Introduction
Post-transcriptional regulation of gene expression
plays a very important role at all stages of normal development,
maintenance and/or pathology of cells, tissues and organs.
Post-transcriptional regulation depends on various RNA-binding
proteins (RBPs) that determine the translational status of mRNAs,
their structure, localizaton and degradation rate. Regulatory RBPs
can be recruited to their target mRNAs via specific recognition
sequences (such as AU-rich sequences for HuR protein, or CPE
sequence for CPEB family) (1,2).
Alternatively, RBPs have been shown to bind to specifically
structured elements within target RNAs, regardless of their
nucleotide sequence. The IMPs/IGF2BPs (IGF-2 mRNA-binding proteins
1–3) are likely to belong to this latter group. IMP target
sequences within RNAs vary to a large extent in different
experimental models (3–6), and this complexity in target
recognition might be due to the presence of numerous RNA binding
domains in the IMPs, as well as by the variety of RNP complexes
where they have been shown to participate (7). A large number of RNA-binding
regulatory proteins are also recruited to their target mRNAs via
short non-coding RNAs, such as miRNAs (reviewed in ref. 8). Cooperation or competition between
different RNA-binding protein complexes determines the rate of
protein expression from a large number of mRNAs (9).
Post-transcriptional regulation of gene expression
is particularly important in the case of proteins characterized by
a short half-life, which are often critical for the control of cell
cycle, signal transduction pathways, circadian rhythm, antigen
processing and other processes (10). We have shown that IMP-3/IGF2BP3
protein binds to and positively regulates the expression of cyclins
D1, D3 and G1 in a number of human cancer cell lines (11). This regulatory mechanism controls
the cell cycle and proliferation. In the present study, we explore
the molecular mechanisms underlying the regulation of the
expression of cyclins by IMP-3 in human rhabdomyosarcoma cells.
Materials and methods
Cell culture, miRNAs, plasmids and
constructs
RD embryonic rhabdomyosarcoma (eRMS) were purchased
from the American Type Culture Collection (ATCC; Manassas, VA, USA)
and cultured as indicated on the ATCC website. miRNA precursors
were purchased from Life Technologies (Carlsbad, CA, USA) and
transfected at the final concentration of 50 nM using Lipofectamine
RNAiMAX (Invitrogen Carlsbad, CA, USA), according to the
manufacturer’s instructions. FLAG-HA-IMP3 expression construct and
stable cell line was previously described (11). psiCHECK-2 reporter constuct was
purchased from Promega (Madison, WI, USA) miRNA target site
blockers were designed by Exiqon, and the sequences were:
TSB-CCND1-let-7g: TGAGGTAAGCGTGAGC; TSB-CCND3-miR-15:
CATGAGGTATTGTGAAAC and the negative control A. TSBs were
transfected at final concentration of 20 nM 24 h after the
transfection of relevant miRNAs.
RNA interference
Transient transfection of siRNA was performed using
Lipofectamine RNAiMAX. Cells (5×105) were plated in
6-well plates, siRNA duplexes were transfected at 20 nM final
concentration for 48 h. The efficiency of the siRNA-mediated
knockdown of gene expression was evaluated by quantitative reverse
transcription PCR (qRT-PCR) and/or western blotting. Unless
otherwise indicated, all experiments were performed 48 h after
siRNA transfection.
Transient transfection of plasmids was performed
using Lipofectamine 2000 (Invitrogen), according to the
manufacturer’s instructions. Cells (2×105) were plated
in 6-well plates; 24 h later the cells were transfected with 1 μg
of plasmid for 48 h. Efficiency of transfection was evaluated by
immunofluorescence (IF) and/or western blotting.
siRNA target sequences were: IMP 3
ggauucuccuaguagcauucau; IMP 3_2 auggaucauuccucaugua; GW182/TNRC6A
gaaaugcucugguccgcuauu; GW182/TNRC6A_2 gcagccucca gcacaaccucuu;
TNRC6B caucugggacaaggugauuguagacg; TNRC6C
ggaauggagacacugugaacucagc; AGO1 ggaguuacuuucauagcauuu; AGO2
gcacggaaguccaucugaa; PTBP1 aacaaugacaagagccgugac; ILF3
ccccagaggacgacaguaaa; ILF3/NF110 gcggauccgacuacaacuacg; ILF3/NF90
cuuccuagagcgucuaaaagu; HuR gaggcuccagucaaaaacca; HNRNPA2B1
ccaggggcucauguaacugu; Control siRNA uagcaaugacgaaugcgua.
Luciferase reporter assays
The 3′UTR of human CCND1 or CCND3 mRNA were cloned
into a psiCHECK2-reporter vector (Promega) downstream of the
reporter gene (Renilla luciferase). HeLa cells were seeded
at 20,000 cells/well in a 96-well plate. Twenty-four hours later,
10 ng of psiCHECK2-E2F5-3′UTR was co-transfected with 50 nM of
miRNA mimic and/or 20 nM siRNA. Co-transfection was performed with
Lipofectamine 2000 (Life Technologies). Forty-eight hours after
transfection, the relative levels of Renilla vs. firefly
luciferase activity (control of transfection efficiency) were
measured with Dual-luciferase reporter assay system (Promega)
according to the manufacturer’s instructions. The luminescence
signal was quantified on a Mithras LB 940 multilabel reader and
analyzed with MikroWin software (Berthold Technologies).
Antibodies
The following antibodies were used for western
blotting. IMP-3 (N-19) sc-47893, CCND1 (DCS-6) sc-20044, CCND3
(D-7) sc-6283, PTBP1/HNRNPI sc-16547, ILF3 sc-136197 and HuR
sc-5261 were obtained from Santa Cruz Biotechnology (Santa Cruz,
CA, USA). GW182 rabbit poly-clonal (A302-329A) was obtained from
Bethyl Laboratories (Montgomery, TX, USA). Rat anti-AGO1
(SAB4200084), AGO2 (SAB4200085), actin (A5441, clone AC-15),
anti-goat IgG (I9140) anti-rabbit IgG (A0545) and anti-mouse IgG
(I8765) were purchased from Sigma-Aldrich.
Quantitative real-time RT-PCR
The qRT-PCR primers for the detection of IMP-3,
CCND1, CCND3 and Cyclo A were as previously described (11,12).
qRT-PCR was performed using a LightCycler (Roche).
Cell lysis and sucrose gradients
RIPA lysis for western blotting. The cell pellets
were incubated in 5 volumes of RIPA buffer [50 mM Tris-HCl (pH
7.5), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1
mM EDTA, protease inhibitors] for 15 min on ice followed by
sonication for 7.5 min (30 sec on, 1 min off) at high intensity.
After centrifugation at 16,000 × g for 15 min at 4°C, the
supernatant was recovered for western blotting.
Total lysate from RD cells (3×107 cells)
was prepared as previously described (13), treated or not with 10 mM puromycin
for 15 min at 37°C, and applied to a 21–47% sucrose gradient in 20
mM Tris-HCl (pH 8.0); 140 mM KCl; 5 mM MgCl2.
Centrifugation was carried out at 40,000 rpm for 2 h and 15 min
using a Beckman SW 41 rotor. Fractions (0.8 ml) were collected,
adsorbance at 260 nm was measured, and each fraction was
ethanol-precipitated, treated with DNAse (Promega), and RNA was
extracted with phenol-chloroform and used for qRT-PCR analysis.
Immunoprecipitation of IMP-3
complexes
Protein complexes were immunoprecipitated from whole
cell extracts [lysis buffer: 10% glycerol, 20 mM Tris-HCl, pH 8,
0.2 mM EDTA, 0.1% NP-40, 0.5 M KCl, protease inhibitors (cOmplete,
Roche)], pre-cleared for 1 h with protein A/G-agarose (Thermo
Fisher Scientific), treated or not with 1 mg/ml protease-free RNAse
A (Roche), using IMP-3 antibody (sc-47893; Santa Cruz
Biotechnology) or goat IgG (Sigma-Aldrich) and protein A/G-agarose.
Complexes were separated on a 4–12% poly-acrylamide gel
(Invitrogen), and stained using the SilverQuest kit from
Invitrogen, according to the manufacturer’s instructions. Mass
spectrometry identification of proteins was carried out by Dr R.
Tomaino, Harvard Medical School.
Drug treatment
DRB treatment
RD cells (4×105) were plated in 6-well
plates and transfected with control or IMP-3 siRNAs; DRB
(Sigma-Aldrich D1916) was added to a final concentration of 100 μM
and the cells were collected at indicated time-points.
Cycloheximide treatment
RD cells (4×105) were plated in 6-well
plates and transfected with control or IMP-3 siRNAs; 48 h later,
cycloheximide (Sigma-Aldrich C 19881G) was added to a final
concentration of 20 μg/ml for 2 h.
In situ hybridization
Probes used were: CCND1 sense,
TAATACGACTCACTATAGGGAGACCCTCGGTGTCCT ACTTCAA and CCND1 antisense,
ATTTAGGTGACACT ATAGGGGATGGTCTCCTTCATCT; CCND3 sense, TAAT
ACGACTCACTATAGGGAGATGGATGCTGGAGGTATG TGA and CCND3 antisense,
ATTTAGGTGACACTATAGA ATGAAGGCCAGGAAATCA.
Cells were seeded on poly-Lysine-coated glass cover
slides and transfected as previously described. Cells were fixed
with 4% paraformaldehyde in phosphate buffered saline (PBS) for 15
min at RT, washed with PBS, incubated with 0.3%
H2O2 dissolved in methanol for 30 min at RT
and washed with PBS. Afterwards, cells were incubated with
prehybridization solution (50% formamide, 2X SSC, 10 mM
Na2HPO4) for 90°C at RT in a humidified
chamber followed by incubation in hybridization solution (1.5 μl
salmon sperm DNA, 1.5 μl tRNA and 200–500 ng of DIG-labelled mRNA
probes pre-heated for 2 min at 90°C and placed on ice for 2 min,
then mixed with 15 μl of 60% formamide, 10 mM
Na2HPO4 and 15 μl of 20% dextran sulphate, 4X
SSC, 0.4% BSA) in a humidified chamber at 37°C overnight. Cells
were washed twice with 2 ml of prehybridization solution at 37°C
for 30 min, then twice with 2X SSC, 0.1% Triton at 37–50°C for 5
min, twice with 1X SSC, 0.1% Triton at 37°C for 5 min, and 5 times
with PBS-Tween at 37°C for 5 min. Cells were incubated for 1 h in
blocking buffer (2% sheep serum, 2 mg/ml BSA, in PBS-Tween) at RT
followed by incubation with anti-DIG-biotin antibody (Roche)
(diluted 1:500 in blocking buffer) overnight in a humidified
chamber at 4°C. To visualize the CCND1 and D3 mRNAs, the TSA Biotin
Systems (Perkin-Elmer) and the Enhanced Liquid Substrate system
(Sigma-Aldrich 3,3-Diaminobenzidine) were used according to the
manufacturer’s instructions.
Protein pull-down by biotinylated
RNA
RT reactions were performed using the
SuperScript® III First-Strand Synthesis system (Life
Technologies) and total RNA from RD cells. Phusion™ High-Fidelity
DNA Polymerase kit for PCR (Thermo Fisher Scientific) was used for
PCR amplification of desired fragments. All forward primers had a
T7 promoter sequence: TAATACGACTCACTATAGGGAGA. The primers are
listed in Table I.
 | Table IPrimers used in the present study. |
Table I
Primers used in the present study.
| Name | Region | Sequence | Location |
|---|
| CCND1:
NM_053056.2 |
| 1 D1 F | 3′UTR |
ggacgtggacatctgagggc | 1082 |
| 1 D1 R | 3′UTR |
ctcccccaccgctcagggtt | 1362 |
| 2 D1 F | 3′UTR |
taaccctgagcggtggggga | 1342 |
| 2 D1 R | 3′UTR |
gctttatcaggaaaagcaca | 1622 |
| 3 D1 F | 3′UTR |
ttgtgcttttcctgataaag | 1602 |
| 3 D1 R | 3′UTR |
tgctacgctggctggtgccc | 1882 |
| 4 D1 F | 3′UTR |
cgggcaccagccagcgtagc | 1862 |
| 4 D1 R | 3′UTR |
tatttcctacacctattgga | 2142 |
| 5 D1 F | 3′UTR |
ccaataggtgtaggaaatag | 2122 |
| 5 D1 R | 3′UTR |
actttcaaacaccagttggc | 2402 |
| 6 D1 F | 3′UTR |
tgccaactggtgtttgaaag | 2382 |
| 6 D1 R | 3′UTR |
aaaataaactgtattaaatc | 2662 |
| 7 D1 F | 3′UTR |
agatttaatacagtttattt | 2642 |
| 7 D1 R | 3′UTR |
cacttcctaaataaaaatta | 2922 |
| 8 D1 F | 3′UTR |
gtaatttttatttaggaagt | 2902 |
| 8 D1 R | 3′UTR |
acatggcagtatatgacaca | 3182 |
| 9 D1 F | 3′UTR |
caatgtcatatactgccatg | 3162 |
| 9 D1 R | 3′UTR |
tctctggggacaccggcgcg | 3457 |
| 10 D1 F | 3′UTR |
ccgcgccggtgtccccagag | 3437 |
| 10 D1 R | 3′UTR |
aaacagaacactagtacata | 3732 |
| 11 D1 F | 3′UTR |
ttatgtactagtgttctgtt | 3712 |
| 11 D1 R | 3′UTR |
tggttcagacagacgccgca | 4007 |
| 12 D1 F | 3′UTR |
tgcggcgtctgtctgaacca | 3987 |
| 12 D1 R | 3′UTR |
ttaccagttttatttctaga | 4282 |
| CCND3:
NM_001760.3 |
| 6 D3 F | 3′UTR |
gccctctggagtggccacta | 1055 |
| 6 D3 R | 3′UTR |
tcccatcagcctggcccacc | 1320 |
| 7 D3 F | 3′UTR |
gccaggctgatgggacagaa | 1305 |
| 7 D3 R | 3′UTR |
tctaggagcagctgtcagca | 1570 |
| 3 D3 F | 3′UTR |
acagctgctcctagagggag | 1555 |
| 3 D3 R | 3′UTR |
tatagcagctccttggccac | 1820 |
| 4 D3 F | 3′UTR |
caaggagctgctatagcctg | 1805 |
| 4 D3 R | 3′UTR |
tttttccaagaagccaaagc | 2055 |
MEGAscript® T7 Transcription kit (Life
Technologies) was used for RNA in vitro transcription and
RNA 3′ End Biotinylation kit (Pierce) for biotinylation of RNA
fragments, all of the above according to the manufacturer’s
instructions.
Biotin pull-down assay was performed as follows: the
cell pellet was lysed in buffer containing 25 mM Tris-HCl, pH 7.4,
150 mM NaCl, 1 mM EDTA, 1% NP-40 and 5% glycerol + protease
inhibitor (cOmplete, Roche) for 15 min on ice, and supernanant was
collected. Biotinylated RNA probes were incubated with cell lysate
in TENT buffer [50 mM Tris-Cl (pH 8.0), 2 mM EDTA, 150 mM NaCl, 1%
Triton X-100] for 30 min at RT, immobilized on streptavidin-agarose
beads, washed three times with TENT buffer and analysed by western
blot analysis to reveal the proteins bound to RNA probes.
Results and Discussion
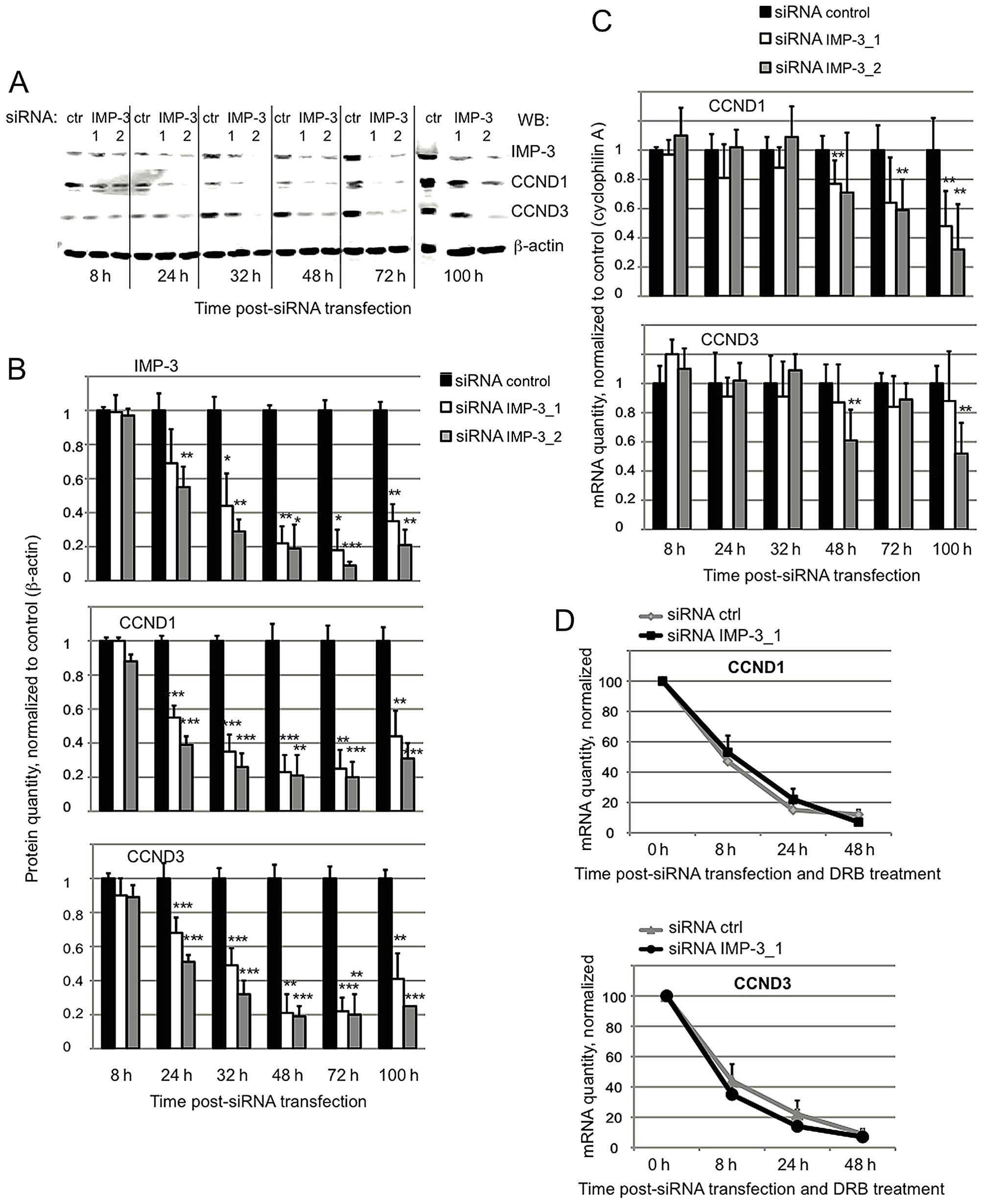
The expression of CCND1 and D3 is
regulated on post-transcriptional level in IMP-3 KD cells
To elucidate the molecular mechanisms of the
regulation of expression of the cyclins by IMP-3, we started by
comparing the levels of CCND1 and D3 mRNA and protein in IMP-3 KD
cells, compared to control. The short half-life of CCND1 and D3
proteins (30–40 min) allows to perform these studies on endogenous
proteins. We have not included CCNG1 in the present study, because
although its expression is regulated by IMP-3, this atypical cyclin
is very stable (with a half-life of over 48 h). A clear decrease of
the levels of CCND1 and D3 proteins becomes visible as early as
24–32 h post-transfection of IMP-3 siRNA, whereas the corresponding
mRNAs do not vary significantly at these time-points (Fig. 1A–C). These results suggest that the
expression of the cyclins is initially downregulated by a
post-transcriptional mechanism which does not depend on the mRNA
levels. Next, we have addressed the question of the mRNA stability
of cyclins by blocking the transcription in IMP-3 KD vs. control
cells with 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole (DRB).
As shown in Fig. 1D, the stability
of mRNAs of CCND1 and D3 does not depend on the presence of IMP-3.
Moreover, the subcellular localization of the cyclin mRNAs did not
appear to change when IMP-3 was decreased. As we have previously
shown (11), both in control and
IMP-3 KD cells, the CCN mRNAs are efficiently exported to the
cytoplasm, even though in IMP-3 KD cells they had a more
perinuclear localization.
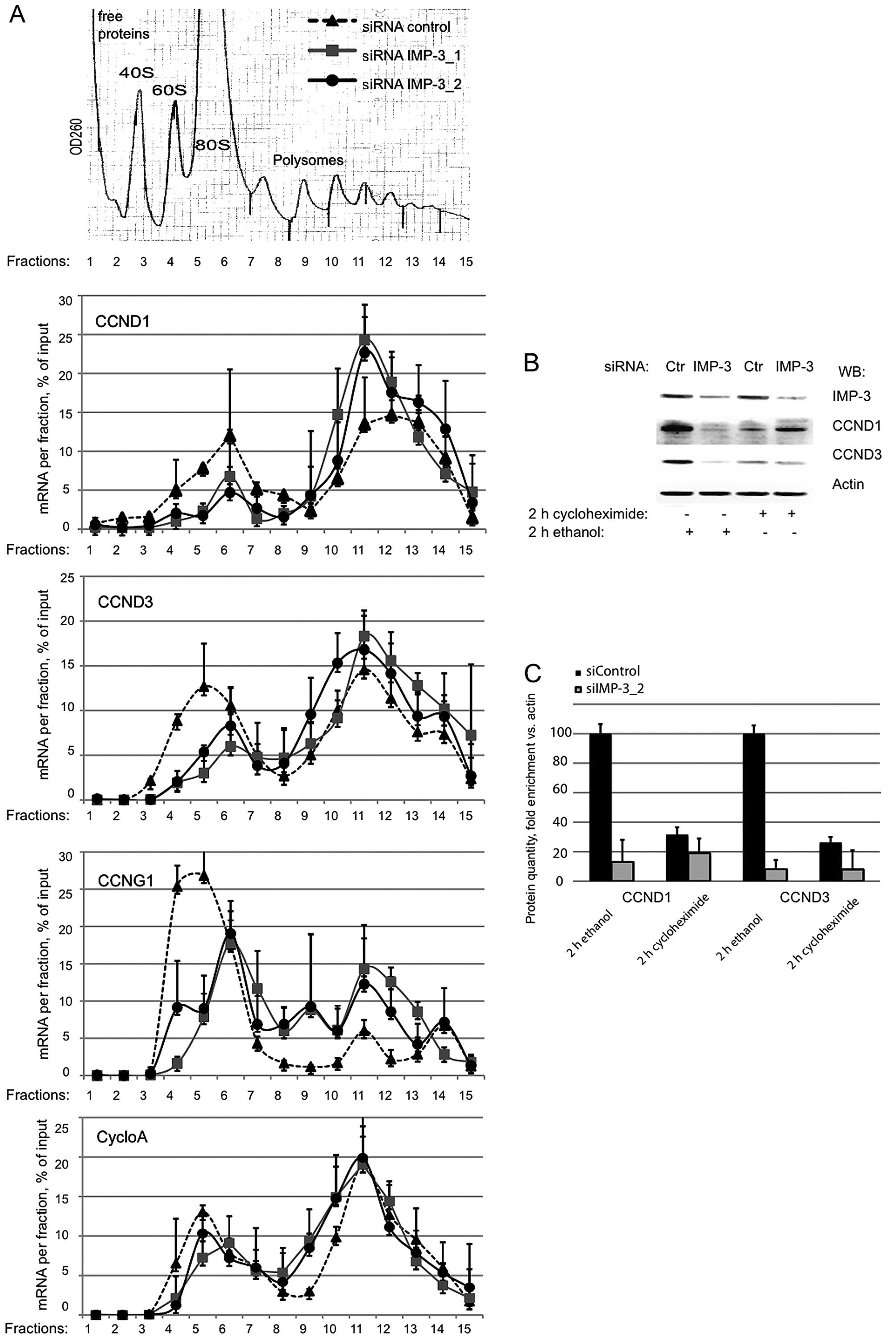
The mRNAs of CCND1 and D3 are associated
with polyribosomes in IMP-3 KD cells, but their translation is
repressed
To address the translational status of the cyclin
mRNAs in IMP-3 KD cells, we performed sucrose gradient separation
of polyribosomes, followed by RNA isolation from each fraction.
Subsequently, we used RT-qPCR to evaluate the relative amounts of
the mRNAs of CCND1 and D3 in polyribosomal vs. monosomal fractions
in IMP-3 KD and control cells. The association of the mRNAs of
CCND1 and D3 with polyribosomes did not decrease in the absence of
IMP-3 (Fig. 2A). On the contrary,
a slight accumulation of these mRNAs in heavy polyribosomal
fractions was observed for the mRNAs of CCND1 and D3, but not for
the control mRNA of cyclophilin A. Taken together with the decrease
of protein levels of the cyclins in IMP-3 KD cells, these
observations suggested that the translation of the mRNAs of CCND1
and D3 can be slowed down in the absence of IMP-3, but this event
does not involve the dissociation of the mRNAs from the ribosomes.
In agreement with this hypothesis, when we inhibited the protein
translation in IMP-3 KD or control cells using the mRNA-ribosome
‘freezing’ drug cycloheximide, we observed a strong decrease of the
cyclins in control cells, whereas in IMP-3 KD cells, these was no
further decrease of the cyclin levels when the translation was
arrested by the drug. We conclude that in IMP-3 KD cells, the
translation of the mRNAs of CCND1 and D3 is already strongly
repressed by a mechanism that does not involve mRNA degradation,
erroneous mRNA localization or disassembly of polyribosomes as an
initial event.
The expression of CCND1 and D3 in IMP-3
KD cells can be fully restored by inactivating the RISC
complex
The mechanisms of the rapid translational arrest
occurring on the mRNAs of CCND1 and D3 in IMP-3 KD cells were
unclear. However, the observed phenomenon strongly resembled the
translational repression caused by miRNA-dependent recruitment of
argonaute (AGO) proteins and/or of GW182/TNRC6A on target mRNAs
(reviewed in ref. 14). Multiple
publications have earlier identified the mRNAs of CCND1 and D3 as
miRNA targets (15,16). In addition, the perinuclear
localization of the mRNAs of CCND1 and D3 in IMP-3 KD cells
observed in our experiments (Fig.
1D) strongly resembled the typical subcellular localization of
activated RISC complexes, as previously reported (17). Therefore, we attempted to
simultaneously knock down key RISC components GW182, AGO1 or AGO2
in our IMP-3 KD cells, and to study the expression levels of CCND1
and D3 proteins. The results presented in Fig. 3A and B clearly show that a KD of
GW182 and of AGO2, but not of AGO1, fully restored the levels of
CCND1 and D3, even when IMP-3 remained downregulated. The KD of
other GW182 family members, TNRC6B and C, which are usually
expressed at low levels, did not rescue the expression of the
cyclins in IMP-3 KD cells (Fig.
3C). We concluded that IMP-3 could protect the mRNAs of CCND1
and D3 from RNAi in human cancer cells. To find out whether this
regulation was direct, we cloned the 3′UTRs of CCND1 and D3 into
the psiCHECK-2 luciferase reporter vector, and first studied the
roles of AGO1, AGO2, GW182 and IMP-3 in the regulation of the
luciferase expression. Consistent with the results observed on
endogenous CCND1 and D3, the expression of the luciferase under the
control of the 3′UTRs of the cyclins was dramatically downregulated
by a KD of IMP-3, upregulated by a KD of GW182, and slightly
increased by a KD of AGO2, but not of AGO1 (Fig. 3D and E). A KD of GW182 in IMP-3
depleted cells released the inhibition of the luciferase in these
conditions (Fig. 3F and G). These
results clearly indicated that IMP-3 and the RNAi machinery were
competing on the 3′UTRs of CCND1 and D3, thus, regulating the
expression of the cyclins. We have subsequently co-transfected
miRNAs let-7g and miR-15, known to regulate CCND1 and D3,
respectively, and have further confirmed that while a KD of IMP-3
increased the repressive effect of endogenous or ectopic miRNAs on
the cyclins, a KD of GW182 fully reversed this effect (Fig. 3H and I).
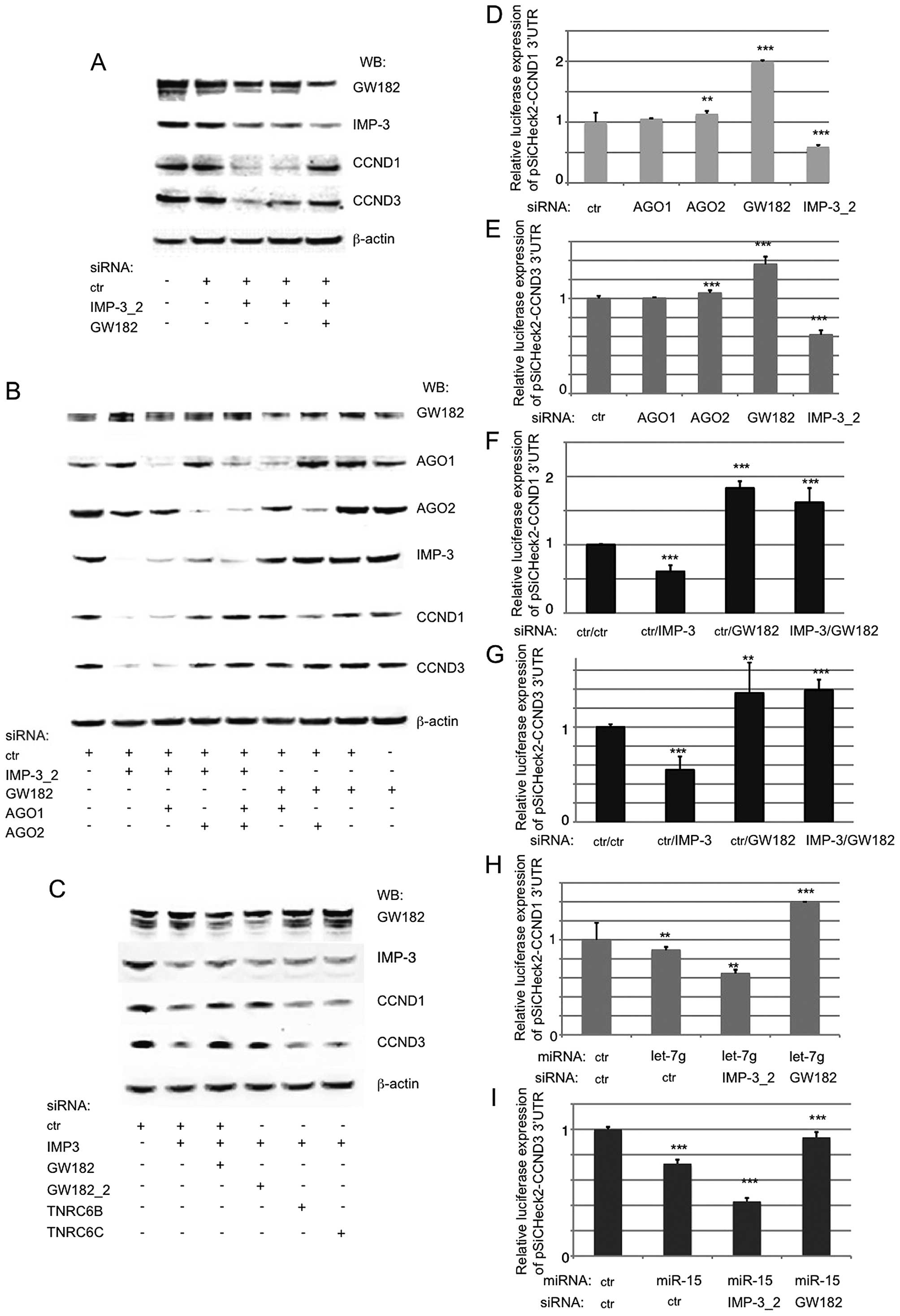 | Figure 3The expression of CCND1 and D3 in
IMP-3 KD cells can be fully restored by inactivating the RISC
complex. (A) A western blot analysis shows a rescue of CCND1 and D3
expression in IMP-3 KD cells when GW182 is knocked down. (B) The
expression of CCND1 and D3 in IMP-3 cells can be rescued by a KD of
AGO2, but not of AGO1 (lanes 1–5). In control cells, the KD of
AGO1, AGO2 or GW182 has no effect on the levels of CCND1 and D3
(lanes 5–9). (C) Only a KD of GW182/TNRC6A, but not of other family
members, TNRC6B and C, can rescue the expression of CCND1 and D3 in
IMP-3 KD cells. (D–I) The regulation of luciferase expression
(psiCHECK-2 reporter vector) under the control of 3′UTRs of CCND1
(D, F and H) and CCND3 (E, G and I) depends on IMP-3, GW182, AGO2
and miRNAs let-7g (CCND1) and miR-15 (CCND3). The luciferase
expression of each vector co-transfected with control siRNA used
was set to 1 and used for normalization of all the respective
assays. Error bars represent SEM from 3 independent experiments.
P-values (above each bar) were calculated using the Mann-Whitney
test and R software: **P<0.01;
***P<0.001. |
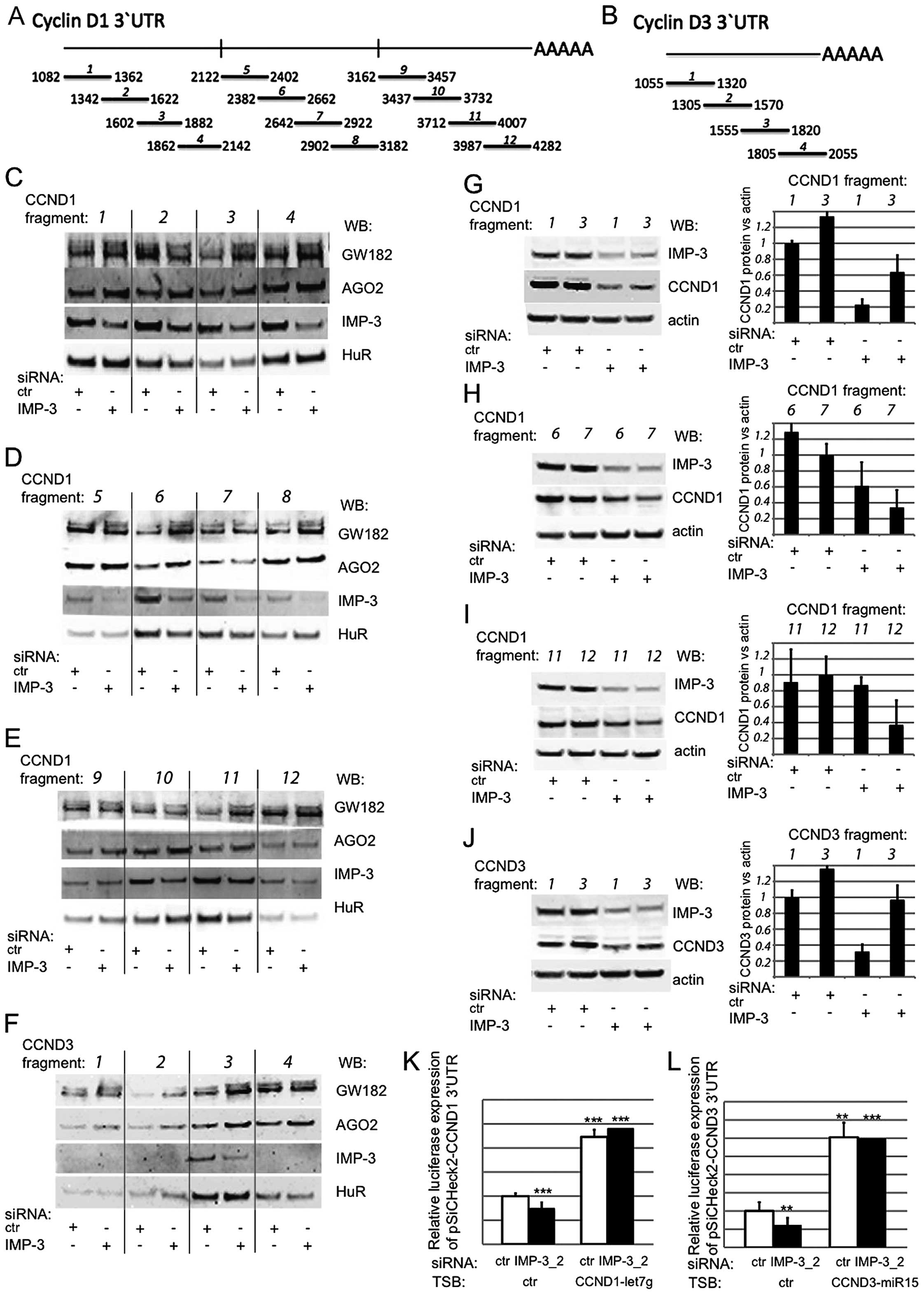
IMP-3 and GW182/AGO2 bind to and compete
within the regions of the 3′UTRs of the cyclins that are critical
for their expression
The IMP proteins do not have a universal binding
motif, and were reported to bind to various RNA sequences depending
on the cell type and experimental model used in different studies.
Various miRNA-binding sites, both experimentally proven or
predicted, are numerous throughout the 3′UTR sequences of CCND1 and
D3 (see Introduction for details). Therefore, no prediction was
possible as to the functionally important binding sites of IMP-3
protein within these mRNAs. We decided to identify these regions by
a two-step experimental approach (Fig.
4). First, we synthesized a number of short, partially
overlapping mRNA fragments covering the 3′UTRs of the cyclins
(Fig. 4A and B), biotinylated
them, and used them in an in vitro pull-down assay together
with total cell extracts from control or IMP-3 KD cells (Fig. 4C–F). In order to test our
hypothesis that IMP-3 could hinder the binding of RISC complex to
the mRNAs of CCND1 and D3, we were looking for RNA fragments where
IMP-3 binding would compete with GW182 and/or AGO2. We used the HuR
protein, a very well-characterized post-transcriptional regulator
of the expression of CCND1 and D3, as an internal control for the
assay, because the binding of HuR to target RNAs depends on the
presence of AU-rich sequences, but not on IMP-3 (1,18).
We were able to identify three fragments within the 3′UTR of CCND1
where the binding of RISC complex components was increased in the
absence of IMP-3: fragments 3, 6 and 11. In the case of CCND3,
IMP-3 competed with GW182 and AGO2 within fragment 3 of 3′UTR.
In the second step of the assay, we attempted to
evaluate the functional importance of the identified binding
regions of IMP-3 within the 3′UTRs of CCND1 and D3, where it
competed with RISC in vitro. To this end, we have
transfected the relevant RNA fragments, or the flanking regions of
3′UTRs, where no competition between IMP-3 and RISC had been
observed, and quantified the expression of CCND1 and D3 proteins
under the control of IMP-3 KD conditions (Fig. 4G–J). If our hypothesis is correct
and the depletion of IMP-3 leads to excessive presence of RISC on
the identified regions of the 3′UTRs of the cyclins, and to
translational repression, then the transfection of the
corresponding RNA fragments in molar excess should lead to the
binding of RISC to the transfected fragments, and to a complete of
partial release of the repression. These were the results we have
observed in at least three independent experiments (an example of
each experiments is shown on the left, and a quantification of
three experiments is shown on the right). The transfection of the
relevant RNA fragments, identified in Fig. 4C–F, led to a partial (Fig. 4G and H) or complete (Fig. 4I and J) release of the expression
of the cyclins, even in IMP-3 KD cells. Moreover, even in control
cells, the transfection of these fragments led to a slight increase
of the level of CCND1 and D3, respectively. Contrary to our
previous observations concerning the binding sites for all three
members of IMP family within the 3′UTRs of the cyclins, obtained
with GST-tagged recombinant proteins in vitro (11), the region 1064–1252 of the CCND3
3′UTR was very weakly bound by endogenous IMP-3 in cell lysate, and
was not functionally important for the competition between IMP-3
and RISC for the regulation of CCND3 translation (Fig. 4F and J).
Notably, there is a binding site for the miRNA let-7
within the fragment 11 of CCND1 3′UTR, (positions 3955–3961), and
miR-15 binds within fragment 3 of CCND3 3′UTR (positions
1813–1819). Blocking these miRNA target sites by specific LNA
antisense inhibitors led to a release of luciferase expression
under the control of the 3′UTRs of CCND1 and D3, and in this case,
a KD of IMP-3 did not change the luciferase expression (Fig. 4K and L).
Therefore, we have experimentally proven that IMP-3
can protect the mRNAs of CCND1 and D3 from RISC-induced
translational repression, and have identified a number of regions
of 3′UTRs targeted by these regulatory mechanisms. The other
regions, such as fragments 1, 7 or 12 of CCND1 3′UTR, might titrate
the RISC complex to some extent when transfected into live cells,
but they will equally titrate it in the presence or in the absence
of IMP-3. Therefore, no rescue of the cyclin expression is observed
following the transfection of these fragments in IMP-3 KD
cells.
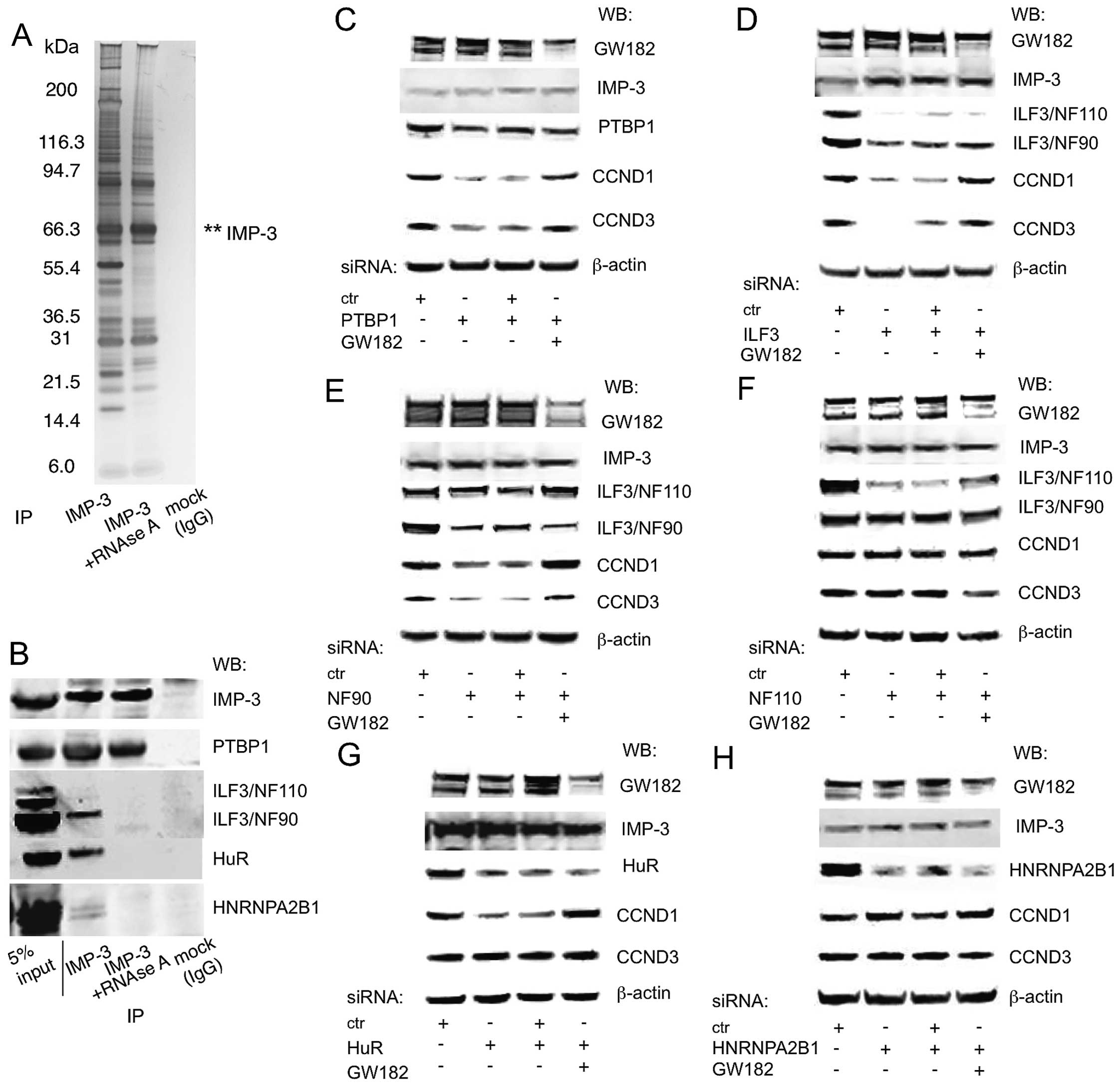
The expression of CCND1 and D3 is
co-regulated by IMP-3 protein partners
A competition between IMP-3 and miRNA-guided GW182
or AGO2 was a novel finding. However, IMP proteins interact with a
large amount of other proteins in the cell, both RNA-binding and
non-RNA binding (Fig. 5A)
(11,12). Thus, it was interesting to
determine the exact composition of the IMP-3 subcomplex that was
functionally relevant to this new regulatory mechanism. In our
previous study, we identified IMP-3 partners that were important or
not for the expression of CCND1, D3 and G1 (11). Among them, PTBP1 was a direct IMP-3
partner, and ILF3, HuR and HNRNPA2B1 depend on the presence of RNA
to co-precipitate with IMP-3 (Fig.
5B). Now, we have systematically tested for the involvement of
these proteins in competition with GW182. IMP-3 partners
PTBP1/HNRNPI and ILF3 were shown to be necessary for the expression
of CCND1 and D3, and their KD was compensated by a simultaneous KD
of GW182 (Fig. 5C and D). ILF3
gene gives rise to two distinct RNA-binding proteins, NF110 and
NF90, which are results of alternative splicing and do not have
similar functions in translational regulation (19). Therefore, we have used
isoform-specific siRNAs and have identified the known regulator of
cell growth NF90, but not NF110, as the partner of IMP-3 that
regulates the expression of CCND1 and D3 in GW182-dependent manner
(Fig. 5E and F). HuR/ELAVL1
interacts with IMP-3 in an RNA-dependent manner, and regulates the
expression of CCND1, but not CCND3 (Fig. 5G). However, the majority of IMP-3
interacting RNA-binding proteins, such as HNRNPA2B1, do not
regulate the expression of the cyclins (Fig. 5H; data not shown). Neither of these
IMP-3 protein partners regulated the protein levels of IMP-3
(Fig. 5C–H). The protein partners
of IMP-3 that have been shown to be important for the expression of
the cyclins, NF90 and PTBP1, demonstrate strong binding along the
3′UTRs of CCND1 and D3, and this binding is dependent on PTBP1 or
independent of NF90 in the presence of IMP-3, especially within the
functionally important fragments we have described above (Fig. 5I). These results have highlighted
the specificity of our experiments and confirmed the existence of a
distinct specific subcomplex of IMP-3 that contains PTBP1 and
ILF3/NF90, and is critical for the protection of the mRNAs of CCND1
and D3 from RNAi.
IMP proteins are known to interact with multiple
other proteins and to regulate the fate of numerous mRNAs, most
frequently by regulation of mRNA stability, but also by impacting
on such diverse processes as mRNA translation, mRNA subcellular
localization and others (3,13,20–24).
These observations suggest the existence of multiple, functionally
diverse IMP-dependent RNP complexes, and highlight the necessity to
study these complexes individually and target-wise. We have
recently demonstrated that the mRNAs of CCNs D1, D3 and G1 are
direct and functionally important targets of IMP-3, as these mRNAs
were found within IMP-3 RNP complexes, and in IMP-3 KD cells the
expression of these cyclins on protein level was sharply
downregulated. The proliferation of IMP-3 KD cells was arrested due
to accumulation of the cells in G1 phase of the cell cycle. This
function is specific to IMP-3, depends on the nuclear-cytoplasmic
shuttling of IMP-3 and its partner HNRNP M, and is not shared by
IMP-1 and IMP-2 (11). Now, we
have identified the molecular mechanisms of this regulation, which
does not depend on regulation of mRNA stability, but involves miRNA
and RISC-dependent, reversible translational arrest in the absence
of IMP-3 and/or its partners PTBP1 and ILF3/NF90. The capacity of
IMP-3 protein complex to bind the cyclin mRNAs in the nucleus
(11) might give a specific
advantage to these mRNAs that are subsequently translocated to the
cytoplasm, already protected from miRNA-RISC complexes, and are
thus translationally active (Fig.
5J). This ability to bind the cyclin mRNAs and to protect them
from RISC is a novel function for a member of the IMP family,
showing an important function for IMP-3 in rapid, sensitive and
reversible cell cycle regulation of human cancer cells.
Acknowledgements
The authors would like to thank Dr A. Harel-Bellan,
Dr F. Dautry and Dr Y. Vassetzky for their helpful discussions. The
present study was a PhD project of E.D., helped by J.K. and M.V. in
a number of experiments, and by G.P. with the statistical analysis
of the results. The study was based on previous unpublished results
obtained by T.R.V. and performed under the direction of A.P.
References
|
1
|
Brennan CM and Steitz JA: HuR and mRNA
stability. Cell Mol Life Sci. 58:266–277. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Richter JD: CPEB: A life in translation.
Trends Biochem Sci. 32:279–285. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jønson L, Vikesaa J, Krogh A, Nielsen LK,
Hansen T, Borup R, Johnsen AH, Christiansen J and Nielsen FC:
Molecular composition of IMP1 ribonucleoprotein granules. Mol Cell
Proteomics. 6:798–811. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Munro TP, Kwon S, Schnapp BJ and St
Johnston D: A repeated IMP-binding motif controls oskar mRNA
translation and anchoring independently of Drosophila melanogaster
IMP. J Cell Biol. 172:577–588. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chao JA, Patskovsky Y, Patel V, Levy M,
Almo SC and Singer RH: ZBP1 recognition of beta-actin zipcode
induces RNA looping. Genes Dev. 24:148–158. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hafner M, Landthaler M, Burger L, Khorshid
M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC,
Munschauer M, et al: Transcriptome-wide identification of
RNA-binding protein and microRNA target sites by PAR-CLIP. Cell.
141:129–141. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nielsen FC, Nielsen J, Kristensen MA, Koch
G and Christiansen J: Cytoplasmic trafficking of IGF-II
mRNA-binding protein by conserved KH domains. J Cell Sci.
115:2087–2097. 2002.PubMed/NCBI
|
|
8
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ciafrè SA and Galardi S: microRNAs and
RNA-binding proteins: A complex network of interactions and
reciprocal regulations in cancer. RNA Biol. 10:935–942. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Glickman MH and Ciechanover A: The
ubiquitin-proteasome proteolytic pathway: Destruction for the sake
of construction. Physiol Rev. 82:373–428. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rivera Vargas T, Boudoukha S, Simon A,
Souidi M, Cuvellier S, Pinna G and Polesskaya A:
Post-transcriptional regulation of cyclins D1, D3 and G1 and
proliferation of human cancer cells depend on IMP-3 nuclear
localization. Oncogene. 33:2866–2875. 2014. View Article : Google Scholar
|
|
12
|
Boudoukha S, Cuvellier S and Polesskaya A:
Role of the RNA-binding protein IMP-2 in muscle cell motility. Mol
Cell Biol. 30:5710–5725. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nielsen J, Christiansen J, Lykke-Andersen
J, Johnsen AH, Wewer UM and Nielsen FC: A family of insulin-like
growth factor II mRNA-binding proteins represses translation in
late development. Mol Cell Biol. 19:1262–1270. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Carthew RW and Sontheimer EJ: Origins and
mechanisms of miRNAs and siRNAs. Cell. 136:642–655. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang Q, Feng MG and Mo YY: Systematic
validation of predicted microRNAs for cyclin D1. BMC Cancer.
9:1942009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bandi N and Vassella E: miR-34a and
miR-15a/16 are co-regulated in non-small cell lung cancer and
control cell cycle progression in a synergistic and Rb-dependent
manner. Mol Cancer. 10:552011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stalder L, Heusermann W, Sokol L, Trojer
D, Wirz J, Hean J, Fritzsche A, Aeschimann F, Pfanzagl V, Basselet
P, et al: The rough endoplasmatic reticulum is a central nucleation
site of siRNA-mediated RNA silencing. EMBO J. 32:1115–1127. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lal A, Mazan-Mamczarz K, Kawai T, Yang X,
Martindale JL and Gorospe M: Concurrent versus individual binding
of HuR and AUF1 to common labile target mRNAs. EMBO J.
23:3092–3102. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Castella S, Bernard R, Corno M, Fradin A
and Larcher JC: Ilf3 and NF90 functions in RNA biology. Wiley
Interdiscip Rev RNA. 6:243–256. 2015. View Article : Google Scholar
|
|
20
|
Nielsen FC, Nielsen J and Christiansen J:
A family of IGF-II mRNA binding proteins (IMP) involved in RNA
trafficking. Scand J Clin Lab Invest (Suppl). 234:93–99. 2001.
View Article : Google Scholar
|
|
21
|
Nielsen J, Adolph SK, Rajpert-De Meyts E,
Lykke-Andersen J, Koch G, Christiansen J and Nielsen FC: Nuclear
transit of human zipcode-binding protein IMP1. Biochem J.
376:383–391. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Oleynikov Y and Singer RH: Real-time
visualization of ZBP1 association with beta-actin mRNA during
transcription and localization. Curr Biol. 13:199–207. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bell JL, Wächter K, Mühleck B, Pazaitis N,
Köhn M, Lederer M and Hüttelmaier S: Insulin-like growth factor 2
mRNA-binding proteins (IGF2BPs): Post-transcriptional drivers of
cancer progression? Cellular and molecular life sciences. Cell Mol
Life Sci. 70:2657–2675, 012.
|
|
24
|
Jønson L, Christiansen J, Hansen TV,
Vikeså J, Yamamoto Y and Nielsen FC: IMP3 RNP safe houses prevent
miRNA-directed HMGA2 mRNA decay in cancer and development. Cell
Rep. 7:539–551. 2014. View Article : Google Scholar : PubMed/NCBI
|