
(Figs. 4F and 6A). Although NF-κB is primarily
considered a transcriptional activator, recently its role in
downregulating gene expression has been reported (58–60).
Thus, we propose that NF-κB could act on CA9 promoter in DEX
treated cells as a transcriptional repressor, however, precise
mechanism of NF-κB action on CA9 regulation during hypoxia
and in GCs presence remains to be elucidated.
Our results showed a dose-dependent reduction of
mRNA and protein levels of CA IX in spheroids after dexamethasone
treatment (Fig. 4E and F). The
size of treated spheroids was significantly decreased in a
concentration-dependent manner (Fig.
4C and D). This reduction could be at least partially caused by
lowered levels of CA IX after DEX treatment which can lead to
impaired pH regulation in the hypoxic core, resulting in slower
proliferation and worsened cell survival. Bladder carcinoma RT112
spheroids expressing surface CA IX activity were more successful in
removing surface-to-core difference in intracellular pH (61). Spheroids formed from HCT116
colorectal cancer cells transfected with CA IX were shown to better
suppress intracellular acidity in the core than their control
counterparts lacking CA IX (62).
The use of membrane-impermeant CA inhibitors linked this effect to
CA IX activity. Research of tumor cell clusters loaded with pH
sensitive dye carboxy-SNARF-1 demonstrated that hypoxia-insensitive
Na-dependent bicarbonate ion transport is important for setting and
stabilizing of resting pH at a mildly alkaline level, promoting
growth (63). CA IX facilitates
this process by its cooperation with bicarbonate transporters NBC1
and AE2 by forming a bicarbonate transport metabolon (5). Experiments with carnosine which
disrupts bicarbonate transport metabolon and thus impairs CA IX
activity also proved the importance of CA IX for spheroid growth
(64). After a hypoxic gradient
was established CA9 silencing in LS174 colon adenocarcinoma
spheroids led to a diminished proliferation (65). CA IX expression was linked with
increased growth rate in spheroids formed from colorectal tumor
cells (66). Silencing of
CA9 significantly decreased number of spheroid-forming cells
in various breast cancer cell lines in hypoxia (67). Above mentioned data obtained from
different tumor 3D models point to the role of CA IX in promoting
3D cell growth.
However, dexamethasone action must also be taken
into account when assessing spheroid growth. Glucocorticoids were
shown to exert anti-proliferative effects mediated by a G1-block in
cell cycle progression (68).
Dexamethasone inhibited proliferation of tumor cells and suppressed
the activity of transcription factor NF-κB, which controls the
expression of genes such as cyclooxygenase COX-2 and cyclin D1,
involved in proliferation control of tumor cells (69). Anti-proliferative action of GC/GR
is correlated to high concentration of GC (31), and our results from the
proliferation assay in hypoxia confirmed a bimodal effect of
dexamethasone on RKO cell monolayer (Fig. 1B). Interestingly, in MCF-7 cells
even higher DEX dose did not inhibit proliferation (data not
shown). Our exploration of hypoxia related pathways (data not
shown) showed that 100 μM dexamethasone dose led to a decrease of
c-myc, Nanog and E2F that play a role in the cycle progression,
apoptosis, and regulation of genes involved in cell proliferation
(70–72). Downregulation of proto-oncogenes
cyclin D1 and c-myc was also demonstrated in head and neck tumor
cells after the inhibition of NF-κB signaling by 10 μM
dexamethasone (73). Akt kinase is
also implicated in cancer progression as it stimulates
proliferation and suppresses apoptosis. This is in agreement with
our findings showing reduced activation of PI3K signaling in the
presence of 100 μM DEX (Fig. 6A).
Taken together, the effect of dexamethasone on 3D growth seems to
be an interplay between impaired pH-regulation in the hypoxic core
due to lower expression of CA IX and the influence of dexamethasone
itself on hypoxic signaling pathways.
By its catalytical as well as PG-domain related
action, carbonic anhydrase IX is involved in various processes
leading to tumor cell survival and dissemination. Clinical data
show that strong CA IX expression often correlates with high tumor
grade, advanced stage, necrosis or poor prognosis in various types
of tumors [including glioblastoma (74), breast carcinoma (75–77),
lung carcinoma (78), head and
neck cancer (79), brain tumors
(80), gastric cancer (81), and non-small cell lung cancer
(82)]. CA IX expression seems to
relate to resistance to antitumor therapy, including chemotherapy,
radiotherapy and anti-angiogenic treatment. Studies of in
vivo xenograft models of various human tumor cells showed that
high CA IX expression was linked with worse radio- and
chemo-sensitivity, but the response to therapy was improved after
the catalytic activity of CA IX was inhibited (33,83).
Similar results were obtained for anti-angiogenic therapy aimed
against VEGF (66). These data are
in accordance with the observation that extracellular acidosis
impairs intake of chemotherapeutic agents and affects radiation
damage (84). At present, the
association between CA IX level in tumor tissue and worse response
to therapy has been proved also in patients suffering from various
types of carcinoma [e.g. head and neck (85), rectal (85,86),
breast (87,88) and non-small cell lung cancer
(82,89)]. Synthetic glucocorticoid
dexamethasone is often prescribed in cancer-related clinical
settings. In the present study, we confirmed that dexamethasone
decreases HIF-1α level and transcriptional activity by
post-transcriptional action. For the first time, we demonstrated
that dexamethasone treatment can reduce mRNA and protein levels of
CA IX in 2D and 3D tumor models. Following dexamethasone dose,
CA9 promoter activity was inhibited by several mechanisms:
as a consequence of decreased activity and level of HIF-1α and also
directly by signaling of activated glucocorticoid receptors. We
propose an additional way of transcriptional downregulation of
CA9: via protein-protein interactions between activated GR
and transcriptional factors (such as AP-1 or NF-κB) (Fig. 7). These interactions could lead to
sequestration of transcriptional factors or to their action in a
repressive mode but the elucidation of precise mechanisms requires
further study. However, as the important fact remains that the
final effect of dexamethasone treatment is the reduction of CA IX
level. As CA IX represents an important component of pro-survival
and pro-migratory machinery of tumors, linked to chemo- and
radiotherapy resistance, our research generated important knowledge
with possible implications for dexamethasone treatment regimens in
cancer patients.
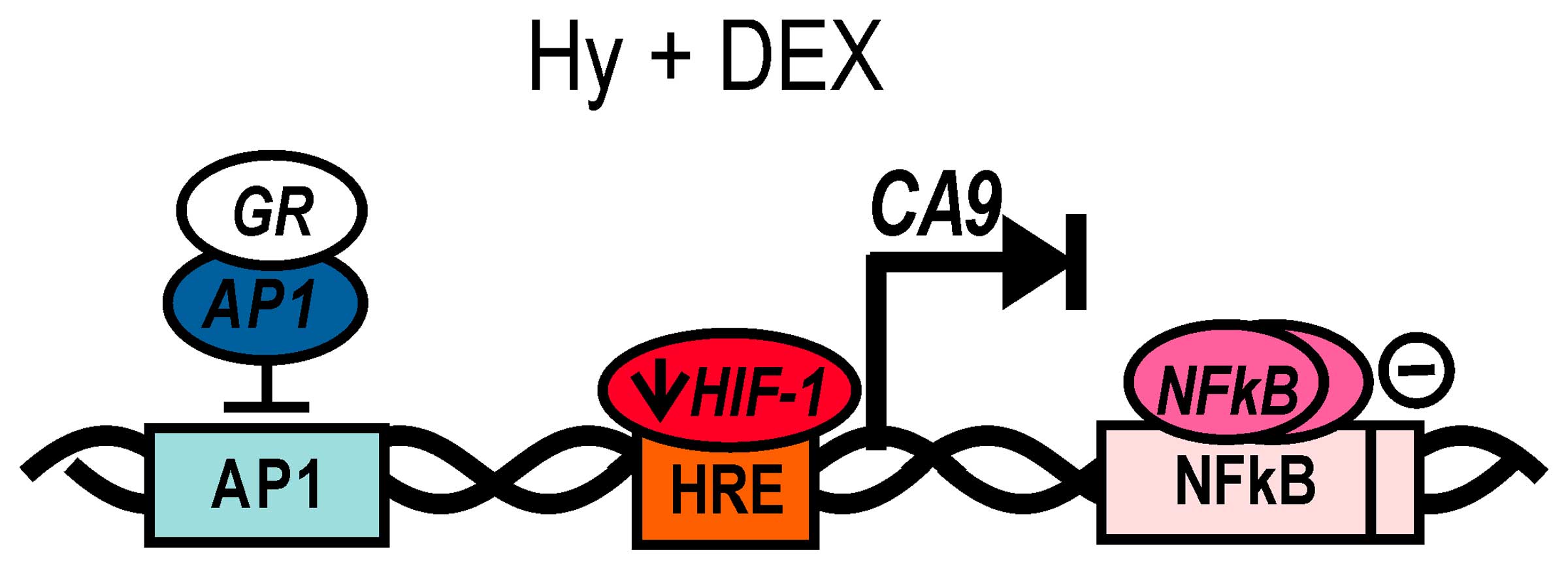 | Figure 7Proposed modes of DEX-mediated
transcriptional regulation of the CA9 gene. In the present
study, we showed that dexamethasone treatment causes reduction of
hypoxia-induced HIF-1α, which in turn leads to a decreased
expression of tumor-associated carbonic anhydrase IX. However, our
data indicate also the existence of other regulatory mechanisms
with the repressive mode of action on CA9 transcription. In
response to GCs, glucocorticoid receptors function as main
transcriptional mediators with ability to mediate gene
transactivation as well as transrepression, depending on target
promoter arrangement. This mechanism mostly includes the
interaction of GR with pro-inflammatory AP-1 and NF-κB, which
prevents binding to their respective sites, both present in
CA9 promoter, or results in masking of their transactivation
domain. As the suppression of NF-κB resulted in a smaller effect of
DEX on the CA9 promoter activity we assume a possible binding of
NF-κB on the CA9 promoter and its role in CA9
regulation. Although NF-κB is primarily considered a
transcriptional activator, several studies reported a direct
down-regulation of gene expression by NF-κB. We also propose that
NF-κB could act on the CA9 promoter as a transcriptional
repressor, however, a precise mechanism of NF-κB action on
CA9 regulation during basal hypoxia and in GCs presence
remains to be elucidated. |
Acknowledgements
The present study was supported by the grants
APVV-0893-11, Biomed ITMS 26240220087, VEGA 2/0081/14 and VEGA
2/0122/16.
Abbreviations:
|
CA IX
|
carbonic anhydrase IX
|
|
DEX
|
dexa-methasone
|
|
GC
|
glucocorticoid
|
|
GR
|
glucocorticoid receptor
|
|
GRE
|
glucocorticoid response element
|
|
HD
|
Huntington's disease
|
|
HIF-1
|
hypoxia-inducible factor-1
|
|
HRE
|
hypoxia response element
|
|
Hy
|
hypoxia
|
|
LDHa
|
lactate dehydrogenase A
|
|
NF-κB
|
nuclear factor-kappaB
|
|
TIS
|
transcription initiation site
|
|
VEGF
|
vascular endothelial growth
factor
|
References
|
1
|
Semenza GL: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lendahl U, Lee KL, Yang H and Poellinger
L: Generating specificity and diversity in the transcriptional
response to hypoxia. Nat Rev Genet. 10:821–832. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pastorek J and Pastorekova S:
Hypoxia-induced carbonic anhydrase IX as a target for cancer
therapy: From biology to clinical use. Semin Cancer Biol. 31:52–64.
2015. View Article : Google Scholar
|
|
4
|
Ditte P, Dequiedt F, Svastova E, Hulikova
A, Ohradanova-Repic A, Zatovicova M, Csaderova L, Kopacek J,
Supuran CT, Pastorekova S, et al: Phosphorylation of carbonic
anhydrase IX controls its ability to mediate extracellular
acidification in hypoxic tumors. Cancer Res. 71:7558–7567. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Svastova E, Witarski W, Csaderova L, Kosik
I, Skvarkova L, Hulikova A, Zatovicova M, Barathova M, Kopacek J,
Pastorek J, et al: Carbonic anhydrase IX interacts with bicarbonate
transporters in lamellipodia and increases cell migration via its
catalytic domain. J Biol Chem. 287:3392–3402. 2012. View Article : Google Scholar :
|
|
6
|
Csaderova L, Debreova M, Radvak P, Stano
M, Vrestiakova M, Kopacek J, Pastorekova S and Svastova E: The
effect of carbonic anhydrase IX on focal contacts during cell
spreading and migration. Front Physiol. 4:2712013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wykoff CC, Beasley NJ, Watson PH, Turner
KJ, Pastorek J, Sibtain A, Wilson GD, Turley H, Talks KL, Maxwell
PH, et al: Hypoxia-inducible expression of tumor-associated
carbonic anhydrases. Cancer Res. 60:7075–7083. 2000.
|
|
8
|
Zatovicova M, Jelenska L, Hulikova A,
Csaderova L, Ditte Z, Ditte P, Goliasova T, Pastorek J and
Pastorekova S: Carbonic anhydrase IX as an anticancer therapy
target: Preclinical evaluation of internalizing monoclonal antibody
directed to catalytic domain. Curr Pharm Des. 16:3255–3263. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dehne N and Brüne B: HIF-1 in the
inflammatory microenvironment. Exp Cell Res. 315:1791–1797. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eltzschig HK and Carmeliet P: Hypoxia and
inflammation. N Engl J Med. 364:656–665. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Barnes PJ and Karin M: Nuclear
factor-kappaB: A pivotal transcription factor in chronic
inflammatory diseases. N Engl J Med. 336:1066–1071. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Taylor CT: Interdependent roles for
hypoxia inducible factor and nuclear factor-kappaB in hypoxic
inflammation. J Physiol. 586:4055–4059. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen ZJ, Parent L and Maniatis T:
Site-specific phosphorylation of IkappaBalpha by a novel
ubiquitination-dependent protein kinase activity. Cell. 84:853–862.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Israël A: The IKK complex: An integrator
of all signals that activate NF-kappaB? Trends Cell Biol.
10:129–133. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Karin M: Nuclear factor-kappaB in cancer
development and progression. Nature. 441:431–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rutz HP: Effects of corticosteroid use on
treatment of solid tumours. Lancet. 360:1969–1970. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rutz HP and Herr I: Interference of
glucocorticoids with apoptosis signaling and host-tumor
interactions. Cancer Biol Ther. 3:715–718. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schaaf MJ and Cidlowski JA: Molecular
mechanisms of glucocorticoid action and resistance. J Steroid
Biochem Mol Biol. 83:37–48. 2002. View Article : Google Scholar
|
|
19
|
Kim H, Lee JM, Park JS, Jo SA, Kim YO, Kim
CW and Jo I: Dexamethasone coordinately regulates angiopoietin-1
and VEGF: A mechanism of glucocorticoid-induced stabilization of
blood-brain barrier. Biochem Biophys Res Commun. 372:243–248. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hata Y, Sassa Y, Kita T, Miura M, Kano K,
Kawahara S, Arita R, Nakao S, Shih JL and Ishibashi T: Vascular
endothelial growth factor expression by hyalocytes and its
regulation by glucocorticoid. Br J Ophthalmol. 92:1540–1544. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Iwai A, Fujii Y, Kawakami S, Takazawa R,
Kageyama Y, Yoshida MA and Kihara K: Down-regulation of vascular
endothelial growth factor in renal cell carcinoma cells by
glucocorticoids. Mol Cell Endocrinol. 226:11–17. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Machein MR, Kullmer J, Rönicke V, Machein
U, Krieg M, Damert A, Breier G, Risau W and Plate KH: Differential
downregulation of vascular endothelial growth factor by
dexamethasone in normoxic and hypoxic rat glioma cells. Neuropathol
Appl Neurobiol. 25:104–112. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yano A, Fujii Y, Iwai A, Kageyama Y and
Kihara K: Glucocorticoids suppress tumor angiogenesis and in vivo
growth of prostate cancer cells. Clin Cancer Res. 12:3003–3009.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu Y, Lucia K, Lange M, Kuhlen D, Stalla
GK and Renner U: Hypoxia inducible factor-1 is involved in growth
factor, glucocorticoid and hypoxia mediated regulation of vascular
endothelial growth factor-A in human meningiomas. J Neurooncol.
119:263–273. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bao Y, Lv F and Ma Y: Effect of
dexamethasone on expression of hypoxia inducible factor-1α and
vascular endothelial growth factor in hypoxic mice. Zhongguo Fei Ai
Za Zhi. 9:143–146. 2006.(In Chinese). PubMed/NCBI
|
|
26
|
Leonard MO, Godson C, Brady HR and Taylor
CT: Potentiation of glucocorticoid activity in hypoxia through
induction of the glucocorticoid receptor. J Immunol. 174:2250–2257.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kopacek J, Barathova M, Dequiedt F,
Sepelakova J, Kettmann R, Pastorek J and Pastorekova S: MAPK
pathway contributes to density- and hypoxia-induced expression of
the tumor-associated carbonic anhydrase IX. Biochim Biophys Acta.
1729:41–49. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kaluz S, Kaluzová M, Chrastina A, Olive
PL, Pastoreková S, Pastorek J, Lerman MI and Stanbridge EJ: Lowered
oxygen tension induces expression of the hypoxia marker MN/carbonic
anhydrase IX in the absence of hypoxia-inducible factor 1 alpha
stabilization: A role for phosphatidylinositol 3′-kinase. Cancer
Res. 62:4469–4477. 2002.PubMed/NCBI
|
|
29
|
Cartharius K, Frech K, Grote K, Klocke B,
Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M and Werner T:
MatInspector and beyond: Promoter analysis based on transcription
factor binding sites. Bioinformatics. 21:2933–2942. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Quandt K, Frech K, Karas H, Wingender E
and Werner T: MatInd and MatInspector: New fast and versatile tools
for detection of consensus matches in nucleotide sequence data.
Nucleic Acids Res. 23:4878–4884. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Abrahám IM, Meerlo P and Luiten PG:
Concentration dependent actions of glucocorticoids on neuronal
viability and survival. Dose Response. 4:38–54. 2006. View Article : Google Scholar
|
|
32
|
Cianchi F, Vinci MC, Supuran CT, Peruzzi
B, De Giuli P, Fasolis G, Perigli G, Pastorekova S, Papucci L, Pini
A, et al: Selective inhibition of carbonic anhydrase IX decreases
cell proliferation and induces ceramide-mediated apoptosis in human
cancer cells. J Pharmacol Exp Ther. 334:710–719. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dubois L, Peeters S, Lieuwes NG, Geusens
N, Thiry A, Wigfield S, Carta F, McIntyre A, Scozzafava A, Dogné
JM, et al: Specific inhibition of carbonic anhydrase IX activity
enhances the in vivo therapeutic effect of tumor irradiation.
Radiother Oncol. 99:424–431. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Morris JC, Chiche J, Grellier C, Lopez M,
Bornaghi LF, Maresca A, Supuran CT, Pouysségur J and Poulsen SA:
Targeting hypoxic tumor cell viability with carbohydrate-based
carbonic anhydrase IX and XII inhibitors. J Med Chem. 54:6905–6918.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ikeda Y, Carson BS and Long DM: The
effects of topical dexamethasone on experimental brain tumors and
peritumoral brain edema. Acta Neurochir Suppl (Wien). 60:397–399.
1994.
|
|
36
|
Maxwell PH, Wiesener MS, Chang GW,
Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER and
Ratcliffe PJ: The tumour suppressor protein VHL targets
hypoxia-inducible factors for oxygen-dependent proteolysis. Nature.
399:271–275. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wiesener MS, Turley H, Allen WE, Willam C,
Eckardt KU, Talks KL, Wood SM, Gatter KC, Harris AL, Pugh CW, et
al: Induction of endothelial PAS domain protein-1 by hypoxia:
Characterization and comparison with hypoxia-inducible
factor-1alpha. Blood. 92:2260–2268. 1998.PubMed/NCBI
|
|
38
|
Culver C, Sundqvist A, Mudie S, Melvin A,
Xirodimas D and Rocha S: Mechanism of hypoxia-induced NF-kappaB.
Mol Cell Biol. 30:4901–4921. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Koong AC, Chen EY and Giaccia AJ: Hypoxia
causes the activation of nuclear factor kappa B through the
phosphorylation of I kappa B alpha on tyrosine residues. Cancer
Res. 54:1425–1430. 1994.PubMed/NCBI
|
|
40
|
Badruddoja MA, Krouwer HG, Rand SD, Rebro
KJ, Pathak AP and Schmainda KM: Antiangiogenic effects of
dexamethasone in 9L gliosarcoma assessed by MRI cerebral blood
volume maps. Neuro Oncol. 5:235–243. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
van Uden P, Kenneth NS and Rocha S:
Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem
J. 412:477–484. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Iyer NV, Leung SW and Semenza GL: The
human hypoxia-inducible factor 1alpha gene: HIF1A structure and
evolutionary conservation. Genomics. 52:159–165. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Minet E, Ernest I, Michel G, Roland I,
Remacle J, Raes M and Michiels C: HIF1A gene transcription is
dependent on a core promoter sequence encompassing activating and
inhibiting sequences located upstream from the transcription
initiation site and cis elements located within the 5′UTR. Biochem
Biophys Res Commun. 261:534–540. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Niu G, Briggs J, Deng J, Ma Y, Lee H,
Kortylewski M, Kujawski M, Kay H, Cress WD, Jove R, et al: Signal
transducer and activator of transcription 3 is required for
hypoxia-inducible factor-1alpha RNA expression in both tumor cells
and tumor-associated myeloid cells. Mol Cancer Res. 6:1099–1105.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bhattacharya S, Michels CL, Leung MK,
Arany ZP, Kung AL and Livingston DM: Functional role of p35srj, a
novel p300/CBP binding protein, during transactivation by HIF-1.
Genes Dev. 13:64–75. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hudson CC, Liu M, Chiang GG, Otterness DM,
Loomis DC, Kaper F, Giaccia AJ and Abraham RT: Regulation of
hypoxia-inducible factor 1alpha expression and function by the
mammalian target of rapamycin. Mol Cell Biol. 22:7004–7014. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Treins C, Giorgetti-Peraldi S, Murdaca J,
Semenza GL and Van Obberghen E: Insulin stimulates
hypoxia-inducible factor 1 through a phosphatidylinositol
3-kinase/target of rapamycin-dependent signaling pathway. J Biol
Chem. 277:27975–27981. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang H, Kubica N, Ellisen LW, Jefferson LS
and Kimball SR: Dexamethasone represses signaling through the
mammalian target of rapamycin in muscle cells by enhancing
expression of REDD1. J Biol Chem. 281:39128–39134. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhou J, Schmid T, Frank R and Brüne B:
PI3K/Akt is required for heat shock proteins to protect
hypoxia-inducible factor 1alpha from pVHL-independent degradation.
J Biol Chem. 279:13506–13513. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Katschinski DM, Le L, Schindler SG, Thomas
T, Voss AK and Wenger RH: Interaction of the PAS B domain with
HSP90 accelerates hypoxia-inducible factor-1alpha stabilization.
Cell Physiol Biochem. 14:351–360. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Maheshwari M, Bhutani S, Das A, Mukherjee
R, Sharma A, Kino Y, Nukina N and Jana NR: Dexamethasone induces
heat shock response and slows down disease progression in mouse and
fly models of Huntington's disease. Hum Mol Genet. 23:2737–2751.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lim W, Park C, Shim MK, Lee YH, Lee YM and
Lee Y: Glucocorticoids suppress hypoxia-induced COX-2 and hypoxia
inducible factor-1α expression through the induction of
glucocorticoid-induced leucine zipper. Br J Pharmacol. 171:735–745.
2014. View Article : Google Scholar :
|
|
53
|
Leis H, Page A, Ramírez A, Bravo A,
Segrelles C, Paramio J, Barettino D, Jorcano JL and Pérez P:
Glucocorticoid receptor counteracts tumorigenic activity of Akt in
skin through interference with the phosphatidylinositol 3-kinase
signaling pathway. Mol Endocrinol. 18:303–311. 2004. View Article : Google Scholar
|
|
54
|
Box AH and Demetrick DJ: Cell cycle kinase
inhibitor expression and hypoxia-induced cell cycle arrest in human
cancer cell lines. Carcinogenesis. 25:2325–2335. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Mottet D, Dumont V, Deccache Y, Demazy C,
Ninane N, Raes M and Michiels C: Regulation of hypoxia-inducible
factor-1alpha protein level during hypoxic conditions by the
phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta
pathway in HepG2 cells. J Biol Chem. 278:31277–31285. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
De Bosscher K, Vanden Berghe W and
Haegeman G: The interplay between the glucocorticoid receptor and
nuclear factor-kappaB or activator protein-1: Molecular mechanisms
for gene repression. Endocr Rev. 24:488–522. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kaluz S, Kaluzová M, Opavský R,
Pastoreková S, Gibadulinová A, Dequiedt F, Kettmann R and Pastorek
J: Transcriptional regulation of the MN/CA 9 gene coding for the
tumor-associated carbonic anhydrase IX. Identification and
characterization of a proximal silencer element. J Biol Chem.
274:32588–32595. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ashburner BP, Westerheide SD and Baldwin
AS Jr: The p65 (RelA) subunit of NF-kappaB interacts with the
histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to
negatively regulate gene expression. Mol Cell Biol. 21:7065–7077.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Baetz D, Regula KM, Ens K, Shaw J, Kothari
S, Yurkova N and Kirshenbaum LA: Nuclear factor-kappaB-mediated
cell survival involves transcriptional silencing of the
mitochondrial death gene BNIP3 in ventricular myocytes.
Circulation. 112:3777–3785. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Datta De D, Datta A, Bhattacharjya S and
Roychoudhury S: NF-kappaB mediated transcriptional repression of
acid modifying hormone gastrin. PLoS One. 8:e734092013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Swietach P, Wigfield S, Cobden P, Supuran
CT, Harris AL and Vaughan-Jones RD: Tumor-associated carbonic
anhydrase 9 spatially coordinates intracellular pH in
three-dimensional multicellular growths. J Biol Chem.
283:20473–20483. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Swietach P, Patiar S, Supuran CT, Harris
AL and Vaughan-Jones RD: The role of carbonic anhydrase 9 in
regulating extracellular and intracellular ph in three-dimensional
tumor cell growths. J Biol Chem. 284:20299–20310. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Hulikova A, Harris AL, Vaughan-Jones RD
and Swietach P: Regulation of intracellular pH in cancer cell lines
under normoxia and hypoxia. J Cell Physiol. 228:743–752. 2013.
View Article : Google Scholar
|
|
64
|
Ditte Z, Ditte P, Labudova M, Simko V,
Iuliano F, Zatovicova M, Csaderova L, Pastorekova S and Pastorek J:
Carnosine inhibits carbonic anhydrase IX-mediated extracellular
acidosis and suppresses growth of HeLa tumor xenografts. BMC
Cancer. 14:3582014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Chiche J, Ilc K, Laferrière J, Trottier E,
Dayan F, Mazure NM, Brahimi-Horn MC and Pouysségur J:
Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell
growth by counteracting acidosis through the regulation of the
intracellular pH. Cancer Res. 69:358–368. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
McIntyre A, Patiar S, Wigfield S, Li JL,
Ledaki I, Turley H, Leek R, Snell C, Gatter K, Sly WS, et al:
Carbonic anhydrase IX promotes tumor growth and necrosis in vivo
and inhibition enhances anti-VEGF therapy. Clin Cancer Res.
18:3100–3111. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Ivanova L, Zandberga E, Siliņa K, Kalniņa
Z, Ābols A, Endzeliņš E, Vendina I, Romanchikova N, Hegmane A,
Trapencieris P, et al: Prognostic relevance of carbonic anhydrase
IX expression is distinct in various subtypes of breast cancer and
its silencing suppresses self-renewal capacity of breast cancer
cells. Cancer Chemother Pharmacol. 75:235–246. 2015. View Article : Google Scholar
|
|
68
|
Mattern J, Büchler MW and Herr I: Cell
cycle arrest by glucocorticoids may protect normal tissue and solid
tumors from cancer therapy. Cancer Biol Ther. 6:1345–1354. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Takada Y, Bhardwaj A, Potdar P and
Aggarwal BB: Nonsteroidal anti-inflammatory agents differ in their
ability to suppress NF-kappaB activation, inhibition of expression
of cyclooxygenase-2 and cyclin D1, and abrogation of tumor cell
proliferation. Oncogene. 23:9247–9258. 2004.PubMed/NCBI
|
|
70
|
Wang ML, Chiou SH and Wu CW: Targeting
cancer stem cells: Emerging role of Nanog transcription factor.
Onco Targets Ther. 6:1207–1220. 2013.PubMed/NCBI
|
|
71
|
Dang CV: c-Myc target genes involved in
cell growth, apoptosis, and metabolism. Mol Cell Biol. 19:1–11.
1999. View Article : Google Scholar
|
|
72
|
Lavia P and Jansen-Dürr P: E2F target
genes and cell-cycle checkpoint control. BioEssays. 21:221–230.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Meyer C, Pries R and Wollenberg B:
Established and novel NF-κB inhibitors lead to downregulation of
TLR3 and the proliferation and cytokine secretion in HNSCC. Oral
Oncol. 47:818–826. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Mayer A, Schneider F, Vaupel P, Sommer C
and Schmidberger H: Differential expression of HIF-1 in
glioblastoma multiforme and anaplastic astrocytoma. Int J Oncol.
41:1260–1270. 2012.PubMed/NCBI
|
|
75
|
Hussain SA, Ganesan R, Reynolds G, Gross
L, Stevens A, Pastorek J, Murray PG, Perunovic B, Anwar MS,
Billingham L, et al: Hypoxia-regulated carbonic anhydrase IX
expression is associated with poor survival in patients with
invasive breast cancer. Br J Cancer. 96:104–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Wykoff CC, Beasley N, Watson PH, Campo L,
Chia SK, English R, Pastorek J, Sly WS, Ratcliffe P and Harris AL:
Expression of the hypoxia-inducible and tumor-associated carbonic
anhydrases in ductal carcinoma in situ of the breast. Am J Pathol.
158:1011–1019. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Chu CY, Jin YT, Zhang W, Yu J, Yang HP,
Wang HY, Zhang ZJ, Liu XP and Zou Q: CA IX is upregulated in
CoCl2-induced hypoxia and associated with cell invasive
potential and a poor prognosis of breast cancer. Int J Oncol.
48:271–280. 2016.
|
|
78
|
Konno H, Ishii G, Nagai K, Yoshida J,
Nishimura M, Nara M, Fujii T, Murata Y, Miyamoto H and Ochiai A:
Carbonic anhydrase IX expression is associated with tumor
progression and a poor prognosis of lung adenocarcinoma. Lung
Cancer. 54:409–418. 2006. View Article : Google Scholar
|
|
79
|
Le QT, Kong C, Lavori PW, O'byrne K, Erler
JT, Huang X, Chen Y, Cao H, Tibshirani R, Denko N, et al:
Expression and prognostic significance of a panel of tissue hypoxia
markers in head-and-neck squamous cell carcinomas. Int J Radiat
Oncol Biol Phys. 69:167–175. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Haapasalo J, Hilvo M, Nordfors K,
Haapasalo H, Parkkila S, Hyrskyluoto A, Rantala I, Waheed A, Sly
WS, Pastorekova S, et al: Identification of an alternatively
spliced isoform of carbonic anhydrase XII in diffusely infiltrating
astrocytic gliomas. Neuro Oncol. 10:131–138. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Chen J, Röcken C, Hoffmann J, Krüger S,
Lendeckel U, Rocco A, Pastorekova S, Malfertheiner P and Ebert MP:
Expression of carbonic anhydrase 9 at the invasion front of gastric
cancers. Gut. 54:920–927. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Stewart DJ, Nunez MI, Behrens C, Liu D,
Lin YH, Lee JJ, Roth J, Heymach J, Swisher SG, Hong WK, et al:
Membrane carbonic anhydrase IX expression and relapse risk in
resected stage I–II non-small-cell lung cancer. J Thorac Oncol.
9:675–684. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Dubois L, Peeters SG, van Kuijk SJ,
Yaromina A, Lieuwes NG, Saraya R, Biemans R, Rami M, Parvathaneni
NK, Vullo D, et al: Targeting carbonic anhydrase IX by
nitroimidazole based sulfamides enhances the therapeutic effect of
tumor irradiation: A new concept of dual targeting drugs. Radiother
Oncol. 108:523–528. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Wojtkowiak JW, Verduzco D, Schramm KJ and
Gillies RJ: Drug resistance and cellular adaptation to tumor acidic
pH microenvironment. Mol Pharm. 8:2032–2038. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Koukourakis MI, Giatromanolaki A,
Danielidis V and Sivridis E: Hypoxia inducible factor (HIf1alpha
and HIF2alpha) and carbonic anhydrase 9 (CA9) expression and
response of head-neck cancer to hypofractionated and accelerated
radiotherapy. Int J Radiat Biol. 84:47–52. 2008. View Article : Google Scholar
|
|
86
|
Lee-Kong SA, Ruby JA, Chessin DB,
Pucciarelli S, Shia J, Riedel ER, Nitti D and Guillem JG:
Hypoxia-related proteins in patients with rectal cancer undergoing
neoadjuvant combined modality therapy. Dis Colon Rectum.
55:990–995. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Span PN, Bussink J, Manders P, Beex LV and
Sweep CG: Carbonic anhydrase-9 expression levels and prognosis in
human breast cancer: Association with treatment outcome. Br J
Cancer. 89:271–276. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Tan EY, Yan M, Campo L, Han C, Takano E,
Turley H, Candiloro I, Pezzella F, Gatter KC, Millar EK, et al: The
key hypoxia regulated gene CAIX is upregulated in basal-like breast
tumours and is associated with resistance to chemotherapy. Br J
Cancer. 100:405–411. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Giatromanolaki A, Koukourakis MI, Sivridis
E, Pastorek J, Wykoff CC, Gatter KC and Harris AL: Expression of
hypoxia-inducible carbonic anhydrase-9 relates to angiogenic
pathways and independently to poor outcome in non-small cell lung
cancer. Cancer Res. 61:7992–7998. 2001.PubMed/NCBI
|