Introduction
Radiotherapy, following surgery and chemotherapy, is
one of the most important therapies for colorectal cancer (CRC) in
the advanced stage. Multiple clinical studies have verified that
radiotherapy can, for example, lower preoperative staging and
improve the operative resection rate and progression-free survival
of patients (1). However, due to
the indigenous biological properties of CRC, the tumor tissue
itself shows low radiosensitivity. Moreover, radiotolerance is
limited during the radiotherapy for CRC because of many normal
risks to organs (such as small intestine and kidney) in its
anatomical environment. Although radiotherapy instruments and
technologies are constantly being upgraded, they exhibit restricted
capacity of simultaneously increasing the dosage at the tumor
region and decreasing the exposure to normal organs, thus limiting
their application in gastrointestinal tumors (2). Therefore, enhancing the
radiosensitivity of CRC cells based on the indigenous biological
properties of tumors has become a critically urgent challenge to be
addressed.
Cellular responses to external damage (due to
radiotherapy and chemotherapy) depend on the self-repairing ability
of cells, such as the ability to repair DNA double-strand breaks
(DSBs) (3). Multiple signaling
pathways are involved in the process of DSB repair, including
homologous recombination repair, nonhomologous end joining, cell
cycle regulation and apoptotic pathways (4,5). p53
binding protein 1 (53BP1) has been confirmed to contribute to
apoptosis induction in DNA damage repair (DDR) and helps in
determining the cellular response to different therapies (6,7).
53BP1 was originally believed to be the binding protein of tumor
suppressor gene p53 and thought to be involved in p53-mediated
apoptosis. 53BP1 has an important function as a significant
reaction medium at the early stage in signal transduction pathways
following DNA damage (6). Also,
tumor tissues differ in their degree of 53BP1 deletion, which is
closely related to the malignant staging of tumor cells and
histological staging, and affects the sensitivity to drug, which is
manifested as resistance to treatment (7,8).
Moreover, 53BP1 can affect tumor (breast cancer and glioma)
responses to radiotherapy (9,10),
although it has been rarely reported how abnormal expression of
53BP1 disturbs the response of CRC to radiotherapy. A previous
study showed that epidermal growth factor receptor-tyrosine kinase
inhibitor-icotinib hydrochloride enhanced the sensitivity of CRC to
radiotherapy by increasing the expression of 53BP1 (11). In contrast, the radiosensitization
effect of icotinib hydrochloride was notably eliminated when the
expression of 53BP1 of CRC cells was decreased by shRNA, which
implied that the abnormal expression of 53BP1 could influence the
radiosensitivity of CRC. However, the exact effects and potential
underlying mechanisms remain to be resolved.
Recent studies showed that on external damage, cells
may activate multiple signaling pathways, which may contribute to
cellular injury repair and induce apoptosis. The
ataxia-telangiectasia mutated kinase (ATM)-checkpoint kinase-2
(CHK2)-p53 pathway plays a pivotal role in the DDR process. When
activated, it may induce cell cycle arrest, leading to the
activation of p53-related apoptotic pathways, which trigger
apoptosis (12,13). 53BP1 is closely related to the
ATM-CHK2-p53 pathway because of its special structure. It possesses
two Tudor domains, which promote it to interact with ATM to involve
in DDR (8,14,15),
and a C-terminal BRTC domain, which activates CHK1 and CHK2 of the
cell cycle to regulate the G1/S, S and G2/M checkpoints,
phosphorylate p53, and then induce p53-dependent apoptosis
(15,16). Thus, considering the
mediator-featured structure of 53BP1, we speculated that the
abnormal expression of 53BP1 could affect the tumor cell
proliferation, apoptosis, and cell cycle distribution through
interfering the ATM-CHK2-p53 pathway.
Previous studies have found that the expression of
53BP1 was deficient in CRC tissue, which was an early factor of
poor prognosis. Therefore, we hypothesized that this deficiency may
be an intrinsic factor that influences the response of colorectal
tumors to radiotherapy. The present study aimed to verify the
correlation between the expression of 53BP1 and radiosensitivity in
various CRC cell lines and to investigate how 53BP1 affects the
response to radiotherapy by disturbing the expression of 53BP1 in
CRC cells in vitro. Moreover, the expression of proteins
related to the ATM-CHK2-p53 pathway was also detected to explore
the intrinsic factors of 53BP1 affecting the radiotherapy efficacy
mechanically.
Materials and methods
Cell culture and animal care
HCT116, SW620, Caco2 and LoVo cells were cultured in
an RPMI-1640 medium containing 10% fetal bovine serum (FBS) at
37°C, 5% CO2, and saturated humidity. Passages were
performed to maintain monolayer growth. Cells were collected at the
exponential growth phase for subsequent experiments. Female athymic
nude mice (nu/nu; body weight, 20–25 g; 6–10 weeks of age) were
purchased from Beijing HFK Bioscience Co., Ltd. (Beijing, China).
The care and treatment of all experimental mice was in accordance
with the institutional guidelines.
Cell transfection
This study used GCACAAGAACTTATGG AAAGT as the shRNA
sequence of 53BP1 and TTCTCCGA ACGTGTCACGT as the shRNA sequence of
the control group. The lentivirus plasmids containing the shRNA and
green fluorescent protein lentivirus vector containing 53BP1 shRNA
were purchased from Shanghai Genechem Co., Ltd. (Shanghai, China).
The concentrated virus solution and HCT116 cells were co-cultured,
and their fluorescence was observed by optical microscopy to
confirm successful transfection. The transfection rates were
verified by western blot analysis and reverse
transcription-polymerase chain reaction (RT-PCR).
Clone formation assay
The cells at the exponential growth phase were
digested into single-cell suspensions. After counting, the living
cells were inoculated onto a 6-well plate at 100–5,000 cells/well.
When cells grew in an adherent manner after a 24-h incubation, they
were exposed to different radiation dosages (0, 2, 4, 6, 8 and 10
Gy) with a dosage rate of 2 Gy/min. Incubation was terminated after
10–14 days when >50 cells formed a clone in a visible cell mass.
The cells were fixed with methanol and stained with Giemsa;
colonies containing at least 50 cells were counted, and cell
survival curves were plotted using the multitarget click model to
compare the difference of radiosensitivity. Relative parameters,
such as mean lethal dose (D0), quasi-threshold
dose (Dq), extrapolation number (N),
surviving fraction at 2 Gy (SF2), and a ratio of
SF2, termed as sensitization enhancing ratio (SER), were
calculated. Three parallel tests were set.
Evaluation of tumor proliferation by
immunofluorescent staining of Ki-67
The proliferation antigen Ki-67 was used to assess
the proliferation rate of tumor. At the exponential growth phase,
2×105 cells/ml were plated in chamber slides. After 24 h
of irradiation (at 0, 2 and 4 Gy), the cells were fixed and
permeabilized with 0.2% Triton X-100 (Wuhan Boster Biological
Technology, Ltd., Wuhan, China). They were then incubated with
anti-Ki-67 antibody (1:50, 19972-1-AP; Proteintech Group, Rosemont,
IL, USA), and then with Alexa Fluor 488-conjugated goat anti-rabbit
(Proteintech Group) at a dilution of 1:200. Finally, the sections
were counterstained with 6-diamidino-2-phenylindole dihydrochloride
(Vector Laboratories, Inc., Burlingame, CA, USA). The sections were
examined on a confocal laser scanning microscope (Olympus, Tokyo,
Japan) equipped with a camera. To determine the percentage of
positive cells with Ki-67, at least 500–1,000 tumor cells per slide
were counted, and the number of Ki-67-positive cells was scored and
the positive rate was counted.
Cell cycle detection by flow
cytometry
A total of 2×105 cells/ml at the
exponential growth phase were collected and inoculated onto a
6-well plate. When the cells attached to the wall after a 24-h
incubation, they were exposed to different irradiation doses (at 0,
2 and 4 Gy). The cells were then digested with trypsin 24 h after
irradiation. After overnight fixation with 70% ethanol, the cells
were digested again with 1% RNase for 30 min at 37°C, followed by
30-min staining with 20 mg/ml propidium iodide (PI). Finally, the
cell cycle was detected using a flow cytometer (BD Biosciences, San
Jose, CA, USA).
Apoptosis detection by flow
cytometry
The cells were routinely digested, washed, suspended
gently with 500 μl combining solution, gently blended with 5 μl
Annexin V-FITC, and finally blended with 5 μl PI using an Annexin
V-FITC apoptosis detection kit, according to the manufacturer’s
instructions. Then, after 10-min incubation at room temperature
(20–25°C) in darkness, cells were analyzed using a flow cytometer
(BD Biosciences). Ten thousand cells in each sample were analyzed,
and the CellQuest software (BD Biosciences) was employed to analyze
the data.
Western blot analysis
The cells were lysed in a radio immunoprecipitation
assay lysis buffer for 30 min at 4°C and centrifuged at 12,000 × g
for 5 min. The supernatant was collected, combined with sodium
dodecyl sulfate (SDS) buffer, and heated to 100°C for 5 min. The
proteins were separated by 10% SDS-PAGE and blotted to the
polyvinylidene fluoride membranes, which were incubated with
primary antibodies against 53BP1 (ab175933, 1:2,000; Abcam,
Cambridge, MA, USA), ATM (#21147, 1:1,000; Signalway), ATMpS1981
(5883, 1:1,000; Cell Signaling Technology, Danvers, MA, USA), CHK2
(AP4999a, 1:1,000; Abgent, San Diego, CA, USA), CHK2pT68 (2197,
1:1,000; Cell Signaling Technology), p53 (AM2244B, 1:1,000;
Abgent), p-P53 (9286, 1:1,000; Cell Signaling Technology),
caspase-9 (10380-1-AP, 1:1,000; Proteintech Group), caspase-3
(BS1518, 1:1,000; Bioworl, Dublin, OH, USA), Bax (1063, 1:1,000;
EpiGentek, Farmingdale, NY, USA), Bcl-2 (2870-P, 1:1,000; Cell
Signaling Technology), or β-actin antibody (1:1,000; Santa Cruz
Biotechnology, Santa Cruz, CA, USA) at 4°C overnight. The membranes
were washed with Tris-buffered saline and Tween-20 three times,
incubated with secondary antibodies for 2 h at room temperature,
and visualized by enhanced chemiluminescence. Band intensities were
analyzed by the Gel-Pro analysis program.
Quantitative RT-PCR
The total RNA was extracted with a TRIzol reagent.
Then, an RT-PCR kit (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) was used to reversely transcribe RNA into cDNA. StepOne and
StepOnePlus Real-Time PCR Systems (Applied Biosystems) were adopted
for amplification analysis. More than three parallel tests were set
for all trials.
Establishing the tumor-bearing nude mouse
model
Cultured tumor cells were collected to prepare tumor
cell suspensions with an RPMI-1640 (1:1) at a concentration of
approximately 2×107 cells/ml for the following animal
experiments. Tumor cells (0.1 ml) (HCT116 and HCT116 with 53BP1
silencing) were subcutaneously injected into the posterior limbs of
the mice and observed for 5–7 days. When tumors reached a diameter
of 10 mm, all nude mice were randomly divided into different groups
(n=3) to receive different treatments as observation by
radiotherapy. For the radiotherapy group, irradiation was performed
using a 6 MV linear accelerator (MDX, Siemens) with an exposure
field of 5×5 cm2, and the total dose was 10 Gy (2 Gy/day
for 5 days). Since the beginning of the treatment, the caliper was
used to measure the longest diameter L and the shortest diameter W
of each tumor every other day, and the tumor volume was calculated
based on the following formula: Tumor volume = (L ×
W2)/2 mm3. The growth time and volume of each
tumor were recorded.
Statistical analysis
SPSS 13.0 software was used for the statistical
analysis. Quantitative data were expressed as mean 13.0. Mean value
comparison among multiple groups was performed by one-way analysis
of variance, and the comparison between the two groups was carried
out by applying the Q-test. P≤0.05 indicated statistical
significance.
Results
Expression of 53BP1 is closely related to
the radiosensitivity of CRC cells
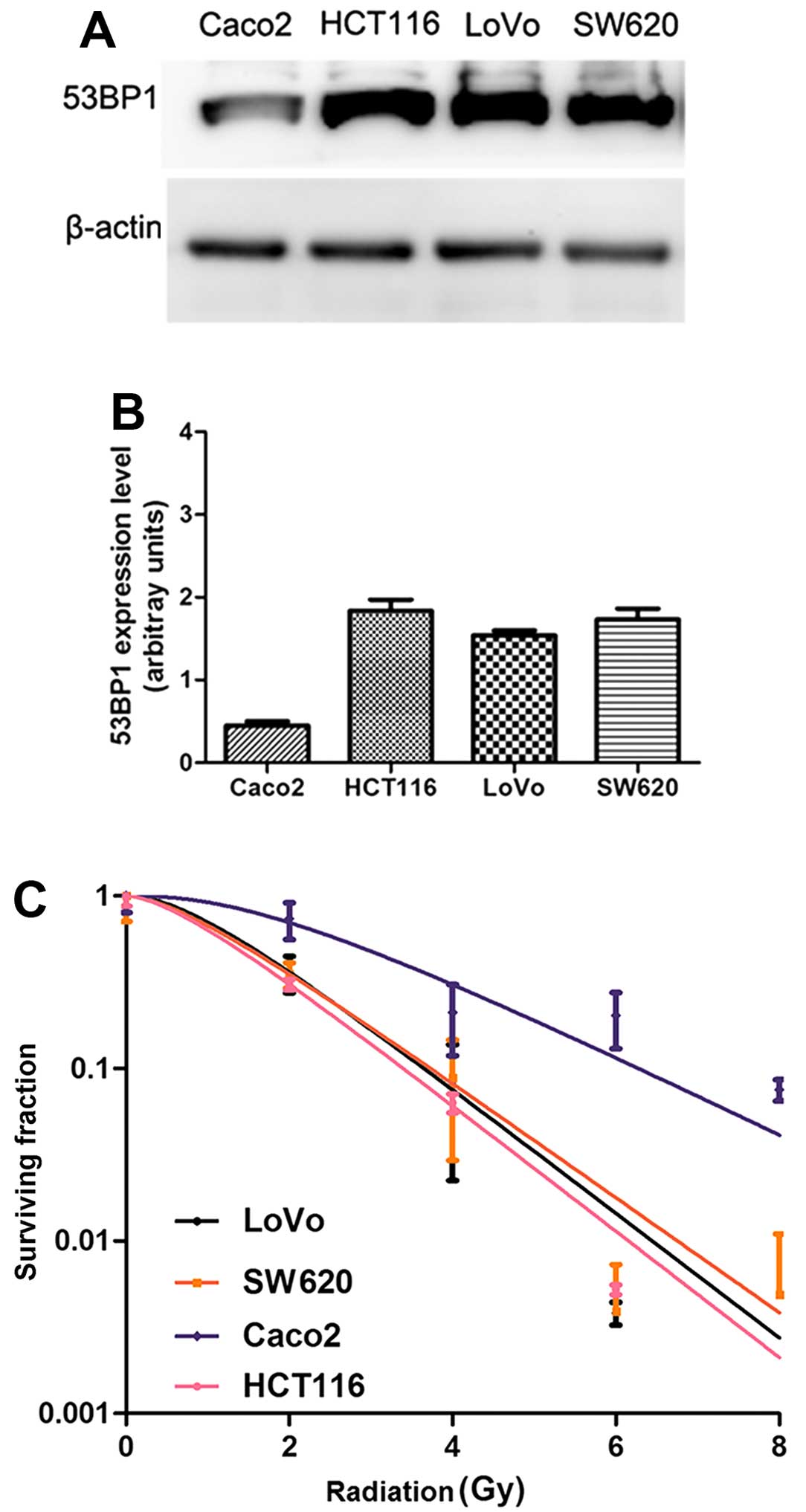
To investigate the correlation between the
expression of 53BP1 and radiosensitivity of CRC cells, the
expression of 53BP1 in the four CRC cell lines (HCT116, SW620,
Caco2 and LoVo cells) was detected in vitro by the western
blot analysis (Fig. 1A and B), and
the radiosensitivity of these intestinal cancer cell lines was
simultaneously detected by the clone formation assay (Fig. 1C). The results showed that the
expression of 53BP1 was closely related to the radiosensitivity of
CRC cells. The HCT116 cell line with the high expression of 53BP1
was relatively sensitive to irradiation with lower values of
SF2, D0, Dq, and
N in the cell survival curve analysis, while the Caco2 cell
line with the low expression of 53BP1 was relatively tolerant to
irradiation with higher values of SF2,
D0, Dq, and N in the
cell survival curve analysis (Table
I).
 | Table IThe relative parameters of cell
survival curves after irradiation. |
Table I
The relative parameters of cell
survival curves after irradiation.
| Parameter/cell
lines | Caco2 | LoVo | SW620 | HCT116 |
|---|
|
D0 | 1.90 | 1.20 | 1.30 | 1.18 |
|
Dq | 3.44 | 1.37 | 1.03 | 0.93 |
| N | 2.812 | 2.137 | 1.792 | 1.786 |
| SF2 | 0.74 | 0.36 | 0.35 | 0.31 |
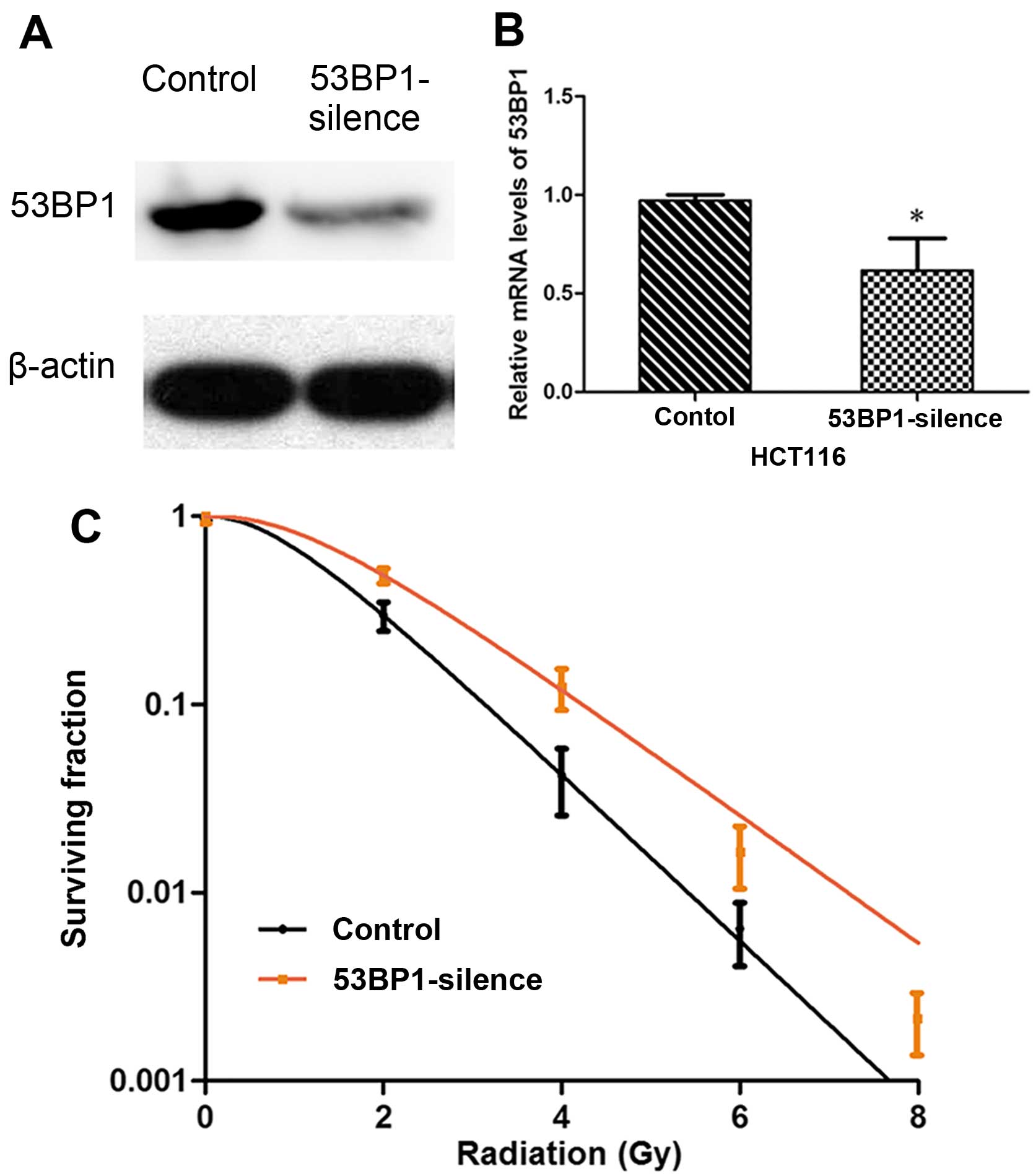
Decreased expression of 53BP1 inhibits
the radiosensitivity of HCT116 cell line
The HCT116 cell line with relatively high expression
of 53BP1 was selected, and stable HCT116 cell stains were
constructed by RNA-intervened lentiviral vector transfection by
decreasing the expression of 53BP1 via shRNA. The effective
transfection rate was verified by western blot analysis and RT-PCR
(Fig. 2A and B). Then, the effect
of transfection on the cell radiosensitivity was detected, which
showed that the decrease in the expression of 53BP1 increased
SF2, D0, Dq, and
N values, and decreased the value of SER. It indicated that
the decreased expression of 53BP1 obviously inhibited the
radiosensitivity of the HCT116 cells (Table II and Fig. 2C).
| Table IIEffect of 53BP1 silencing on
radiosensitivity in HCT116 cells. |
Table II
Effect of 53BP1 silencing on
radiosensitivity in HCT116 cells.
| Parameter/cell
lines | Control | 53BP1 silence |
|---|
|
D0 | 0.98 | 1.28 |
|
Dq | 1.50 | 2.35 |
| N | 1.53 | 1.84 |
| SF2 | 0.30 | 0.49 |
| SER | | 0.77 |
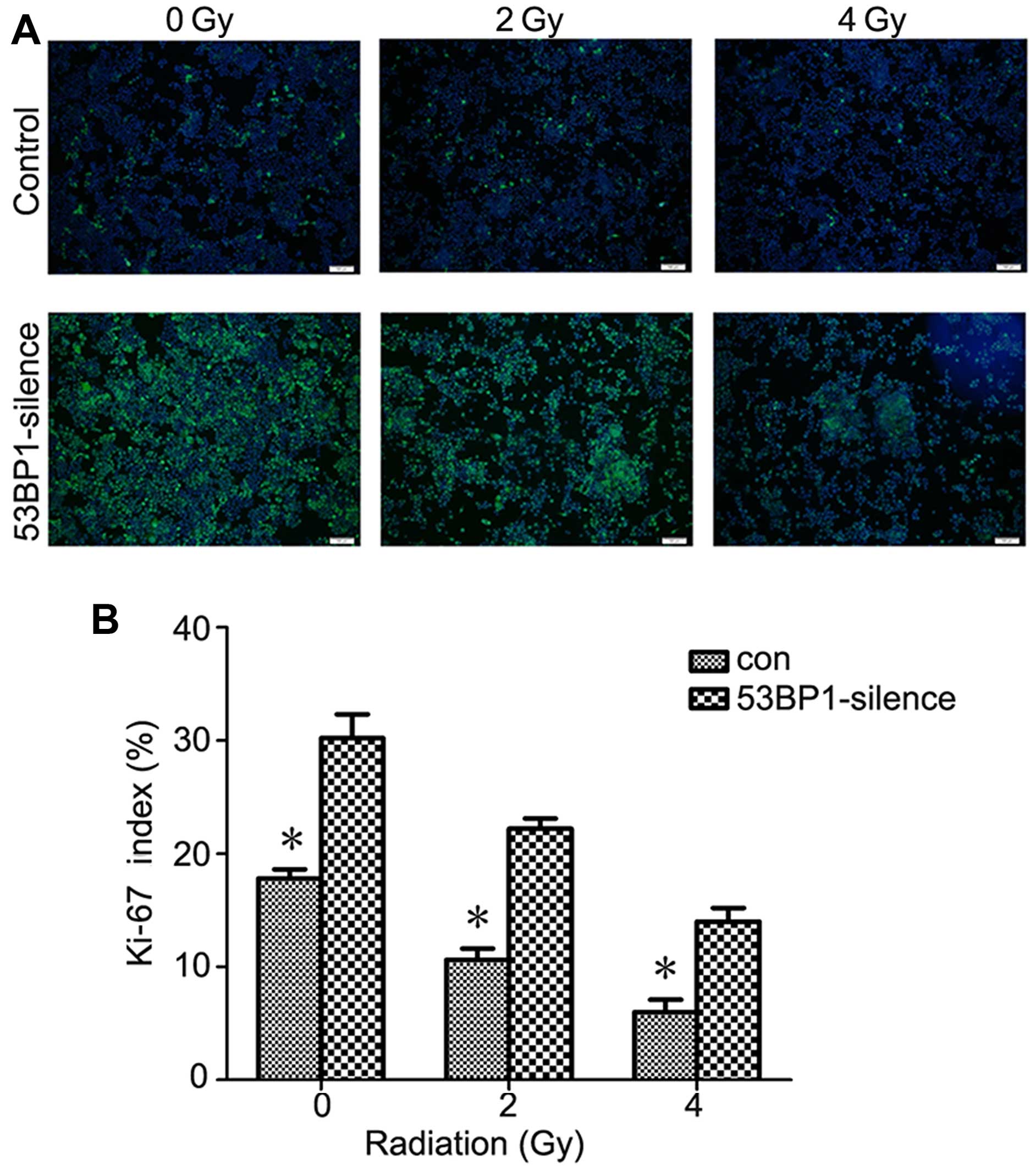
Decreased expression of 53BP1 increases
the proliferation rate of HCT116 cells after irradiation
To determine the association between the
radiosensitivity of HCT116 cells after 53BP1 silencing and tumor
proliferation, the expression of Ki-67 was investigated by
immunofluorescent staining. As shown in Fig. 3, the results revealed that the
decrease in the expression of 53BP1 obviously increased the
expression of Ki-67 compared with the control group. The
proliferation rate was 31±4 vs. 18±4% before irradiation
(P<0.05). After irradiation of 2 and 4 Gy, the 53BP1-silenced
group still revealed stronger capacity to induce tumor
proliferation compared with the control group. The proliferation
rate was 22±3 vs. 11±4% for 2 Gy and 14±5 vs. 6±2% for 4 Gy. The
difference between the two groups was obvious (P<0.05).
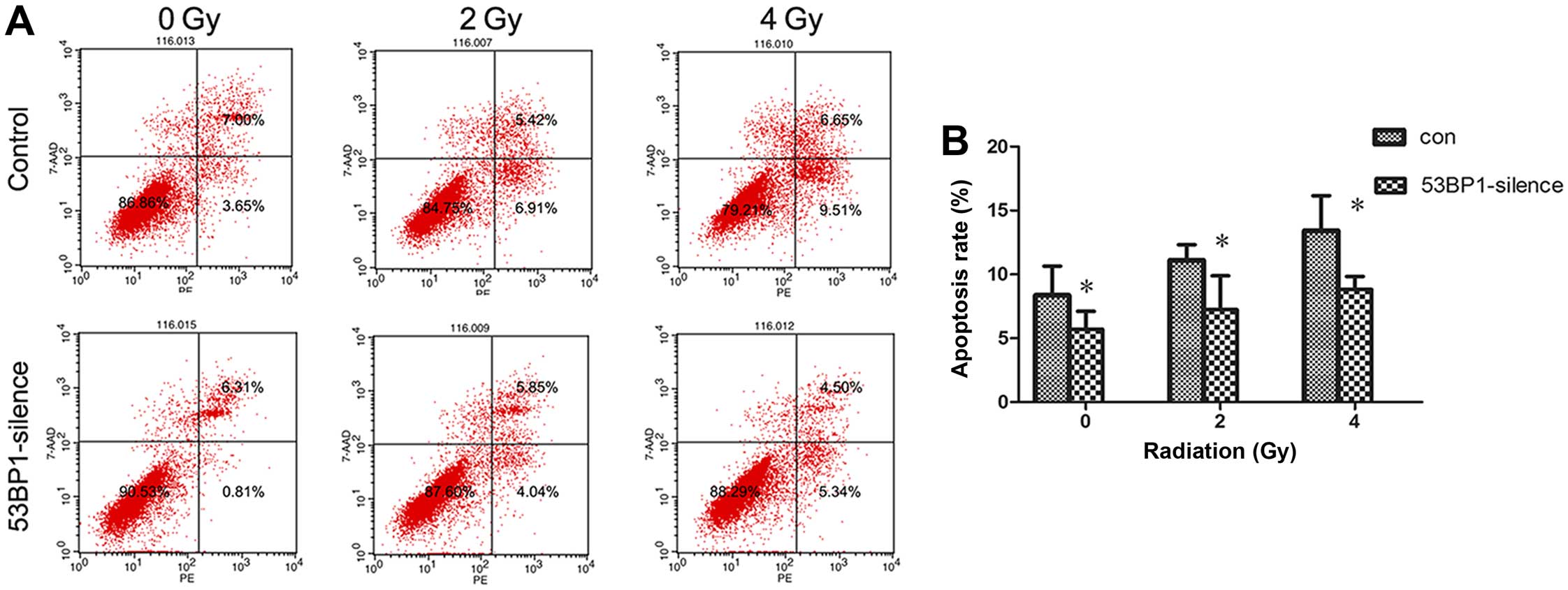
Decreased expression of 53BP1 reduces the
apoptotic rate of HCT116 cells after radiotherapy
The effect of the decreased expression of 53BP1 on
tumor cell apoptosis was investigated after radiotherapy. The
apoptotic rate of tumor cells before and after intervention at 24 h
after exposure to irradiation doses of 0, 2 and 4 Gy was detected
using flow cytometry (Fig. 4A).
The results showed that the decrease in the expression of 53BP1
significantly reduced apoptosis of HCT116 cells (8.49±1.88 vs.
4.19±1.08%; P<0.05); the decrease in the expression of 53BP1
reduced apoptosis after irradiation (10.58±1.16 vs. 5.91±2.65% for
2 Gy, 13.43±3.15 vs. 8.82±1.17% for 4 Gy; P<0.05) (Fig. 4B).
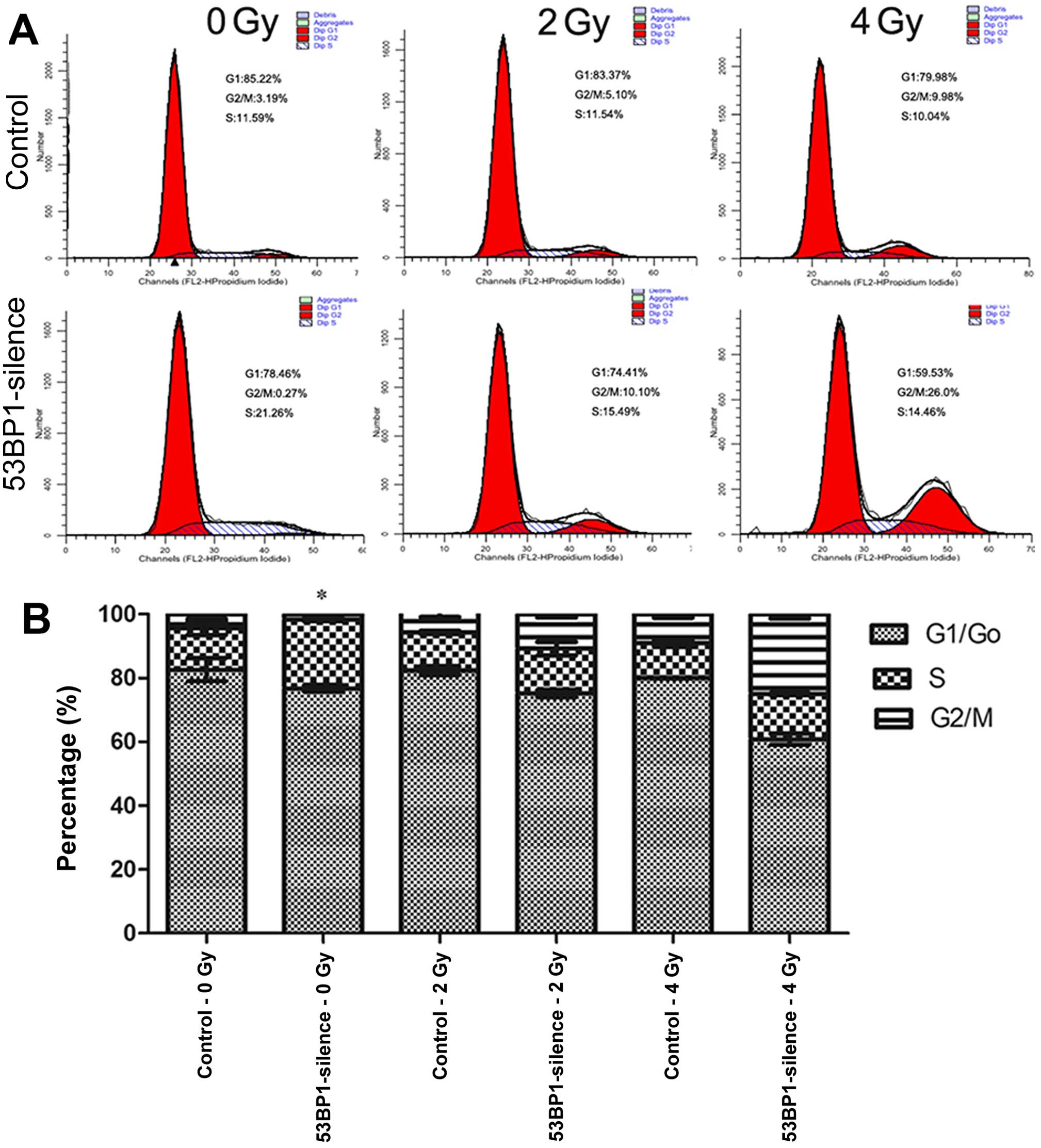
Decreased expression of 53BP1 increases
the percentage of S-phase of HCT116 cells after irradiation
The effect of the decreased expression of 53BP1 on
the cell cycle of HCT116 cells after irradiation was investigated
using the flow cytometer (Fig.
5A). The results showed that the decrease in the expression of
53BP1 remarkably increased the percentage of the S-phase in tumor
cells (21.78±0.73 vs. 13.09±2.12%; P<0.05), indicating that the
decreased expression of 53BP1 ushered tumor cells into the
proliferation cycle with an accelerated proliferation rate. This
result was consistent with the results of previous studies. After
irradiation with an exposure to doses 2 and 4 Gy, the two 53BP1
silencing groups exhibited relatively higher percentages of S-phase
compared with the control groups (14.92±0.71 vs. 11.91±0.53% for 2
Gy, 14.23±0.32 vs. 10.80±1.07% for 4 Gy) although the difference
between these two exposure groups was not significant (Fig. 5B). This indicates that the decrease
in the expression of 53BP1 ushers tumor cells into a
high-proliferation status, which inhibits the efficacy of regular
radiotherapy.
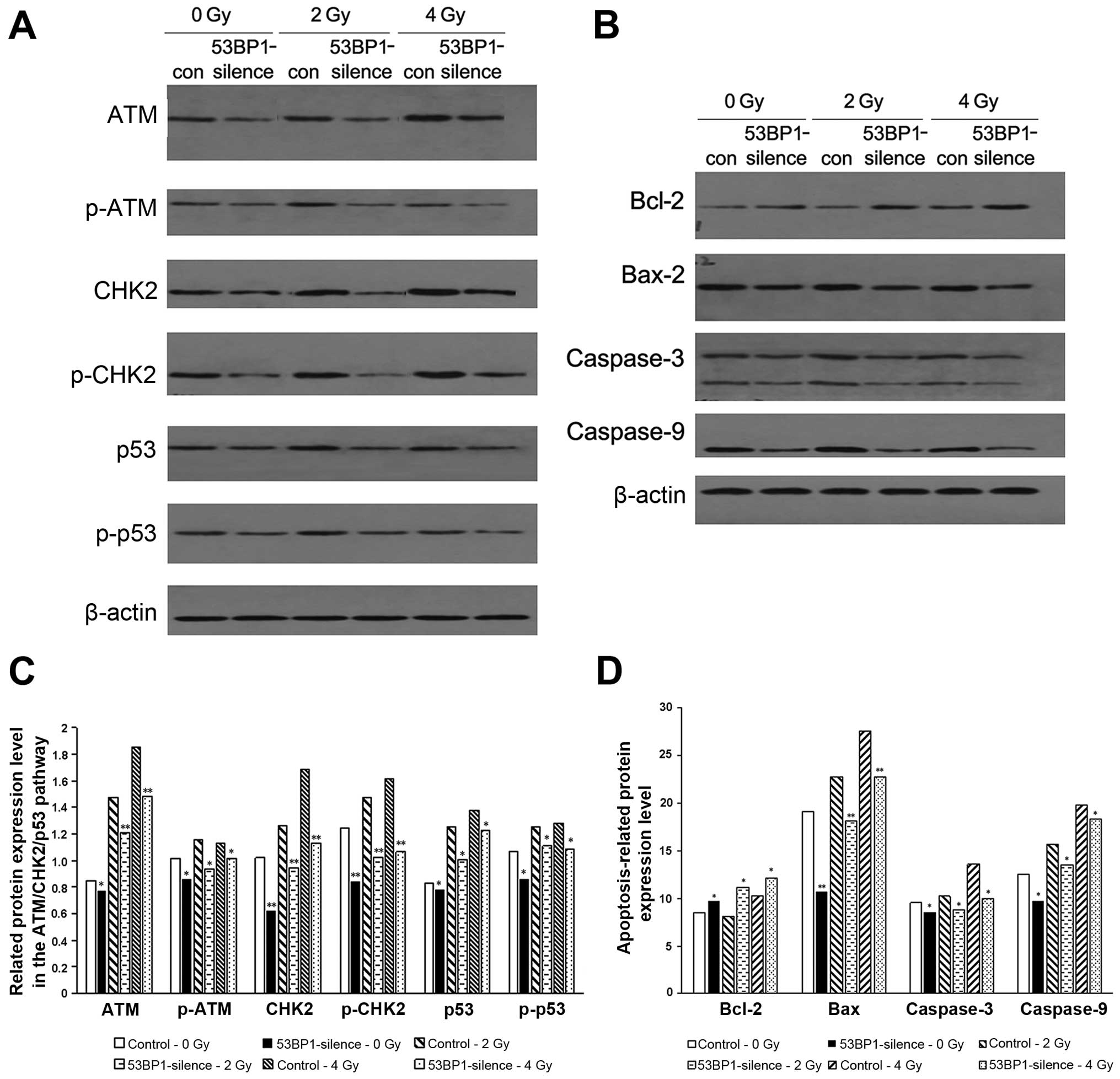
Decreased expression of 53BP1 inhibits
the expression of relevant proteins in the ATM-CHK2-p53
pathway
The ATM-CHK2-p53 pathway plays a pivotal role in the
DNA injury repair process and that activation of this pathway may
induce the cell cycle arrest, aggravate the DNA injury, and trigger
the expression of p53-related apoptotic proteins. Therefore,
western blot analysis was used to detect the expression of relevant
proteins and their phosphorylated products in the ATM-CHK2-p53
pathway; apoptosis-related proteins caspase-9, caspase-3 and Bax;
and the anti-apoptosis protein Bcl-2 before and after the decreased
expression of 53BP1 and with or without radiation of 2 and 4 Gy.
The results showed that the decrease in the expression of 53BP1
decreased the expression of apoptotic pathway-related proteins ATM,
CHK2 and p53 as well as their phosphorylated products (Fig. 6A and C); reduced the
apoptosis-related proteins caspase-9, caspase-3 and Bax; and
increased the expression of anti-apoptosis protein Bcl-2 (Fig. 6B and D), which may be the imminent
cause of the decreased expression of 53BP1, resulting in an active
proliferation of tumor cells. Similarly, the 53BP1-silenced group
showed the reduced expression of the afore-mentioned
apoptosis-related proteins and enhanced expression of the
anti-apoptosis protein Bcl-2 compared with the control group after
exposure to different radiation doses. This indicated that the
decrease in the expression of 53BP1 led to the tolerance of tumor
cells to irradiation via inhibiting the apoptosis-related
ATM-CHK2-p53 pathway and the expression of relevant pro-apoptosis
proteins and anti-apoptosis proteins.
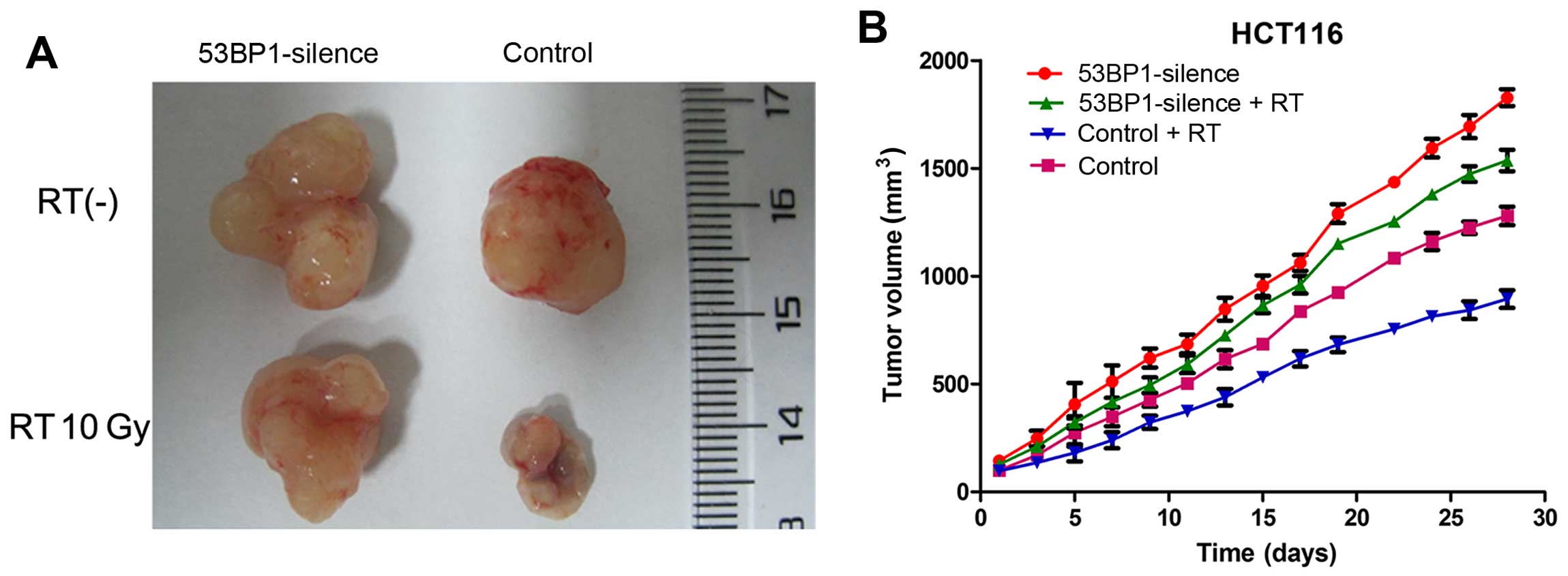
Silencing 53BP1 in vivo induces the
proliferation of tumor cells and inhibits reactivity to
radiotherapy
To investigate the effect of 53BP1 on tumor
proliferation and radiotherapy efficacy in vivo, the
tumor-bearing nude mouse model was established. HCT116 and HCT116
with genetically silenced 53BP1 were subcutaneously injected into
the right posterior limb of the mice. Tumor nodes were palpated
after 7–10 days, and reached a diameter of 10 mm after 2 weeks.
Subsequently, radiotherapy was performed (10 Gy/5F). The size of
each tumor was recorded every other day starting from the beginning
of treatment. When the tumors of a certain group reached a volume
of 2 cm3, all nude mice were sacrificed to record the
final tumor volume. The results showed that the tumors of the
53BP1-silenced group showed a relatively higher proliferation
activity and reached a size of nearly 2 cm3 on the 28th
day of the treatment. Then, all nude mice were sacrificed and the
final volume of each tumor was recorded. The final tumor volumes of
the 53BP1-silenced group without radiotherapy and the control group
were 1.82±0.06 and 1.28±0.06 cm3, respectively, with a
significant difference (P<0.01). The 53BP1-silenced group with
radiotherapy had a markedly higher proliferation velocity compared
with the control group (final tumor volume: 1.54±0.07 vs. 0.89±0.06
cm3; P=0.01), with a significant difference (Fig. 7). The results showed that the
decrease in the expression of 53BP1 notably induced the growth of
the transplanted colorectal tumor, which exhibited resistance to
radiotherapy.
Discussion
Radiotherapy against CRC has now hit a bottleneck.
Decades of radiosensitization studies have only realized a limited
efficacy, which is primarily caused by the limited radiosensitivity
of CRC cells. Therefore, the analysis of indigenous factors that
affect tumor radiosensitivity is of crucial importance and has
become an urgent problem that needs to be resolved. Multiple
studies have shown that genetic stability is the necessary
condition under which cells retain their normal function and
maintain basic survival (8,17).
However, DNA is quite vulnerable to various factors, and this
injury may lead to gene mutations, genomic instability, chromosomal
loss and reorganization, apoptosis, or even cancerization if timely
and precise repair is unavailable (18,19).
The DDR pathway plays an essential role in triggering DDR and
maintaining the genomic stability of normal cells. Dysfunction of
critical factors in the DDR pathway is closely related to the
onset, development, and therapy resistance of multiple tumors
(20–24). 53BP1 is a recently found cancer
suppressor gene, which is also an important member of the DDR
pathway family. It is involved in DDR, maintains genomic stability,
and regulates apoptosis in coordination with p53 and ATM (25,26).
The deficiency of 53BP1 may lead to failed anchoring of broken
chromosome ends, which lays the foundation for chromosomal
aberration (22). Studies have
shown that different degrees of 53BP1 deletion exist during the
onset and development of multiple tumors and that this deletion is
closely related to tumor staging, malignancy grading, and even
therapy resistance (8,17). Neboori et al (9) found that 53BP1 deletion resulted in
an increased local recurrence rate of breast cancer after
radiotherapy. Similarly, Han et al (27) found that the decrease in the
expression of 53BP1 could enhance the response to DNA damaging
agents. The deletion of the expression of 53BP1 in CRC tissue has
been detected in previous studies, which was found to be related to
the location of the tumors: the right half of the colon exhibited a
higher 53BP1 deletion rate compared with the left half of the
colon. Furthermore, 53BP1 deficiency was also an early risk factor
of poor prognosis (28).
Therefore, this study hypothesized that the deficiency of 53BP1 in
CRC tissue probably played a part in influencing the response to
radiotherapy.
The present study performed in vivo and in
vitro trials to verify this hypothesis. The expression of 53BP1
in various CRC cell lines was detected by western blot analysis
together with differences in their radiosensitivity. The results
showed that the expression of 53BP1 was closely related to the
radiosensitivity of CRC cells. HCT116 cells with the high
expression of 53BP1 were relatively sensitive to radioactivity,
while Caco2 cells with low expression of 53BP1 were relatively
tolerant to radioactive rays. In this study, the expression of
53BP1 was further silenced via gene interference to investigate the
effects of 53BP1 on the radiosensitivity of CRC. The HCT116 cell
line with the high expression of 53BP1 was selected, and its stable
cell line was constructed with an RNA-intervened lentiviral vector
to decrease the expression of 53BP1 by shRNA. The effects of 53BP1
on radiosensitivity were detected by the clone formation assay
before and after 53BP1 silencing. The results showed that the
decrease in the expression of 53BP1 inhibited the radiosensitivity
of the HCT116 cell line. Furthermore, the effects of decreased
expression of 53BP1 on proliferation and apoptosis were
investigated, which revealed that the exposure to radiation doses
of 0, 2 and 4 Gy remarkably induced tumor proliferation and
inhibited tumor cell apoptosis. Then, the effect on the cell cycle
was investigated using flow cytometry, which showed that the
decreased expression of 53BP1 remarkably increased the percentage
of the S-phase in HCT116 cells; the two exposure groups
(irradiation of 2 and 4 Gy) had higher percentages of S-phase
compared with the control groups, although the difference between
these two groups was not significant. The previous studies also
showed that the decreased expression of 53BP1 can increase the
percentage of S-phase (28), which
is correlated with cell proliferation (29,30).
The results indicated that the decreased expression of 53BP1
prominently enhanced tumor proliferation, inhibited apoptosis, and
ushered cells into the proliferation stage, which was associated
with radioresistance.
However, the potential mechanisms of 53BP1 impacting
the radiosensitivity of CRC is still uncertain. Han et al
(27) found that the increased
expression of 53BP1 can affect the expression of certain proteins
in the DDR pathway to inhibit DDR, which was related to an enhanced
sensitivity to therapy. Recent studies have recognized that
ATM-CHK2-p53 as the primary pathway related to apoptosis is
involved in the process of DDR repair, and the deletion of critical
elements in this pathway results in the resistance to radiotherapy
(17). 53BP1 has been found to be
critically related to this pathway in the DDR process, thus, this
study speculated that 53BP1 participates in regulating apoptosis
and cell cycle distribution probably via affecting the ATM-CHK2-p53
pathway and then influencing the response of tumor tissue to
radiotherapy. Therefore, after the expression of 53BP1 was
decreased in HCT116 cells, mediated by shRNA interference, the
proteins and their phosphorylated variants in the ATM-CHK2-p53
pathway were detected by western blot analysis, along with
apoptosis-related protein caspase-9, caspase-3, Bax and Bcl-2. The
results revealed that the decrease in the expression of 53BP1
decreased the expression of proteins related to the ATM-CHK2-p53
pathway (ATM and CHK2, as well as their phosphorylated products);
decreased the expression of apoptosis-related proteins caspase-9,
caspase-3 and Bax; and increased the expression of the
anti-apoptosis protein Bcl-2. This indicated that 53BP1 affected
the response of CRC to radiotherapy through impacting the
ATM-CHK2-p53 pathway and that 53BP1 played an important role in
affecting the response of tumor cells to radiotherapy. The
tumor-bearing nude mouse model was then established to investigate
the effects of 53BP1 silencing on radiotherapy in vivo,
which showed that 53BP1 silencing notably induced tumor
proliferation and that the tumor had a relatively
high-proliferation rate even after radiotherapy. Therefore, 53BP1
silencing was verified in vivo to inhibit the efficacy of
radiotherapy.
Oncotherapy has now entered the era of precise
treatment. Precise radiotherapy is required to maximize efficacy
and minimize toxicity. However, no clear factors have been
specified to predict the efficacy of radiotherapy. The in
vitro and in vivo studies showed that 53BP1 deletion
inhibited the ATM-CHK2-p53 pathway for inducing cell proliferation,
inhibiting apoptosis, and then inhibiting the radiosensitivity of
intestinal cancer. These results provide new perspectives for
future studies in this field, but further clinical studies remain
to be performed.
Abbreviations:
|
53BP1
|
p53 binding protein 1
|
|
CRC
|
radiosensitivity of colorectal
cancer
|
|
DSBs
|
double-strand breaks
|
|
DDR
|
damage repair
|
|
ATM
|
ataxia-telangiectasia mutated
kinase
|
|
CHK2
|
checkpoint kinase-2
|
|
RT-PCR
|
reverse transcription-polymerase chain
reaction
|
|
SER
|
sensitization enhancing ratio
|
References
|
1
|
Shin SJ, Yoon HI, Kim NK, Lee KY, Min BS,
Ahn JB, Keum KC and Koom WS: Upfront systemic chemotherapy and
preoperative short-course radiotherapy with delayed surgery for
locally advanced rectal cancer with distant metastases. Radiat
Oncol. 6:992011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang T, Liang ZW, Han J, Bi JP, Yang ZY
and Ma H: Double-arc volumetric modulated therapy improves dose
distribution compared to static gantry IMRT and 3D conformal
radiotherapy for adjuvant therapy of gastric cancer. Radiat Oncol.
10:1142015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang M, Kern AM, Hülskötter M, Greninger
P, Singh A, Pan Y, Chowdhury D, Krause M, Baumann M, Benes CH, et
al: EGFR-mediated chromatin condensation protects KRAS-mutant
cancer cells against ionizing radiation. Cancer Res. 74:2825–2834.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Di Micco R, Fumagalli M, Cicalese A,
Piccinin S, Gasparini P, Luise C, Schurra C, Garre’ M, Nuciforo PG,
Bensimon A, et al: Oncogene-induced senescence is a DNA damage
response triggered by DNA hyper-replication. Nature. 444:638–642.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bartkova J, Rezaei N, Liontos M,
Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E,
Niforou K, Zoumpourlis VC, et al: Oncogene-induced senescence is
part of the tumorigenesis barrier imposed by DNA damage
checkpoints. Nature. 444:633–637. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Carr SM, Munro S, Zalmas LP, Fedorov O,
Johansson C, Krojer T, Sagum CA, Bedford MT, Oppermann U and La
Thangue NB: Lysine methylation-dependent binding of 53BP1 to the
pRb tumor suppressor. Proc Natl Acad Sci USA. 111:11341–11346.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bouwman P, Aly A, Escandell JM, Pieterse
M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni
A, Yang Q, et al: 53BP1 loss rescues BRCA1 deficiency and is
associated with triple-negative and BRCA-mutated breast cancers.
Nat Struct Mol Biol. 17:688–695. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nuciforo PG, Luise C, Capra M, Pelosi G
and d’Adda di Fagagna F: Complex engagement of DNA damage response
pathways in human cancer and in lung tumor progression.
Carcinogenesis. 28:2082–2088. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Neboori HJ, Haffty BG, Wu H, Yang Q, Aly
A, Goyal S, Schiff D, Moran MS, Golhar R, Chen C, et al: Low p53
binding protein 1 (53BP1) expression is associated with increased
local recurrence in breast cancer patients treated with
breast-conserving surgery and radiotherapy. Int J Radiat Oncol Biol
Phys. 83:e677–e683. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Squatrito M, Vanoli F, Schultz N, Jasin M
and Holland EC: 53BP1 is a haploinsufficient tumor suppressor and
protects cells from radiation response in glioma. Cancer Res.
72:5250–5260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ma H, Bi J, Liu T, Ke Y, Zhang S and Zhang
T: Icotinib hydrochloride enhances the effect of radiotherapy by
affecting DNA repair in colorectal cancer cells. Oncol Rep.
33:1161–1170. 2015.PubMed/NCBI
|
|
12
|
Shi Y, Felley-Bosco E, Marti TM, Orlowski
K, Pruschy M and Stahel RA: Starvation-induced activation of
ATM/Chk2/p53 signaling sensitizes cancer cells to cisplatin. BMC
Cancer. 12:5712012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chung YM, Park SH, Tsai WB, Wang SY, Ikeda
MA, Berek JS, Chen DJ and Hu MC: FOXO3 signalling links ATM to the
p53 apoptotic pathway following DNA damage. Nat Commun. 3:10002012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bartkova J, Horejsí Z, Sehested M, Nesland
JM, Rajpert-De Meyts E, Skakkebaek NE, Stucki M, Jackson S, Lukas J
and Bartek J: DNA damage response mediators MDC1 and 53BP1:
Constitutive activation and aberrant loss in breast and lung
cancer, but not in testicular germ cell tumours. Oncogene.
26:7414–7422. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Morales JC, Franco S, Murphy MM, Bassing
CH, Mills KD, Adams MM, Walsh NC, Manis JP, Rassidakis GZ, Alt FW,
et al: 53BP1 and p53 synergize to suppress genomic instability and
lymphomagenesis. Proc Natl Acad Sci USA. 103:3310–3315. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ausborn NL, Wang T, Wentz SC, Washington
MK, Merchant NB, Zhao Z, Shyr Y, Chakravarthy AB and Xia F: 53BP1
expression is a modifier of the prognostic value of lymph node
ratio and CA 19-9 in pancreatic adenocarcinoma. BMC Cancer.
13:1552013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Squatrito M, Brennan CW, Helmy K, Huse JT,
Petrini JH and Holland EC: Loss of ATM/Chk2/p53 pathway components
accelerates tumor development and contributes to radiation
resistance in gliomas. Cancer Cell. 18:619–629. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ward IM, Difilippantonio S, Minn K,
Mueller MD, Molina JR, Yu X, Frisk CS, Ried T, Nussenzweig A and
Chen J: 53BP1 cooperates with p53 and functions as a
haploinsufficient tumor suppressor in mice. Mol Cell Biol.
25:10079–10086. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gorgoulis VG, Vassiliou LV, Karakaidos P,
Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr,
Kastrinakis NG, Levy B, et al: Activation of the DNA damage
checkpoint and genomic instability in human precancerous lesions.
Nature. 434:907–913. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Khanna A: DNA damage in cancer
therapeutics: A boon or a curse? Cancer Res. 75:2133–2138. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Karanika S, Karantanos T, Li L, Corn PG
and Thompson TC: DNA damage response and prostate cancer: Defects,
regulation and therapeutic implications. Oncogene. 34:2815–2822.
2015. View Article : Google Scholar :
|
|
22
|
Tian H, Gao Z, Li H, Zhang B, Wang G,
Zhang Q, Pei D and Zheng J: DNA damage response - a double-edged
sword in cancer prevention and cancer therapy. Cancer Lett.
358:8–16. 2015. View Article : Google Scholar
|
|
23
|
Lai TC, Chow KC, Lin TY, Chiang IP, Fang
HY, Chen CY and Ho SP: Expression of 53BP1 as a cisplatin-resistant
marker in patients with lung adenocarcinomas. Oncol Rep.
24:321–328. 2010.PubMed/NCBI
|
|
24
|
Li X, Kong X, Kong X, Wang Y, Yan S and
Yang Q: 53BP1 sensitizes breast cancer cells to 5-fluorouracil.
PLoS One. 8:e749282013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Clarke AR, Jones N, Pryde F, Adachi Y and
Sansom OJ: 53BP1 deficiency in intestinal enterocytes does not
alter the immediate response to ionizing radiation, but leads to
increased nuclear area consistent with polyploidy. Oncogene.
26:6349–6355. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cao L, Xu X, Bunting SF, Liu J, Wang RH,
Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A, et al: A selective
requirement for 53BP1 in the biological response to genomic
instability induced by Brca1 deficiency. Mol Cell. 35:534–541.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Han X, Zhang L, Chung J, Mayca Pozo F,
Tran A, Seachrist DD, Jacobberger JW, Keri RA, Gilmore H and Zhang
Y: UbcH7 regulates 53BP1 stability and DSB repair. Proc Natl Acad
Sci USA. 111:17456–17461. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bi J, Huang A, Liu T, Zhang T and Ma H:
Expression of DNA damage checkpoint 53BP1 is correlated with
prognosis, cell proliferation and apoptosis in colorectal cancer.
Int J Clin Exp Pathol. 8:6070–6082. 2015.PubMed/NCBI
|
|
29
|
Sun CC, Chiu HT, Lin YF, Lee KY and Pang
JH: Y-27632, a ROCK inhibitor, promoted limbal epithelial cell
proliferation and corneal wound healing. PLoS One. 10:e01445712015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li T, Shi HY, Hua YX, Gao C, Xia Q, Yang G
and Li B: Effects of allicin on the proliferation and cell cycle of
chondrocytes. Int J Clin Exp Pathol. 8:12525–12532. 2015.
|