Introduction
Mitomycin C (MC) is a bioreductive anticancer drug
produced by Streptomyces casespitosus (1,2) and
commonly used to treat many cancers, such as stomach, anal, and
lung cancers (3,4). However, the response rates are only
~15–20% (4). Although its mode of
action has been extensively examined (5), it is still the center of numerous
research endeavors (6–10). In particular, this drug is used
intensively to investigate the mechanisms involved in DNA repair
(8–10). Studies showed that mitomycin C
induces DNA damage via DNA alkylation to produce DNA mono-adducts,
intrastrand cross-links and interstrand cross-links (ICLs)
(10,11). The main toxicity of MC is due to
these interstrand MC-DNA crosslinks (12).
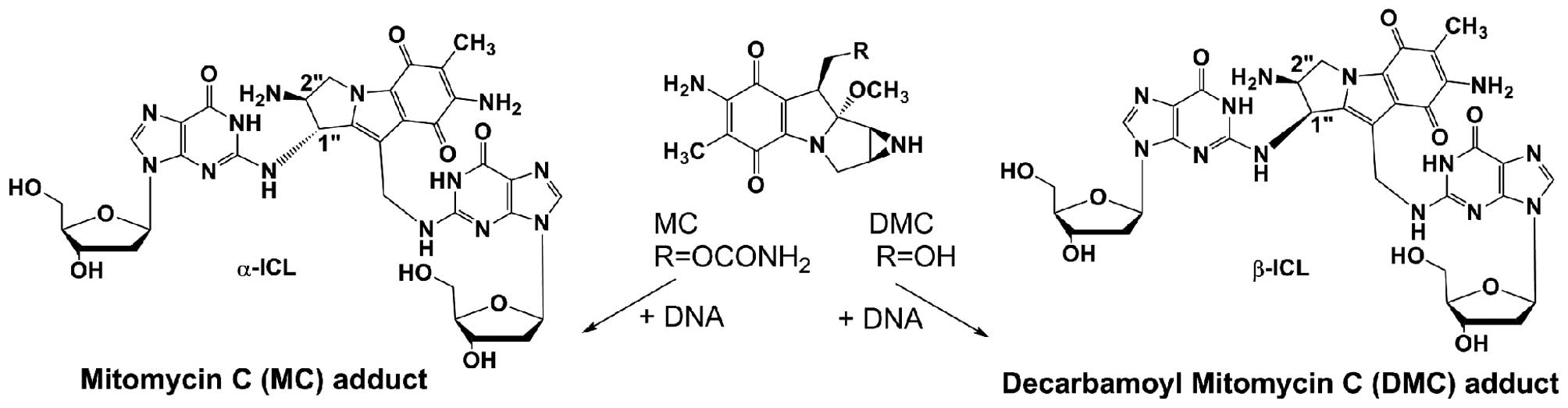
The MC analog, 10-decarbamoyl mitomycin C (DMC), has
not been fully investigated. It has recently been found that the
major ICLs produced by MC and DMC have opposite stereochemistry. MC
generates an ICL with trans stereochemistry (α-ICL) and DMC
generates an ICL with cis stereochemistry (β isomer, β-ICL)
(Fig. 1). Experimental evidence
points to crosslinks α-ICL and β-ICL as the lesions primarily
responsible for the cytotoxicity of the mitomycins (13). When the cellular and molecular
response of human cancer cells treated with MC and DMC were
compared, it was found that DMC was more toxic in human cancer
cells with or without a functioning p53 (14) and that DMC provokes a strong
p53-independent cell death (7).
Xiao et al (15) further
suggested that DMC could enhance Chk1 checkpoint activation and
Rad51 chromatin recruitment via a p53-independent disassociation of
ATR chromatin eviction.
The study of anticancer drug induced DNA damage
mechanisms has mainly focused on cell cycle and checkpoint control
proteins. However, there are other mechanisms which can regulate
cell cycle progression, such as p21WAF1/CIP1. Esposito
et al (16) demonstrated
that nucleolar stressors, 5-fluouracil (5-FU) and oxaliplatin
(L-OHP), trigger cell cycle arrest and apoptosis by altering
rpL3-regulated p21 expression. Moreover, this alteration of p21
expression by rpL3 could occur in a p53-independent manner
(17). In many human cancers,
abnormal expressions of cyclin D1 and cyclin E (which can promote
the transition of G1/S phase) have been observed (18). This accelerated G1/S transition
effect triggered by cyclin D1 and cyclin E can be inhibited by
p21WAF1/CIP1 when anticancer drugs induce DNA damage
(19,20). Choi et al (20) observed that MC inhibited the G1/S
transition by p53-dependent p21WAF1/CIP1, but at
sublethal MC concentrations, the accumulation of cyclin E with a
delayed increase of p21WAF1/CIP1 promoted G1/S
transition. It suggests that the cell cycle G1/S transition is
controlled by cyclin E and p21WAF1/CIP1 in a MC
dose-dependent manner.
The p53 tumor suppressor protein is one of the key
players for keeping genetic stability following DNA damage and is
the target of many chemotherapeutic drugs (21,22).
However, p53 gene is inactively mutated in more than half of human
cancers (22–24). p21WAF1/CIP1, known as a
protein cyclin-dependent kinase inhibitor, is a major effector of
p53 and is involved in p53-dependent and -independent control of
cell proliferation and death (25). p21WAF1/CIP1 expression
has been linked with irreversible cell cycle arrest in both G1 and
G2/M. In this study, MCF-7 (p53-proficient cell line) and K562
(p53-deficient cell line) were used to elucidate the role of
p21WAF1/CIP1 in the signaling mechanism of MC and DMC
and their effects on cell cycle. An oligonucleotide (18 mer)
bearing the major MC-ICL (α-ICL) was also synthesized and
transfected into cells to unveil the effect of the α-ICL on the
regulation of p21WAF1/CIP1.
Materials and methods
Cell culture and reagents
Human breast cancer cells (MCF-7) and leukemia
cancer cells (K562) were obtained from the American Type Tissue
Culture (Manassas, VA, USA). Both cell lines have been used for
mitomycin C studies (7,26). MCF-7 cell line is a p53-proficient
cell line. K562 cell line is a p53-deficient cell line with an
inactivation mutation in exon 5 (27). Dulbecco's modified Eagle's medium
(DMEM), RPMI-1640, fetal bovine serum (FBS), heat-inactivated horse
serum, gentamicin (50 mg/ml) were obtained from Invitrogen
(Gaithersburg, MD, USA). MCF-7 cells were cultured within DMEM
supplemented with 10% FBS and 50 μg/ml gentamicin. K562 cells were
cultured within RPMI-1640 supplemented with 10% FBS, 2 mM glutamine
and 50 μg/ml gentamicin. Both cells were maintained in a humidified
atmosphere at 37°C and 5% CO2. For chemical treatment,
cells were cultured in 60-mm Petri dishes one day prior to the
experiment. Cells were grown until medium density (~80% confluence)
before chemical treatments.
Mitomycin C was a generous gift from Dr Maria Tomasz
(Hunter College, City University of New York, New York, USA).
Decarbamoyl mitomycin C was synthesized based on the protocol of
Kinoshita et al (28).
β-actin antibody was obtained from Sigma-Aldrich
(St. Louis, MO, USA). p145 p21WAF1/CIP1 and p146
p21WAF1/CIP1, and p21WAF1/CIP1 antibodies
were from Santa Cruz. Bio-Rad DC (detergent compatible) protein
assay reagents were purchased from Bio-Rad (Hercules, CA, USA). The
Super Signal West Pico chemiluminescent kit, secondary antibodies
containing anti-rabbit IgG or anti-mouse IgG conjugated to
horseradish peroxidase, mammalian protein extraction reagent
(M-PER) lysis buffer, Halt protease inhibitor cocktail, and
stripping buffer were obtained from Pierce (Rockford, IL, USA).
SuperFect™ transfection reagent was purchased from Qiagen
(Valencia, CA, USA). All other reagents were purchased from
Sigma-Aldrich.
α-interstrand cross-link (α-ICL)
synthesis
The synthesis protocol was adapted from
Borowy-Borowski et al (5)
with minor modifications. Briefly, a 1:1 molar mixture of the 18
mer and its complementary strand (0.415 μmol of oligo 1:
ACTGATCTCGTTAGTCAT and 0.415 μmol of oligo 2 -complementary-:
ATGACTAACGAGATCAGT) were mixed with 50 μmol of MC (from a
20-μmol/ml solution in 70% water and 30% methanol). The mixture was
lyophilized and redisolved in 4.5 ml of Tris buffer (0.1 M, pH
7.4). The mixture was heated to 55°C for 10 min and then cooled
down to 4°C over the course of 5 h. The mixture was then deaerated
for 30 min by bubbling argon. An equal amount of a freshly prepared
sodium dithionite solution (100 μl of the same Tris buffer and 2.78
mg of sodium dithionite i.e., 16 μmol of sodium dithionite) was
added to the solution every 10 min. The addition was repeated 4
times. The last addition was made as a titration i.e., sodium
dithionite was added until the purple color disappeared. The
solution was then stirred open to air for 20 min to reoxidize the
hydroquinones. The cross-link was pre-purified by sep-pak
chromatography and ethanol precipitation. Final purification was
achieved by 20% urea polyacrylamide gel (29).
Evidence of crosslink formation was obtained from
enzymatic digestion of the cross-linked oligonucleotide and HPLC
analysis of the digest (Buffer A, 20 mM ammonium acetate; Buffer B,
30% acetonitrile and 70% Buffer A; 0.8 ml/min flow rate. Gradient,
20% B to 60% B in 60 min. Elution times: dC, 12 min; dG, 17 min;
dT, 21 min; dA, 24 min; dG-MC-dG, 27 min) (data not shown). Thermal
denaturation analysis was conducted to confirm cross-link formation
and showed Tm of the unmodified duplex of 46.72°C and
Tm of MC-α-ICL of 61.29°C.
Chemical treatments
MC and DMC were dissolved in 30% methanol and
freshly diluted with cell culture media to obtain tested
concentrations. Dosages were chosen based on previous studies
(6). Cells were treated with
chemicals and incubated at 37°C for different periods of incubation
time as indicated. The final methanol concentration in the
treatment was <0.15%, which is not toxic to the cells.
Cell cycle analysis
The cells were washed twice with ice cold
phosphate-buffered saline (PBS) and fixed in 70% ethanol at 4°C
overnight. Then the fixed cells were stained with a 50
μg/ml propidium iodide solution containing 100 μg/ml
RNase A and glucose buffer at room temperature for 30 min, and the
cell cycle distribution was analyzed by Attune® Acoustic
Focusing Cytometer using Attune® Cytometric
Software.
Western blot analysis
After 24 h of chemical exposure, cells were lysed
with a mixture of M-PER, Halt protease inhibitor, and a phosphatase
inhibitor cocktail. The concentration of protein samples was
determined by using Bio-Rad DC (detergent compatible) protein assay
reagents (Bio-Rad).
Protein samples (40 μg) from each treatment
condition were resolved via 8% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred to PVDF membranes. Membranes were blocked in PBS/0.05%
Tween-20, containing 5% BSA, and were then probed overnight with
primary antibody. The antibody was detected with corresponding
horseradish peroxidase-linked secondary antibody. Blots were
developed using Super Signal West Pico Chemiluminescent Substrate
detection reagents. Membranes were then stripped with stripping
buffer for 15 min at room temperature and re-probed with β-actin (1
μg/μl) or p21WAF1/CIP1 antibody as the loading control.
Chemiluminescent signals were captured using the Geliance 600
imaging system (Perkin-Elmer, Shelton, CT, USA) and analyzed by
GeneTools software (Syngene, Frederick, MD, USA).
Transfection
p53 shRNA plasmid from Santa Cruz was transiently
transfected into MCF-7 cells to knock down the expression of p53 by
using SuperFect™ transfection reagent 24 h before chemical
treatments. Briefly, p53 shRNA plasmid was mixed with SuperFect in
a ratio of 1 μg DNA to 2 μl transfection reagent according to the
manufacturer's instructions. The mixtures were added in 30 μl of
serum-free DMEM for 10 min at room temperature. The mixtures were
then mixed with 0.6 ml of complete cell culture media and
transferred to the cells for a 3-h incubation at 37°C in humidified
air containing 5% CO2. The transfection mixtures were
then removed and replaced with fresh media after washing the cells
twice with PBS. The cells were then incubated for an additional 24
h at 37°C in humidified air containing 5% CO2 prior to
chemical treatments.
The isolated α-interstrand cross-link was also
transfected into cells by SuperFect transfection reagent using the
same protocol as p53 shRNA plasmid transfection.
Statistical analyses
All experiments were performed at least in
triplicate, and results are reported as means ± SEM. Statistical
significance was determined using one-way analysis of variance
(ANOVA) followed by Dunnett's post hoc test (p<0.05, with
control at 100%) by using GraphPad PRISM® 6
software.
Results
Cell cycle distribution patterns of MCF-7
and K562 cells treated with MC and DMC are shifted to G1/S phase
and to S phase, respectively
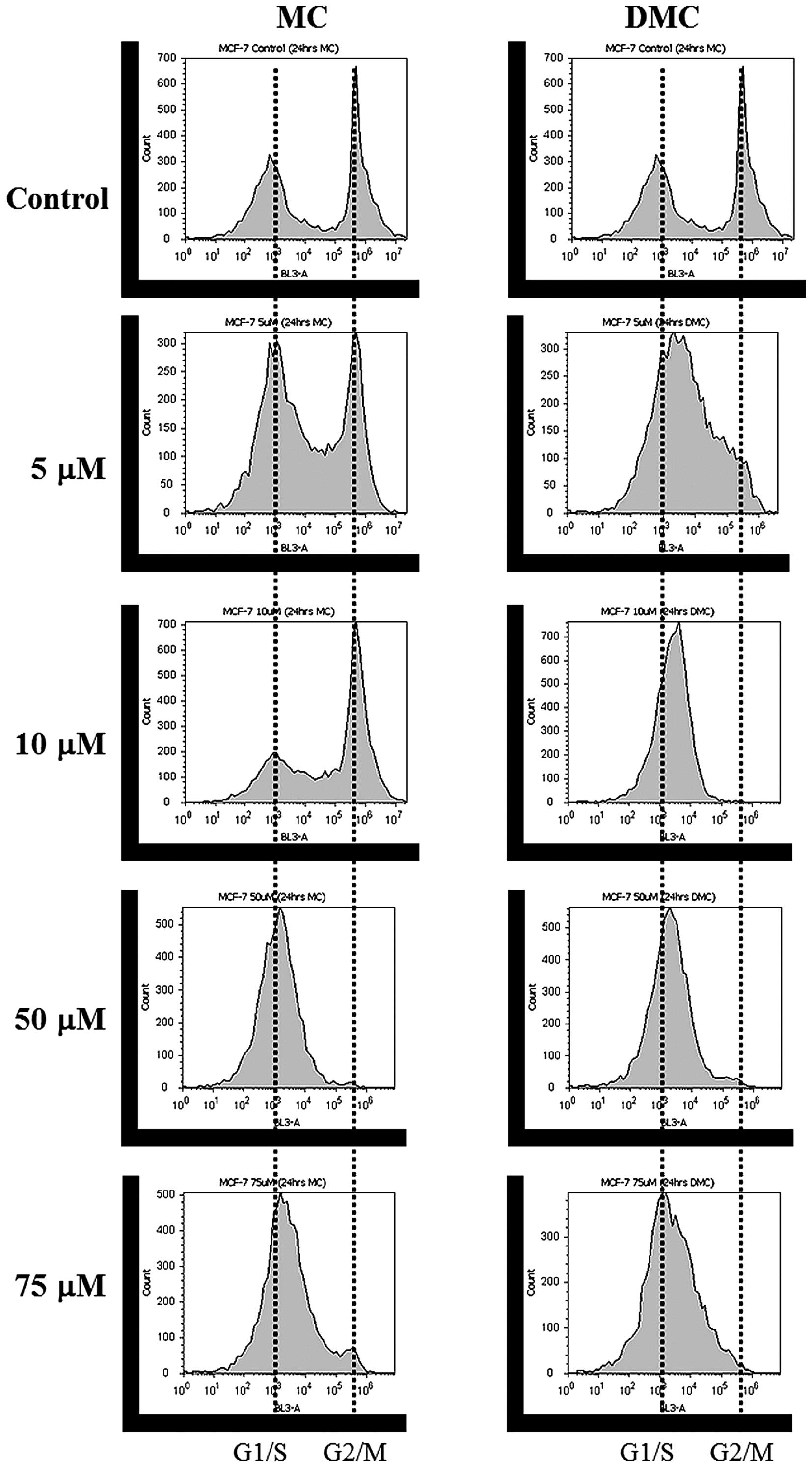
DNA content distribution profiles of MCF-7 treated
with various concentrations of MC and DMC for 24 h are shown in
Fig. 2 and Tables I and II. The cell cycle pattern of MCF-7
control cells appeared normal, with most cells halted at the G1/G0
phase (49.4% cell population at G1/G0 phase, 10.1% at S phase and
32.6% at G2/M phase). MC exposure induced a shift of MCF-7 cell
cycle distribution pattern from G2/M to G1/S phase as MC
concentration increased. When MCF-7 cells were treated with 50 μM
and 75 μM of MC, >90% of cells halted at the G1/S phase
(Fig. 2, left panel and Table I). These results concur with
previous studies which suggest that MC shifts cell cycles to G1/S
or S phase (30,31). DMC exposure also induced a shift of
MCF-7 cell cycle distribution pattern from G2/M to G1/S as DMC
concentration increased (Fig. 2,
right panel and Table II).
Furthermore, DMC induced a shift to G1/S (43.5±39.5% = 83%) at the
concentration as low as 5 μM and at lower concentration range as
compared to MC.
 | Table ICell cycle profiles of MCF-7 treated
with various concentrations of mitomycin C (MC) for 24 h. |
Table I
Cell cycle profiles of MCF-7 treated
with various concentrations of mitomycin C (MC) for 24 h.
| Cell cycle
phase |
|---|
|
|
|---|
| Concentration of
MC | G1/G0 (%) | S (%) | G2/M (%) |
|---|
| Control | 49.4 | 10.1 | 32.6 |
| 5 μM | 55.8 | 14.8 | 24.6 |
| 10 μM | 37.7 | 5.9 | 50.3 |
| 50 μM | 94.0 | 3.5 | 0 |
| 75 μM | 90.3 | 4.4 | 0 |
| Table IICell cycle profiles of MCF-7 treated
with various concentrations of 10-decarbamoyl mitomycin C (DMC) for
24 h. |
Table II
Cell cycle profiles of MCF-7 treated
with various concentrations of 10-decarbamoyl mitomycin C (DMC) for
24 h.
| Cell cycle
phase |
|---|
|
|
|---|
| Concentration of
DMC | G1/G0 (%) | S (%) | G2/M (%) |
|---|
| Control | 49.4 | 10.1 | 32.6 |
| 5 μM | 43.5 | 39.5 | 8.7 |
| 10 μM | 95.7 | 1.1 | 0 |
| 50 μM | >90.0 | <10 |
| 75 μM | >90.0 | <10 |
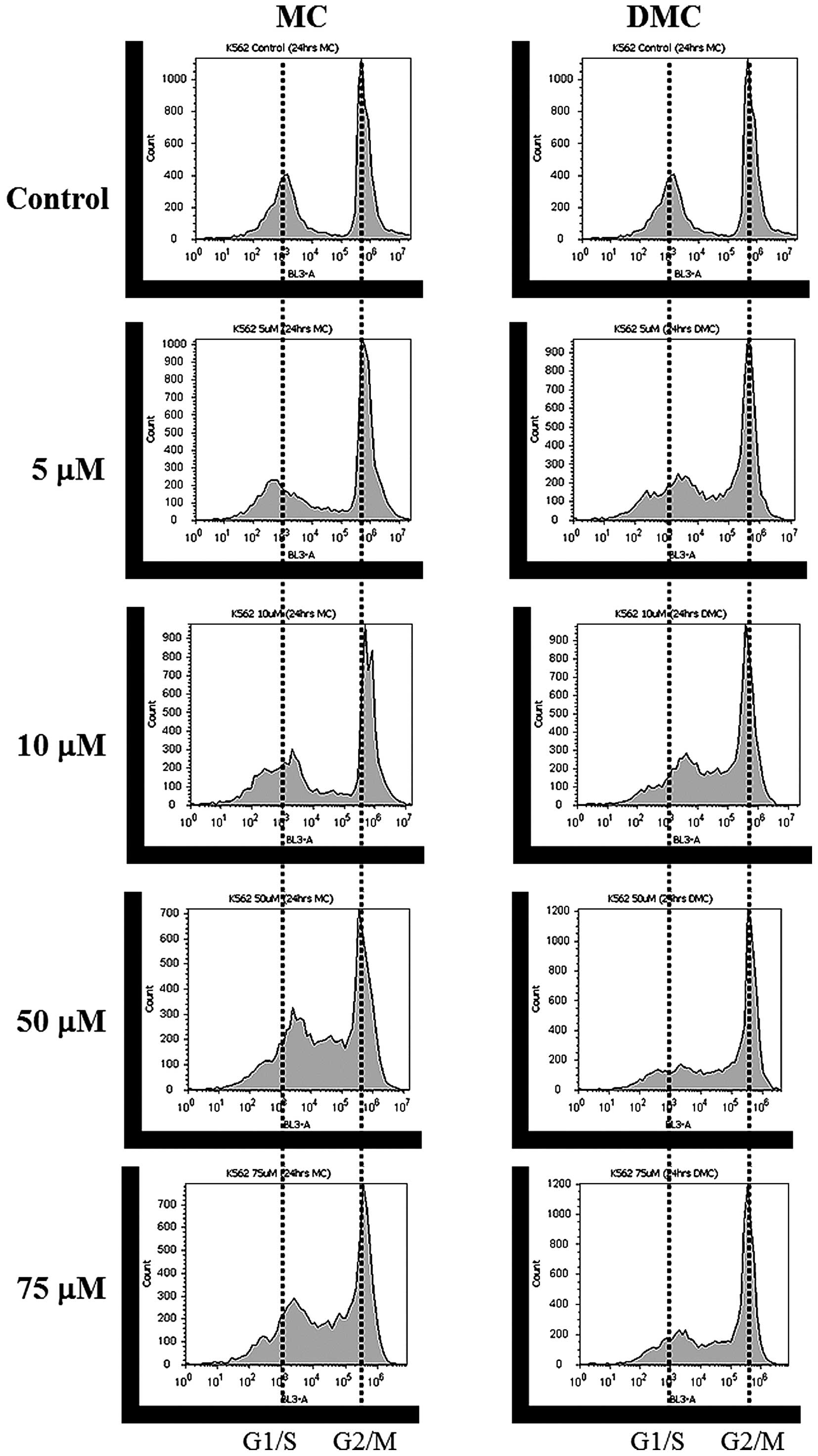
For K562 cells, DNA content distribution profiles of
cells treated with various concentrations of MC and DMC for 24 h
are shown in Fig. 3 and Table III and IV. Most of K562 control cells were
halted at the G2/M phase (41.4% of the cell population at G1/G0
phase, 4.2% at S phase and 52.5% at G2/M phase). MC exposure
increased cells halted at the S phase for cells treated with 50 μM
and 100 μM MC (Fig. 3, left panel
and Table III). The cell
population of K562 at G2/M was also reduced at 50 and 100 μM. DMC
exposure not only increased cells halted in the S phase, but also
decreased cells arrested in the G1/G0 phase as DMC concentration
increased (Fig. 3, right panel and
Table IV). DMC exposure increased
cells halted at the S phase at as low as 5 μM and at lower
concentration range as compared to MC.
| Table IIICell cycle profiles of K562 treated
with various concentrations of mitomycin C (MC) for 24 h. |
Table III
Cell cycle profiles of K562 treated
with various concentrations of mitomycin C (MC) for 24 h.
| Cell cycle
phase |
|---|
|
|
|---|
| Concentration of
MC | G1/G0 (%) | S (%) | G2/M (%) |
|---|
| Control | 41.4 | 4.2 | 52.5 |
| 5 μM | 33.9 | 6.0 | 53.1 |
| 10 μM | 47.2 | 3.8 | 46.1 |
| 50 μM | 40.5 | 13.1 | 43.5 |
| 75 μM | 42.8 | 18.8 | 35.2 |
| Table IVCell cycle profiles of K562 treated
with various concentrations of 10-decarbamoyl mitomycin C (DMC) for
24 h. |
Table IV
Cell cycle profiles of K562 treated
with various concentrations of 10-decarbamoyl mitomycin C (DMC) for
24 h.
| Cell cycle
phase |
|---|
|
|
|---|
| Concentration of
DMC | G1/G0 (%) | S (%) | G2/M (%) |
|---|
| Control | 41.4 | 4.2 | 52.5 |
| 5 μM | 40.8 | 9.7 | 47.0 |
| 10 μM | 34.8 | 12.5 | 50.2 |
| 50 μM | 30.6 | 10.0 | 57.8 |
| 75 μM | 33.9 | 17.5 | 48.1 |
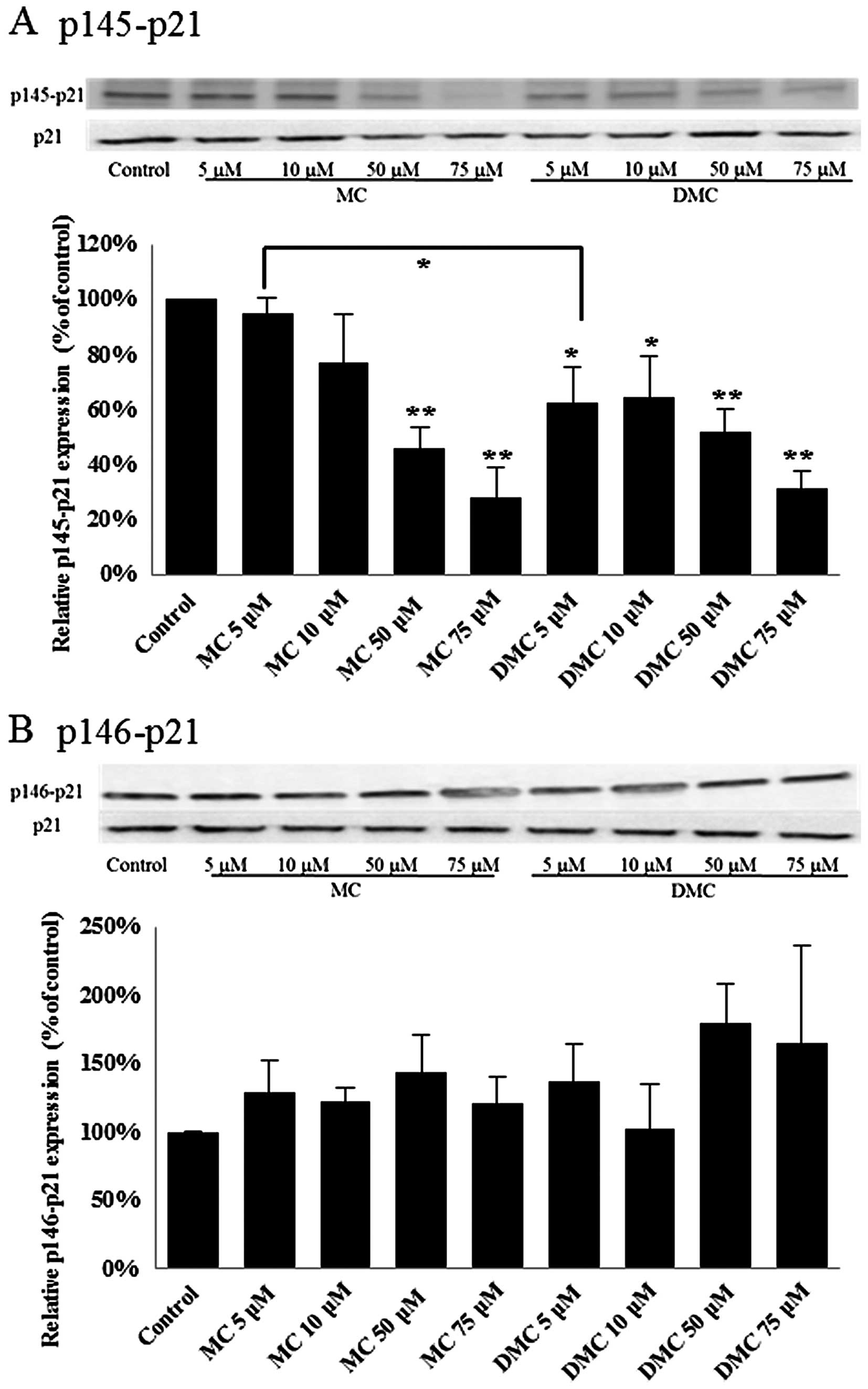
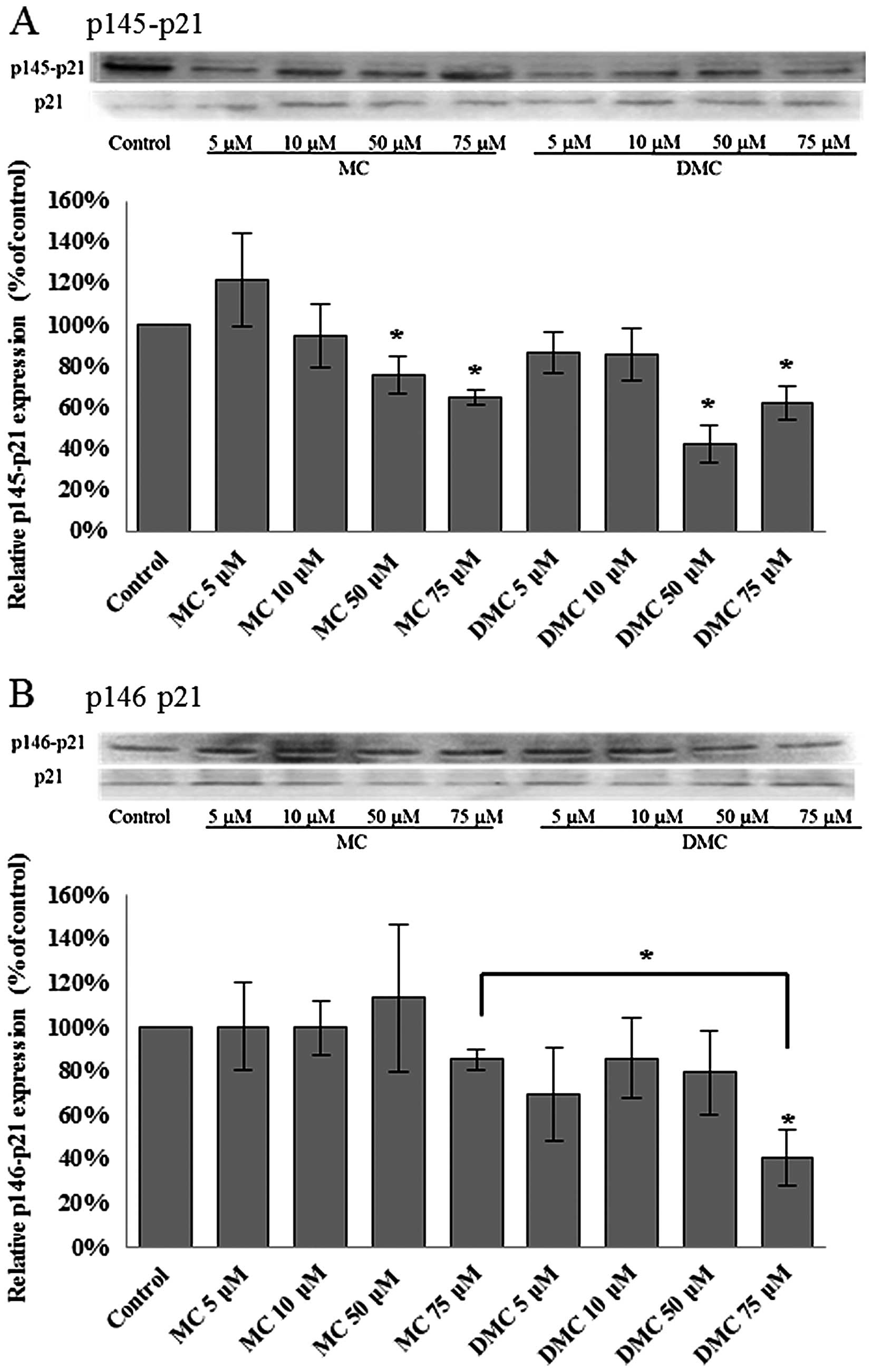
p21WAF1/CIP1 in MCF-7 and K562
cells is activated by MC and DMC
p21WAF1/CIP1 has been linked with
irreversible cell cycle arrest in both G1 and G2/M. Phosphorylation
of p21WAF1/CIP1 at Thr145 (p145-p21) induces its
cytoplasmic accumulation to promote cell proliferation (32). In addition, phosphorylation
p21WAF1/CIP1 at Ser146 (p146-p21) leads to the stability
of p21WAF1/CIP1 and cell survival (33). Dephosphorylation of
p21WAF1/CIP1 will increase the translocation of active
p21WAF1/CIP1 to nuclei and subsequently trigger gene
expression for cell proliferation suppression. In order to study
the p21WAF1/CIP1 activation, the levels of
phosphorylated p21WAF1/CIP1 at Thr145 and Ser146 were
detected for cells treated with MC and DMC by western blot
analysis.
After 24 h of chemical exposure, MC significantly
dephosphorylated the p21WAF1/CIP1 at Thr145 (p145-p21)
in 50 μM and 75 μM treated MCF-7 cells (Fig. 4A). DMC significantly
dephosphorylated p21WAF1/CIP1 at Thr145 (p145-p21) in
MCF-7 cells exposed with the dosage as low as 5 μM (Fig. 4A). DMC at 5 μM reduced more
p145-p21 in MCF-7 cells as compared with cells treated with 5 μM
MC. However, there was no significant statistical change on the
level of phosphorylation of p21WAF1/CIP1 at Ser146
(p146-p21) in MCF-7 cells treated with MC and DMC for 24 h
(Fig. 4B).
For K562 cells, MC and DMC significantly reduced the
level of phosphorylated p21WAF1/CIP1 at Thr145
(p145-p21) in K562 cells treated with 50 μM and 75 μM of chemicals
for 24 h (Fig. 5A). The level of
phosphorylated p21WAF1/CIP1 at Ser146 (p146-p21) was
only significantly decreased when K562 cells were treated with 75
μM of DMC for 24 h (Fig. 5B). DMC
at 75 μM reduced more p146-p21 in K562 cells as compared with cells
treated with 75 μM MC.
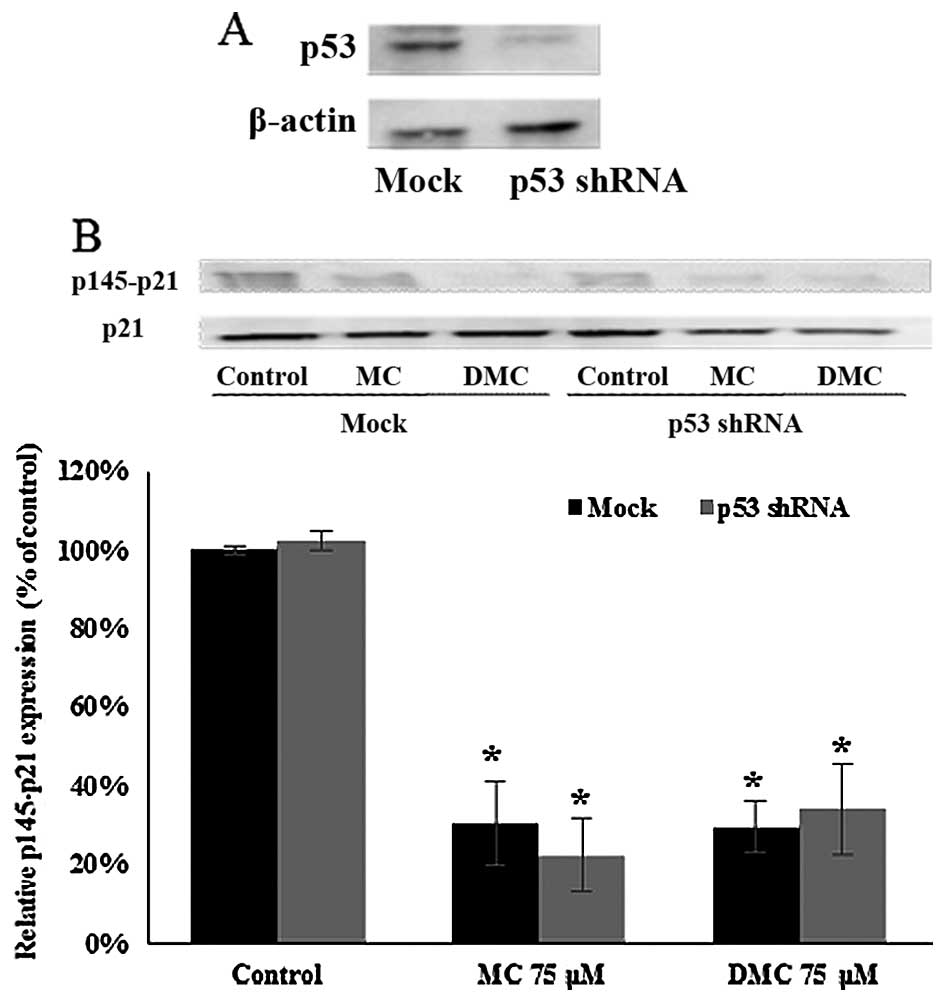
Activation of p21WAF1/CIP1 by
MC and DMC in MCF-7 is p53-independent
p21WAF1/CIP1 expression can be regulated
p53-dependently and -independently. In order to investigate the
role of p53 in MC and DMC induced p21WAF1/CIP1
activation, p53 shRNA plasmid was transfected into MCF-7 cells to
knock down p53 expression. p53 expression was reduced by 73.6%
after 24 h of p53 shRNA knockdown (Fig. 6A). After 24 h of p53 shRNA
knockdown, cells were treated with 75 μM of MC and DMC for 24 h.
The level of phosphorylated p21WAF1/CIP1 at Thr145
(p145-p21) in p53 knockdown cells (no chemical exposure) was the
same as one in normal MCF-7 cells (no chemical exposure). The
levels of p145-p21 in p53 knockdown cells treated with MC and DMC
for 24 h were the same as the one in normal MCF-7 cells treated
under the same condition, with ~70% reduction as compared with
control cells (Fig. 6B).
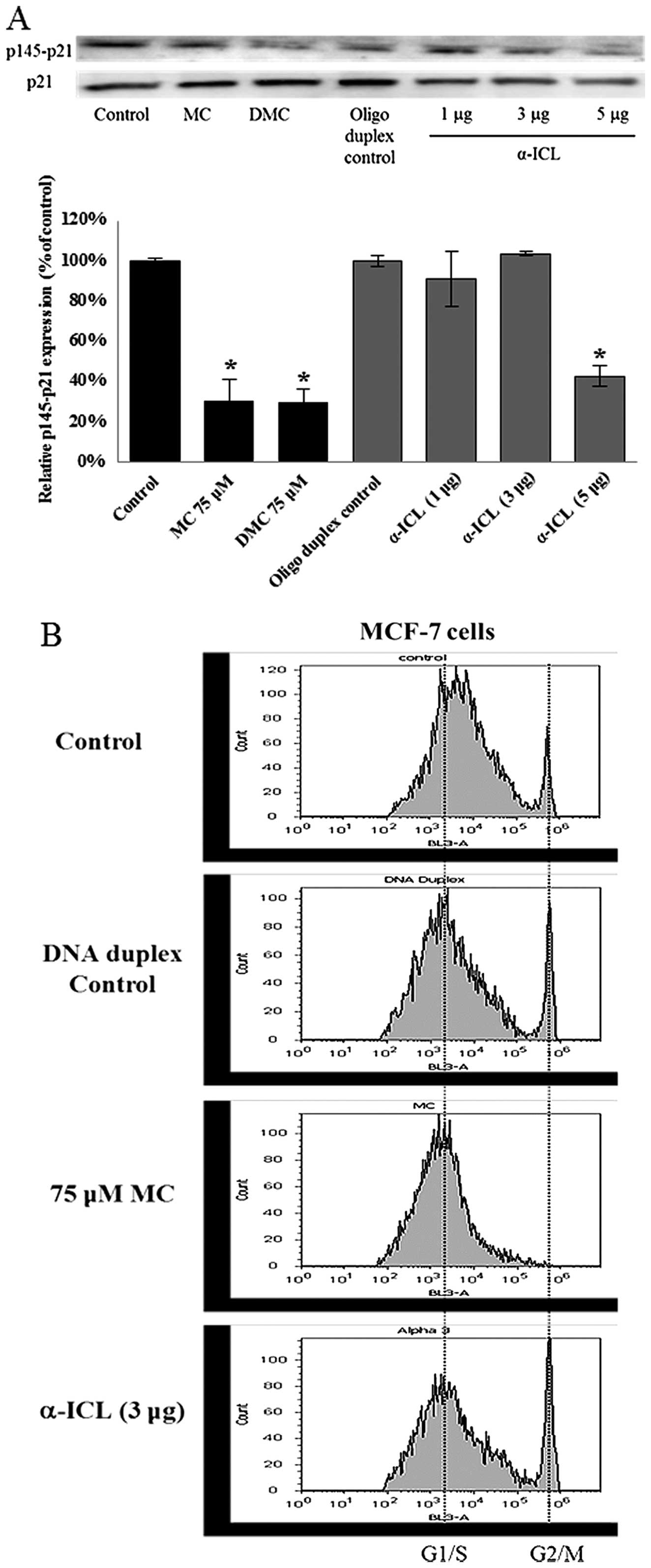
α-ICL itself can activate
p21WAF1/CIP1, but not halt the cell cycle at G1/S phase
in MCF-7
The α-ICL produced by MC plays a major role in MC
induced cytotoxicity. In order to study whether the α-ICL itself
can induce p21WAF1/CIP1 activation as MC did, MCF-7
cells were transfected with the α-ICL (1, 3 or 5 μg) and with DNA
duplex (5 μg) as transfection control. Concomitantly some MCF-7
cells (no transfection) were treated with 75 μM MC or DMC. After
24-h exposure to the α-ICL, MC, and DMC, the levels of
phosphorylated p21WAF1/CIP1 at Thr145 (p145-p21) were
decreased by ~60% for the 5 μg α-ICL transfected group (Fig. 7A) as compared to DNA duplex
control. However, no significant change in the cell cycle
distribution pattern was observed between the α-ICL transfected
group and the DNA duplex control group (Fig. 7B).
Discussion
Mitomycin C (MC) is a commonly used chemotherapy
agent. Both MC and its analog, DMC (decarbamoyl mitomycin C), cause
various DNA lesions, such as α DNA interstrand cross-links (β-ICL)
for MC and β DNA interstrand cross-links (β-ICL) for DMC. These
ICLs are among the most toxic DNA lesions (13). However, only ~20% of cancers
respond to MC treatment (4). DMC
is more toxic in human cancer cells with or without functioning p53
(14,34) as compared to MC. Both MC and DMC
also regulate cell cycle by different mechanisms to cause cell
cycle arrest at different phases (20,34).
MC at 5 μM has been known to trigger cell cycle arrest at S phase
in human hepatocellular carcinoma cells, HepG2 (p53 wild-type
cells) and Hep3B (p53 null cells) (20). Boamah et al (34) also demonstrated that 1 μM DMC
halted cell proliferation at the S and G2/M phases and MC at 10 μM
arrested cells at S phase in p53-null DLD1 colon cancer cells. The
present cell cycle analysis is in agreement with those studies and
showed that both MC and DMC increased the cell population at the
G1/S phase in MCF-7 (p53 wild-type) cells and at the S phase in
K562 (p53-deficient) cells. p53, also known as cell cycle check
protein, can mediate a G1 arrest to preserve the genetic stability
under DNA damage (35). p53
deficiency in K562 could possibly lead cells to lose the regulation
of G1 progression. Thus, there was no significant change on G1
phase cell population observed in this study.
Abbas et al (7) and Boamah et al (34) indicated that DMC triggers stronger
p53-independent cell damage. This suggests that another signaling
pathway besides p53 could be responsible for DMC induced cell
death. p21WAF1/CIP1 is a p53 downstream effector and
inhibits the G1-S transition. However, p21WAF1/CIP1
expression can also be regulated by a p53-independent mechanism
(36). p21WAF1/CIP1 has
been shown to be upregulated in ML-1 p53 wild-type cells by MC and
DMC (7) and in HepG2 p53 wild-type
cells by MC (20). In this study,
the status of p21WAF1/CIP1 activation induced by MC and
DMC in MCF-7 and K562 cells was measured by the level of
phosphorylated p21WAF1/CIP1. Dephosphorylation of
p21WAF1/CIP1 will activate p21WAF1/CIP1 and
translocate p21WAF1/CIP1 to nuclei. Our results showed
that p21WAF1/CIP1 was dephosphorylated by MC and DMC in
both MCF-7 and K562 cells. DMC showed stronger
p21WAF1/CIP1 activation as compared to MC.
Since p21WAF1/CIP1 can be regulated by
p53-dependent and -independent mechanisms (36) and p21WAF1/CIP1 was also
activated in K562 (p53 deficient) cells after MC and DMC exposures,
it is possible that p21WAF1/CIP1 activation by MC and
DMC in MCF-7 could be regulated by a p53-independent pathway. Russo
et al (17) and Esposito
et al (16) suggested
p21WAF1/CIP1 regulates cell cycle arrest and apoptosis
in p53-independent mechanism in response to ribosome protein rpL3.
In this study, p53 shRNA knockdown did lower the expression of p53
in MCF-7, but did not attenuate MC and DMC induced
p21WAF1/CIP1 dephosphorylation. This indicates that
activation of p21WAF1/CIP1 in MCF-7 cells by MC and DMC
did not require p53. DNA lesions formed by MC and DMC have been
linked to their cytotoxic mechanisms. In this study, the major DNA
lesion of MC (α-ICL) was introduced into MCF-7 cells. The
activation of p21WAF1/CIP1 was observed to a similar
extent when compared to cells treated with MC and DMC. However,
there was no significant change in cell cycle distribution pattern
after the α-ICL was introduced into MCF-7 cells when compared with
DNA duplex control. This indicates that the α-ICL itself is
sufficient to trigger p21WAF1/CIP1 activation, but not
to set the cell cycle at rest. Several reasons may explain this
phenomenon. ICLs are extremely toxic to cells (37). MC generates a total of 6 different
covalent DNA lesions (38,39) and other DNA adducts that are not
examined in this study may contribute to MC-induced cell cycle
arrest. Secondly, the transfected oligonucleotide bearing the ICL
may not set the cell cycle at rest because it is not part of a
replicating machinery i.e., DNA itself or part of a replicating
plasmid. Moreover, another CIP/KIP family member, p27, could play a
role in this regulation. p27 is also a cyclin-dependent kinase
inhibitor as p21WAF1/CIP1 and has similar cellular
function as p21WAF1/CIP1. Rao et al (40) demonstrated that
p21WAF1/CIP1 and p27 are involved in lovastatin
triggered G1 cell cycle arrest in a p53-independent manner.
Therefore, it would be of interest to compare p27 expression in
response to MC, DMC, and ICLs in cells with or without functioning
p53.
In conclusion, mitomycin C and its derivative
10-decar-bamoyl mitomycin C triggered cell cycle arrest at G1/S
phase in MCF-7 cells and at S phase in K562 cells. DMC induced the
arrest at lower concentrations as compared with MC. MC and DMC both
decreased the phosphorylated p21WAF1/CIP1 at Thr145 in
MCF-7 and K562 cells. DMC showed stronger effect on
dephosphorylation of p21WAF1/CIP1 at Thr145 in MCF-7
cells. Knocking down p53 did not change the effect of MC and DMC on
p21WAF1/CIP1 activation. The major DNA lesion produced
by MC, α-ICL, did activate p21WAF1/CIP1, but did not
halt the cell cycle. This study demonstrated that MC and DMC
activated p21WAF1/CIP1 in p53-independent manner and
α-ICL can trigger the activation of p21WAF1/CIP1. In the
future, p21WAF1/CIP1 and other p53-independent signaling
pathways in response to the β-ICL will require further
investigation.
Acknowledgements
This study was supported by NIH grant 5SC3GM105460
and the PRISM program at John Jay. Special thanks to John Jay
College of Criminal Justice Start-up Fund, PRISM fund, and PSC CUNY
grant. PRISM is the Program for Research Initiatives for Science
Majors at John Jay College and funded by the Title V, HSI-STEM and
MSEIP programs within the US Department of Education; the PAESMEM
program through the National Science Foundation; and New York
State's Graduate Research and Teaching Initiative.
Abbreviations:
|
MC
|
mitomycin C
|
|
DMC
|
10-decarbamoyl mitomycin C
|
|
α-ICL
|
α interstrand cross-link
|
References
|
1
|
Hata T, Hoshi T, Kanamori K, Matsumae A,
Sano Y, Shima T and Sugawara R: Mitomycin, a new antibiotic from
Streptomyces. I. J Antibiot (Tokyo). 9:141–146. 1956.
|
|
2
|
Sartorelli AC, Hodnick WF, Belcourt MF,
Tomasz M, Haffty B, Fischer JJ and Rockwell S: Mitomycin C: A
prototype bioreductive agent. Oncol Res. 6:501–508. 1994.PubMed/NCBI
|
|
3
|
Verweij J and Pinedo H: Cancer
Chemotherapy and Biological Response Modifiers. Annual 11. Pinedo
HM, Chabner BA and Longo DL: 67. Elsevier Science Publishers B.V;
Amsterdam: 1990
|
|
4
|
Bradner WT: Mitomycin C: A clinical
update. Cancer Treat Rev. 27:35–50. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Borowy-Borowski H, Lipman R and Tomasz M:
Recognition between mitomycin C and specific DNA sequences for
cross-link formation. Biochemistry. 29:2999–3006. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Boamah EK, White DE, Talbott KE, Arva NC,
Berman D, Tomasz M and Bargonetti J: Mitomycin-DNA adducts induce
p53-dependent and p53-independent cell death pathways. ACS Chem
Biol. 2:399–407. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Abbas T, Olivier M, Lopez J, Houser S,
Xiao G, Kumar GS, Tomasz M and Bargonetti J: Differential
activation of p53 by the various adducts of mitomycin C. J Biol
Chem. 277:40513–40519. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ben-Yehoyada M, Wang LC, Kozekov ID, Rizzo
CJ, Gottesman ME and Gautier J: Checkpoint signaling from a single
DNA interstrand crosslink. Mol Cell. 35:704–715. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Räschle M, Knipscheer P, Enoiu M, Angelov
T, Sun J, Griffith JD, Ellenberger TE, Schärer OD and Walter JC:
Mechanism of replication-coupled DNA interstrand crosslink repair.
Cell. 134:969–980. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Weng MW, Zheng Y, Jasti VP, Champeil E,
Tomasz M, Wang Y, Basu AK and Tang MS: Repair of mitomycin C mono-
and inter-strand cross-linked DNA adducts by UvrABC: A new model.
Nucleic Acids Res. 38:6976–6984. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shinohara K, Bando T, Sasaki S, Sakakibara
Y, Minoshima M and Sugiyama H: Antitumor activity of
sequence-specific alkylating agents: Pyrolle-imidazole CBI
conjugates with indole linker. Cancer Sci. 97:219–225. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Palom Y, Suresh Kumar G, Tang LQ, Paz MM,
Musser SM, Rockwell S and Tomasz M: Relative toxicities of DNA
cross-links and monoadducts: New insights from studies of
decarbamoyl mitomycin C and mitomycin C. Chem Res Toxicol.
15:1398–1406. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kaspárková J and Brabec V: Recognition of
DNA interstrand cross-links of cis-diamminedichloroplatinum(II) and
its trans isomer by DNA-binding proteins. Biochemistry.
34:12379–12387. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Patrick SM, Tillison K and Horn JM:
Recognition of cisplatin-DNA interstrand cross-links by replication
protein A. Biochemistry. 47:10188–10196. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xiao G, Kue P, Bhosle R and Bargonetti J:
Decarbamoyl mitomycin C (DMC) activates p53-independent ataxia
telangiectasia and rad3 related protein (ATR) chromatin eviction.
Cell Cycle. 14:744–754. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Esposito D, Crescenzi E, Sagar V, Loreni
F, Russo A and Russo G: Human rpL3 plays a crucial role in cell
response to nucleolar stress induced by 5-FU and L-OHP. Oncotarget.
5:11737–11751. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Russo A, Esposito D, Catillo M,
Pietropaolo C, Crescenzi E and Russo G: Human rpL3 induces G1/S
arrest or apoptosis by modulating p21 (waf1/cip1) levels in a
p53-independent manner. Cell Cycle. 12:76–87. 2013. View Article : Google Scholar :
|
|
18
|
Resnitzky D, Gossen M, Bujard H and Reed
SI: Acceleration of the G1/S phase transition by expression of
cyclins D1 and E with an inducible system. Mol Cell Biol.
14:1669–1679. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
He G, Kuang J, Huang Z, Koomen J,
Kobayashi R, Khokhar AR and Siddik ZH: Upregulation of p27 and its
inhibition of CDK2/cyclin E activity following DNA damage by a
novel platinum agent are dependent on the expression of p21. Br J
Cancer. 95:1514–1524. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Choi SY, Shen YN, Woo SR, Yun M, Park JE,
Ju YJ, Jeong J, Shin HJ, Joo HY, Park ER, et al: Mitomycin C and
doxorubicin elicit conflicting signals by causing accumulation of
cyclin E prior to p21WAF1/CIP1 elevation in human
hepatocellular carcinoma cells. Int J Oncol. 40:277–286. 2012.
|
|
21
|
Willers H, Dahm-Daphi J and Powell SN:
Repair of radiation damage to DNA. Br J Cancer. 90:1297–1301. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Harris SL and Levine AJ: The p53 pathway:
Positive and negative feedback loops. Oncogene. 24:2899–2908. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Petitjean A, Achatz MI, Borresen-Dale AL,
Hainaut P and Olivier M: TP53 mutations in human cancers:
Functional selection and impact on cancer prognosis and outcomes.
Oncogene. 26:2157–2165. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Faulhaber O and Bristow RG: Basis of cell
kill following clinical radiotherapy. Application of Apoptosis to
Cancer Treatment. Sluyser M: Springer; Amsterdam: pp. 293–320.
2005, View Article : Google Scholar
|
|
25
|
Fang L, Igarashi M, Leung J, Sugrue MM,
Lee SW and Aaronson SA: p21Waf1/Cip1/Sdi1 induces permanent growth
arrest with markers of replicative senescence in human tumor cells
lacking functional p53. Oncogene. 18:2789–2797. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sugiyama K, Shimizu M, Akiyama T, Tamaoki
T, Yamaguchi K, Takahashi R, Eastman A and Akinaga S: UCN-01
selectively enhances mitomycin C cytotoxicity in p53 defective
cells which is mediated through S and/or G(2) checkpoint
abrogation. Int J Cancer. 85:703–709. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Law JC, Ritke MK, Yalowich JC, Leder GH
and Ferrell RE: Mutational inactivation of the p53 gene in the
human erythroid leukemic K562 cell line. Leuk Res. 17:1045–1050.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kinoshita S, Uzu K, Nakano K and Takahashi
T: Mitomycin derivatives. 2. Derivatives of decarbamoylmitosane and
decar-bamoylmitosene. J Med Chem. 14:109–112. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Summer H, Grämer R and Dröge P: Denaturing
urea polyacrylamide gel electrophoresis (Urea PAGE). J Vis Exp.
32:e14852009.
|
|
30
|
Zhou QM, Wang XF, Liu XJ, Zhang H, Lu YY
and Su SB: Curcumin enhanced antiproliferative effect of mitomycin
C in human breast cancer MCF-7 cells in vitro and in vivo. Acta
Pharmacol Sin. 32:1402–1410. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yu J, Zhao L, Li Y, Li N, He M, Bai X, Yu
Z, Zheng Z, Mi X, Wang E, et al: Silencing of Fanconi anemia
complementation group F exhibits potent chemosensitization of
mitomycin C activity in breast cancer cells. J Breast Cancer.
16:291–299. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rössig L, Jadidi AS, Urbich C, Badorff C,
Zeiher AM and Dimmeler S: Akt-dependent phosphorylation of
p21(Cip1) regulates PCNA binding and proliferation of endothelial
cells. Mol Cell Biol. 21:5644–5657. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li Y, Dowbenko D and Lasky LA: AKT/PKB
phosphorylation of p21Cip/WAF1 enhances protein
stability of p21Cip/WAF1 and promotes cell survival. J
Biol Chem. 277:11352–11361. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Boamah EK, Brekman A, Tomasz M, Myeku N,
Figueiredo-Pereira M, Hunter S, Meyer J, Bhosle RC and Bargonetti
J: DNA adducts of decarbamoyl mitomycin C efficiently kill cells
without wild-type p53 resulting from proteasome-mediated
degradation of checkpoint protein 1. Chem Res Toxicol.
23:1151–1162. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Di Leonardo A, Linke SP, Clarkin K and
Wahl GM: DNA damage triggers a prolonged p53-dependent G1 arrest
and long-term induction of Cip1 in normal human fibroblasts. Genes
Dev. 8:2540–2551. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Macleod KF, Sherry N, Hannon G, Beach D,
Tokino T, Kinzler K, Vogelstein B and Jacks T: p53-dependent and
independent expression of p21 during cell growth, differentiation,
and DNA damage. Genes Dev. 9:935–944. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lawley PD and Phillips DH: DNA adducts
from chemotherapeutic agents. Mutat Res. 355:13–40. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bargonetti J, Champeil E and Tomasz M:
Differential toxicity of DNA adducts of mitomycin C. J Nucleic
Acids. 2010:6989602010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tomasz M: Mitomycin C: Small, fast and
deadly (but very selective). Chem Biol. 2:575–579. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rao S, Lowe M, Herliczek TW and Keyomarsi
K: Lovastatin mediated G1 arrest in normal and tumor breast cells
is through inhibition of CDK2 activity and redistribution of p21
and p27, independent of p53. Oncogene. 17:2393–2402. 1998.
View Article : Google Scholar : PubMed/NCBI
|