1. Telomeres and telomerase
Each round of DNA replication results in the
shortening of DNA strands due to the inability of the replication
complex to completely replicate the lagging strand. This process in
turn eventually results in genomic instability and loss of genetic
information after multiple rounds of replication. To solve this
end-replication problem, chromosome ends are capped by telomeres,
which consist of six nucleotide repeats and specialized binding
proteins that buffer replication losses (1). The repeats can be regenerated by the
telomerase enzyme complex, which consists of a non-coding RNA, hTR,
serving as the hexamer repeat template, and the catalytic subunit,
reverse transcriptase (hTERT). By counteracting the telomere
shortening incurred by the DNA replication process, the telomerase
complex lengthens telomeres, thereby prolonging cell survival and
allowing continued proliferation (2).
2. Telomerase regulation
Telomerase activity is low to absent in somatic
cells, though highly expressed in embryonic and stem cells.
Telomerase is also upregulated in cancer, as over 90% of human
malignancies show telomerase expression, considered an early event
in cancer progression (3,4). In cancer, the main mechanism of
telomerase activation is through regulation of hTERT transcription,
through genetic changes such as mutations altering transcription
factor binding, by epigenetic changes such as histone modification
and chromatin remodeling or promoter methylation, and by
alternative splicing of the transcript (2,5–7). As
explored below, a connection between the promoter methylation and
the alternative splicing is emerging in the field as a means by
which cancer cells turn on hTERT expression, resulting both in
active telomerase and telomere elongation.
3. Gene regulation by methylation
In higher eukaryotes, DNA methylation at CpG sites
in and around gene promoter regions controls and regulates gene
expression. CpG methylation is directed by DNA methyltransferases
(DNMTs) which methylate the fifth carbon of the pyrimidine ring of
cytosine (8). The principal
methyltransferase, DNMT1, adds methyl groups during DNA replication
and de novo methylates DNA in cancer (9). Many CpG sites are clustered into CpG
islands that are typically 1,000 base pairs long and have high GC
content. Approximately 80% of CpG sites are methylated in mammals,
largely in intergenic regions known as heterochromatin, while most
sites in promoters and first exons remain unmethylated (10,11).
The promoter and transcription start site (TSS) tends to be
unmethylated in actively transcribed genes, since methylated DNA is
associated with gene silencing through both interference of
transcription factor binding and, by affecting chromatin
architecture (12). Importantly,
aberrant DNA methylation is a hallmark of cancer cells, and tends
to occur early in cancer development (13). In cancer, characteristic changes in
methylation patterns involve both genome-wide CpG hypomethylation,
which occurs predominantly in intergenic regions, and
hypermethylation of CpG islands at promoters. Promoter
hypermethylation may result in silencing of tumor suppressors, and
promoter hypomethylation can result in activation of
proto-oncogenes (14). Intergenic
hypomethylation may also lead to expression of dormant non-coding
RNA species and otherwise suppressed genetic elements transcribed
from normally silent regions of the genome (15–17).
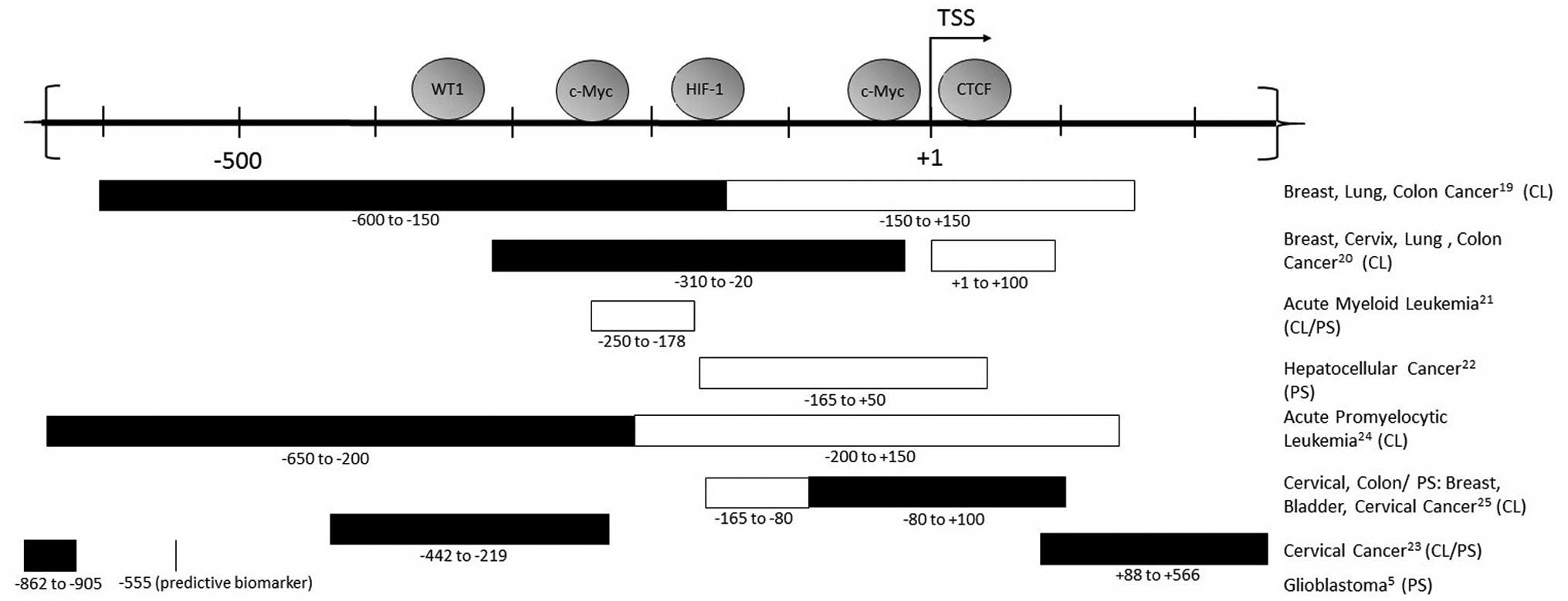
4. hTERT promoter methylation in cancer
The hTERT promoter is located in a 4 kb CpG island
−1800 to +2200 (relative to TSS), and has a GC content of 70%
(18). The precise pattern of
promoter methylation that results in activation of hTERT in cancer
is still under investigation. However, a methylation pattern does
emerge from many studies of the promoter, including extensive
bisulfite sequencing of telomerase positive cancer cell lines. The
promoter region of the actively transcribed hTERT is demethylated
at the TSS [−200 to +100], while the promoter region further
upstream of the TSS [−650 to −200] is hypermethylated (19). Studies examining specific sections
of the hTERT promoter corroborate this in a myriad of cancer cell
lines as well as hematological malignancies and solid tumor types,
as depicted in Fig. 1 (5,19–26).
Its methylation status is also considered a biomarker; in pediatric
brain tumors, one methylated CpG site in the promoter (cg11625005)
is used as a reliable marker for tumor progression and prognosis
(5).
5. Transcription factor regulation by
methylation of hTERT promoter
It is known that the hTERT methylation pattern plays
a major role in transcription factor binding, which in turn alters
the expression of hTERT. This is demonstrated by experimental
evidence pointing to a ‘minimal promoter’ corresponding to
transcription factor binding sites, e.g. c-Myc for hTERT at −258 to
−78, that must be unmethylated in order for hTERT expression to
occur (27). Methylation also
plays a significant role further upstream in the hTERT promoter
where many repressor binding sites are located. These sites are
hypermethylated in cancer to prevent binding of repressors such as
the Wilms’ tumor protein (WT1) and the transcriptional repressor
CCCTC-binding factor (CTCF) that binds to CpG rich regions at the
TSS (21,25,27–29).
Perturbing the methylation status of the promoter with
5aza-2′-deoxycytidine (DAC), which globally reduces DNA methylation
by DNMT inhibition, results in reduced levels of hTERT
transcription, which may be due to demethylation of the repressor
binding sites (30).
c-Myc
Hypomethylation at the minimal promoter of hTERT
allows c-Myc, a key positive regulator of hTERT expression, to bind
242 bases upstream of the TSS at the E-box. Studies have shown that
c-Myc binding is methylation-sensitive and that binding is greatly
reduced or absent when the site is methylated (31). This control of hTERT expression by
E-box methylation is also seen in human embryonic teratocarcinoma
cells. The undifferentiated embryonic cells have higher expression
of hTERT and telomerase activity with hypomethylation of the
minimal promoter. Conversely, during the process of differentiation
of these cells there is a significant increase in methylation at
the E-box, resulting in inactivation of hTERT expression. This mode
of repression of hTERT can be reversed by treatment of late
differentiating cells with DAC, highlighting the importance of
c-Myc binding and regulation by methylation (27).
WT1
In contrast to the minimal promoter being
hypomethylated to allow c-Myc binding, the promoter region further
upstream, corresponding to the binding sites of multiple
repressors, is hypermethylated in cancer. For example, the
repressor WT1, with its binding site from −358 to −349, is known to
suppress hTERT transcription (27). This is supported by a study in
clear cell renal cell carcinoma, in which WT1 is overexpressed, and
direct binding of WT1 to the promoter results in repression of
hTERT (32). WT1 binding is also
known to be methylation sensitive, with binding interference assays
showing reduced binding when one or more methylated bases are
present in the binding sequence (33). The WT1 binding site of hTERT
exhibits increased CpG methylation in cancer, resulting in blocking
the repressive effects of the factor for hTERT expression (20,34).
CTCF
Similar to WT1, as a repressor of hTERT, CTCF binds
adjacent to the transcription start site, near the beginning of
exon one (+4 to +39), and near the beginning of exon two (+422 to
+440). Studies have shown that CTCF binding at the first site
represses hTERT transcription and that this binding is blocked in
cancer (35). Blocking is
established by methylation as CTCF’s binding affinity is inversely
correlated with the degree of methylation; CTCF is unable to bind
fully methylated DNA. Cancer cells have aberrant methylation in the
first CTCF binding site, typified by HPV-transformed cervical
cancer cells showing increased methylation and activated hTERT
expression (23). Conversely, in
colon cancer cell lines, downregulating DNTM1, and the resultant
demethylation of the CTCF binding site, causes increased CTCF
binding and repression of hTERT (36,37).
Furthermore, in breast cancer cells, hTERT transcript levels
increase with a concomitant decrease in cellular apoptosis when
CTCF is downregulated by siRNA (38).
6. Role of transcription factor binding on
splicing
While it is known that transcription factors binding
to the promoter regulate gene expression, it is becoming evident
that such binding also affects splicing. Proteins with a cis
regulatory role in both transcription and splicing have been
demonstrated in multiple genes. The regulation is believed to act
through the kinetic coupling model, where the rate of transcription
elongation affects alternative splicing (39,40).
Experiments assessing alternative splicing in a promoter swapping
system have established promoter specificity in controlling
alternative splicing (41). This
splicing role is consistent with splicing occurring
co-transcriptionally as splicing factor assembly arises during
transcription. As detailed below, manipulation of splicing through
transcription factors can be accomplished either by the factor
directly influencing the spliceosome or, by recruitment of
additional factors (42,43).
Direct splicing role of WT1 with U2AF and
RBM4
WT1, a negative regulator of hTERT, promotes
splicing of multiple transcripts by both interacting directly with
several splicing factors and through incorporation into
spliceosomes. In the nucleus, the regulator co-localizes with the
splicing machinery, specifically with U2AF and RBM4. The U2AF
heterodimer is essential for binding upstream of the splice site
and helps the U2 snRNA anneal at the branch point. Interaction with
WT1 affects U2AF binding and therefore splice site selection
(44). Also in splice site
selection, RBM4 influences alternative splicing during selection of
the 5′ exon splice site, and interacts with WT1 in nuclear
speckles, thought to be compartments for spliceosome assembly
(45). Recruitment of these
splicing factors by WT1 highlights the link between the
transcription factor binding and spliceosome assembly.
Indirect splicing role of WT1 with
SRPK1
While WT1 interacts with the splicing machinery as
discussed above, it also plays an indirect role in splicing by
transcriptionally repressing SRPK1, a splicing factor kinase. As
shown in acute myeloid leukemia, where it is often overexpressed,
WT1 causes alternative exon usage events commonly seen in the
disease (46). Conversely, when
WT1 is knocked down in hematopoietic progenitor cells, vascular
endothelial growth factor (VEGF) exhibits an atypical splicing
pattern (47). In studying the
relationship between WT1 and VEGF, an important growth factor in
cancer, loss of WT1 results in increased abundance of isoform
VEGF-a120, while transfection of WT1 results in exon inclusion and
loss of the 120 isoform. This demonstrates that WT1 is essential
for controlling the splicing of VEGF-a (48). Studies of the VEGF-b isoform have
also shown alteration of splicing patterns with WT1 manipulation.
Transfection of various cell lines with WT1 results in an increase
of VEGF165b. Furthermore, inclusion of the WT1 KTS sequence, which
plays a role in RNA binding, shows no effect on splicing, while a
single nucleotide change that alters DNA binding of WT1 changes the
splicing of VEGF. Thus, splicing correlates with the ability of the
transcription factor WT1 to stably bind DNA, rather than WT1 acting
post-transcriptionally (49).
Splicing role of HIF-1 and hypoxia on
hTERT
Hypoxia is known to enhance hTERT expression through
activation of the hTERT promoter in stem cells as well as in cancer
(50,51). In hypoxic tumor conditions, HIF-1
is overexpressed and activates the hTERT promoter by binding at two
sites between −165 and +51. If the HIF-1 binding sites are mutated,
hTERT promoter activity decreases even in normoxic conditions and
under hypoxic conditions, the HIF-1 induced overexpression of hTERT
is lost. It has been shown that in hypoxia, cell survival is
increased through maintenance of an undifferentiated state, and
this correlates with increased hTERT expression and telomerase
activity compared to normoxic conditions (51,52).
Furthermore, the splicing pattern of hTERT is quite variable at
differing levels of oxygenation. Overall, the hTERT splicing
pattern is altered in hypoxia, with an increased expression of the
full length active form, while the β deletion and α/β double
deletion splice variants remain unchanged. Additionally, blocking α
and β splicing results in spontaneous human embryonic stem cell
differentiation (50). In hypoxia,
HIF-1 increases the association of RNA Pol II with transcription
initiation factors, resulting in more effective and efficient
transcription. It has been proposed that modulation of hTERT
transcription by HIF-1 in hypoxia controls splicing by affecting
Pol II rates of transcription and enhancing expression of the
active full length transcript (53).
Direct splicing role of CTCF intragenic
binding with Pol II pausing
The direct role of CTCF modulating alternative
splicing in a co-transcriptional manner has recently emerged.
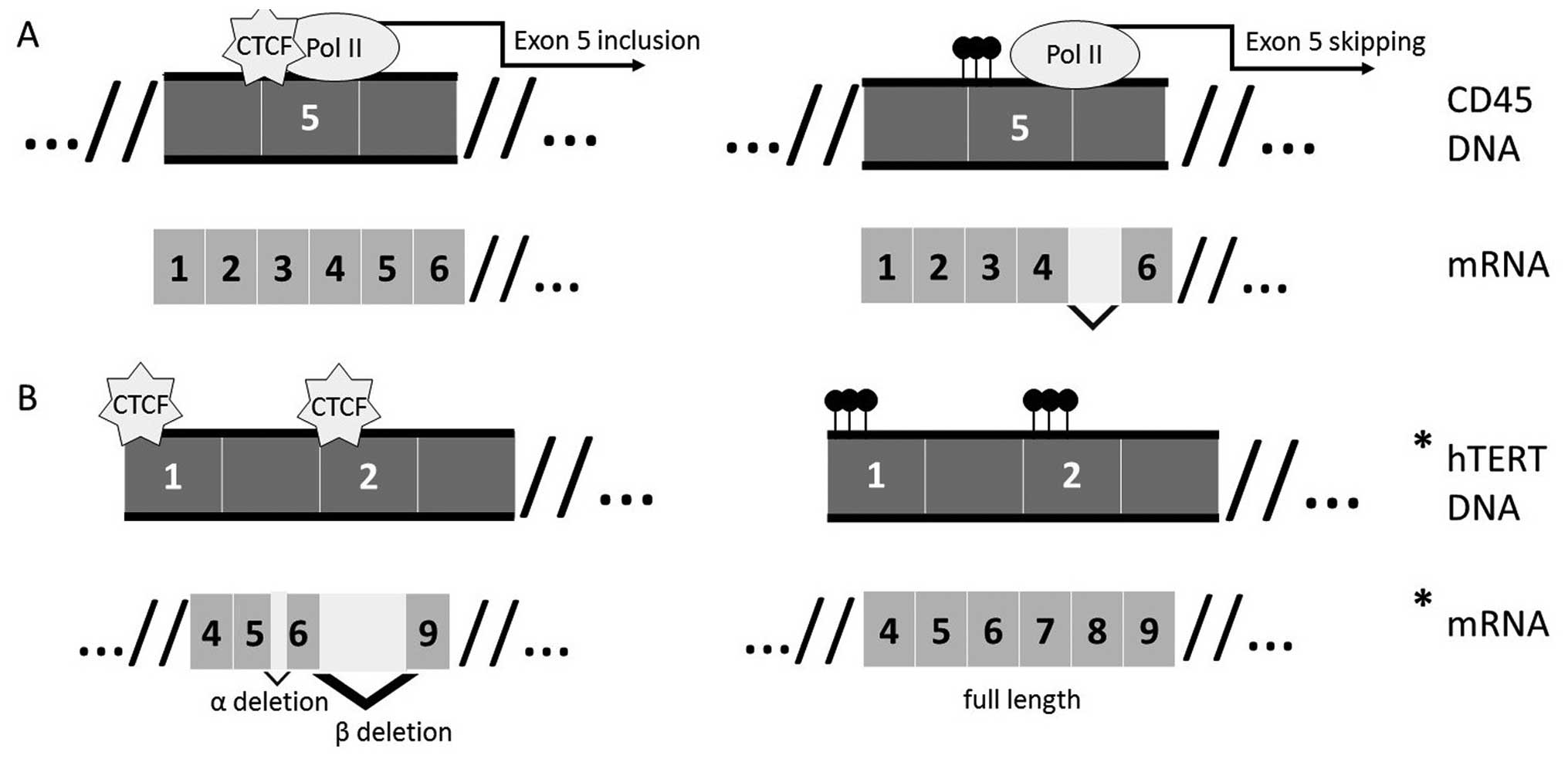
Shukla and colleagues (56) showed
that CTCF binding at actively transcribed DNA causes RNA polymerase
II pausing, resulting in recognition of weak splice site signals
and therefore inclusion of ‘weak’ exons in CD45. They also
demonstrated that the pausing was methylation dependent; when CTCF
cannot bind the methylated site, weak exons are not included in the
final spliced transcript (Fig. 2A)
(55,56). Multiple additional genes have been
shown to have promoter-proximal CTCF binding (100 bases downstream
of the TSS) that results in longer Pol II pausing compared to genes
without promoter-proximal binding sites. This effect is dependent
on the binding site position relative to the TSS, with less pausing
associated with sites at greater distance from the TSS (57). RNA polymerase II pausing due to
CTCF results in decreased processivity and contributes to promoter
proximal pausing, thereby directly modulating the dynamics of
transcription and splicing (57,58).
Indirect splicing role of CTCF by
facilitation of long range interactions
CTCF is well known for its architectural role in
establishing boundaries based on topologically associating domains
(TADs) that link distant enhancers with promoters and other
regulatory sequences. This chromatin looping by CTCF allows for
architectural reorganization resulting in co-location of factors to
influence transcription. Indeed, approximately 15% of CTCF binding
sites are located in promoters, while over 40% of CTCF binding
sites are located in the 5′UTR, introns, and other intragenic
regions. For example, the murine Myb locus has a CTCF binding site
in the first intron, which loops to the promoter during
differentiation, and a second CTCF site in a regulatory element
further upstream. This CTCF interaction allows for juxtaposition of
necessary transcription factors to regulate Pol II and modulate
expression of Myb. After differentiation, the expression of the
gene is halted due to CTCF architecture reorganization. This
highlights the ability of CTCF to regulate initiation of
transcription as well as elongation and Pol II pausing during
transcription by looping together promoter, upstream enhancer
elements and intronic sequences. Influencing the elongation of Pol
II, as well as the juxtaposition of regulatory factors, are known
to be important for splicing, as discussed above regarding the
elongation rate of Pol II and CD45 splicing (59,60).
7. Telomerase regulation by alternative
splicing in hTERT
While in many genes transcription factor binding has
been shown to play a role in splicing, the mechanism of hTERT
splicing is not as clearly defined. hTERT splicing plays a crucial
role in dictating the activity of telomerase, since only the
full-length transcript is catalytically active in the telomerase
ribonucleoprotein complex. Furthermore, many cancers show changes
in hTERT splicing patterns.
hTERT splicing switch in cancer
Modulation of alternative splicing is detected in
multiple types of cancers, where cancer cells utilize an
alternative splicing switch that results in discernible isoform
signatures (61). This switching
is nonrandom and also seen in tissue development, including the
hTERT splice switch, well characterized in kidney development and
various cancer cell types (62–74).
This mode of hTERT regulation has been proposed to be necessary due
to the difficulty to completely cease transcription, since even
very small amounts of transcript may have significant cellular
effects, given the limited number of cellular targets, i.e., 92
telomeres in the normal human cell (2). Cancer cells with active telomerase
have an average of 20 hTERT transcripts per cell resulting in
100–500 active telomerase complexes (75). Alternative splicing yielding
inactive and/or inhibitory forms of hTERT allows for downregulation
of telomerase activity without complete repression of
transcription.
Mechanism of hTERT alternative
splicing
Studies on the mechanisms of alternative splicing of
hTERT so far have revealed regulation primarily by long range
interactions, not at nearby splice sites as seen in other genes.
One proposed mechanism is via variable number tandem repeats
(VNTR), that are located in intronic regions over 1 kb from the
splice site, regulating splicing of the β deletion form. The
mechanism of VNTR control over splicing is not understood, but
could involve recruitment of RNA binding factors to the repeats.
These RNA binding factors would then interact and form a specific
landscape with other proteins conducive to spliceosome recruitment
(2,75). The mechanism for the dependence on
VNTR for splicing of the β deletion needs further examination, as
do additional splicing mechanisms for the other isoforms.
Over twenty different isoforms of hTERT have so far
been reported, with the most common being various deletions in the
reverse transcriptase domain, such as the α deletion, β deletion
and α/β deletion shown in Fig. 2B
(76). Other important isoforms
have been identified, such as intron 2 and 14 retention in lung and
colon cancer as well as exclusion of exon 2 in normal cells
(77,78). All known isoforms result in an
inactive telomerase complex. For example, the α deletion is a
dominant negative variant while the β deletion results in a
truncated protein targeted for nonsense-mediated decay (75). While normal cells express mainly
inactive hTERT isoforms, a splicing switch occurs in cancer cells,
resulting in production of the full length active transcript
(79). The cause of the splicing
switch is unknown, but is likely due to changes of binding factors,
possibly similar to changes to long range interactions through VNTR
in the case of the β deletion or, as stated above, from the ability
of certain transcription factors to bind and affect transcription
elongation rates.
8. Concluding remarks
Human telomerase reverse transcriptase expression
has many facets of regulation, including promoter methylation and
alternative splicing, as outlined above. These two methods of
regulation can become intertwined by the co-transcriptional nature
of mammalian pre-mRNA splicing. This allows transcription factors
bound to the gene, including the promoter, to influence alternative
splicing of the transcript. Understanding the regulation of hTERT
is crucial to understanding telomerase activity in normal cells as
well as in cancer. Elucidating the mechanism of cellular control of
hTERT transcription will further our knowledge of the intricate
instructions directing activation of this essential genome
maintenance machinery.
Study of hTERT transcription is a unique opportunity
to understand the control of a low abundance transcript with a
prominent role in cancer. Insight into how cancer commandeers the
regulatory control, be it through methylation or through the
splicing machinery, to produce full length hTERT and resulting in
active telomerase would be highly beneficial clinically. Currently,
therapeutic telomerase inhibitors are not well tolerated by
patients. The therapy must be continually administered for multiple
replication cycles to have an effect, resulting in the need for
extended periods of treatment, which can be challenging due to
significant drug toxicity. Promising anticancer therapeutics such
as imetelstat, an antisense oligonucleotide binding to hTR, target
the active telomerase complex, or consist of small molecule
inhibitors inhibiting active TERT binding to the RNA (80). Alternatively, therapies inhibiting
TERT from being activated before the telomerase complex forms, by
altered expression or splicing, could provide a new mechanistic
avenue for effective anticancer therapeutics.
References
|
1
|
Lü MH, Liao ZL, Zhao XY, Fan YH, Lin XL,
Fang DC, Guo H and Yang SM: hTERT-based therapy: A universal
anticancer approach (Review). Oncol Rep. 28:1945–1952.
2012.PubMed/NCBI
|
|
2
|
Wong MS, Chen L, Foster C, Kainthla R,
Shay JW and Wright WE: Regulation of telomerase alternative
splicing: A target for chemotherapy. Cell Rep. 3:1028–1035. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Koziel JE, Fox MJ, Steding CE, Sprouse AA
and Herbert BS: Medical genetics and epigenetics of telomerase. J
Cell Mol Med. 15:457–467. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Umbricht CB, Sherman ME, Dome J, Carey LA,
Marks J, Kim N and Sukumar S: Telomerase activity in ductal
carcinoma in situ and invasive breast cancer. Oncogene.
18:3407–3414. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Castelo-Branco P, Choufani S, Mack S,
Gallagher D, Zhang C, Lipman T, Zhukova N, Walker EJ, Martin D,
Merino D, et al: Methylation of the TERT promoter and risk
stratification of childhood brain tumours: An integrative genomic
and molecular study. Lancet Oncol. 14:534–542. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu X, Bishop J, Shan Y, Pai S, Liu D,
Murugan AK, Sun H, El-Naggar AK and Xing M: Highly prevalent TERT
promoter mutations in aggressive thyroid cancers. Endocr Relat
Cancer. 20:603–610. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kyo S, Takakura M, Fujiwara T and Inoue M:
Understanding and exploiting hTERT promoter regulation for
diagnosis and treatment of human cancers. Cancer Sci. 99:1528–1538.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Adams RL: Eukaryotic DNA
methyltransferases - structure and function. BioEssays. 17:139–145.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jair KW, Bachman KE, Suzuki H, Ting AH,
Rhee I, Yen RW, Baylin SB and Schuebel KE: De novo CpG island
methylation in human cancer cells. Cancer Res. 66:682–692. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wan J, Oliver VF, Wang G, Zhu H, Zack DJ,
Merbs SL and Qian J: Characterization of tissue-specific
differential DNA methylation suggests distinct modes of positive
and negative gene expression regulation. BMC Genomics. 16:492015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Deaton AM and Bird A: CpG islands and the
regulation of transcription. Genes Dev. 25:1010–1022. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Baylin SB and Jones PA: A decade of
exploring the cancer epigenome - biological and translational
implications. Nat Rev Cancer. 11:726–734. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stirzaker C, Millar DS, Paul CL, Warnecke
PM, Harrison J, Vincent PC, Frommer M and Clark SJ: Extensive DNA
methylation spanning the Rb promoter in retinoblastoma tumors.
Cancer Res. 57:2229–2237. 1997.PubMed/NCBI
|
|
14
|
Smith IM, Glazer CA, Mithani SK, Ochs MF,
Sun W, Bhan S, Vostrov A, Abdullaev Z, Lobanenkov V, Gray A, et al:
Coordinated activation of candidate proto-oncogenes and cancer
testes antigens via promoter demethylation in head and neck cancer
and lung cancer. PLoS One. 4:e49612009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002.PubMed/NCBI
|
|
16
|
Tahira AC, Kubrusly MS, Faria MF, Dazzani
B, Fonseca RS, Maracaja-Coutinho V, Verjovski-Almeida S, Machado MC
and Reis EM: Long noncoding intronic RNAs are differentially
expressed in primary and metastatic pancreatic cancer. Mol Cancer.
10:1412011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hangauer MJ, Vaughn IW and McManus MT:
Pervasive transcription of the human genome produces thousands of
previously unidentified long intergenic noncoding RNAs. PLoS Genet.
9:e10035692013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Choi JH, Park SH, Park J, Park BG, Cha SJ,
Kong KH, Lee KH and Park AJ: Site-specific methylation of CpG
nucleotides in the hTERT promoter region can control the expression
of hTERT during malignant progression of colorectal carcinoma.
Biochem Biophys Res Commun. 361:615–620. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zinn RL, Pruitt K, Eguchi S, Baylin SB and
Herman JG: hTERT is expressed in cancer cell lines despite promoter
DNA methylation by preservation of unmethylated DNA and active
chromatin around the transcription start site. Cancer Res.
67:194–201. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guilleret I and Benhattar J: Unusual
distribution of DNA methylation within the hTERT CpG island in
tissues and cell lines. Biochem Biophys Res Commun. 325:1037–1043.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pettigrew KA, Armstrong RN, Colyer HA,
Zhang SD, Rea IM, Jones RE, Baird DM and Mills KI: Differential
TERT promoter methylation and response to 5-aza-2′-deoxycytidine in
acute myeloid leukemia cell lines: TERT expression, telomerase
activity, telomere length, and cell death. Genes Chromosomes
Cancer. 51:768–780. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Iliopoulos D, Satra M, Drakaki A,
Poultsides GA and Tsezou A: Epigenetic regulation of hTERT promoter
in hepatocellular carcinomas. Int J Oncol. 34:391–399.
2009.PubMed/NCBI
|
|
23
|
de Wilde J, Kooter JM, Overmeer RM,
Claassen-Kramer D, Meijer CJ, Snijders PJ and Steenbergen RD: hTERT
promoter activity and CpG methylation in HPV-induced
carcinogenesis. BMC Cancer. 10:2712010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Azouz A, Wu YL, Hillion J, Tarkanyi I,
Karniguian A, Aradi J, Lanotte M, Chen GQ, Chehna M and
Ségal-Bendirdjian E: Epigenetic plasticity of hTERT gene promoter
determines retinoid capacity to repress telomerase in
maturation-resistant acute promyelocytic leukemia cells. Leukemia.
24:613–622. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Renaud S, Loukinov D, Abdullaev Z,
Guilleret I, Bosman FT, Lobanenkov V and Benhattar J: Dual role of
DNA methylation inside and outside of CTCF-binding regions in the
transcriptional regulation of the telomerase hTERT gene. Nucleic
Acids Res. 35:1245–1256. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kumari A, Srinivasan R, Vasishta RK and
Wig JD: Positive regulation of human telomerase reverse
transcriptase gene expression and telomerase activity by DNA
methylation in pancreatic cancer. Ann Surg Oncol. 16:1051–1059.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lopatina NG, Poole JC, Saldanha SN, Hansen
NJ, Key JS, Pita MA, Andrews LG and Tollefsbol TO: Control
mechanisms in the regulation of telomerase reverse transcriptase
expression in differentiating human teratocarcinoma cells. Biochem
Biophys Res Commun. 306:650–659. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu J, Zhao Y and Wang S: Chromatin and
epigenetic regulation of the telomerase reverse transcriptase gene.
Protein Cell. 1:22–32. 2010. View Article : Google Scholar
|
|
29
|
Guilleret I and Benhattar J: Demethylation
of the human telomerase catalytic subunit (hTERT) gene promoter
reduced hTERT expression and telomerase activity and shortened
telomeres. Exp Cell Res. 289:326–334. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tsujioka T, Yokoi A, Itano Y, Takahashi K,
Ouchida M, Okamoto S, Kondo T, Suemori S, Tohyama Y and Tohyama K:
Five-aza-2′-deoxycytidine-induced hypomethylation of cholesterol
25-hydroxylase gene is responsible for cell death of
myelodysplasia/leukemia cells. Sci Rep. 5:167092015. View Article : Google Scholar
|
|
31
|
Prendergast G and Ziff E:
Methylation-sensitive sequence-specific DNA binding by the c-Myc
basic region. Science. 251:186–189. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sitaram RT, Degerman S, Ljungberg B,
Andersson E, Oji Y, Sugiyama H, Roos G and Li A: Wilms’ tumour 1
can suppress hTERT gene expression and telomerase activity in clear
cell renal cell carcinoma via multiple pathways. Br J Cancer.
103:1255–1262. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Drummond IA, Rupprecht HD, Rohwer-Nutter
P, Lopez-Guisa JM, Madden SL, Rauscher FJ III and Sukhatme VP: DNA
recognition by splicing variants of the Wilms’ tumor suppressor,
WT1. Mol Cell Biol. 14:3800–3809. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shin KH, Kang MK, Dicterow E and Park NH:
Hypermethylation of the hTERT promoter inhibits the expression of
telomerase activity in normal oral fibroblasts and senescent normal
oral keratinocytes. Br J Cancer. 89:1473–1478. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Renaud S, Loukinov D, Bosman FT,
Lobanenkov V and Benhattar J: CTCF binds the proximal exonic region
of hTERT and inhibits its transcription. Nucleic Acids Res.
33:6850–6860. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Feldmann A, Ivanek R, Murr R, Gaidatzis D,
Burger L and Schübeler D: Transcription factor occupancy can
mediate active turnover of DNA methylation at regulatory regions.
PLoS Genet. 9:e10039942013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Choi JH, Min NY, Park J, Kim JH, Park SH,
Ko YJ, Kang Y, Moon YJ, Rhee S, Ham SW, et al: TSA-induced DNMT1
downregulation represses hTERT expression via recruiting CTCF into
demethylated core promoter region of hTERT in HCT116. Biochem
Biophys Res Commun. 391:449–454. 2010. View Article : Google Scholar
|
|
38
|
Meeran SM, Patel SN and Tollefsbol TO:
Sulforaphane causes epigenetic repression of hTERT expression in
human breast cancer cell lines. PLoS One. 5:e114572010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kornblihtt AR: Promoter usage and
alternative splicing. Curr Opin Cell Biol. 17:262–268. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nieto Moreno N, Giono LE, Cambindo Botto
AE, Muñoz MJ and Kornblihtt AR: Chromatin, DNA structure and
alternative splicing. FEBS Lett. 589:3370–3378. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cramer P, Cáceres JF, Cazalla D, Kadener
S, Muro AF, Baralle FE and Kornblihtt AR: Coupling of transcription
with alternative splicing: RNA pol II promoters modulate SF2/ASF
and 9G8 effects on an exonic splicing enhancer. Mol Cell.
4:251–258. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Schor IE, Gómez Acuña LI and Kornblihtt
AR: Coupling between transcription and alternative splicing. Cancer
Treat Res. 158:1–24. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Naftelberg S, Schor IE, Ast G and
Kornblihtt AR: Regulation of alternative splicing through coupling
with transcription and chromatin structure. Annu Rev Biochem.
84:165–198. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Davies RC, Calvio C, Bratt E, Larsson SH,
Lamond AI and Hastie ND: WT1 interacts with the splicing factor
U2AF65 in an isoform-dependent manner and can be incorporated into
spliceosomes. Genes Dev. 12:3217–3225. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Markus MA, Heinrich B, Raitskin O, Adams
DJ, Mangs H, Goy C, Ladomery M, Sperling R, Stamm S and Morris BJ:
WT1 interacts with the splicing protein RBM4 and regulates its
ability to modulate alternative splicing in vivo. Exp Cell Res.
312:3379–3388. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mohamed AM, Balsat M, Thenoz M, Koering C,
Payen-Gay L, Cheok M, Mortada H, Auboeuf D, Pinatel C, El-Hamri M,
et al: Oncogene- and drug resistance-associated alternative exon
usage in acute myeloid leukemia (AML). Oncotarget. 7:2889–2909.
2016.
|
|
47
|
Katuri V, Gerber S, Qiu X, McCarty G,
Goldstein SD, Hammers H, Montgomery E, Chen AR and Loeb DM: WT1
regulates angiogenesis in Ewing Sarcoma. Oncotarget. 5:2436–2449.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cunningham TJ, Palumbo I, Grosso M, Slater
N and Miles CG: WT1 regulates murine hematopoiesis via maintenance
of VEGF isoform ratio. Blood. 122:188–192. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Amin EM, Oltean S, Hua J, Gammons MV,
Hamdollah-Zadeh M, Welsh GI, Cheung MK, Ni L, Kase S, Rennel ES, et
al: WT1 mutants reveal SRPK1 to be a downstream angiogenesis target
by altering VEGF splicing. Cancer Cell. 20:768–780. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Radan L, Hughes CS, Teichroeb JH, Vieira
Zamora FM, Jewer M, Postovit LM and Betts DH: Microenvironmental
regulation of telomerase isoforms in human embryonic stem cells.
Stem Cells Dev. 23:2046–2066. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yatabe N, Kyo S, Maida Y, Nishi H,
Nakamura M, Kanaya T, Tanaka M, Isaka K, Ogawa S and Inoue M:
HIF-1-mediated activation of telomerase in cervical cancer cells.
Oncogene. 23:3708–3715. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Nishi H, Nakada T, Kyo S, Inoue M, Shay JW
and Isaka K: Hypoxia-inducible factor 1 mediates upregulation of
telomerase (hTERT). Mol Cell Biol. 24:6076–6083. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Anderson CJ, Hoare SF, Ashcroft M,
Bilsland AE and Keith WN: Hypoxic regulation of telomerase gene
expression by transcriptional and post-transcriptional mechanisms.
Oncogene. 25:61–69. 2006.
|
|
54
|
Kechris K, Yang YH and Yeh RF: Prediction
of alternatively skipped exons and splicing enhancers from exon
junction arrays. BMC Genomics. 9:5512008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kornblihtt AR: CTCF: From insulators to
alternative splicing regulation. Cell Res. 22:450–452. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Shukla S, Kavak E, Gregory M, Imashimizu
M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R and
Oberdoerffer S: CTCF-promoted RNA polymerase II pausing links DNA
methylation to splicing. Nature. 479:74–79. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Paredes SH, Melgar MF and Sethupathy P:
Promoter-proximal CCCTC-factor binding is associated with an
increase in the transcriptional pausing index. Bioinformatics.
29:1485–1487. 2013. View Article : Google Scholar :
|
|
58
|
Adelman K and Lis JT: Promoter-proximal
pausing of RNA polymerase II: Emerging roles in metazoans. Nat Rev
Genet. 13:720–731. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ong CT and Corces VG: CTCF: An
architectural protein bridging genome topology and function. Nat
Rev Genet. 15:234–246. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Stadhouders R, Thongjuea S, Andrieu-Soler
C, Palstra RJ, Bryne JC, van den Heuvel A, Stevens M, de Boer E,
Kockx C, van der Sloot A, et al: Dynamic long-range chromatin
interactions control Myb proto-oncogene transcription during
erythroid development. EMBO J. 31:986–999. 2012. View Article : Google Scholar :
|
|
61
|
Sebestyén E, Zawisza M and Eyras E:
Detection of recurrent alternative splicing switches in tumor
samples reveals novel signatures of cancer. Nucleic Acids Res.
43:1345–1356. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ulaner GA, Hu JF, Vu TH, Giudice LC and
Hoffman AR: Tissue-specific alternate splicing of human telomerase
reverse transcriptase (hTERT) influences telomere lengths during
human development. Int J Cancer. 91:644–649. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lincz LF, Mudge LM, Scorgie FE, Sakoff JA,
Hamilton CS and Seldon M: Quantification of hTERT splice variants
in melanoma by SYBR green real-time polymerase chain reaction
indicates a negative regulatory role for the β deletion variant.
Neoplasia. 10:1131–1137. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Mavrogiannou E, Strati A, Stathopoulou A,
Tsaroucha EG, Kaklamanis L and Lianidou ES: Real-time RT-PCR
quantification of human telomerase reverse transcriptase splice
variants in tumor cell lines and non-small cell lung cancer. Clin
Chem. 53:53–61. 2007. View Article : Google Scholar
|
|
65
|
Listerman I, Sun J, Gazzaniga FS, Lukas JL
and Blackburn EH: The major reverse transcriptase-incompetent
splice variant of the human telomerase protein inhibits telomerase
activity but protects from apoptosis. Cancer Res. 73:2817–2828.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kolquist KA, Ellisen LW, Counter CM,
Meyerson M, Tan LK, Weinberg RA, Haber DA and Gerald WL: Expression
of TERT in early premalignant lesions and a subset of cells in
normal tissues. Nat Genet. 19:182–186. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Meyerson M, Counter CM, Eaton EN, Ellisen
LW, Steiner P, Caddle SD, Ziaugra L, Beijersbergen RL, Davidoff MJ,
Liu Q, et al: hEST2, the putative human telomerase catalytic
subunit gene, is up-regulated in tumor cells and during
immortalization. Cell. 90:785–795. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Yi X, Shay JW and Wright WE: Quantitation
of telomerase components and hTERT mRNA splicing patterns in
immortal human cells. Nucleic Acids Res. 29:4818–4825. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kotoula V, Hytiroglou P, Pyrpasopoulou A,
Saxena R, Thung SN and Papadimitriou CS: Expression of human
telomerase reverse transcriptase in regenerative and precancerous
lesions of cirrhotic livers. Liver. 22:57–69. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ohyashiki JH, Hisatomi H, Nagao K, Honda
S, Takaku T, Zhang Y, Sashida G and Ohyashiki K: Quantitative
relationship between functionally active telomerase and major
telomerase components (hTERT and hTR) in acute leukaemia cells. Br
J Cancer. 92:1942–1947. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Barclay JY, Morris AG and Nwokolo CU:
HTERT mRNA partially regulates telomerase activity in gastric
adenocarcinoma and adjacent normal gastric mucosa. Dig Dis Sci.
50:1299–1303. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Rha SY, Jeung HC, Park KH, Kim JJ and
Chung HC: Changes of telomerase activity by alternative splicing of
full-length and β variants of hTERT in breast cancer patients.
Oncol Res. 18:213–220. 2009. View Article : Google Scholar
|
|
73
|
Liu Y, Wu BQ, Zhong HH, Tian XX and Fang
WG: Quantification of alternative splicing variants of human
telomerase reverse transcriptase and correlations with telomerase
activity in lung cancer. PLoS One. 7:e388682012. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Wang Y, Meeker AK, Kowalski J, Tsai HL,
Somervell H, Heaphy C, Sangenario LE, Prasad N, Westra WH, Zeiger
MA, et al: Telomere length is related to alternative splice
patterns of telomerase in thyroid tumors. Am J Pathol.
179:1415–1424. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wong MS, Wright WE and Shay JW:
Alternative splicing regulation of telomerase: A new paradigm?
Trends Genet. 30:430–438. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kilian A, Bowtell DD, Abud HE, Hime GR,
Venter DJ, Keese PK, Duncan EL, Reddel RR and Jefferson RA:
Isolation of a candidate human telomerase catalytic subunit gene,
which reveals complex splicing patterns in different cell types.
Hum Mol Genet. 6:2011–2019. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Saebøe-Larssen S, Fossberg E and
Gaudernack G: Characterization of novel alternative splicing sites
in human telomerase reverse transcriptase (hTERT): Analysis of
expression and mutual correlation in mRNA isoforms from normal and
tumour tissues. BMC Mol Biol. 7:262006. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Withers JB, Ashvetiya T and Beemon KL:
Exclusion of exon 2 is a common mRNA splice variant of primate
telomerase reverse transcriptases. PLoS One. 7:e480162012.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Khosravi-Maharlooei M, Jaberipour M,
Hosseini Tashnizi A, Attar A, Amirmoezi F and Habibagahi M:
Expression pattern of alternative splicing variants of human
telomerase reverse Transcriptase (hTERT) in cancer cell lines was
not associated with the origin of the cells. Int J Mol Cell Med.
4:109–119. 2015.PubMed/NCBI
|
|
80
|
Jafri MA, Ansari SA, Alqahtani MH and Shay
JW: Roles of telomeres and telomerase in cancer, and advances in
telomerase-targeted therapies. Genome Med. 8:692016. View Article : Google Scholar : PubMed/NCBI
|