Introduction
Head and neck cancers consistently rank among the
six most frequently diagnosed types of cancer in the world
(1). Over 90% of head and neck
cancers are squamous cell carcinomas (2). Laryngeal squamous cell carcinoma
(LSCC) is a common type of head and neck malignancy worldwide.
Despite the advance in the conventional therapies, the overall
survival rate for LSCC has not significantly improved (3). Epidermal growth factor receptor
(EGFR) is a member of the HER tyrosine kinase growth factor
receptor family and is invariably involved in signaling pathways
affecting cellular growth, differentiation and proliferation
(4). It has been well established
that overexpression of EGFR promotes tumor growth and progression,
including maturation, angiogenesis, invasion, metastasis and
inhibition of apoptosis (5).
Several research groups have confirmed that LSCC shows
significantly higher EGFR expression compared with normal
epithelium (6). Our group had also
detected the EGFR gene amplification in human laryngeal squamous
carcinoma HEp-2 cells and in 11 laryngeal carcinoma tissues by
fluorescence in situ hybridization (FISH) (7,8).
High expression levels of EGFR contribute to oncogenesis and tumor
progression in LSCC (9). Taken
together, these reports indicate that EGFR is a promising target
for LSCC.
Cetuximab (Cet) is a mouse-human chimeric anti-EGFR
monoclonal antibody. Cet binds specifically to the extracellular
domain of EGFR and induces an internalization of the receptor
leading to downregulation of EGFR (10). Cet has been shown to inhibit the
proliferation of a variety of cultured malignant human cell lines
that overexpress EGFR (11,12).
In 2004, Cet was approved as an intravenous infusion for the
treatment of head and neck cancers by the United States Food and
Drug Administration (FDA) (13).
However, Cet when used as a single agent exhibits limited efficacy
with lower response rates (<15%) in head and neck squamous cell
carcinoma (HNSCC) patients (14).
It is, therefore, essential to benchmark antitumor activity against
the data accumulated with Cet when given as part of a doublet or
triplet combination (15).
Anticancer agents derived from herbs and plants
continue to attract attention globally due to their purported
better efficiency. Rabdosia rubescens, the widely used herb
in traditional Chinese medicine, is shown to suppress tumor
progression, prolong survival and has low adverse effect in
patients with esophageal, gastric and hepatic carcinoma (16,17).
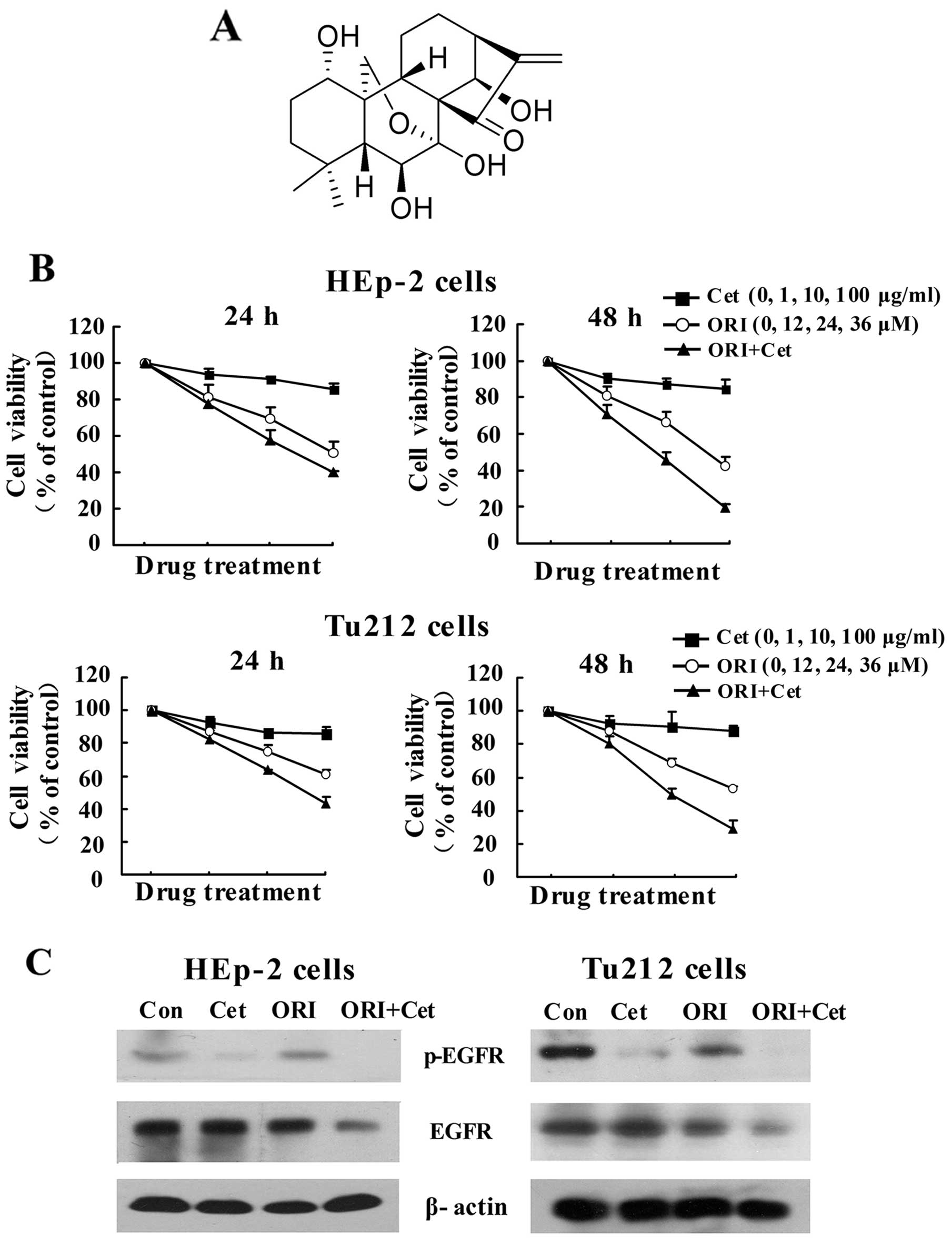
Oridonin (ORI) (Fig. 1A), a
natural kaurene diterpenoid, is an essential antitumor component
from Rabdosia rubescens. ORI exhibits significant antitumor
activity in a variety of cancer cells (18,19)
and has low cytotoxicity against normal cells and tissues (20,21).
Our previous studies also showed that ORI inhibits cell growth in
HEp-2 cells through suppression of EGFR expression (22). Since combination of Cet with other
cytotoxic drugs could be an effective management for HNSCC, it
would be interesting to delineate and characterize the antitumor
effects of the combined treatment with Cet and ORI on LSCC
cells.
In the present study, we show that combination of
ORI with Cet at lower doses synergistically increased the antitumor
effects in LSCC cells as compared with either single agent.
Furthermore, our results demonstrated that ORI plus Cet
significantly induced apoptosis and cell cycle arrest in
vitro and in vivo through inhibition of EGFR
phosphorylation and activation of ROS-mediated JNK signaling
pathway.
Materials and methods
Reagents
Cet was obtained from Merck KGaA (Darmstadt,
Germany). ORI was obtained from the Beijing Institute of Biological
Products (Beijing, China). RPMI-1640 medium and fetal bovine serum
(FBS) were obtained from Gibco-BRL (Gaithersburg, MD, USA). The
primary antibodies for western blotting and immunohistochemical
studies were purchased from Cell Signaling Technology (Beverly, MA,
USA). All of the other chemicals were purchased from Sigma-Aldrich
(St. Louis, MO, USA).
Cell culture
HEp-2 and Tu212 cells were obtained from the
American Type Culture Collection (ATCC; Manassas, VA, USA). Cell
lines were maintained at 37°C and 5% CO2 in RPMI-1640
medium supplemented with 10% FBS and 10 μg/ml streptomycin and 100
μg/ml penicillin.
Cell viability assay
Cell viability of ORI and Cet on LSCC cells were
measured by MTT assay. HEp-2 and Tu212 cells were seeded onto
96-well culture plates at a density of 0.5 and 1×104
cells/well. Following treatment with the indicated concentrations
of ORI (12, 24 and 36 μM) and/or Cet (1, 10 and 100 μg/ml) for 24
and 48 h, the cells were washed twice with PBS. Subsequently, MTT
was then added at a final concentration of 0.5 μg/ml, and the cells
were further incubated for 2.5 h. The medium was removed, and the
formazan crystals were dissolved in dimethyl sulfoxide (DMSO) (150
μl). The optical density (OD) was measured at 490 nm using a
microplate reader (BioTek Laboratories, Winooski, VT, USA). The
percentage of cell viability was calculated as follows: Cell
viability (%) = [(A490, sample − A490,
blank)/(A490, control − A490,
blank)]x100.
The combined effects of ORI and Cet on cell growth
inhibition were analyzed using the software CalcuSyn (Biosoft,
Ferguson, MO, USA), which applies the median-effect equation of
Chou and the CI (combination index) equation of Chou and Talalay
(23).
Fluorescence microscopy examination
The apoptotic nuclear morphology was assessed by
staining the cells with the fluorescent DNA-binding dye acridine
orange (AO). After treatment with ORI and/or Cet for 48 h, LSCC
cells were stained with 20 μg/ml AO for 15 min, and then the
nuclear morphology was observed under a fluorescence microscope
(Olympus, Tokyo, Japan).
Flow cytometric analysis of apoptosis and
cell cycle distribution
Cell apoptosis was analyzed using an Annexin
V-PE/7-AAD apoptosis kit (BD Biosciences, San Diego, CA, USA)
according to the manufacturer's instructions. After treatment with
ORI and/or Cet for 48 h, LSCC cells were washed with binding buffer
and centrifuged. The cell pellet was resuspended in binding buffer,
and 5 μl Annexin-V-PE and 5 μl 7-AAD were added, mixed, and the
preparation was incubated for 15 min in the dark at room
temperature. The apoptotic cells were measured using a BD
FACSCalibur flow cytometer (Becton-Dickinson, Franklin Lakes, NJ,
USA).
To evaluate the cell cycle distribution, LSCC cells
were treated with ORI and/or Cet for 48 h. Cell cycle distribution
was conducted according to a previous report (22). All data were recorded and analyzed
using the FlowJo software version 7.6 (Tree Star, Inc., Ashland,
OR, USA).
Detection of intracellular ROS
accumulation
Intracellular ROS accumulation was monitored using
DCF-DA, which is a specific probe for the presence of hydrogen
peroxide. The experiments were conducted by flow cytometry as
previously described (24).
HEp-2 xenografts in nude mice
All of the experimental procedures were approved,
and the mice were maintained and treated in accordance with the
institutional guidelines of Animal Care and Use Committee of
Tianjin International Joint Academy of Biotechnology and Medicine.
Six-to-eight-week-old female BALB/c athymic
(nu+/nu+) mice were purchased from Vital
River Laboratories, Co., Ltd. (Beijing, China). Logarithmically
growing HEp-2 cells were harvested by trypsinization, and each
mouse was given injections of 1×106 cells subcutaneously
into the right flank. Tumor growth was assessed every day by
caliper measurement. Tumor volume (mm3) was calculated
by the formula: π/6 × larger diameter × (smaller
diameter)2. In these experiments, all the mice were
randomly divided into 4 groups and injected intraperitoneally
(i.p.) with vehicle, Cet (1 mg/mice) alone, ORI (20 mg/kg) alone,
or ORI and Cet in combination (n=10 per group). Animals in the
control group were treated with DMSO and sterile PBS given by
i.p.
In the study, all of the mice were sacrificed at the
end of the treatment, and their tumors were harvested for
immunohistochemical analysis. ELISA was used to test the ROS
production of tumor sample, as previously described (25).
Immunohistochemistry and TUNEL assay
Immunohistochemical analysis of tumor tissue from
nude mouse xenografts was performed following the standard
protocol. Terminal deoxynucleotidyl transferase-mediated dUTP nick
end labeling assay (TUNEL) was carried out according to the
manufacturer's instructions (Promega, Madison, WI, USA) as
previously described (26).
Western blot assays
Culture cells and subcutaneous tumors were lysed in
cell lysis buffer, and the protein concentrations were determined
using the Bradford absorbance assay. Protein expression was
analyzed by western blot as previously described (22).
Data analysis
All data were analyzed using SPSS 17.0 software.
One-way ANOVA was employed to analyze the differences between sets
of data. For all analysis, P<0.05 was considered to indicate a
statistically significant result.
Results
Combined treatment with ORI and Cet
induces LSCC cells death via inhibition of EGFR
The anti-proliferative effects of combination of ORI
and Cet were examined using the MTT assay in LSCC cell lines. As
shown in Fig. 1B, Cet alone only
had a moderate inhibitory effect on the growth of LSCC cells, while
ORI alone caused a time- and concentration-dependent inhibition of
proliferation. Importantly, ORI in combination with Cet augmented
growth inhibition of LSCC cells. The CI value of the combination of
24 μM ORI and 10 μg/ml Cet for 48 h was 0.59 for HEp-2 cells, while
the CI value of the combination of 36 μM ORI and 10 μg/ml Cet for
48 h was 0.50 for Tu212 cells (Table
I). The CI values <1 indicate that ORI and Cet exhibit
synergistic growth inhibition of LSCC cells.
 | Table IIn vitro combination study of
Cet with ORI in HEp-2 and Tu212 cells. |
Table I
In vitro combination study of
Cet with ORI in HEp-2 and Tu212 cells.
| Combining Cet with
ORI | CI of HEp-2
cells | CI of Tu212
cells |
|---|
|
|
|---|
| 24 h | 48 h | 24 h | 48 h |
|---|
| 1 μg/ml Cet |
| 12 μM ORI | 0.75 | 0.71 | 1.51 | 0.66 |
| 24 μM ORI | 0.81 | 0.67 | 0.72 | 0.61 |
| 36 μM ORI | 0.66 | 0.63 | 0.55 | 0.51 |
| 10 μg/ml Cet |
| 12 μM ORI | 0.78 | 0.73 | 1.23 | 0.78 |
| 24 μM ORI | 0.70 | 0.59 | 0.83 | 0.663 |
| 36 μM ORI | 0.60 | 0.62 | 0.50 | 0.50 |
| 100 μg/ml Cet |
| 12 μM ORI | 0.99 | 1.02 | 1.89 | 0.68 |
| 24 μM ORI | 0.73 | 0.60 | 1.57 | 0.69 |
| 36 μM ORI | 0.56 | 0.64 | 0.67 | 0.54 |
To characterize the expression of EGFR that might
correlate with the observed growth inhibition, the effect of
ORI/Cet combination on the levels of phosphorylated and total EGFR
was examined by western blotting. Cet alone reduced p-EGFR
expression in two LSCC cells, whereas ORI induced moderate
inhibition. Combination treatment further decreased the expression
of p-EGFR and total EGFR (Fig.
1C).
Combination of ORI and Cet induces
apoptosis through activation of Fas-mediated extrinsic apoptotic
pathway
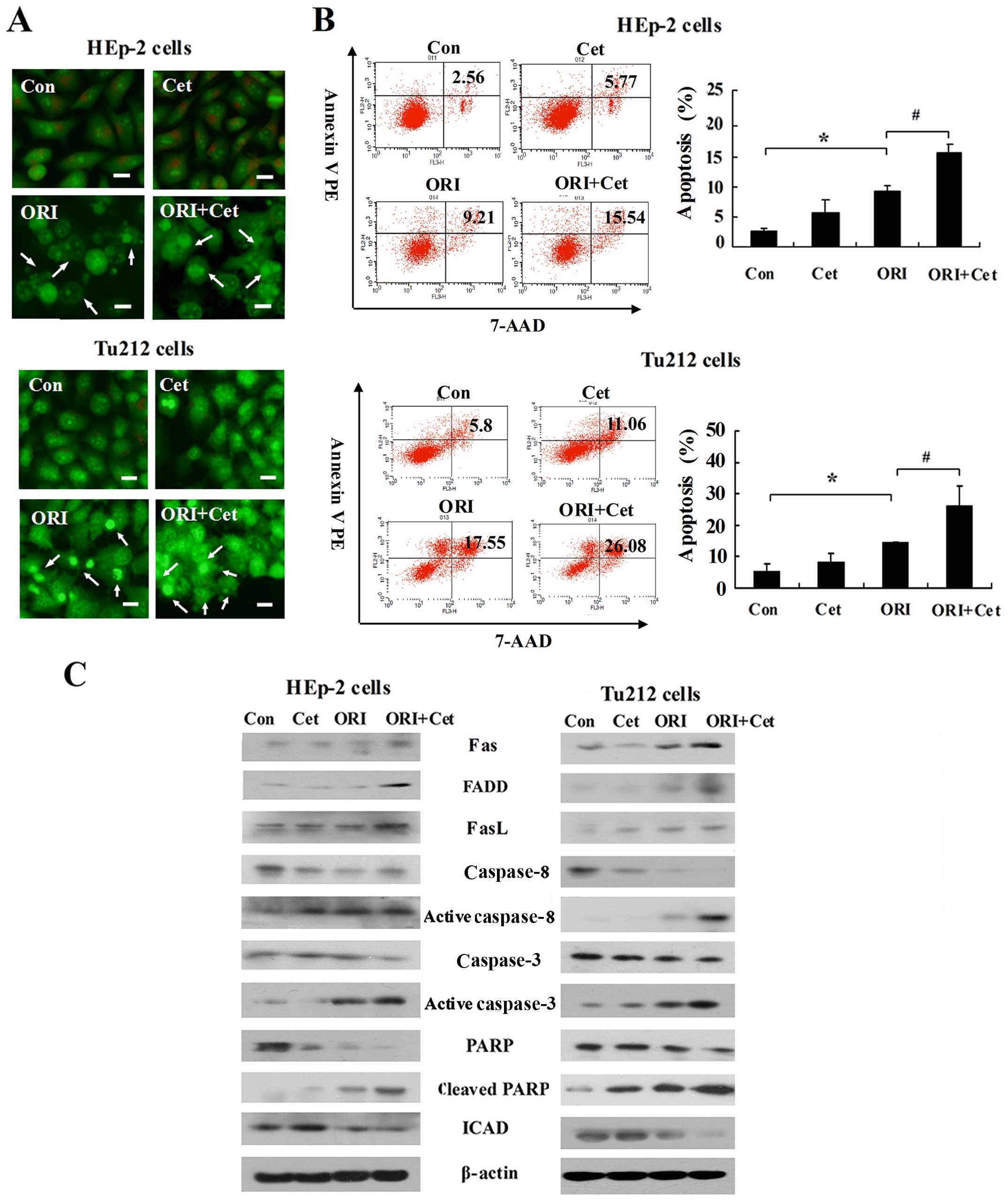
Next, we verified whether ORI and Cet exert an
anticancer effect via induction of apoptosis. As shown in Fig. 2A, compared with the control group,
more apoptotic cells were observed in ORI-treated and combination
groups. ORI plus Cet caused strong apoptotic cell death in HEp-2
and Tu212 cells, which was higher than that caused by either agent
alone (Fig. 2B).
Since the Fas/FasL system is an important apoptosis
signal transduction pathway (27),
we investigated whether the Fas/FasL-mediated pathway is related to
the ORI/Cet combination-induced apoptosis. As shown in Fig. 2C, LSCC cells exposed to ORI alone
increased the expression levels of Fas and FasL when compared to
control cells. Importantly, the levels of Fas, FasL and FADD were
significantly enhanced by ORI plus Cet. Moreover, after 48-h
exposure to ORI, we found that caspase-8, caspase-3 and PARP were
cleaved to their active forms. Cleaved PARP, activated caspase-3
and activated caspase-8 were further increased as a result of the
treatment with a combination of ORI and Cet. The expressions of
ICAD were dramatically lower when both agents were combined
compared with each agent alone.
Combined treatment with ORI and Cet
induces G2/M phase cell cycle arrest
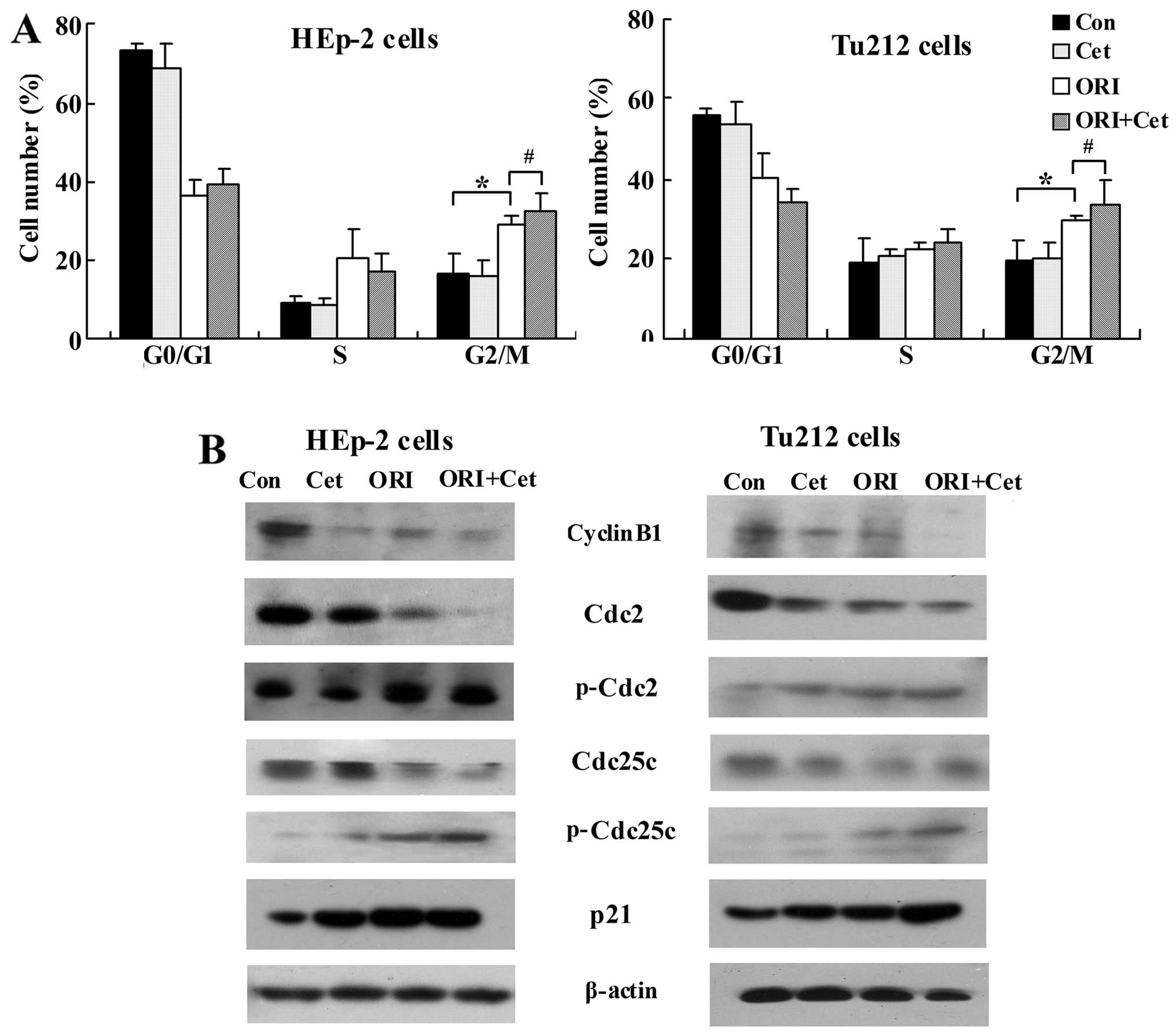
We further characterized the effects of ORI and Cet
combination on the cell cycle distribution. As shown in Fig. 3A, Cet treatment did not show cell
cycle arrest, whereas ORI caused G2/M arrest in LSCC cells compared
to the control. Moreover, combination treatment with Cet and ORI
resulted in a significant G2/M phase arrest compared with ORI given
alone.
Next, the effects of ORI/Cet on the modulation of
molecular events associated with the G2/M phase were investigated
in LSCC cells. As shown in Fig.
3B, ORI plus Cet resulted in a significant suppression of
cyclin B1, which is essential for the transition from the S to the
M-phase. Moreover, the combination of ORI and Cet decreased in
Cdc25C and Cdc2 protein levels, but strongly increased the levels
of p-Cdc2 and p-Cdc25c compared with either agent alone or control
group. On the other hand, compared with each agent alone, ORI plus
Cet moderately increased the protein levels of p21 (Fig. 3B), an upstream kinase associated
with Cdc25C-cyclin B1/Cdc2 (28).
ROS-mediated JNK pathway regulates
ORI/Cet-induced apoptosis and cell cycle arrest
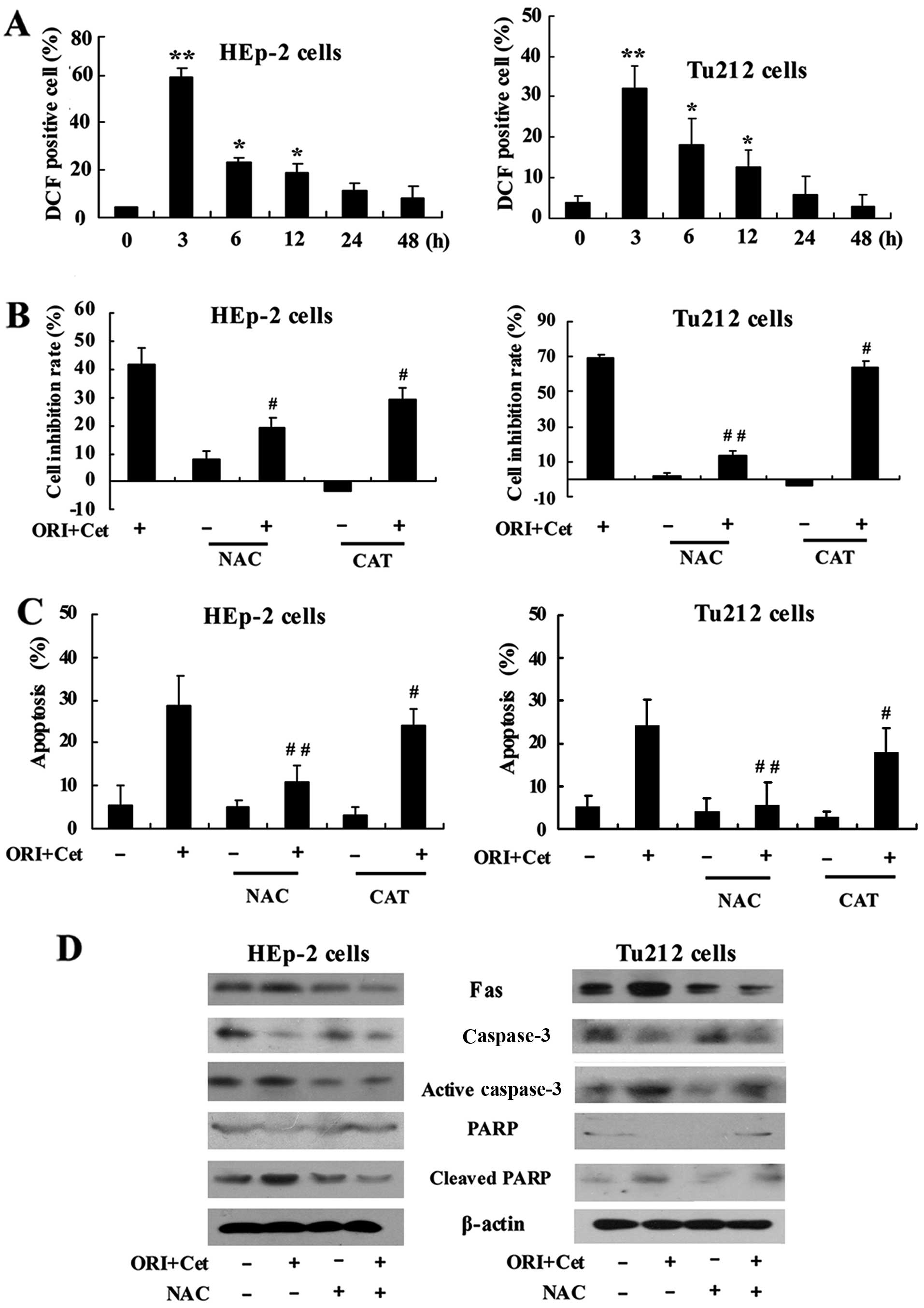
We investigated whether the cell growth inhibition
induced by the combination of ORI and Cet was triggered by ROS
accumulation. As shown in Fig. 4A,
the treatment of LSCC cells with the combination of ORI and Cet
could rapidly induce ROS production. The highest generation of ROS
was observed after 3 h of exposure to the two agents.
To determine whether ROS is involved in cell growth
inhibition induced by the combination of ORI and Cet, NAC
(N-acetylcysteine) and CAT (catalase), two general free radical
scavengers (29,30), were introduced. NAC and CAT showed
significant inhibitory effects on ORI/Cet-induced cell growth
inhibition (Fig. 4B). Moreover,
the percentage of apoptotic cells of combination groups were
significantly reduced after pretreatment with NAC and CAT (Fig. 4C). NAC could partly inhibit the
expression of the cleaved PARP and activated caspase-3 induced by
ORI/Cet (Fig. 4D). On the other
hand, the proportion of LSCC cells treated with ORI/Cet plus NAC or
CAT in G2/M phase was significantly lower than that in cells
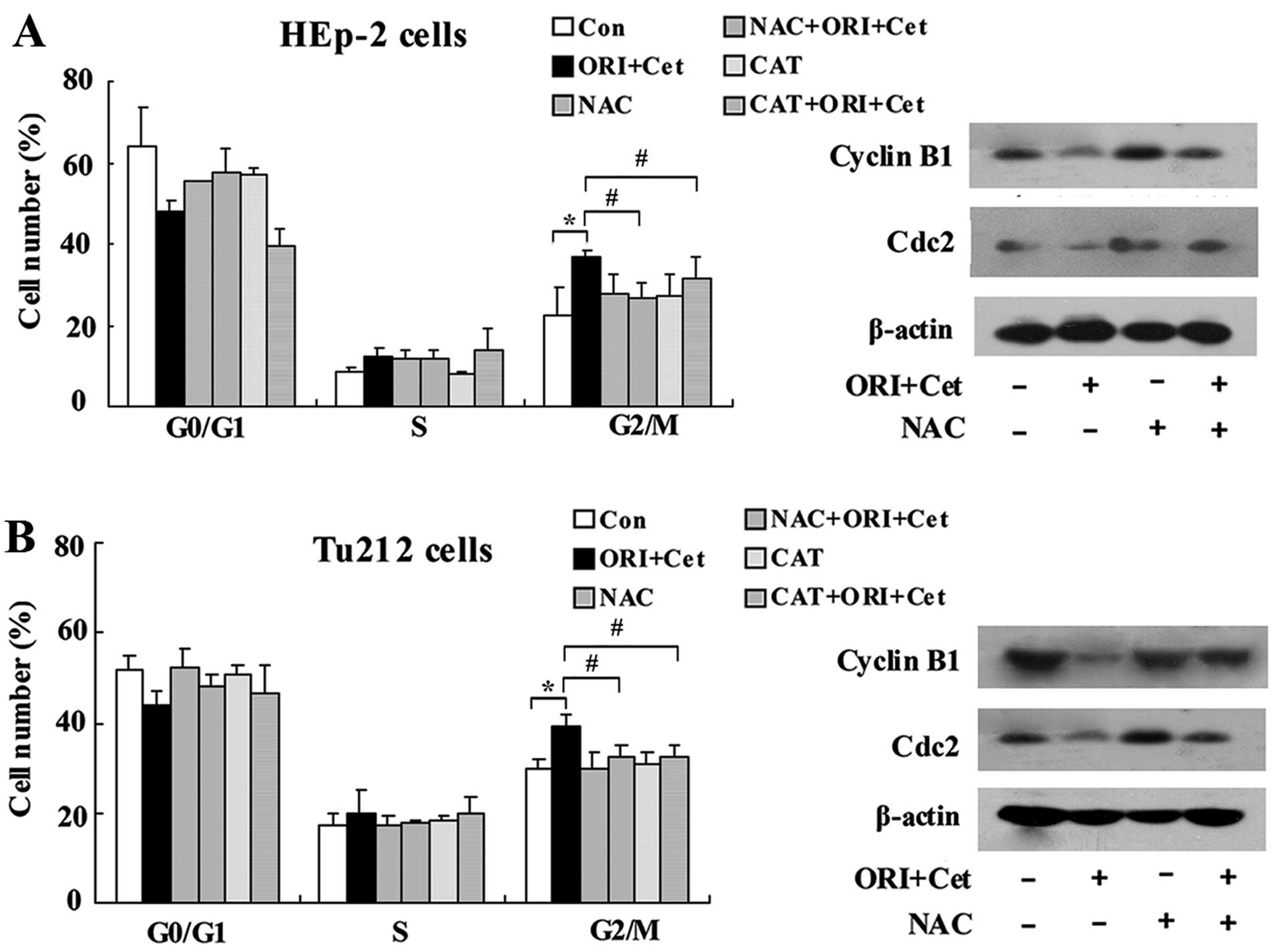
treated with ORI/Cet (Fig. 5).
Pretreatment with NAC showed a significant protection against
ORI/Cet-induced Cdc2 and cyclin B1 degradation (Fig. 5).
Next, we explored the contribution of MAPK
(mitogen-activated protein kinase) family proteins ERK, JNK and p38
to the ORI/Cet-induced cell apoptosis and G2/M phase arrest. As
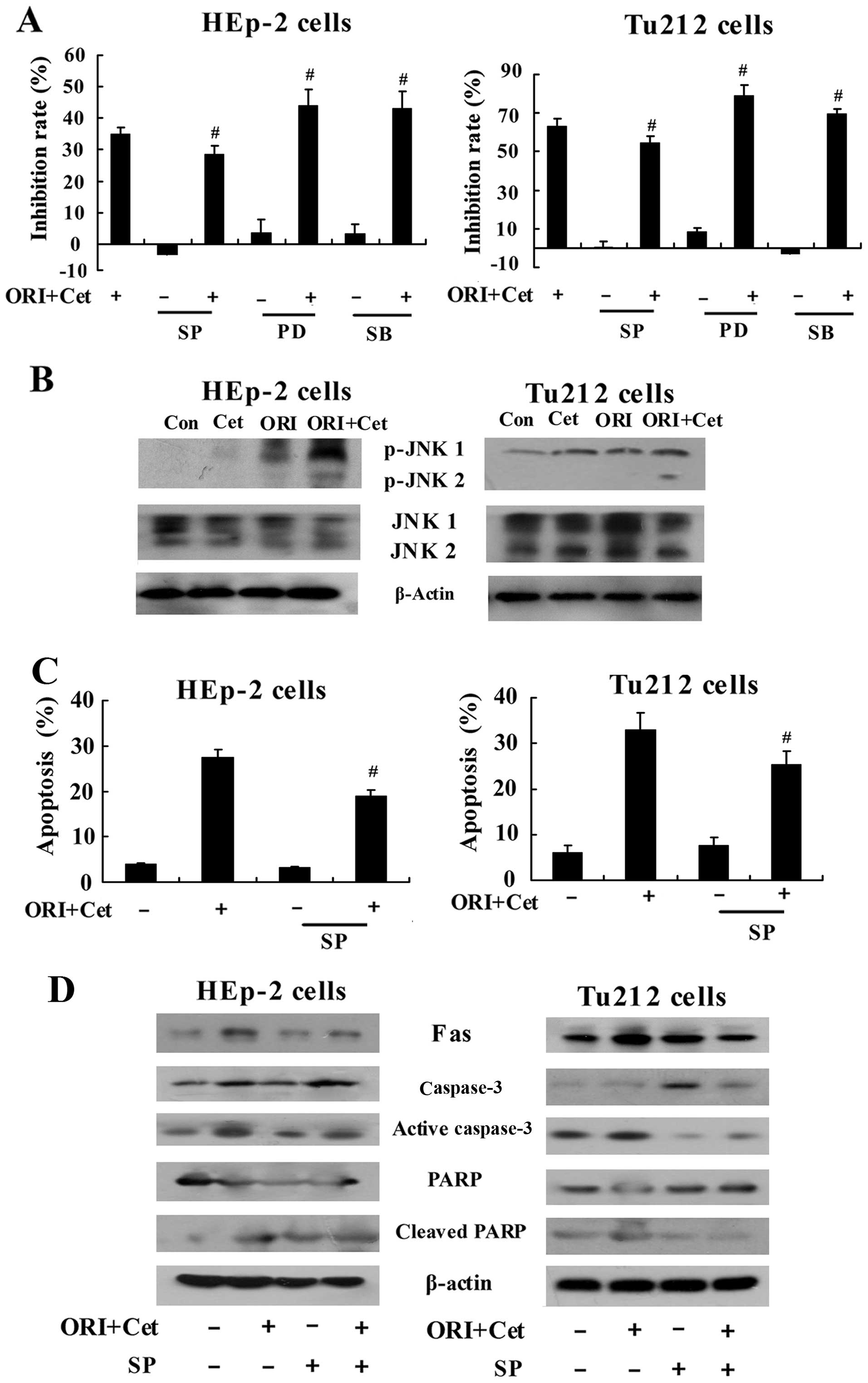
shown in Fig. 6A, pretreatment
with JNK inhibitor SP600125 (31)
exhibited a significant inhibitory effect on cytotoxicity of the
combination of ORI and Cet in LSCC cells, while pretreatment with
ERK inhibitor PD98059 and p38 inhibitor SB203580 markedly increased
ORI/Cet-induced cytotoxicity. Moreover, both ORI and Cet
upregulated the level of p-JNK in the LSCC cells. Treatment of
cells with ORI combined with Cet strongly increased the expression
of p-JNK (Fig. 6B). In addition,
compared with the combination group, inhibiting JNK resulted in
decreased apoptosis in ORI/Cet-treated LSCC cells (Fig. 6C). The levels of Fas, cleaved PARP
and activated caspase-3 were decreased by SP600125 (Fig. 6D). On the other hand, the
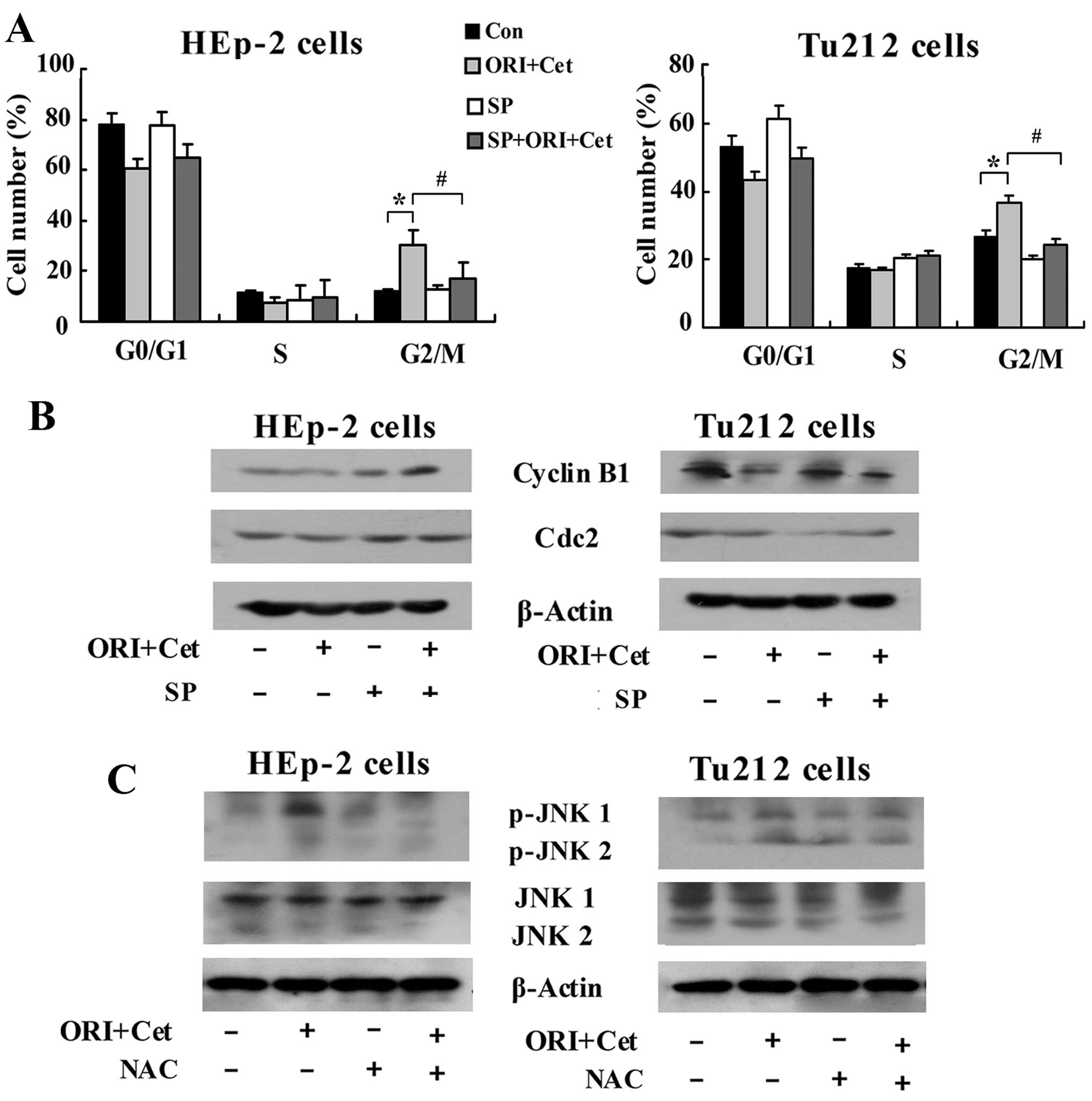
inhibition of JNK was associated with obvious abolition of
ORI/Cet-induced G2/M arrest (Fig.
7A). Addition of SP600125 abolished ORI/Cet-induced
downregulation of cyclin B1 and Cdc2 (Fig. 7B). Notably, western blot results
indicated that inhibiting the generation of ROS partly inhibited
the ORI/Cet-induced upregulation of p-JNK both in HEp-2 cells and
Tu212 cells (Fig. 7C). All these
results suggested that ROS-mediated JNK pathway participated in the
regulation of G2/M phase arrest and apoptosis induced by ORI/Cet
treatment.
Combined treatment of ORI with Cet leads
to in vivo tumor regression through inhibiting the activation of
EGFR
To characterize the in vivo effects of the
combination of ORI and Cet, a series of experiments were conducted
in HEp-2 tumor xenografts. As shown in Fig. 8A, ORI and Cet alone produced
significant growth inhibition in HEp-2 xenografts. Combined
treatment of Cet with ORI further inhibited tumor growth and
resulted in substantial growth delay in the HEp-2 xenografts. All
of the treatments were well tolerated, and there were no signs of
toxicity or body weight loss during therapy.
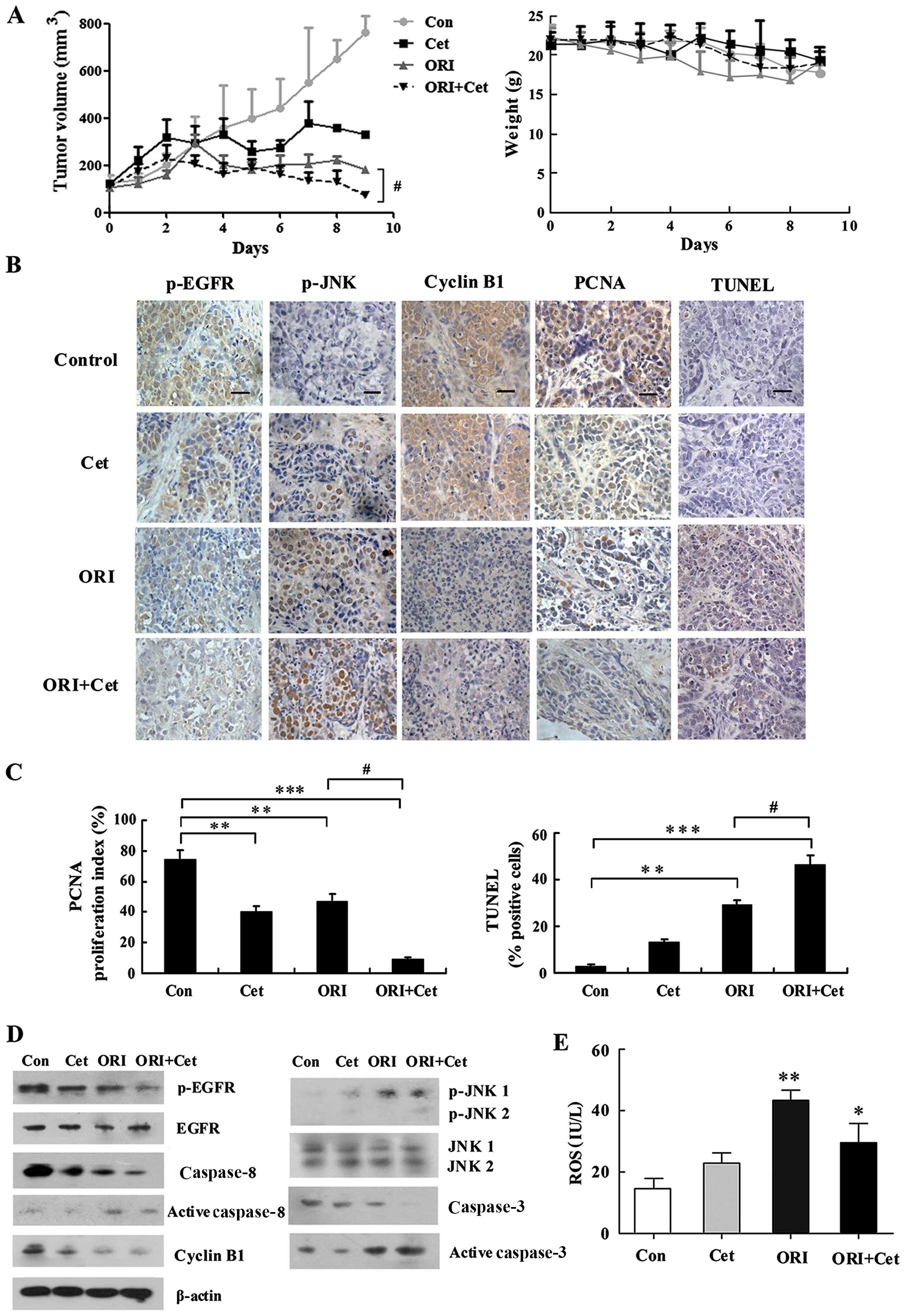 | Figure 8ORI/Cet significantly inhibits HEp-2
growth and induces apoptosis in a xenograft mouse model. (A) Mice
bearing HEp-2 xenografts were treated with vehicle, ORI (20
mg/kg/day), Cet (1 mg/kg twice for 5 days) or their combination for
10 days. Each group included 10 mice. Tumor volume was measured
every day. After 10 days, the mice were sacrificed and the tumors
were removed and analyzed. Data are expressed as mean (± SEM) tumor
volume. Body weight of mice with HEp-2 xenografts was measured
every day during treatment with vehicle control, ORI, Cet or their
combination. (B) Representative images of HEp-2 tumors
immunohistochemically stained with antibodies to p-EGFR, p-JNK,
cyclin B1 and PCNA, and representative images of apoptotic cell
death in HEp-2 tumors as measured by the TUNEL assay. Bar
represents 10 μm. (C) Quantification of proliferation index (PCNA)
and apoptosis index (TUNEL). (D) Expression levels of p-EGFR, EGFR,
p-JNK, JNK, cyclin B1 and caspases in tumor tissues from various
treatment groups were analyzed by western blot analysis. (E) ELISA
measurement of the ROS level in tumors tissues treated by ORI
and/or Cet. n=4, Mean ± SD. *P<0.05,
**P<0.01, ***P<0.001 compared to the
control group; #P<0.05 compared to the ORI alone
group. |
We also elucidated the molecular mechanisms of the
treatment effects with ORI plus Cet on mouse HEp-2 xenografts. Cet
alone had a minimum effect on p-EGFR expression, whereas ORI alone
effectively reduced p-EGFR expression levels. The levels of p-EGFR
in the combined treatment group were significantly lower than that
in the ORI group (Fig. 8B and
D).
We further evaluated tumor cell proliferation by
PCNA staining. Treatment with ORI or Cet alone decreased the
percentage of PCNA-positive proliferating tumor cells, with
proliferation indices of 40 and 47%, respectively. Combined
treatment with ORI and Cet markedly decreased the percentage of
PCNA-positive proliferating tumor cells to 9% (Fig. 8B and C). We also examined whether
treatment with ORI and/or Cet induced apoptosis in vivo in
HEp-2 tumors. Combined treatment significantly increased the
percentage of apoptotic cells to 46.3% compared with the cells of
mono-treated (ORI 29.3 and Cet 13%) or control mice (3%) (Fig. 8B and C). In addition, ORI plus Cet
increased the activation of caspase-8 and caspase-3 (Fig. 8D).
Next, we detected the expression of cyclin B1 in the
tumor tissue. As shown in Fig. 8B and
D, Cet and ORI treatments decreased the percentage of mitotic
cells as indicated by a decrease in the number of cyclin B1
positive cells. Similar to in vitro findings, there were
fewer cells positive for cyclin B1 in the combination group than in
ORI-treated group.
Combining ORI and Cet induces ROS
production and JNK activation in HEp-2 xenografts
To investigate whether ORI plus Cet induced ROS
generation in vivo, the levels of ROS in the different
treatment tumor tissues were examined. As shown in Fig. 8E, compared with the control group,
both ORI and combination treatment resulted in a more significant
elevation in ROS levels. However, no statistical difference in ROS
levels was observed between the ORI and combined treatment
group.
To substantiate the activation of JNK induced by the
combination of ORI and Cet in vivo, p-JNK in xenograft
tissues was revealed by immunohistochemistry and western blotting.
As shown in Fig. 8B and D, p-JNK
was detected in ORI and Cet-treated xenograft tissues compared with
the control group, and combined treatment with ORI and Cet
significantly increased the expression levels of p-JNK in
tumors.
Discussion
In comparison with normal tissues, LSCC shows higher
expression of EGFR, which promotes processes responsible for tumor
growth and progression (6). Cet,
an anti-EGFR monoclonal antibody, has failed to bring the expected
antitumor outcome (14).
Additionally, Cet is not sufficient to induce apoptosis and
generally has less cytostatic effects on cell growth in many tumor
cell lines (32,33), including LSCC cells. However, in
combination with several anticancer agents from natural products,
Cet has been shown to increase the incidence of human tumor cell
apoptosis in a number of model systems. Park et al (34) observed marked antitumor activity
with the combination of Cet and genistein, a natural isoflavonoid
derived from soy, in mice bearing an oral squamous cell carcinoma
(OSCC) xenograft. Cet in combination with docetaxel, a taxoid
derived from the needles of the European yellow tree, could inhibit
the proliferation of non-small cell lung cancer PC9/G2 cells in
vitro and in vivo (35). Leeman-Neill et al (36) reported that guggulsterone, a
naturally-occurring compound used in traditional Indian medicine,
augments Cet-induced cell growth inhibition of HNSCC. Consistent
with these observations, our results firstly demonstrated that a
combination of Cet and ORI, an active component from Rabdosia
rubescens, is an effective way to achieve synergistic antitumor
effects against LSCC in vitro and in vivo. The
present study also explored the potential mechanisms underlying the
observed synergy of the combined treatment.
ORI was reported to exhibit potent antitumor effects
against many cell lines through different mechanisms including
MAPK, mTOR and NF-κB (37,38). In addition, ORI led to apoptosis in
A431 cells (39), H1975 cells
(40) and HEp-2 cells (22) through blockage of EGFR
phosphorylation. Herein, our results also demonstrated that ORI
could inhibit EGFR phosphorylation both in vitro and in
vivo. Cet alone showed stronger suppression on EGFR
phosphorylation than ORI alone in vitro, whereas ORI had
stronger anti-proliferation effect on LSCC cells compared to Cet
alone, suggesting that except EGFR pathway, the growth inhibitory
effect of ORI appears to be associated with other signaling
pathways. Importantly, combination of ORI with Cet has synergistic
anti-proliferative activity in vitro with significant
inhibition of p-EGFR expression. In addition, we observed that the
combination treatment with ORI and Cet significantly suppressed
levels of p-EGFR in tumor tissue and caused tumor regression of
HEp-2 xenograft in vivo, compared with ORI or Cet alone. We
did not observe any serious side-effects in the treatment groups
during the entire experimental period. These results suggest that
the two agents given together had a potent anticancer effect
against LSCC through downregulation of p-EGFR.
Decreased activity of EGFR leads to downregulation
of several downstream signaling cascades resulting in apoptosis of
cancer cells (41). Consistent
with this report, our results demonstrated that the inhibitory
effects of the combined treatment with ORI and Cet on the EGFR
phosphorylation was accompanied by a marked apoptosis in
vitro and in vivo. It was reported that anti-EGFR
inhibitors decreased phosphorylation of EGFR and its downstream
effector Akt and amplified the induction of Fas-mediated apoptosis
(42). Here, LSCC cells when
co-treated with ORI and Cet showed higher expressions of Fas, FasL
and activated caspase-8 than those in ORI alone-treated cells.
Furthermore, in the HEp-2 tumor xenografts, combined treatment
resulted in significant apoptosis as well as marked increase in
activated caspase-8 and -3, suggesting that Fas-mediated extrinsic
pathway plays an important role in ORI/Cet-induced tumor cell
death.
Previous studies have demonstrated that Cet inhibits
cell cycle progression in non-small cell lung cancer lines, causing
cells to arrest in the G1 gap phase that occurs prior to DNA
synthesis (43). Nevertheless, Cet
did not induce G0/G1 phase arrest in our models, which might be
attributed to our use of different cell lines or the dose of Cet.
The present results were in agreement with previous reports that
ORI could induce G2/M arrest in HEp-2 (44), HCT116 (45) and MCF-7 cells (46). Interestingly, Cet strongly enhanced
ORI-caused G2/M arrest in vitro and in vivo. It is
now established that p21 is a universal inhibitor of cyclin kinases
which controls cell cycle by activating and/or inactivating the
cyclin-dependent kinases (28).
Cdc25C is a critical regulator of Cdc2/cyclin B1 kinase activity
and controls cell cycle progression by dephosphorylating and
activating CDKs (47). In the
present study, the combined treatment of Cet and ORI moderately
increased p21 levels, which phosphorylated and inactivated Cdc25c,
resulting in the inactivation of cyclin B1/Cdc2 complex. Hence, our
findings support that p21-Cdc25C-cyclin B1/Cdc2 pathway could be
one of the possible underlying molecular events associated with the
strong synergistic effect of the ORI/Cet combination on G2/M arrest
in LSCC cells.
ROS act as secondary messengers and are essential
for cell signaling and various cellular processes, including
apoptosis and cell cycle arrest (48). It is reported that ORI induced
apoptosis and G2/M arrest via ROS activation in L929 cells
(49) and HEp-2 cells (44). The present study demonstrated that
ORI induced apoptosis and G2/M arrest via ROS generation not only
in HEp-2 cells but also in Tu212 cells, another type of LSCC cell
line. In addition, the combination group also induced significant
ROS production both in vitro and in vivo. However,
combined treatment did not induce more ROS generation compared with
the ORI alone group in vivo, indicating that other mediators
might be involved in the antitumor effect of combination treatment
with ORI and Cet in vivo. Furthermore, NAC or CAT
pretreatment resulted in the marked inhibition of ORI/Cet-induced
cell growth inhibition, apoptosis and Fas expression, indicating
that combination treatment induced Fas-dependent apoptosis through
ROS generation in LSCC cells. Moreover, pretreatment with NAC or
CAT significantly decreased ORI/Cet-induced G2/M arrest, suggesting
that combination of ORI and Cet caused ROS generation which is
involved with combined treatment induced G2/M arrest. Taken
together, these results suggest that the combined treatment with
ORI and Cet could trigger ROS generation, which was related to the
Fas-dependent apoptosis and G2/M phase arrest in LSCC cells.
MAPK cascades are activated by various cellular
stresses and growth factors, which are signaling transduction
molecules in apoptosis and cell cycle arrest (50). JNK pathway, as an important
subgroup of MAPK super family, has been reported to be involved in
tumor cell death induced by different chemotherapeutic agents
(51). Herein, we demonstrated
that combination of ORI and Cet remarkably increased the levels of
p-JNK in vitro and in vivo. Additionally, the
inhibition of JNK by SP600125 decreased, in part, the
ORI/Cet-induced cell death, apoptosis, G2/M arrest and related
protein, suggesting that JNK activation contributed to
ORI/Cet-induced extrinsic apoptosis and G2/M arrest. The
involvement of JNK in ORI/Cet-induced growth inhibition is partial,
and additional studies are still necessary to identify the other
actors involved in this mechanism including the downstream
effectors of EGFR, such as STAT or Akt. In addition, although
diverse stimuli activate MAPK pathways, in many cases, the
generation of ROS is responsible for increases in MAPK activity
(52). In the present study, we
found that NAC significantly inhibited the phosphorylation of JNK
in LSCC cells especially in HEp-2 cells, suggesting that
ORI/Cet-induced JNK activation was mediated through ROS generation.
Together, these findings imply that ROS might be an upstream
mediator of JNK activation, which initiates the series of apoptotic
events and G2/M phase arrest induced by ORI/Cet.
In summary, the present study shows that combined
treatment of ORI and Cet exerts synergistic effects on cell growth
inhibition via downregulation of p-EGFR. ORI plus Cet activates ROS
mediated-JNK pathway, and invokes strong anticancer activity
against LSCC by inducing apoptosis and G2/M cell arrest in
vivo and in vitro. Thus, a combination of ORI and Cet
might be a promising approach for the treatment of laryngeal
cancer.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (no. 81373797), the China
Postdoctoral Science Special Foundation (no. 2014T70224) and the
China Postdoctoral Science Foundation (no. 2013M541192).
References
|
1
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Forastiere A, Koch W, Trotti A and
Sidransky D: Head and neck cancer. N Engl J Med. 345:1890–1900.
2001. View Article : Google Scholar
|
|
4
|
Jorissen RN, Walker F, Pouliot N, Garrett
TP, Ward CW and Burgess AW: Epidermal growth factor receptor:
Mechanisms of activation and signalling. Exp Cell Res. 284:31–53.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rocha-Lima CM, Soares HP, Raez LE and
Singal R: EGFR targeting of solid tumors. Cancer Control.
14:295–304. 2007.PubMed/NCBI
|
|
6
|
Itakura Y, Sasano H, Shiga C, Furukawa Y,
Shiga K, Mori S and Nagura H: Epidermal growth factor receptor
overexpression in esophageal carcinoma. An immunohistochemical
study correlated with clinicopathologic findings and DNA
amplification. Cancer. 74:795–804. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li FC, Li YH, Zhao X, Fu WN, Xu ZM, Li ZG
and Sun KL: Association of homozygous deletion of p15 and p16 gene
and amplification of EGFR gene in laryngeal squamous cell
carcinoma. Yi Chuan Xue Bao. 31:109–113. 2004.(In Chinese).
PubMed/NCBI
|
|
8
|
Li F, Kang N, Fu W, Sun X, Gao H and Sun
K: Detection of epidermal growth factor receptor gene amplification
in human laryngeal carcinomas by means of fluorescence in situ
hybridization. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 17:278–280.
2000.(In Chinese). PubMed/NCBI
|
|
9
|
Moghal N and Sternberg PW: Multiple
positive and negative regulators of signaling by the EGF-receptor.
Curr Opin Cell Biol. 11:190–196. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Prewett M, Rockwell P, Rockwell RF,
Giorgio NA, Mendelsohn J, Scher HI and Goldstein NI: The biologic
effects of C225, a chimeric monoclonal antibody to the EGFR, on
human prostate carcinoma. J Immunother Emphasis Tumor Immunol.
19:419–427. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Matar P, Rojo F, Cassia R, Moreno-Bueno G,
Di Cosimo S, Tabernero J, Guzmán M, Rodriguez S, Arribas J,
Palacios J, et al: Combined epidermal growth factor receptor
targeting with the tyrosine kinase inhibitor gefitinib (ZD1839) and
the monoclonal antibody cetuximab (IMC-C225): Superiority over
single-agent receptor targeting. Clin Cancer Res. 10:6487–6501.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huether A, Höpfner M, Baradari V, Schuppan
D and Scherübl H: EGFR blockade by cetuximab alone or as
combination therapy for growth control of hepatocellular cancer.
Biochem Pharmacol. 70:1568–1578. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Galizia G, Lieto E, De Vita F, Orditura M,
Castellano P, Troiani T, Imperatore V and Ciardiello F: Cetuximab,
a chimeric human mouse anti-epidermal growth factor receptor
monoclonal antibody, in the treatment of human colorectal cancer.
Oncogene. 26:3654–3660. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kohrt HE, Colevas AD, Houot R, Weiskopf K,
Goldstein MJ, Lund P, Mueller A, Sagiv-Barfi I, Marabelle A, Lira
R, et al: Targeting CD137 enhances the efficacy of cetuximab. J
Clin Invest. 124:2668–2682. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Le Tourneau C and Siu LL:
Molecular-targeted therapies in the treatment of squamous cell
carcinomas of the head and neck. Curr Opin Oncol. 20:256–263. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang R, Chen P, Fan QX and Wang R:
Clinical efficacy for the treatment of esophageal cancer with
rabdosia rubescens alone and combining with chemotherapy. Life Sci
J. 4:22–25. 2007.
|
|
17
|
Wang R and Wang L: Efficacy of Rabdosia
rubescens in treating 95 cases with esophageal and gastric cardiac
cancer. Cancer Res Prev Treat. 11:86–87. 1994.
|
|
18
|
Kuo LM, Kuo CY, Lin CY, Hung MF, Shen JJ
and Hwang TL: Intracellular glutathione depletion by oridonin leads
to apoptosis in hepatic stellate cells. Molecules. 19:3327–3344.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li X, Li X, Wang J, Ye Z and Li JC:
Oridonin up-regulates expression of P21 and induces autophagy and
apoptosis in human prostate cancer cells. Int J Biol Sci.
8:901–912. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang CL, Wu LJ, Tashiro S, Onodera S and
Ikejima T: Oridonin induced A375-S2 cell apoptosis via
bax-regulated caspase pathway activation, dependent on the
cytochrome c/caspase-9 apoptosome. J Asian Nat Prod Res. 6:127–138.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou GB, Chen SJ, Wang ZY and Chen Z: Back
to the future of oridonin: Again, compound from medicinal herb
shows potent antileukemia efficacies in vitro and in vivo. Cell
Res. 17:274–276. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kang N, Zhang JH, Qiu F, Tashiro S,
Onodera S and Ikejima T: Inhibition of EGFR signaling augments
oridonin-induced apoptosis in human laryngeal cancer cells via
enhancing oxidative stress coincident with activation of both the
intrinsic and extrinsic apoptotic pathways. Cancer Lett.
294:147–158. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang XJ, Li Y, Luo L, Wang H, Chi Z, Xin
A, Li X, Wu J and Tang X: Oxaliplatin activates the Keap1/Nrf2
antioxidant system conferring protection against the cytotoxicity
of anticancer drugs. Free Radic Biol Med. 70:68–77. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang DT, He J, Wu M, Li SM, Gao Q and Zeng
QP: Artemisinin mimics calorie restriction to trigger mitochondrial
biogenesis and compromise telomere shortening in mice. PeerJ.
3:e8222015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Siegelin MD, Plescia J, Raskett CM,
Gilbert CA, Ross AH and Altieri DC: Networks for therapy of
glioblastoma global targeting of subcellular heat shock. Mol Cancer
Ther. 9:1638–1646. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang D, Torres CM, Bardhan K, Zimmerman M,
McGaha TL and Liu K: Decitabine and vorinostat cooperate to
sensitize colon carcinoma cells to Fas ligand-induced apoptosis in
vitro and tumor suppression in vivo. J Immunol. 188:4441–4449.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xiong Y, Hannon GJ, Zhang H, Casso D,
Kobayashi R and Beach D: p21 is a universal inhibitor of cyclin
kinases. Nature. 366:701–704. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee WY, Liu KW and Yeung JH: Reactive
oxygen species-mediated kinase activation by dihydrotanshinone in
tanshinones-induced apoptosis in HepG2 cells. Cancer Lett.
285:46–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Salcher S, Hagenbuchner J, Geiger K,
Seiter MA, Rainer J, Kofler R, Hermann M, Kiechl-Kohlendorfer U,
Ausserlechner MJ and Obexer P: C10ORF10/DEPP, a transcriptional
target of FOXO3, regulates ROS-sensitivity in human neuroblastoma.
Mol Cancer. 13:2242014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Moon DO, Kim MO, Kang CH, Lee JD, Choi YH
and Kim GY: JNK inhibitor SP600125 promotes the formation of
polymerized tubulin, leading to G2/M phase arrest,
endoreduplication, and delayed apoptosis. Exp Mol Med. 41:665–677.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ciardiello F and Tortora G: A novel
approach in the treatment of cancer: Targeting the epidermal growth
factor receptor. Clin Cancer Res. 7:2958–2970. 2001.PubMed/NCBI
|
|
33
|
Balin-Gauthier D, Delord JP, Rochaix P,
Mallard V, Thomas F, Hennebelle I, Bugat R, Canal P and Allal C: In
vivo and in vitro antitumor activity of oxaliplatin in combination
with cetuximab in human colorectal tumor cell lines expressing
different level of EGFR. Cancer Chemother Pharmacol. 57:709–718.
2006. View Article : Google Scholar
|
|
34
|
Park SJ, Kim MJ, Kim YK, Kim SM, Park JY
and Myoung H: Combined cetuximab and genistein treatment shows
additive anti-cancer effect on oral squamous cell carcinoma. Cancer
Lett. 292:54–63. 2010. View Article : Google Scholar
|
|
35
|
Zhang L, Li XF, Tand L, Zhao YM and Zhou
CC: The effects of cetuximab in combination with docetaxel for the
acquired resistance to EGFR-TKI in non-small cell lung cancer
cells. Tumor. 34:584–590. 2014.
|
|
36
|
Leeman-Neill RJ, Wheeler SE, Singh SV,
Thomas SM, Seethala RR, Neill DB, Panahandeh MC, Hahm ER, Joyce SC,
Sen M, et al: Guggulsterone enhances head and neck cancer therapies
via inhibition of signal transducer and activator of
transcription-3. Carcinogenesis. 30:1848–1856. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang YY, Lv YF, Lu L and Cai L: Oridonin
inhibits mTOR signaling and the growth of lung cancer tumors.
Anticancer Drugs. 25:1192–1200. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bao R, Shu Y, Wu X, Weng H, Ding Q, Cao Y,
Li M, Mu J, Wu W, Ding Q, et al: Oridonin induces apoptosis and
cell cycle arrest of gallbladder cancer cells via the mitochondrial
pathway. BMC Cancer. 14:2172014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li D, Wu LJ, Tashiro S, Onodera S and
Ikejima T: Oridonin inhibited the tyrosine kinase activity and
induced apoptosis in human epidermoid carcinoma A431 cells. Biol
Pharm Bull. 30:254–260. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xiao X, He Z, Cao W, Cai F, Zhang L, Huang
Q, Fan C, Duan C, Wang X, Wang J, et al: Oridonin inhibits
gefitinib-resistant lung cancer cells by suppressing
EGFR/ERK/MMP-12 and CIP2A/Akt signaling pathways. Int J Oncol.
48:2608–2618. 2016.PubMed/NCBI
|
|
41
|
Selvendiran K, Bratasz A, Tong L, Ignarro
LJ and Kuppusamy P: NCX-4016, a nitro-derivative of aspirin,
inhibits EGFR and STAT3 signaling and modulates Bcl-2 proteins in
cisplatin-resistant human ovarian cancer cells and xenografts. Cell
Cycle. 7:81–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Iwase M, Takaoka S, Uchida M, Yoshiba S,
Kondo G, Watanabe H, Ohashi M and Nagumo M: Epidermal growth factor
receptor inhibitors enhance susceptibility to Fas-mediated
apoptosis in oral squamous cell carcinoma cells. Oral Oncol.
44:361–368. 2008. View Article : Google Scholar
|
|
43
|
Kriegs M, Gurtner K, Can Y, Brammer I,
Rieckmann T, Oertel R, Wysocki M, Dorniok F, Gal A, Grob TJ, et al:
Radiosensitization of NSCLC cells by EGFR inhibition is the result
of an enhanced p53-dependent G1 arrest. Radiother Oncol.
115:120–127. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kang N, Zhang JH, Qiu F, Chen S, Tashiro
S, Onodera S and Ikejima T: Induction of G2/M phase
arrest and apoptosis by oridonin in human laryngeal carcinoma
cells. J Nat Prod. 73:1058–1063. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gao FH, Hu XH, Li W, Liu H, Zhang YJ, Guo
ZY, Xu MH, Wang ST, Jiang B, Liu F, et al: Oridonin induces
apoptosis and senescence in colorectal cancer cells by increasing
histone hyperacetylation and regulation of p16, p21, p27 and c-myc.
BMC Cancer. 10:6102010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hsieh TC, Wijeratne EK, Liang JY,
Gunatilaka AL and Wu JM: Differential control of growth, cell cycle
progression, and expression of NF-kappaB in human breast cancer
cells MCF-7, MCF-10A, and MDA-MB-231 by ponicidin and oridonin,
diter-penoids from the chinese herb Rabdosia rubescens. Biochem
Biophys Res Commun. 337:224–231. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tandon M, Salamoun JM, Carder EJ, Farber
E, Xu S, Deng F, Tang H, Wipf P and Wang QJ: SD-208, a novel
protein kinase D inhibitor, blocks prostate cancer cell
proliferation and tumor growth in vivo by inducing G2/M cell cycle
arrest. PLoS One. 10:e01193462015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Clerkin JS, Naughton R, Quiney C and
Cotter TG: Mechanisms of ROS modulated cell survival during
carcinogenesis. Cancer Lett. 266:30–36. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cheng Y, Qiu F, Ye YC, Guo ZM, Tashiro S,
Onodera S and Ikejima T: Autophagy inhibits reactive oxygen
species-mediated apoptosis via activating p38-nuclear factor-kappa
B survival pathways in oridonin-treated murine fibrosarcoma L929
cells. FEBS J. 276:1291–1306. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li JP, Yang YX, Liu QL, Pan ST, He ZX,
Zhang X, Yang T, Chen XW, Wang D, Qiu JX, et al: The
investigational Aurora kinase A inhibitor alisertib (MLN8237)
induces cell cycle G2/M arrest, apoptosis, and autophagy via p38
MAPK and Akt/mTOR signaling pathways in human breast cancer cells.
Drug Des Devel Ther. 9:1627–1652. 2015.PubMed/NCBI
|
|
51
|
Sánchez-Pérez I, Martínez-Gomariz M,
Williams D, Keyse SM and Perona R: CL100/MKP-1 modulates JNK
activation and apoptosis in response to cisplatin. Oncogene.
19:5142–5152. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Pereira L, Igea A, Canovas B, Dolado I and
Nebreda AR: Inhibition of p38 MAPK sensitizes tumour cells to
cisplatin-induced apoptosis mediated by reactive oxygen species and
JNK. EMBO Mol Med. 5:1759–1774. 2013. View Article : Google Scholar : PubMed/NCBI
|