Introduction
Esophageal carcinoma is one of the most common
malignancies of the digestive system. In 2011, esophageal carcinoma
was the fifth and eighth most common cause of death worldwide in
men and women, respectively (1).
Among this group of cancers, esophageal squamous cell carcinoma
(ESCC) has high metastatic potential with frequent initial
dissemination to regional lymph nodes and distant metastases at
diagnosis, characteristics that underlie the majority of
ESCC-related deaths (2,3). Despite recent advances in clinical
treatment, patients with ESCC exhibit a poor prognosis, with an
average 5-year survival rate of <20% worldwide due to distant
metastases (4). Although the
standard treatment for most solid cancers is surgical resection,
most patients with unresectable malignant tumors, including ESCC,
are treated by irradiation-based therapy. In recent years, ~50% of
all patients with solid cancer received radiation therapy. However,
several recent preclinical and clinical studies have demonstrated
that irradiation induces an increase in invasiveness and metastatic
potential of several cancer cell types, including glioma, colon,
breast, and lung cancer (5–7).
Elucidation of the mechanisms underlying irradiation-induced
metastasis is important to overcome failure of radiation
therapy.
Epithelial mesenchymal transition (EMT) has become
recognized as one of the major factors causing metastatic relapse
and resistance to anticancer agents (8,9). EMT
is a crucial process in cancer progression, providing cancer cells
with the ability to escape from the primary site, invade stromal
tissues, and migrate to distant regions of the body. As a result of
EMT epithelial cells lose their defined cell-cell/cell-substratum
contacts and their structural/functional polarity and become
spindle shaped and morphologically similar to activated
fibroblasts. At the molecular level, epithelial cells undergoing
EMT are characterized by downregulation of epithelial makers (such
as E-cadherin and β-catenin) and loss of cell polarity and
intercellular adhesion molecules, concomitant with upregulation of
mesenchymal markers (vimentin, N-cadherin, and fibronectin) and
nuclear localization of β-catenin (10,11).
Cells that gain expression of mesenchymal markers show a front-rear
polarity associated with weak cell-cell contact, increased
motility, and resistance to apoptosis (10). Tumor cells that gain a more
mesenchymal-like phenotype have migratory capacity at the expense
of proliferative potential, and acquire resistance to DNA damage
inducers including chemical agents and irradiation, thus becoming
resistant to chemotherapy and radiotherapy (8,9).
Transforming growth factor-β1 (TGF-β1) is a multifunctional
cytokine that modulates cell proliferation, differentiation,
apoptosis, and extracellular matrix production (12). TGF-β1 is known to play a critical
role in EMT (13). TGF-β1 mediates
its effects through binding to a heteromeric complex of
transmembrane serine/threonine kinases and a type II receptor,
which results in activation of the receptor and initiation of
Smad-dependent and -independent EMT pathways (13,14).
Environmental factors, including nicotine, ultraviolet light, and
irradiation, also promote EMT (15,16).
Irradiation can cause many cellular effects, including apoptosis,
senescence, and genomic instability, that may lead to cancer cell
death. Conversely, irradiation also foments an increase in extra-
and intracellular levels of TGF-β1 in patients and induces
acceleration of metastatic cancer progression (6,17,18).
Recently, irradiation was found to enhance cell migration,
invasion, and metastasis through induction of EMT in various cancer
cell lines, including ESCC cells (7,19).
Histone deacetylases (HDACs) are a family of enzymes
that remove acetyl groups from histone and non-histone proteins and
play an important role in regulating gene transcription and protein
functions (20). HDAC inhibitors
induce cell cycle arrest, differentiation, and apoptosis in
vitro and in vivo (21–23).
Over the last year several HDAC inhibitors have been introduced
into clinical trials with successful results. Most epigenetic
studies in the anticancer field have used valproic acid (VPA), the
most potent HDAC inhibitor (24).
The fact that VPA has been safely used in long-term therapy of
patients with epilepsy over decades is a clear advantage, and phase
I and II clinical trials of VPA in cancer have provided promising
results (25,26). In addition, tests of several
protocols involving the use of VPA against diverse neoplasias are
ongoing (20). VPA is a promising
anticancer agent with effects correlated with the transcriptional
regulation of specific cancer-related genes. We have noted the
effectiveness of VPA as an anticancer agent and its ability to
suppress collagen synthesis. In previous studies, we demonstrated
that VPA enhances irradiation-induced cytotoxicity via chromatin
decondensation and inhibition of DNA double-strand break (DSB)
repair in human ESCC cells (27,28).
VPA also prevents the morphologic changes characteristic of
activation and inhibits the expression of collagen type1 α1 and
TGF-β1 in human hepatic stellate cells (29). Recently, several reports have shown
that HDAC inhibitors suppress metastatic potential in cancer cells
by attenuating EMT (30,31). However, there are no data on the
potential role of VPA in the inhibition of irradiation-induced
EMT.
The aim of this study was to evaluate the inhibitory
effects of VPA on radiation-induced EMT in human ESCC cells and to
reveal the underlying mechanisms.
Materials and methods
Cell lines, cell culture, and
treatment
The TE9 cell line (human ESCC cell line, poorly
differentiated) was kindly provided by Dr Tetsuro Nishihira
(Kenotokorozawa Hospital, Saitama, Japan). Cells were grown in
RPMI-1640 (Invitrogen, Tokyo, Japan) medium supplemented with 2 mM
glutamine, 10% fetal bovine serum (FBS; Nichirei Biosciences, Inc.,
Tokyo, Japan), 100 U/l penicillin, and 100 μg/ml streptomycin
(Invitrogen) and maintained at 37°C in a 5% CO2
incubator. The cells were seeded in gelatin-coated
75-cm2 flasks (BioCoat, BD Biosciences, NJ, USA) and
harvested with 0.25% trypsin-EDTA before use.
Irradiation
Cultures were irradiated using MBR-150R-3 (Hitachi
Medicotechnology, Hitachi, Japan) at a dose rate of 1.5 Gy/min.
Power output of X-ray irradiation was 125 kV, 20 mA.
Forward-scattered radiation, 0.5-mm Al, and 0.2-mm Cu filters were
used.
Reagents and antibodies
VPA was purchased from Sigma-Aldrich Co. (Tokyo,
Japan) and used at concentrations of 0.1, 0.5, 1, 5 and 10 mM. VPA
was dissolved in phosphate-buffered saline (PBS) to a stock
concentration of 100 mM and stored at −20°C. TGF-β1 was purchased
from Sigma-Aldrich and used at a concentration of 10 ng/ml. Mouse
monoclonal antibodies to E-cadherin, vimentin, TGF-β1, Smad2,
Smad3, matrix metalloproteinase 9 (MMP-9), HCAM (CD44), and
β-catenin and rabbit polyclonal antibodies to phosphorylated Smad2
(p-Smad2), phosphorylated Smad3 (p-Smad3), Twist, Snail, Slug, and
MMP-7 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA,
USA). Rabbit polyclonal antibodies to MMP-2 were obtained from
Millipore (Billerica, MA, USA). Mouse monoclonal antibodies to
β-actin and HIF-1α were obtained from Sigma-Aldrich and Thermo
Fisher Scientific (Rockford, IL, USA), respectively.
Cell viability assay
TE9 cells were plated in small dishes at a density
of 5×104/ml in medium with 10% FBS and allowed to adhere
for 24 h before incubation in serum-free medium for 24 h. Cells
were treated with vehicle or VPA (0, 0.1, 0.5, 1, 5, 10 mM) for 48
h and harvested by trypsinization. Cells were stained with a 0.4%
trypan blue solution (Sigma Chemical Co., St. Louis, MO, USA), and
counted on a Cellometer Vision automated cell counter (Nexcelom
Bioscience, Lawrence, MA, USA) according to the manufacturer's
protocol. Cell viabilities were represented as mean percentage
relative to matched vehicle-treated cells for triplicate
experiments with internal triplicates. At least three independent
experiments were performed for statistical analysis.
Cell growth assay
The total number of living cells as a measure of
proliferation was determined using a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT;
Sigma-Aldrich) assay. Cells were seeded in quadruplicate in 95-well
plates (BD BioCoat) with 5×103 cells per well and
incubated for 24 h at 37°C. After incubation, the supernatant was
discarded and replaced with fresh serum-free medium. VPA was
dissolved in PBS and added to the cell culture medium at various
concentrations (0, 0.1, 0.5, 1, 5 and 10 mM). After 48-h exposure
to VPA, the cells were incubated at 37°C for 3 h with MTT solution
(500 μg/ml) in fresh culture medium. The medium was removed and
crystallized formazan dye in the cells was solubilized by addition
of dimethylsulfoxide. The percentage inhibition was determined by
comparing absorbance at 540 nM of VPA-treated cells with that of
untreated controls using a microplate reader (Bio-Rad 550; Bio-Rad,
Japan). Three independent experiments were performed for
statistical analysis.
Fluorescence immunocytochemistry
TE9 cells were grown on a 2-well Lab-Tek Chamber
Slide System (Nalge Nunc International, Rochester, NY, USA) to 80%
confluence and incubated for 24–48 h at 37°C in a humid atmosphere
of 5% CO2/95% air. Subconfluent cells were transferred
to serum-free medium for 24 h, and then treated with 2 Gy
irradiation or 10 ng/ml TGF-β1 with or without 3-h pretreatment
with 1 mM VPA. At 24- to 48 h after exposure to irradiation or
TGF-β1, morphologic changes were observed by phase-contrast
microscopy. The cells were then subjected to immunostaining. Cells
on coverslips were fixed with methanol and acetone (1:1 v/v). After
pretreatment with protein blocking serum for 10 min at room
temperature to block non-specific binding, TE9 cells were incubated
with primary antibody [anti-E-cadherin (1:200), anti-vimentin
(1:200), or anti-β-catenin (1:100)] overnight at 4°C. Following
washes in PBS, cells were incubated with the appropriate Alexa
Fluor® 488 and 592 nm (Molecular Probes/Invitrogen, OR,
USA; 1:400) for 1 h at room temperature. Nuclei were counterstained
with bis-benzimide (Hoechst 33258; Sigma-Aldrich Co.) for 5 min and
cells were visualized with an immunofluorescence microscope
(BX50/BX-FLA; Olympus, Japan).
Invasion assay
Cell invasion ability was analyzed by an invasion
assay using a BioCoat Matrigel Invasion Chamber for 24-well plates
(BD Bioscience). In brief, Matrigels were rehydrated before use.
Cells were treated with TGF-β1 or irradiation as described above
and seeded into the upper chamber system in serum-free media
(5×104 cells/well) with the control or Matrigel
membrane. The lower chamber contained 750 μl of fresh medium with
10% FBS. After 24-h incubation at 37°C in a humid atmosphere of 5%
CO2/95% air, the upper surface of the filter was wiped
with a cotton swab and fixed in 100% methanol. Cells on the lower
surface of the membrane were stained by hematoxylin and counted
using a microscope. The number of cells in three random optical
fields (×40 magnification) was averaged. Percentage invasion was
calculated as the ratio of the number of invaded cells through the
Matrigel membrane relative to that through the control
membrane.
Migration assay
Cells were seeded in 6-well culture plates and grown
to ~80% confluence. The confluent cell monolayer was scraped with a
10-μl pipette tip. Cells were treated with TGF-β1 or irradiation as
described above. Images were captured immediately after wounding (0
h) and after 48-h incubation at 37°C in a humid atmosphere of 5%
CO2/95% air. Percentage of migration was calculated as
the ratio of migrated cells from the leading edge 48 h after
scratching relative to that in control experiments.
Western blotting
Expression of EMT-related proteins was analyzed by
western blotting. First, cells were lysed in RIPA buffer (Wako Pure
Chemical Industries, Ltd., Osaka, Japan) containing a protease and
phosphatase inhibitor cocktail (Sigma-Aldrich). The protein
concentration of each sample was measured using a BCA protein assay
kit (Thermo Scientific, Waltham, MA, USA). For sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), 20 μg of
protein from each sample was separated on a 12.5% gradient
polyacrylamide gel (e-PAGEL, ATTO Corp., Tokyo, Japan) and
transferred to polyvinylidene difluoride membranes (Bio-Rad
Laboratories). After incubation with blocking solution (EZ Block
ATTO Corp.) at room temperature for 30 min, the membranes were
incubated with primary antibody overnight at 4°C, followed by
incubation with horseradish peroxidase-labeled anti-mouse or
anti-rabbit IgG secondary antibody (Santa Cruz Biotechnology) for 1
h at room temperature. Immune complexes were detected using the ECL
Plus Western blotting detection system (GE Healthcare UK, Ltd.,
Buckinghamshire, UK) and the Light Capture system (ATTO Co. Ltd.).
The density of each band was quantified by the CS analyzer program
(Atto Co. Ltd.). We used primary antibodies against the following
proteins: E-cadherin (1:500), vimentin (1:500), β-catenin
(1:1,000), TGF-β1 (1:1,000), Smad2 (1:200), Smad3 (1:200),
phosphorylated-Smad2 (p-Smad2) (1:200), p-Smad3 (1:200), Twist
(1:200), Snail (1:200), Slug (1:200), pro-MMP-2 (1:200), pro-MMP-7
(1:200), pro-MMP-9 (1:200), and HIF-1α (1:500). Antibody against
β-actin (1:10,000) was used as an internal control.
Flow cytometry
To detect CD44, flow cytometry was performed using
standard protocols. Cells were harvested with 0.25% trypsin-EDTA.
Cell suspension and anti-CD44 antibody (1:20) were mixed and
incubated for 15 min at room temperature. After centrifugation at
2,000 rpm for 3 min at 4°C to remove debris, cells were analyzed on
a BD FACS Calibur flow cytometer (Beckman Coulter, Brea, CA,
USA).
Data analysis
All experiments were independently performed at
least three times and the results were expressed as mean ± SD. Data
were analyzed with SPSS statistics 19 (SPSS Inc., Chicago, IL, USA)
by Student's unpaired t-test and χ2 test. P<0.05 was
considered to indicate a statistically significant difference.
Results
In vitro effect of VPA on cytostasis and
cytotoxicity of TE9 cells
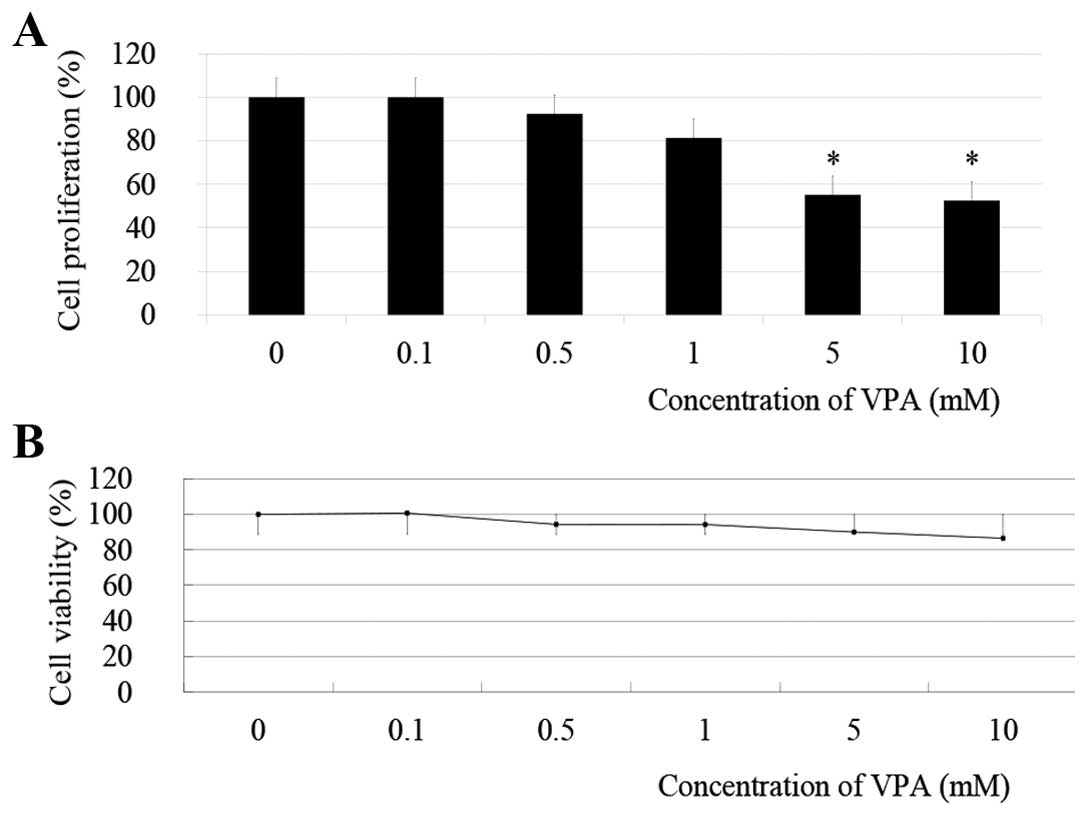
Treatment of TE9 cells with increasing doses (0,
0.5, 1, 5 and 10 mM) of VPA for 48 h induced a significant
dose-dependent decrease in cell proliferation as assayed by MTT
metabolism (Fig. 1A). Treatement
with 5 and 10 mM VPA significantly inhibited TE9 cell proliferation
to 55.2±5.3% (P=0.0045) and 52.5±5.0% (P=0.004) compared with
control, respectively. Trypan blue assays revealed no cytotoxic
effect of VPA against TE9 cells even at the highest concentration
(10 mM) (Fig. 1B).
The effect of VPA on morphologic changes
induced by TGF-β1 or irradiation in TE9 cells
Untreated TE9 cells showed a cobblestone-like
morphology and strong cell-cell adhesion (Fig. 2A). Treatment with 10 ng/ml TGF-β1
or 2 Gy of irradiation induced remarkable morphologic changes after
48 h of stimulation, including loss of cell polarity, appearance of
spindle-shaped cells, and enlargement of the cell space (Fig. 2B and C). However, TE9 cells that
were pretreated with 1 mM VPA did not show obvious morphologic
changes (Fig. 2E and F).
The effect of VPA on changes in
expression of epithelial and mesenchymal markers induced by TGF-β1
or irradiation in TE9 cells
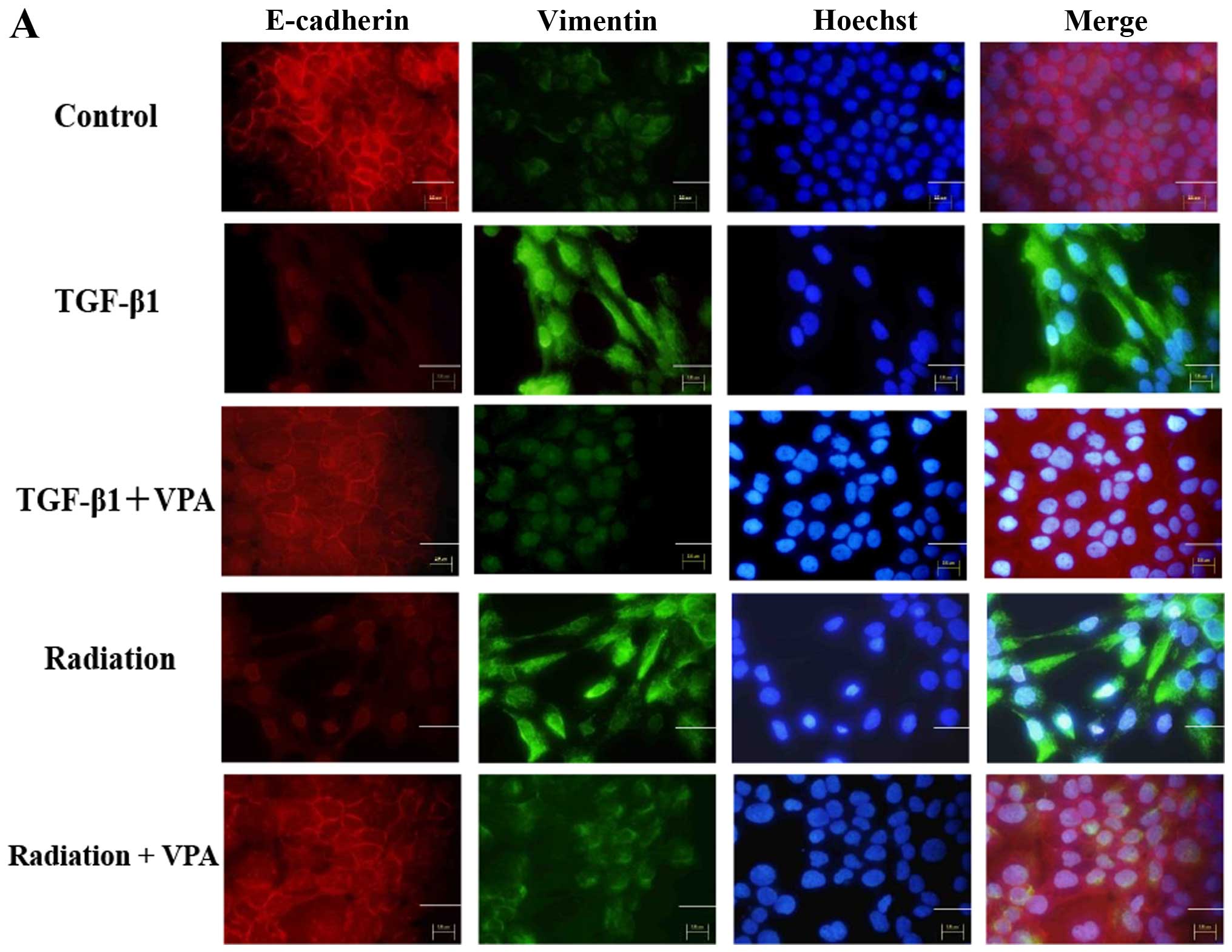
To investigate the inhibitory effect of VPA on EMT
induced by TGF-β1 or irradiation in TE9 cells, we examined the
expression pattern of the epithelial marker E-cadherin and the
mesenchymal marker vimentin with or without VPA treatment under
TGF-β1 or irradiation stimulation by immunocytochemistry and
western blot analysis (Fig. 3).
TE9 cells stimulated by TGF-β1 or irradiation showed a significant
decrease in E-cadherin expression and a concomitant increase in
vimentin expression compared with control cells.
Immunocytochemistry showed that E-cadherin expression at the
membrane was completely lost after TGF-β1 or irradiation
stimulation. In contrast, cells that were pretreated with 1 mM VPA
showed less inhibition of E-cadherin expression than untreated
cells when stimulated by TGF-β1 or irradiation. Vimentin expression
was strongly increased and uniformly distributed under TGF-β1 or
irradiation stimulation. Upregulation of vimentin expression by
TGF-β1 or irradiation stimulation was clearly inhibited by VPA.
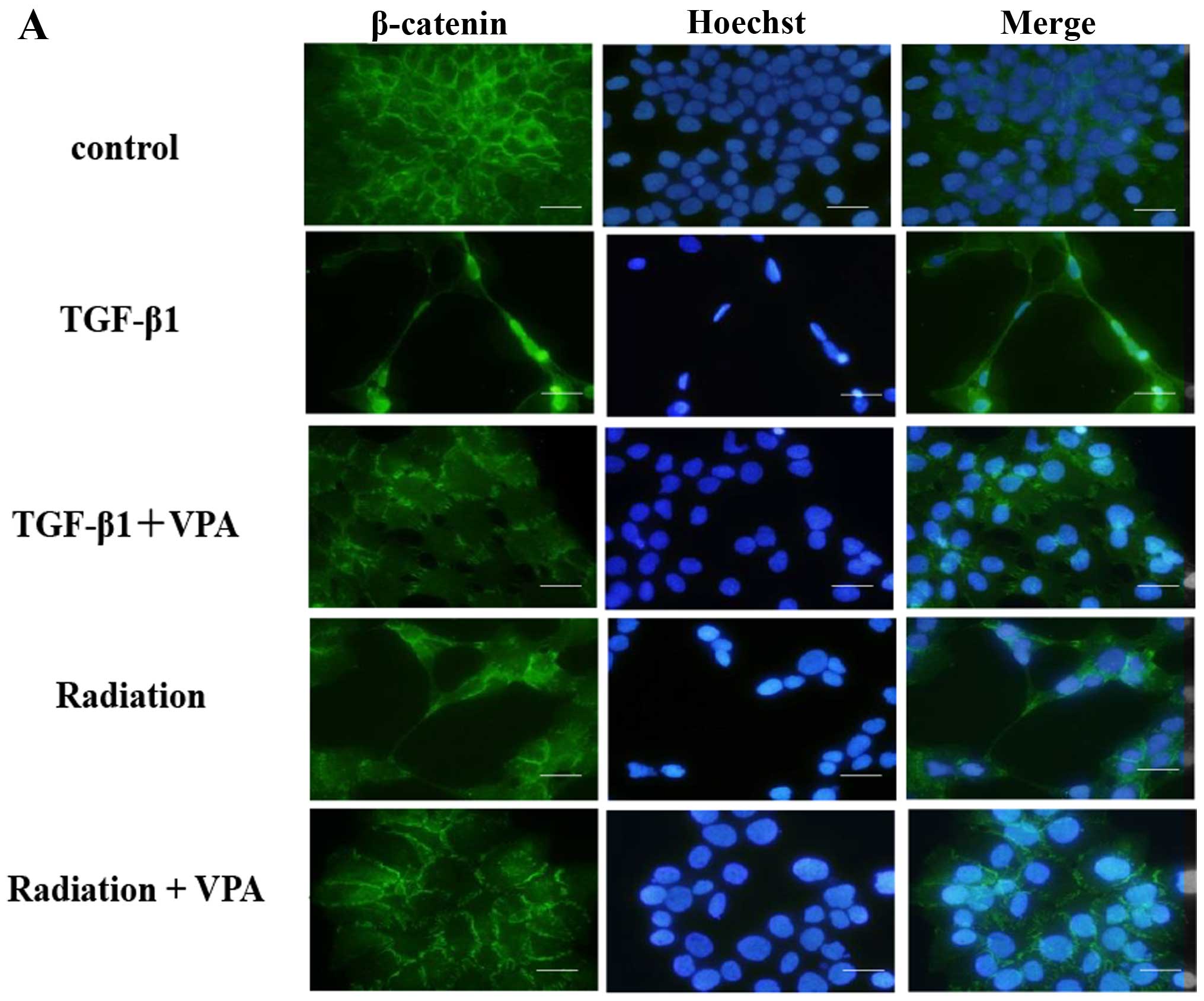
Effect of VPA on β-catenin translocation
promoted by TGF-β1 or irradiation in TE9 cells
One characteristic of EMT is breakdown of the
cytoplasmic cell adhesion complex. Breakdown of this complex causes
a change in the localization of β-catenin from its usual
membrane-bound location. Immunofluorescence studies showed that
β-catenin translocated to the nucleus or cytoplasm from its usual
membrane-bound location 24–48 h after treatment with TGF-β1 or
irradiation. Pretreatment with 1 mM VPA inhibited these changes in
the localization of β-catenin after TGF-β1 or irradiation
stimulation (Fig. 4A). In
contrast, the total amount of β-catenin protein assessed by western
blot analysis was not altered by stimulation with TGF-β1 or
irradiation or by pretreatment with VPA (Fig. 4B).
Effect of VPA on invasion and migration
promoted by TGF-β1 or irradiation stimulation in TE9 cells
To further understand the action of VPA on EMT, we
investigated the effect of VPA on the enhancement of the invasive
and migratory activity of TE9 cells by TGF-β1 or irradiation. Cells
treated with TGF-β1 or irradiation showed 1.55-fold (P=0.041) and
1.69-fold (P=0.038) enhanced invasion ability respectively compared
with the control. Pretreatment with 1 mM VPA decreased the
enhancement of TE9 cell invasiveness by TGF-β1 or irradiation
(0.65-fold, P<0.01 and 0.63-fold enhancement, P<0.05,
respectively) (Fig. 5A and B).
In the migration assay, TGF-β1- or
irradiation-treated TE9 cells migrated almost completely across the
leading edge after 48 h. TGF-β1 or irradiation significantly
increased TE9 cell migration ability (1.49-fold, P<0.001 and
1.31-fold, P=0.03, respectively). Treatment with 1 mM VPA
significantly suppressed the enhancement of TE9 cell migration
ability by TGF-β1 or irradiation stimulation (0.82-fold, P<0.01
and 0.86-fold, P<0.05, respectively) (Fig. 5C and D).
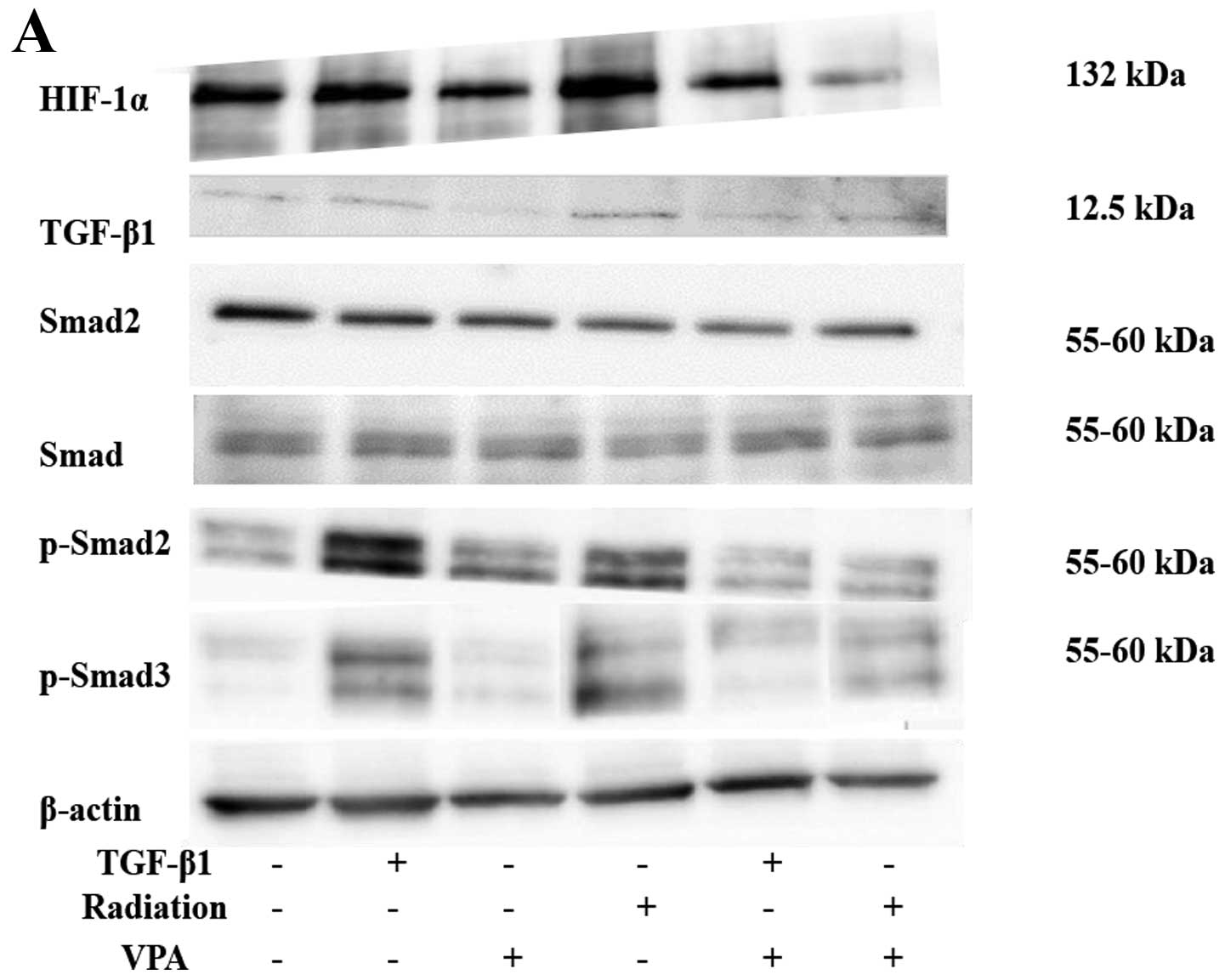
Effect of VPA on the expression of
EMT-related proteins and MMPs in TE9 cells
Interaction of TGF-β1 and TGF-β receptor type I
(TβR-I) leads to phosphorylation of Smad2 and Smad3, the key
mediators of TGF-β1 signaling. Therefore, we examined TGF-β1
expression and Smad2 and Smad3 phosphorylation after stimulation by
TGF-β1 or irradiation. Stimulation by TGF-β1 or irradiation
resulted in increased TGF-β1 expression. Consistent with this
finding, stimulation by TGF-β1 or irradiation also induced Smad2
and Smad3 phosphorylation. Administration of VPA (1 mM) abolished
both responses. Total Smad2 and Smad3 were abundant in control
cells, and expression levels were not affected by TGF-β1,
irradiation, or VPA treatment (Fig.
6). Exposure of serum-starved TE9 cells to TGF-β1 or
irradiation resulted in increased phosphorylation of Smad2 and
Smad3. Incubation with 1 mM VPA suppressed phosphorylation of Smad2
and Smad3 after TGF-β1 or irradiation stimulation, whereas
expression of total Smad2 and Smad3 was not affected (Fig. 6A), confirming that VPA can block
TGF-β1 signaling. We further delineated the link between TGF-β1- or
irradiation-induced HIF-1α expression and EMT progression by
showing that VPA (1 mM) suppressed HIF-1α expression. In addition,
TGF-β1 or irradiation strongly induced expression of the
gelatinases MMP-2, MMP-7, and MMP-9 whereas VPA strongly inhibited
the induction of these enzymes (Fig.
6B).
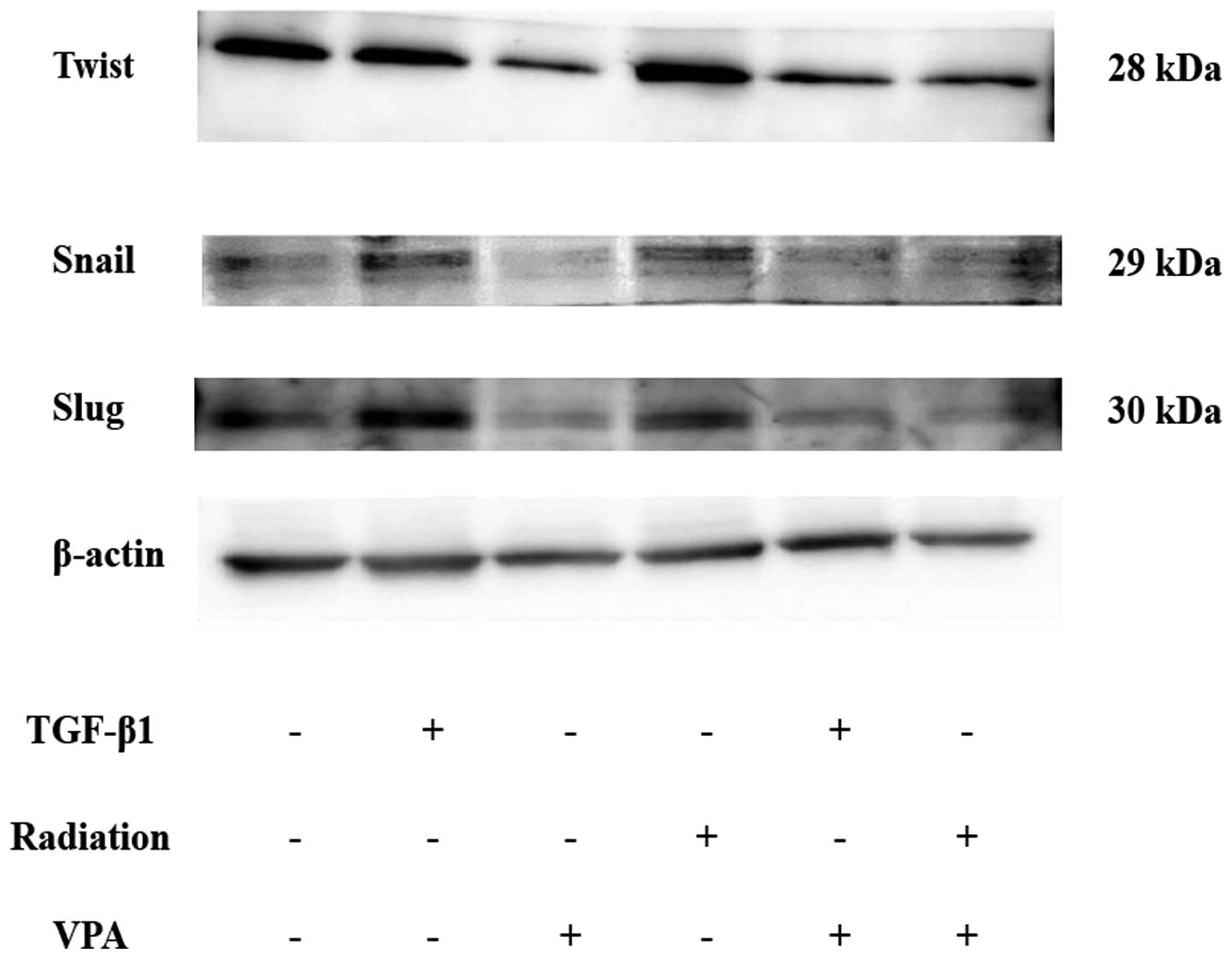
Effect of VPA on induction of
transcription factor expression by TGF-β1 or irradiation
stimulation in TE9 cells
Several key inducers of EMT are transcription
factors such as Twist, Snail, and Slug that repress E-cadherin
expression. Western blot analysis showed that TGF-β1 or irradiation
stimulation induced upregulation of Twist, Snail, and Slug
expression (Fig. 7). Pretreatment
with VPA resulted in inhibition of Twist, Snail, and Slug
expression.
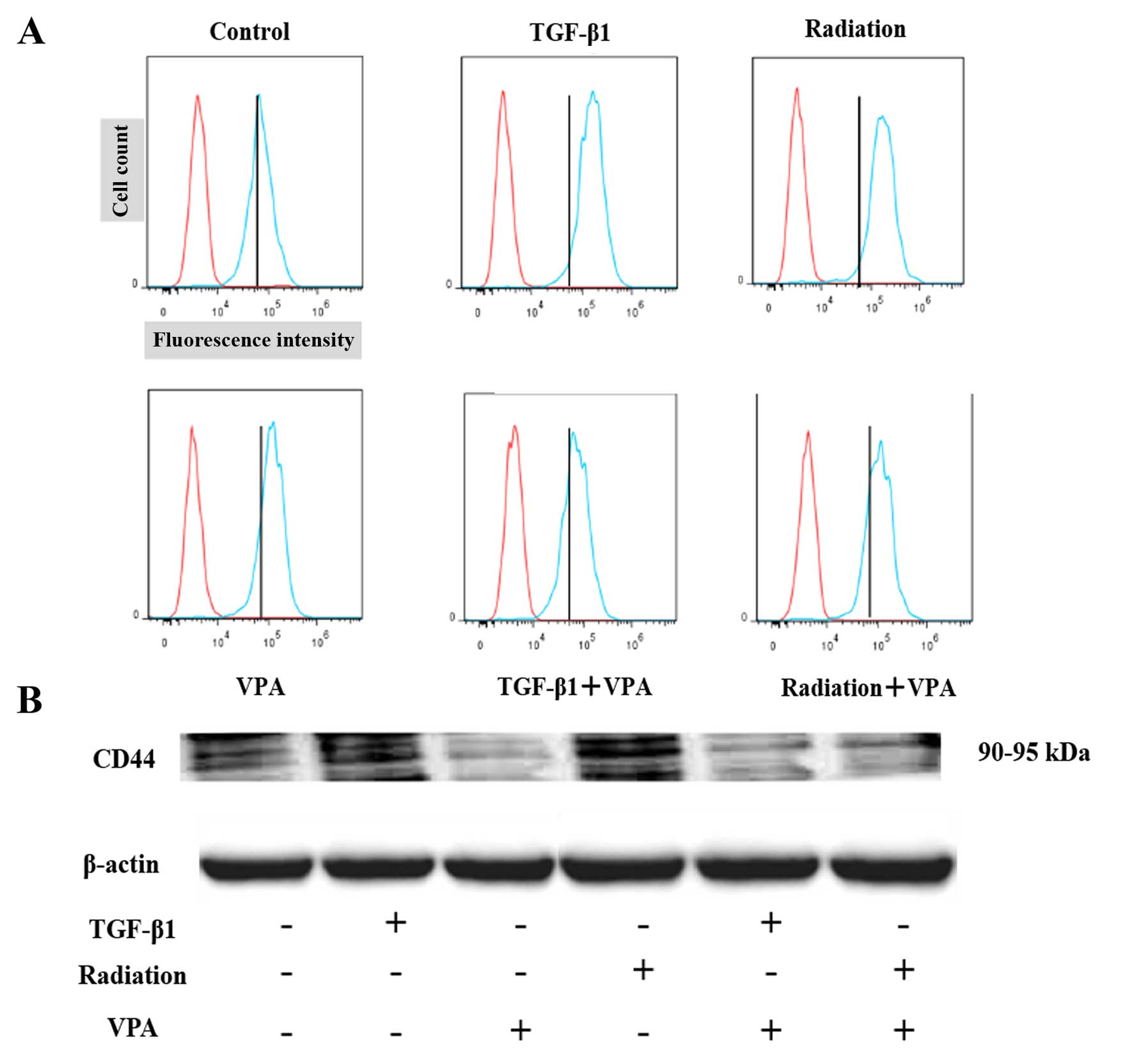
Effect of VPA on the induction of stem
cell markers by TGF-β1 or irradiation stimulation in TE9 cell
By western blotting, the level of total CD44 protein
was increased by TGF-β1 treatment or irradiation and was decreased
by VPA (Fig. 8A). Fluorescence
activated cell sorting (FACS) analysis also showed an increase in
CD44 levels in TGF-β1 or irradiation-treated TE9 cells that was
inhibited by VPA (Fig. 8B). These
results indicate that TGF-β1 or irradiation exposure can alter
cellular expression of markers associated with cancer stem-like
cell properties and that VPA inhibits transformation to a cancer
stem-like phenotype.
Discussion
In this study, we demonstrated that irradiation
induced morphologic and molecular alternations consistent with
acquisition of a mesenchymal-like phenotype in ESCC cells. As a
result of induction of EMT, ESCC cells attained invasive and
migratory potential. In addition, the HDAC inhibitor VPA inhibited
growth, invasion, migration, and the transformation to cancer
stem-like phenotype associated with TGF-β1 or irradiation
stimulation of ESCC cells.
Radiotherapy plays an important clinical role as the
major non-surgical treatment for esophageal cancer. However,
previous studies have reported that irradiation may enhance the
metastatic potential of residual cancer (6,18).
Local failure and distant metastasis are the primary causes of
radiotherapy failure. Therefore, tumor recurrence and metastasis
might be associated with tumor biological behavior, as well as with
EMT induced by irradiation. Recently, examination of a
radio-resistant ESCC cell line obtained after fractionated
radiation treatment showed that fractionated irradiation promoted
EMT (19). In this study, we
examined whether single-dose irradiation can induce EMT in TE9 ESCC
cells. We showed that 2 Gy of irradiation induced spindle cell-like
morphologic changes, decreased expression of membranous E-cadherin,
upregulated vimentin expression, and changed the localization of
β-catenin from its usual membrane-bound location to the cytoplasm.
Furthermore, we showed upregulation of transcription factors Slug,
Snail, and Twist, which regulate EMT (32). These findings indicate that
single-dose irradiation also induces EMT in ESCC cells. We also
found that 2 Gy of irradiation induced EMT in other ESCC cell lines
including TE10 (well differentiated type), TE11 (moderately
differentiated type), and KES (well differentiated type) (data not
shown).
One of the major pathways of EMT is the TGF-β1/Smad
pathway. Components of the TGF-β1/Smad signaling pathway are well
known as an important signal transducers for cell proliferation,
differentiation, and survival, and also serve as key factors in the
regulation of cancer cell invasion and metastasis. In mice bearing
tumor cells, irradiation causes increased circulating levels of
TGF-β1 as well as increased circulating tumor cells and lung
metastasis (17). We similarly
found that irradiation induced intracellular accumulation of TGF-β1
and increased phosphorylation of Smad2 and Smad3, key mediators of
TGF-β1 signaling. Although the molecular basis of a functional
interaction between hypoxia, HIF-1α, and TGF-β1 signaling is not
well understood at this point, the simplest explanation is that
TGF-β1 levels increase in response to hypoxia (33). In this study, irradiation increased
HIF-1α expression at the protein level, and irradiation-induced
HIF-1α further upregulated the expression of MMPs. HIF-1α is known
as the key regulator of the cellular response to hypoxia. Induced
expression of HIF-1α and its target genes plays a critical role in
cell growth, metastasis, and resistance to radiotherapy (33). It is noteworthy that irradiation
enhanced the invasion and migration ability of TE9 cells, possibly
through the upregulation of MMPs. Recent studies indicate that MMPs
can activate EMT (34). MMPs
cleave cell-ECM adhesion proteins and cell-cell junction proteins,
releasing individual epithelial cells from epithelial sheets and
initiating outside-in signaling pathways that lead to widespread
changes in gene transcription patterns (34). Upregulation of HIFs in cancer cells
may also occur in the hypoxic intra-tumoral regions formed within
primary and secondary neoplasms. Cancer cells stimulated by HIF-1α
show enhanced production of TGF-β1, thus the TGF-β1/Smad pathway is
activated and consequent production of transcription factors is
increased, leading to EMT. The expression of TGF-β1 is further
enhanced, stimulating expression of cancer stem cell markers such
as CD44 and CD133, which are considered to play a role in the
acquisition of cancer stem cell-like properties by cancer cells. We
showed that irradiation induced CD44 expression in TE9 cells. It
might therefore be speculated that TE9 cells obtained stem
cell-like characteristics after irradiation. It is likely that
cancer stem cells are responsible for initiation, progression,
recurrence, metastasis, and chemoradiotherapy resistance of cancer
(35). Therefore, fractionated
irradiation might induce radioresistance through the acquisition of
cancer stem cell-like characteristics. In this study, we
administered irradiation at a single dose of 2 Gy, a dose that
reduced TE9 cell survival to 30% in our previous study (27). Although 2 Gy of radiation killed
the majority of the ESCC cells, the cells that survived after
irradiation showed EMT and cancer stem cell-like characteristics
with increased invasive, migration, and radioresistant phenotypes.
These findings are consistent with recent reports that tumor cells
can gain cancer stem cell properties as a result of EMT (36), leading to a higher probability of
metastasis and radiation/drug resistance.
HDAC inhibitors are now considered to be promising
anti-cancer agents and some of these compounds, including VPA, are
near clinical stage or already on the market. Lei et al
(37) showed that HDAC1 is
required for TGF-β1-induced EMT and cell migration in hepatocytes.
Recently, several reports showed that HDAC inhibitor suppress EMT
in various cells, including cancer cells (31,38,39).
HDAC inhibitors suppress metastatic potential and reverse
chemoresistance in cancer cells through suppression of EMT
(30,31). Although the majority of reports
support a suppressive effect of HDAC inhibitors on EMT, two reports
show that the class I and II HDAC inhibitor VPA promotes EMT of
colorectal cancer cells (40,41).
Therefore, we investigated the inhibitory effect of VPA on
irradiation-induced EMT in ESCC cells. We found that VPA clearly
inhibited EMT induced by TGF-β1 or irradiation in these cells.
Thus, VPA inhibits EMT and acquisition of cancer stem cell-like
properties in ESCC cells. VPA might have a mutually exclusive
effect on EMT between squamous cell carcinoma and adenocarcinoma;
further studies are needed to confirm this apparent conflicting
action of VPA on EMT.
We have previously demonstrated the synergistic
effect of VPA on radiation therapy against ESCC (27). VPA enhances the radiosensitivity of
ESCC cells through chromatin decondensation with histone
hyperacetylation and increases the level of radiation-induced DNA
DSBs. VPA inhibits DNA DSB repair by homologous recombination
through the suppression of Rad51 and by non-homologous end joining
through the acetylation of Ku70 (27,28).
As shown in this study, VPA can also suppress radiation-induced
EMT, leading to inhibition of invasion and metastasis, and reducing
resistance to further chemoradiotherapy.
We performed the same experiments using
well-differentiated, moderately differentiated type, poorly
differentiated types of ESCC cell and found that the poorly
differentiated type showed the most remarkable changes in EMT (data
not shown). Clinically, this suggests that cases of poorly
differentiated squamous cell carcinoma are good candidates for
combined VPA treatment with radiation therapy.
EMT is classified into three types depending on its
biologic or pathologic role. Type 2 EMT is associated with
inflammation and fibrosis, and is becoming increasingly recognized
in adult pathologic conditions (42). Some reports show the effectiveness
of chemoradiotherapy as neoadjuvant treatment for esophageal
cancer. Surgery for esophageal cancer after radiotherapy is
sometimes complicated by fibrosis. Concomitant usage of VPA during
neoadjuvant radiotherapy might be useful to avoid fibrosis around
the tumor bed.
In conclusion, irradiation enhances the invasiveness
of ESCC, partially through morphologic and molecular changes such
as EMT and the induction of stem cells. Simultaneous use of the
HDAC inhibitor VPA and radiotherapy might be useful not only to
eradicate local disease, but also to control the systemic
dissemination or metastasis associated with radiotherapy. Our data
suggest that VPA might be an ideal therapeutic agent when combined
with radiation for the treatment of ESCC.
Acknowledgements
This study was supported by JSPS KAKENHI (grant no.
25461909).
Abbreviations:
|
ESCC
|
esophageal squamous cell carcinoma
|
|
EMT
|
epithelial mesenchymal transition
|
|
HDAC
|
histone deacetylase
|
|
DSB
|
double-strand break
|
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tachimori Y, Ozawa S, Numasaki H,
Fujishiro M, Matsubara H, Oyama T, Shinoda M, Toh Y, Udagawa H and
Uno T: Comprehensive registry of esophageal cancer in Japan, 2007.
Esophagus. 12:101–129. 2015. View Article : Google Scholar
|
|
3
|
Osugi H, Takemura M, Higashino M, Takada
N, Lee S, Ueno M, Tanaka Y, Fukuhara K, Hashimoto Y, Fujiwara Y, et
al: Causes of death and pattern of recurrence after esophagectomy
and extended lymphadenectomy for squamous cell carcinoma of the
thoracic esophagus. Oncol Rep. 10:81–87. 2003.
|
|
4
|
Ando N, Ozawa S, Kitagawa Y, Shinozawa Y
and Kitajima M: Improvement in the results of surgical treatment of
advanced squamous esophageal carcinoma during 15 consecutive years.
Ann Surg. 232:225–232. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sofia Vala I, Martins LR, Imaizumi N,
Nunes RJ, Rino J, Kuonen F, Carvalho LM, Rüegg C, Grillo IM, Barata
JT, et al: Low doses of ionizing radiation promote tumor growth and
metastasis by enhancing angiogenesis. PLoS One. 5:e112222010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Park JK, Jang SJ, Kang SW, Park S, Hwang
SG, Kim WJ, Kang JH and Um HD: Establishment of animal model for
the analysis of cancer cell metastasis during radiotherapy. Radiat
Oncol. 7:1532012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Moncharmont C, Levy A, Guy JB, Falk AT,
Guilbert M, Trone JC, Alphonse G, Gilormini M, Ardail D, Toillon
RA, et al: Radiation-enhanced cell migration/invasion process: A
review. Crit Rev Oncol Hematol. 92:133–142. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Haslehurst AM, Koti M, Dharsee M, Nuin P,
Evans K, Geraci J, Childs T, Chen J, Li J, Weberpals J, et al: EMT
transcription factors snail and slug directly contribute to
cisplatin resistance in ovarian cancer. BMC Cancer. 12:912012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wintzell M, Löfstedt L, Johansson J,
Pedersen AB, Fuxe J and Shoshan M: Repeated cisplatin treatment can
lead to a multiresistant tumor cell population with stem cell
features and sensitivity to 3-bromopyruvate. Cancer Biol Ther.
13:1454–1462. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelial-mesenchymal transition: New insights in
signaling, development, and disease. J Cell Biol. 172:973–981.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee MY and Shen MR: Epithelial-mesenchymal
transition in cervical carcinoma. Am J Transl Res. 4:1–13.
2012.PubMed/NCBI
|
|
12
|
Barcellos-Hoff MH and Akhurst RJ:
Transforming growth factor-beta in breast cancer: Too much, too
late. Breast Cancer Res. 11:2022009. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Morrison CD, Parvani JG and Schiemann WP:
The relevance of the TGF-β Paradox to EMT-MET programs. Cancer
Lett. 341:30–40. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nagaraj NS and Datta PK: Targeting the
transforming growth factor-beta signaling pathway in human cancer.
Expert Opin Investig Drugs. 19:77–91. 2010. View Article : Google Scholar
|
|
15
|
Wang M, Hada M, Saha J, Sridharan DM,
Pluth JM and Cucinotta FA: Protons sensitize epithelial cells to
mesenchymal transition. PLoS One. 7:e412492012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu MA, Kiang A, Wang-Rodriguez J, Rahimy
E, Haas M, Yu V, Ellies LG, Chen J, Fan JB, Brumund KT, et al:
Nicotine promotes acquisition of stem cell and
epithelial-to-mesenchymal properties in head and neck squamous cell
carcinoma. PLoS One. 7:e519672012. View Article : Google Scholar
|
|
17
|
Biswas S, Guix M, Rinehart C, Dugger TC,
Chytil A, Moses HL, Freeman ML and Arteaga CL: Inhibition of
TGF-beta with neutralizing antibodies prevents radiation-induced
acceleration of metastatic cancer progression. J Clin Invest.
117:1305–1313. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tsukamoto H, Shibata K, Kajiyama H,
Terauchi M, Nawa A and Kikkawa F: Irradiation-induced
epithelial-mesenchymal transition (EMT) related to invasive
potential in endometrial carcinoma cells. Gynecol Oncol.
107:500–504. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
He E, Pan F, Li G and Li J: Fractionated
ionizing radiation promotes epithelial-mesenchymal transition in
human esophageal cancer cells through PTEN deficiency-mediated Akt
activation. PLoS One. 10:e01261492015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ocker M: Deacetylase inhibitors - focus on
non-histone targets and effects. World J Biol Chem. 1:55–61. 2010.
View Article : Google Scholar
|
|
21
|
Singh TR, Shankar S and Srivastava RK:
HDAC inhibitors enhance the apoptosis-inducing potential of TRAIL
in breast carcinoma. Oncogene. 24:4609–4623. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shankar S, Davis R, Singh KP, Kurzrock R,
Ross DD and Srivastava RK: Suberoylanilide hydroxamic acid
(Zolinza/vorinostat) sensitizes TRAIL-resistant breast cancer cells
ortho-topically implanted in BALB/c nude mice. Mol Cancer Ther.
8:1596–1605. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Marks PA and Xu WS: Histone deacetylase
inhibitors: Potential in cancer therapy. J Cell Biochem.
107:600–608. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Duenas-Gonzalez A, Candelaria M,
Perez-Plascencia C, Perez-Cardenas E, de la Cruz-Hernandez E and
Herrera LA: Valproic acid as epigenetic cancer drug: Preclinical,
clinical and transcriptional effects on solid tumors. Cancer Treat
Rev. 34:206–222. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Atmaca A, Al-Batran SE, Maurer A, Neumann
A, Heinzel T, Hentsch B, Schwarz SE, Hövelmann S, Göttlicher M,
Knuth A, et al: Valproic acid (VPA) in patients with refractory
advanced cancer: A dose escalating phase I clinical trial. Br J
Cancer. 97:177–182. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mohammed TA, Holen KD, Jaskula-Sztul R,
Mulkerin D, Lubner SJ, Schelman WR, Eickhoff J, Chen H and Loconte
NK: A pilot phase II study of valproic acid for treatment of
low-grade neuroendocrine carcinoma. Oncologist. 16:835–843. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shoji M, Ninomiya I, Makino I, Kinoshita
J, Nakamura K, Oyama K, Nakagawara H, Fujita H, Tajima H, Takamura
H, et al: Valproic acid, a histone deacetylase inhibitor, enhances
radiosensitivity in esophageal squamous cell carcinoma. Int J
Oncol. 40:2140–2146. 2012.PubMed/NCBI
|
|
28
|
Makita N, Ninomiya I, Tsukada T, Okamoto
K, Harada S, Nakanuma S, Sakai S, Makino I, Kinoshita J, Hayashi H,
et al: Inhibitory effects of valproic acid in DNA double-strand
break repair after irradiation in esophageal squamous carcinoma
cells. Oncol Rep. 34:1185–1192. 2015.PubMed/NCBI
|
|
29
|
Watanabe T, Tajima H, Hironori H,
Nakagawara H, Ohnishi I, Takamura H, Ninomiya I, Kitagawa H,
Fushida S, Tani T, et al: Sodium valproate blocks the transforming
growth factor (TGF)-β1 autocrine loop and attenuates the
TGF-β1-induced collagen synthesis in a human hepatic stellate cell
line. Int J Mol Med. 28:919–925. 2011.PubMed/NCBI
|
|
30
|
Ruscetti M, Dadashian EL, Guo W, Quach B,
Mulholland DJ, Park JW, Tran LM, Kobayashi N, Bianchi-Frias D, Xing
Y, et al: HDAC inhibition impedes epithelial-mesenchymal plasticity
and suppresses metastatic, castration-resistant prostate cancer.
Oncogene. 35:3781–3795. 2016. View Article : Google Scholar :
|
|
31
|
Sakamoto T, Kobayashi S, Yamada D, Nagano
H, Tomokuni A, Tomimaru Y, Noda T, Gotoh K, Asaoka T, Wada H, et
al: A Histone deacetylase inhibitor suppresses
epithelial-mesenchymal transition and attenuates chemoresistance in
biliary tract cancer. PLoS One. 11:e01459852016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Paoli P, Giannoni E and Chiarugi P:
Anoikis molecular pathways and its role in cancer progression.
Biochim Biophys Acta. 1833.3481–3498. 2013.
|
|
34
|
Radisky ES and Radisky DC: Matrix
metalloproteinase-induced epithelial-mesenchymal transition in
breast cancer. J Mammary Gland Biol Neoplasia. 15:201–212. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schatton T and Frank MH: Cancer stem cells
and human malignant melanoma. Pigment Cell Melanoma Res. 21:39–55.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Castellanos JA, Merchant NB and
Nagathihalli NS: Emerging targets in pancreatic cancer:
Epithelial-mesenchymal transition and cancer stem cells. Onco
Targets Ther. 6:1261–1267. 2013.PubMed/NCBI
|
|
37
|
Lei W, Zhang K, Pan X, Hu Y, Wang D, Yuan
X, Shu G and Song J: Histone deacetylase 1 is required for
transforming growth factor-beta1-induced epithelial-mesenchymal
transition. Int J Biochem Cell Biol. 42:1489–1497. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bruzzese F, Leone A, Rocco M, Carbone C,
Piro G, Caraglia M, Di Gennaro E and Budillon A: HDAC inhibitor
vorinostat enhances the antitumor effect of gefitinib in squamous
cell carcinoma of head and neck by modulating ErbB receptor
expression and reverting EMT. J Cell Physiol. 226:2378–2390. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mateen S, Raina K, Agarwal C, Chan D and
Agarwal R: Silibinin synergizes with histone deacetylase and DNA
methyltransferase inhibitors in upregulating E-cadherin expression
together with inhibition of migration and invasion of human
non-small cell lung cancer cells. J Pharmacol Exp Ther.
345:206–214. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Feng J, Cen J, Li J, Zhao R, Zhu C, Wang
Z, Xie J and Tang W: Histone deacetylase inhibitor valproic acid
(VPA) promotes the epithelial mesenchymal transition of colorectal
cancer cells via up regulation of Snail. Cell Adhes Migr.
9:495–501. 2015. View Article : Google Scholar
|
|
41
|
Ji M, Lee EJ, Kim KB, Kim Y, Sung R, Lee
SJ, Kim DS and Park SM: HDAC inhibitors induce
epithelial-mesenchymal transition in colon carcinoma cells. Oncol
Rep. 33:2299–2308. 2015.PubMed/NCBI
|
|
42
|
Kovacic JC, Mercader N, Torres M, Boehm M
and Fuster V: Epithelial-to-mesenchymal and
endothelial-to-mesenchymal transition: From cardiovascular
development to disease. Circulation. 125:1795–1808. 2012.
View Article : Google Scholar : PubMed/NCBI
|