Introduction
The Forkhead box O (FOXO) family of transcription
factors maintains homeostasis under conditions of, e.g., oxidative
stress, starvation and growth factor deprivation (reviewed in refs.
1,2). By regulating the expression of genes
such as Bcl-2-like protein 11 (BIM), Fas ligand
(FasL), transforming growth factor-β (TGF-β), tumor
necrosis factor-related apoptosis-inducing ligand (TRAIL),
CBP/p300 interacting transactivator with Glu/Asp rich
carboxy-terminal domain 2 (CITED2), manganese superoxide
dismutase (MnSOD), isocitrate dehydrogenase 1 (IDH1)
and survivin (3,4), it controls important cellular
functions, such as survival, cell growth, differentiation,
senescence, oxidative stress resistance and metabolism. First
considered tumor suppressors, FOXOs also exhibit pro-tumoral
functions in a context-dependent manner. On the one hand, FOXO
activity promotes cell differentiation, cell cycle arrest and
apoptosis and is inhibited by the activation of well characterized
oncogenic signaling pathways such as phosphatidylinositol 3-kinase
(PI3K)-protein kinase B (PKB) (5,6). On
the other hand, FOXO factors have been described to be involved in
resistance to anti-cancer drugs, maintenance of leukemia-initiating
cells (7) and colon cancer
metastasis (8).
So far, four FOXO family members have been
identified in humans: FOXO1, FOXO3a, FOXO4 and FOXO6. Although they
all bind to the motif 5′-TTGTTTAC-3′ and may act redundantly, they
also display distinct functions and some of these differences are
attributed to their tissue-specific expression pattern. Especially
FOXO3a has been associated with longevity and diseases such as
cancer and was shown to be activated in response to various stress
stimuli. Function and activity of FOXO transcription factors are
substantially controlled by post-translational modifications,
including phosphorylation, acetylation, methylation and
ubiquitination, and binding protein partners. Their subcellular
localization modified by environmental stimuli is considered of
major importance (9–11). The regulation of
nuclear-cytoplasmic FOXO shuttling is complex and influenced by
multiple signaling pathways. Frequently activated in malignant
glioma (12), PKB phosphorylates
FOXO in the nucleus and thus enables FOXO binding to 143-3
proteins, leading to its nuclear export and inactivation.
In glioma, FOXO factors have been shown to induce
differentiation of glioblastoma stem-like cells (13) and to inhibit tumor growth (14). They can suppress MYC
expression, induce apoptosis, decrease glycolysis and inhibit
glucose uptake (15). However, in
glioblastoma stem cells with functional tumor protein p53 (TP53),
FOXO proteins have recently been demonstrated to be essential for
maintaining stemness and survival after ionizing radiation combined
with dual inhibition of PI3K and mechanistic target of rapamycin
(mTOR) (16).
Reprogramming of metabolism is one of the evolving
hallmarks of cancer (17), and has
received increasing attention during the last decade. Current
treatment strategies for malignant gliomas continue to achieve
little success. The metabolic versatility of glioma cells that
adapt to conditions of recurrent deficiency in nutrients and/or
oxygen may be paramount for this (18,19).
Histologically, glioblastomas are characterized by rapid
proliferation and extensive neo-vascularization. Structurally and
functionally abnormal blood vessels are unable to meet the demands
for nutrients and oxygen. This ends up in fluctuating local hypoxia
that again encourages angiogenesis, invasion and metabolic
alterations (20), including an
increased dependency on glycolysis. Hypoxia-inducible factors
(HIFs) here play substantial roles. Jensen et al found that
FOXO3a was activated in hypoxia as a target gene of HIF1 and that a
knockdown of FOXO3a increased cell death in hypoxia both in
HeLa and in U2OS cells. FOXO3a was required for hypoxic suppression
of mitochondrial mass, oxygen consumption and reactive oxygen
species (ROS) production (21).
The present study sought to expand our understanding
of FOXO3a function in glioma biology. While in glioma cells,
primarily antitumor activities of FOXO3a have been described,
inhibition of the upstream regulator PKB, and thereby activation of
FOXO3a function, confers protection against hypoxia-induced cell
death (22). Therefore, we aimed
to elucidate the function of FOXO3a under starvation conditions
that are commonly found in the glioma microenvironment.
Materials and methods
Reagents
PCR primers and small interfering ribonucleic acids
(siRNAs) were purchased from Sigma-Aldrich (St. Louis, MO, USA) or
Qiagen (Hilden, Germany). siRNA sequences, Sigma-Aldrich: CITED2,
sense r(CCAGGUUUAACAACUC CCA)dTdT, antisense r(UGGGAGUUGUUAAACCUGG)
dTdT and sense r(CCUAAUGGGCGAGCACAUA)dTdT, antisense
r(UAUGUGCUCGCCCAUUAGG)dTdT; FOXO3a, sense
r(GAAUGAUGGGCUGACUGAA)dTdT, antisense r(UUCAGUCAGCCCAUCAUUC)dTdT.
siRNA sequences, Qiagen: AllStars negative control, FOXO1, sense
r(GGA GGU AUG AGU CAG UAU A)dTdT, antisense r(UAUACUGACU
CAUACCUCC)dAdT, sense r(UCAUGUCUUGAUAAGU UAA)dTdT, antisense
r(UUAACUUAUCAAGACAUGA) dGdG and sense r(CCAAGUAGCCUGUUAUCAA)dTdT,
antisense r(UUGAUAACAGGCUACUUGG)dTdT; FOXO3a, sense
r(GAAUGAUGGGCUGACUGAA)dTdT, antisense r(UUCAGUCAGCCCAUCAUUC)dAdG,
sense r(GGAACG UGAUGCUUCGCAA)dTdT, antisense r(UUGCGAAGCAU
CACGUUCC)dGdG and sense r(GAUUCAUGCGGGUCC AGAA)dTdT, antisense
r(UUCUGGACCCGCAUGMUC) dGdA. Metafectene Pro was acquired from
Biontex (Munich, Germany), Attractene from Qiagen, DEVD-amc
(N-acetyl-Asp-Glu-Val-Asp-aminomethyl-coumarin) and
Z-VAD-fmk
(N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone) from
Bachem (Weil am Rhein, Germany) and recombinant human TRAIL from
PeproTech (Rocky Hill, NJ, USA). Antibodies used were anti-actin
(polyclonal goat anti-human, sc1616; Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA), anti-FOXO3a (immunohistochemistry,
monoclonal rabbit anti-human, clone EP1949Y), anti-FOXO3a (western
blot analysis, monoclonal rabbit anti-human, clone EPR1950) (both
from Epitomics, Burlingame, CA, USA), anti-HIF1α (BD Transduction
Laboratories, San Jose, CA, USA), anti-phospho-AKT Ser473
(polyclonal rabbit anti-mouse), anti-phospho-FOXO1 Ser256
(polyclonal rabbit anti-human) (both from Cell Signaling
Technology, Danvers, MA, USA), anti-phospho-FOXO3a Thr32
(polyclonal rabbit anti-human; Upstate, Merck Millipore, Darmstadt,
Germany), anti-phospho-TP53 Ser15 (polyclonal rabbit anti-human;
Cell Signaling Technology) and anti-TP53 (monoclonal mouse
anti-human; Calbiochem, Merck Millipore). The 3HRE-pTK-luc reporter
construct was a generous gift from Berra et al (23). The adenoviral FOXO constructs
(24), plasmids encoding FOXO3a,
FOXO3a-A3 and FOXO3a-green fluorescent protein (GFP) (25), the plasmid encoding FOXO1-GFP
(26) and the constructs FOXO3a-TM
and FOXO3a-TM-ER-dDB (27) were
kindly provided by colleagues from other laboratories. The TP53-luc
construct was purchased from Agilent Technologies (Santa Clara, CA,
USA).
Cell culture
LNT-229 human malignant glioma cells were kindly
provided by N. de Tribolet. Cells were cultured in Dulbecco’s
modified Eagle’s Medium (4,500 mg/l glucose; Sigma-Aldrich),
supplemented with 10% fetal calf serum (FCS; PAA, Pasching,
Austria), 2 mM glutamine, 100 IU/ml penicillin and 100 μg/ml
streptomycin, at 37°C and 5% CO2. For some experiments,
glucose was added to serum- and glucose-free medium to give final
concentrations of 2 or 5 mM. A Binder CB53 incubator (Binder,
Tuttlingen, Germany) was used for experiments in hypoxia.
FOXO3a and HIF1 gene silencing
To silence FOXO3a gene expression, short
hairpin RNA (shRNA) sequences were cloned into the pSUPER-puro
vector (28). FOXO3a shRNA
sequences were sense GATCCCCGGAACGTGATGCTTCGCAATT
CAAGAGATTGCGAAGCATCACGTTCCTTTTTGGAAA, antisense
TCGATTTCCAAAAAGGAACGTGATGCTTCG CAATCTCTTGAATTGCGAAGCATCACGTTCCGGG
and sense GATCCCCGAATGATGGGCTGACTGAATTCAAG
AGATTCAGTCAGCCCATCATTCTTTTTGGAAA, anti-sense
TCGATTTCCAAAAAGAATGATGGGCTGACTGA ATCTCTTGAATTCAGTCAGCCCATCATTCGGG,
respectively. The scrambled shRNA sequence has been published
previously (29). Glioma cells
were transfected using Attractene, stable cell lines were generated
by puromycin selection (5 μg/ml). LNT-229 cells stably transfected
with an shRNA targeting HIF1α and its control (Sima)
as well as HIF2α and HIF1α-HIF2α
double-knockdown LNT-229 cells were a kind gift of Henze et
al (30).
Growth and viability assays
A crystal violet staining assay was used for the
determination of cell density. The dye was resolubilized in 0.1 M
sodium citrate, the absorbance measured at 560 nm (Multiskan™ EX;
Thermo Fisher Scientific, Langenselbold, Germany). Cell
proliferation was quantified based on the incorporation of
bromodeoxyuridine (BrdU Cell Proliferation ELISA; Roche
Diagnostics, Mannheim, Germany). For the analysis of cell death,
after treatment e.g. under hypoxic conditions, adherent and
non-adherent cells were collected, washed in phosphate-buffered
saline (PBS), stained with 1 μg/ml propidium iodide (PI) and
analyzed by flow cytometry (BD Canto II, Heidelberg, Germany).
PI-positive cells were regarded as dead cells. Caspase activity was
assessed using the fluorescent substrate DEVD-amc as previously
described (31) and a Mithras LB
940 microplate reader (Berthold Technologies, Bad Wildbad,
Germany).
ROS analysis
Following treatment e.g. under hypoxic conditions,
cells were washed twice with PBS, incubated for 20 min at 37°C in
the presence of 10 μM H2DCFDA (Invitrogen, Carlsbad, CA,
USA) washed with PBS and analyzed by flow cytometry.
Determination of glucose uptake
Glucose concentrations of cell-free supernatants
were measured on a Hitachi 917 analyzer (Roche Diagnostics).
Furthermore, cells were incubated for 10, 75 and 150 min,
respectively, in medium supplemented with 100 μM 2-NBDG
(Invitrogen) under normoxic or hypoxic conditions. Afterwards,
cells were washed twice with PBS, detached, resuspended in PBS
containing 1 μg/ml PI and analyzed by flow cytometry at 465/540 nm.
PI-positive (dead) cells were excluded from the analysis.
Measurement of oxygen consumption
Cells were seeded in glass dishes. After 24 h medium
was changed to pre-incubated treatment medium and the dishes were
sealed air-tight. After another 48 h, oxygen concentration in the
culture medium was measured in an ABL-80 FLEX Blood Gas Analyzer
(Radiometer, Willich, Germany). Oxygen consumption was then
normalized to protein concentration (32). Furthermore, oxygen consumption was
determined using an XF96 extracellular flux analyzer (Seahorse
Bioscience, North Billerica, MA, USA).
Luciferase reporter assay
Using Metafectene Pro, cells were cotransfected with
a 3HRE-pTK-luc (23) and, for
normalization of transfection efficiency, a pRL-CMV Renilla
luciferase construct. Activities of Firefly and Renilla
luciferase (33) were analyzed in
an Infinite® M200 PRO microplate reader (Tecan,
Maennedorf, Switzerland).
SDS-PAGE and immunoblotting
Cells were lysed in a buffer containing 50 mM
Tris-HCl, 120 mM NaCl, 5 mM EDTA, 0.5% Nonidet P-40, 2 μg/ml
aprotinin, 10 μg/ml leupeptin, 100 μg/ml phenylmethylsulfonyl
fluoride, 50 mM NaF, 200 μM NaVO5 and phosphatase
inhibitor cocktails I and II (Sigma-Aldrich) and protein content
was quantified using a Bradford assay (Bio-Rad, Hercules, CA, USA).
Total protein (20 μg) was separated under reducing conditions by
sodium dodecyl sulfate polyacrylamide gel electrophoresis
(SDS-PAGE) and electroblotted on nitrocellulose (Amersham,
Braunschweig, Germany). Membranes were blocked in Tris-buffered
saline containing 5% skim milk and 0.1% Tween-20 and incubated with
the appropriate primary (dilution 1:1,000) and secondary (dilution
1:3,000) antibodies. Immune complexes were detected by enhanced
chemiluminescence (Pierce, Rockford, IL, USA). Densitometric
analysis was performed using ImageJ according to http://rsb.info.nih.gov/ij/docs/menus/analyze.html#gels
(34).
Real-time quantitative PCR (RT-qPCR)
Extraction of total RNA was performed using TRIzol™
(Invitrogen) and the RNeasy™ system (Qiagen), cDNA synthesis using
1.5 μg of RNA and SuperScript VILO™ (Invitrogen). RNA originating
from normal human brain was used with institutional review board
approval (University Cancer Center Frankfurt). Real-time PCR was
carried out in an iQ5 real-time PCR detection system (Bio-Rad,
Munich, Germany) with ABsolute™ Blue qPCR SYBR-Green Fluorescein
Mix (Thermo Fisher Scientific, Waltham, MA, USA). Gene expression
data were normalized to the internal controls 18S ribosomal RNA
(18S rRNA) and succinate dehydrogenase complex, subunit A,
flavoprotein variant (SDHA) using the ddCT method and the iQ5
software (Bio-Rad). Primer sequences: 18S rRNA, forward
5′-CGGCTACCACATCC AAGGAA-3′ and reverse 5′-GCTGGAATTACCGCGGCT-3′;
BIM (35), CITED2 (36), FOXO1 forward 5′-ATACATATGGCC
AATCCAGCATG-3′ and reverse 5′-CTTCAAGAGTCCAGGC GCAC-3′; FOXO3a
forward 5′-GAAGTTCCCCAGCGACT TGG-3′ and reverse
5′-CCAACCCATCAGCATCCATG-3′; glucose transporter 1 (GLUT1), forward
5′-GATTGGCTCCT T CTCTGTGG-3′ and reverse 5′-TCAAAGGACTTGCCCAG
TTT-3′; manganese superoxide dismutase (MnSOD2) forward
5′-AAGGGAGATGTTACAGCCCAGATA-3′ and reverse 5′-TCC
AGAAAATGCTATGATTGATATGAC-3′ (RTPrimerDB ID 2593); SDHA forward
5′-TGGGAACAAGAGGGCAT CTG-3′ and reverse
5′-CCACCACTGCATCAAATTCATG-3′; solute carrier family 16, member 3
(monocarboxylic acid transporter 4) (SLC16A3) (37), TRAIL (35), vascular endothelial growth factor
(VEGF), forward 5′-CTACCTCCACCATGCCAAGT-3′ and reverse
5′-ATGTTGGACTCCTCAGTGGG-3′.
Immunohistochemistry (IHC)
Glioma tissue specimens were derived from the
University Cancer Center tumor bank (Goethe University, Frankfurt
am Main, Germany) and fixed in 4% paraformaldehyde (formalin). The
use of patient material was endorsed by the local ethics committee
(Goethe University). Immunohistochemistry for FOXO3a and HIF1α was
performed as previously described (38) using freshly cut 3 μm thick slides
from formalin-fixed, paraffin-embedded tissue samples on the
automated IHC staining system Discovery XT (Roche/Ventana, Tucson,
AZ, USA).
Fluorescence microscopy
LNT-229 cells stably expressing FOXO3a-GFP
were seeded on glass coverslips coated with poly-L-lysine and
allowed to adhere overnight. After treatment, cells were washed
with PBS, fixed in 2% paraformaldehyde, washed twice with PBS,
stained with 4′,6-diamidino-2-phenyl-indole (DAPI, 1 μg/ml in PBS)
and analyzed by fluorescence microscopy (BZ-8000; Keyence, Osaka,
Japan).
Statistical analysis
Experiments were performed at least three times with
similar results. Data analysis was carried out with Excel
(Microsoft, Seattle, WA, USA). Significance was tested using the
two-tailed Student’s t-test.
Results
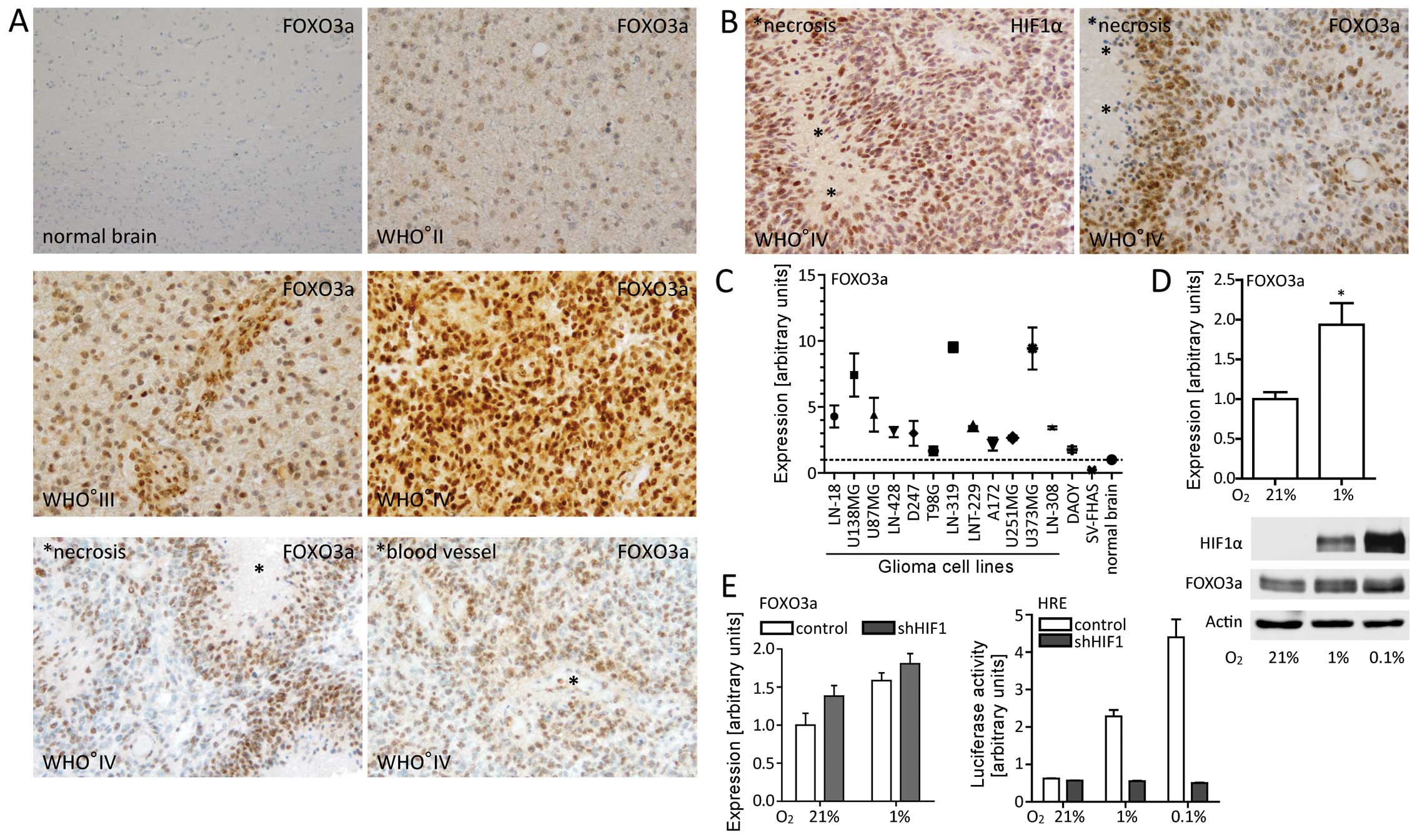
FOXO3a expression increases with glioma
WHO grade
In order to assess expression and localization of
FOXO3a in gliomas, we performed immunohistochemistry on tissue
samples of a panel of gliomas with different WHO grades. We
observed that expression increased with WHO grade and accentuated
in perinecrotic and perivascular tumor regions (Fig. 1A). To identify hypoxic areas, we
additionally performed immunohistochemical staining of HIF1α. This
revealed a similar distribution pattern of FOXO3a and HIF1α
especially in perinecrotic ‘pseudopalisading’ glioma cells
(Fig. 1B) where restricted
availability of oxygen and nutrients is expected (39). Subsequently, we compared FOXO3a
mRNA and protein expression in different established glioma cell
lines and normal brain. All glioma cell lines exhibited elevated
FOXO3a expression compared to normal brain (Fig. 1C) (data not shown).
FOXO3a is upregulated under hypoxic
conditions in a HIF1-independent manner
To test our immunohistochemical observations, we
analyzed FOXO3a expression under hypoxic conditions in
vitro. Both mRNA and protein levels increased in LNT-229 glioma
cells (Fig. 1D). However,
shRNA-mediated silencing of the HIF1α gene did not result in
reduced FOXO3a levels indicating that HIF1α was not the
primary mediator of elevated FOXO3a expression under hypoxic
conditions (Fig. 1E). As HIF2α may
compensate for HIF1α deficiency, we also analyzed FOXO3a
levels in HIF2α knockdown as well as in
HIF1α-HIF2α double-knockdown LNT-229 cells. Both in
normoxia and in hypoxia, depletion of HIF2α or simultaneous
depletion of HIF1α and HIF2α did not affect FOXO3a
expression (data not shown).
FOXO3a overexpression induces cell death
in LNT-229 cells
First, we verified that inhibition of PKB activity
resulted in dephosphorylation of FOXO1 and FOXO3a in our glioma
cell line (Fig. 2A). To monitor
changes in FOXO3a subcellular localization, we carried out
immunofluorescence studies employing FOXO3a-GFP cells (25). Transient expression of FOXO3a-GFP
induced cell death in LNT-229 cells and this cell death was more
pronounced than when introducing FOXO1-GFP (Fig. 2B). As both FOXO3a and FOXO1 alone
may act pro-apoptotically (40,41),
we analyzed FOXO1 expression after transient transfection of
FOXO3a constructs. Enhanced FOXO3a expression did not
affect FOXO1 mRNA levels (Fig.
2C). Using a 4-hydroxytamoxifen (4-OHT) inducible, constitutive
nuclear FOXO3a-TM-construct (27)
and the corresponding control FOXO3a-TM-ER-dDB without DNA-binding
domain, we observed a strong increase in TRAIL expression
induced by FOXO3a (Fig. 2D).
However, cell death occurring upon transfection with constructs
encoding FOXO3a and a constitutive nuclear FOXO3a-A3 was
inhibitable by Z-VAD-fmk only to a lesser extent whereas Z-VAD-fmk
completely reversed TRAIL-induced cell death which served as a
positive control (data not shown). Presumably, like TP53, FOXO3a
may trigger both apoptosis and autophagy (42) and cell death following
FOXO3a overexpression has frequently been described
(41).
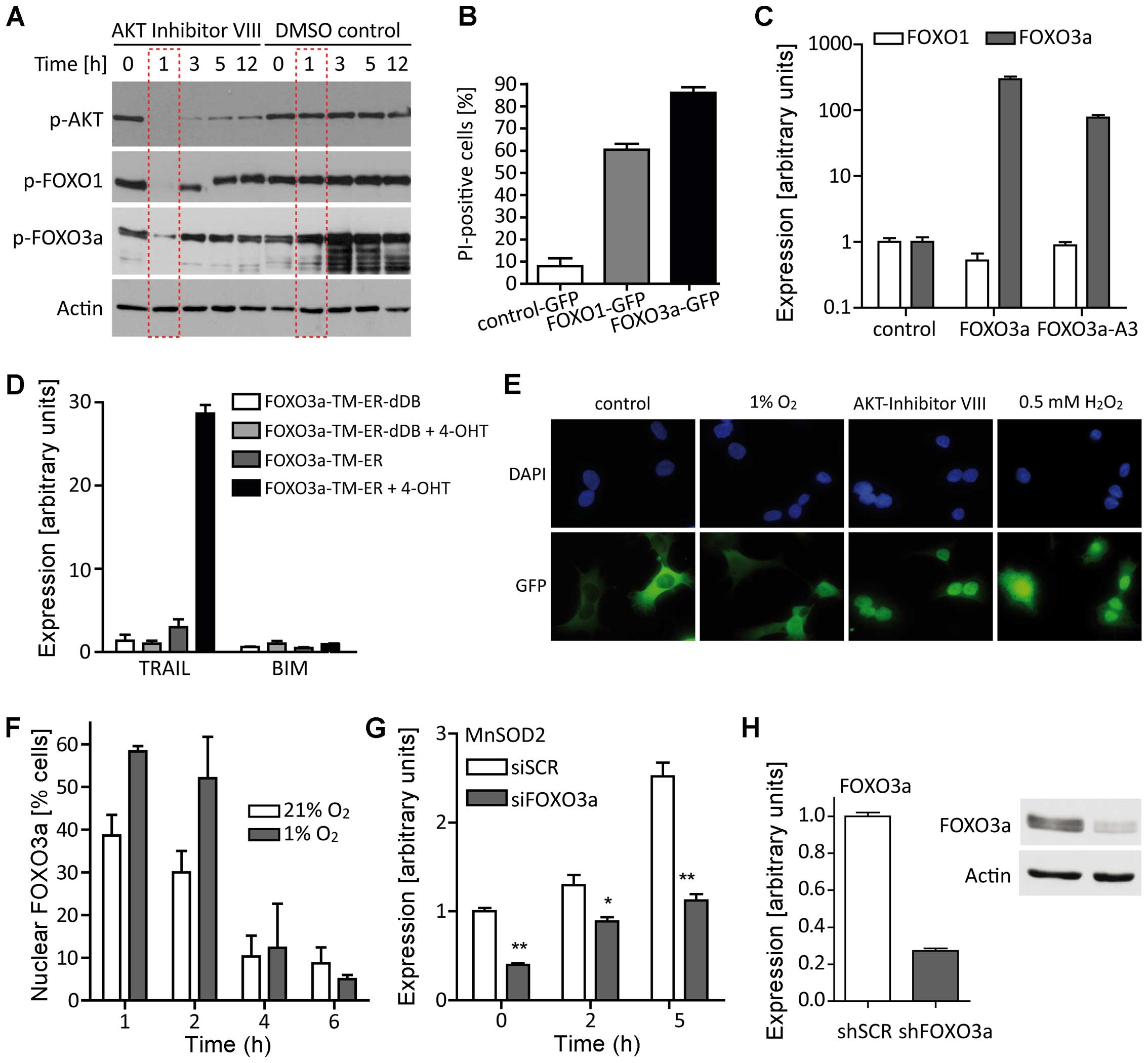 | Figure 2FOXO overexpression provokes
cell death and different stressors result in FOXO3a translocation
from the cytoplasm to the nucleus. (A) LNT-229 cells were treated
with AKT inhibitor VIII (5 μM) or DMSO control for the indicated
periods of time and whole cell lysates were subjected to SDS-PAGE
and immunoblotting against phospho-AKT, phospho-FOXO1 and
phospho-FOXO3a. (B) LNT-229 cells were transiently transfected with
plasmids encoding FOXO1-GFP, FOXO3a-GFP or, as a
control, GFP. After 48 h, cell viability was determined by
PI staining and flow cytometry (mean ± SEM). Equal transfection
efficiency was verified by fluorescence microscopy for GFP. (C)
Transient overexpression of FOXO3a or constitutive nuclear FOXO3a
(FOXO3a-A3) did not increase FOXO1 mRNA levels 24 h
post-transfection (mean ± SEM). (D) LNT-229 cells were transiently
transfected with plasmids encoding a 4-hydroxy-tamoxifen (4-OHT)
inducible, constitutive nuclear FOXO3a-TM-construct or the
corresponding control FOXO3a-TM-ER-dDB without DNA-binding domain.
TRAIL and BIM expression was assessed 24 h after
addition of 4-OHT or DMSO control (mean ± SEM). (E) LNT-229 cells
stably expressing FOXO3a-GFP were serum-starved and exposed
to hypoxia, AKT inhibitor VIII or H2O2.
Subcellular FOXO3a localization was examined by fluorescence
microscopy. (F) Twenty-four hours after seeding, culture medium was
replaced by serum-free medium containing 5 mM glucose. After 1, 2,
4 and 6 h, LNT-229 cells stably expressing FOXO3a-GFP were
fixed, stained with DAPI and analyzed for FOXO3a-GFP localization
by fluorescence microscopy (mean ± SEM). (G) LNT-229 cells were
incubated with 0.5 mM H2O2 in serum-free
medium containing 5 mM glucose for 0 h (control), 2 and 5 h,
respectively. MnSOD2 levels were determined by RT-qPCR after
treatment with H2O2 and siRNA-mediated
knockdown of FOXO3a (50 nM) or scrambled (SCR) control (mean
± SEM, *p<0.05 and **p<0.01). (H)
shRNA-mediated FOXO3a knockdown was verified by RT-qPCR and
western blot analysis. |
LNT-229 cells modulate the subcellular
localization of FOXO3a in response to environmental stress and
pharmacological inhibition of PKB
Counteracted by phosphatase and tensin homolog
(PTEN), insulin and growth factor signaling activates PKB that in
turn phosphorylates FOXO3a at three conserved residues and thereby
causes its cytoplasmic localization (5,43–45).
To study changes in the subcellular localization of FOXO3a, we
generated LNT-229-cells stably expressing FOXO3a-GFP by
repeated selection and fluorescence-activated cell sorting (FACS).
Using these cells, we observed FOXO3a shifting to the nucleus after
implementation of different stressors: serum deprivation, hypoxia
and hydrogen peroxide (H2O2) (Fig. 2E). Extent and duration of this
shift varied depending on the kind of stress. Pharmacological
blockade of PKB via AKT inhibitor VIII caused an immediate and
durable translocation to the nucleus that could partially be
reverted by addition of 10% FCS to the medium (data not shown).
Addition of 0.5 mM H2O2 forced FOXO3a to
permanently stay in the nucleus even in the presence of FCS.
Nuclear shifting of FOXO3a after serum starvation was strongly
enhanced by hypoxia and always a temporary event of short duration
(<4 h) (Fig. 2F).
FOXO3a gene silencing increases
intracellular ROS levels and boosts cell death associated with
oxidative stress
ROS are byproducts of aerobic metabolism and
involved in diverse cellular functions. On the one hand and at
‘lower levels’, they support cell proliferation through the
activation of growth-related signaling pathways, on the other hand
and at ‘higher levels’, they promote apoptosis. To determine
whether FOXO3a regulation contributes to the resistance of
malignant glioma cells under conditions of low glucose, low oxygen
and high ROS levels, we used four different siRNA sequences
targeting FOXO3a and yielding comparable results. MnSOD2,
one of the main antioxidant enzymes and a FOXO3a target gene
(46), was upregulated following
stimulation with H2O2. Transient
siRNA-mediated suppression of FOXO3a significantly reduced
MnSOD2 expression (Fig.
2G). LNT-229-shFOXO3a cells (Fig.
2H) were employed to determine intracellular ROS by flow
cytometric analysis of the ROS-sensitive dye
dichlorodihydrofluorescein diacetate (H2DCFDA). An
increase in intracellular ROS was observed after administration of
0.5–1 mM H2O2 in LNT-229-shFOXO3a cells
compared to the control cells (Fig.
3A). Loss of FOXO3a not only affected the intracellular ROS
homeostasis but also elevated cell death rates after addition of
H2O2. Addition of 10% FCS to the cell culture
medium increased cell death in both LNT-229-shFOXO3a and control
cells. Presumably, more ROS were generated by accelerated metabolic
processes in the presence of serum, exceeding the detoxification
capacity of both cell lines. However, also in this setup, control
cells displayed better survival rates than LNT-229-shFOXO3a cells
(Fig. 3B). LNT-229-shFOXO3a cells
accumulated more intracellular ROS than their control cells, and
this was even more pronounced under hypoxia. Severe hypoxia
amplified ROS production in both cell lines, again more in
LNT-229-shFOXO3a than in control cells (Fig. 3C). Addition of serum also boosted
ROS production (data not shown).
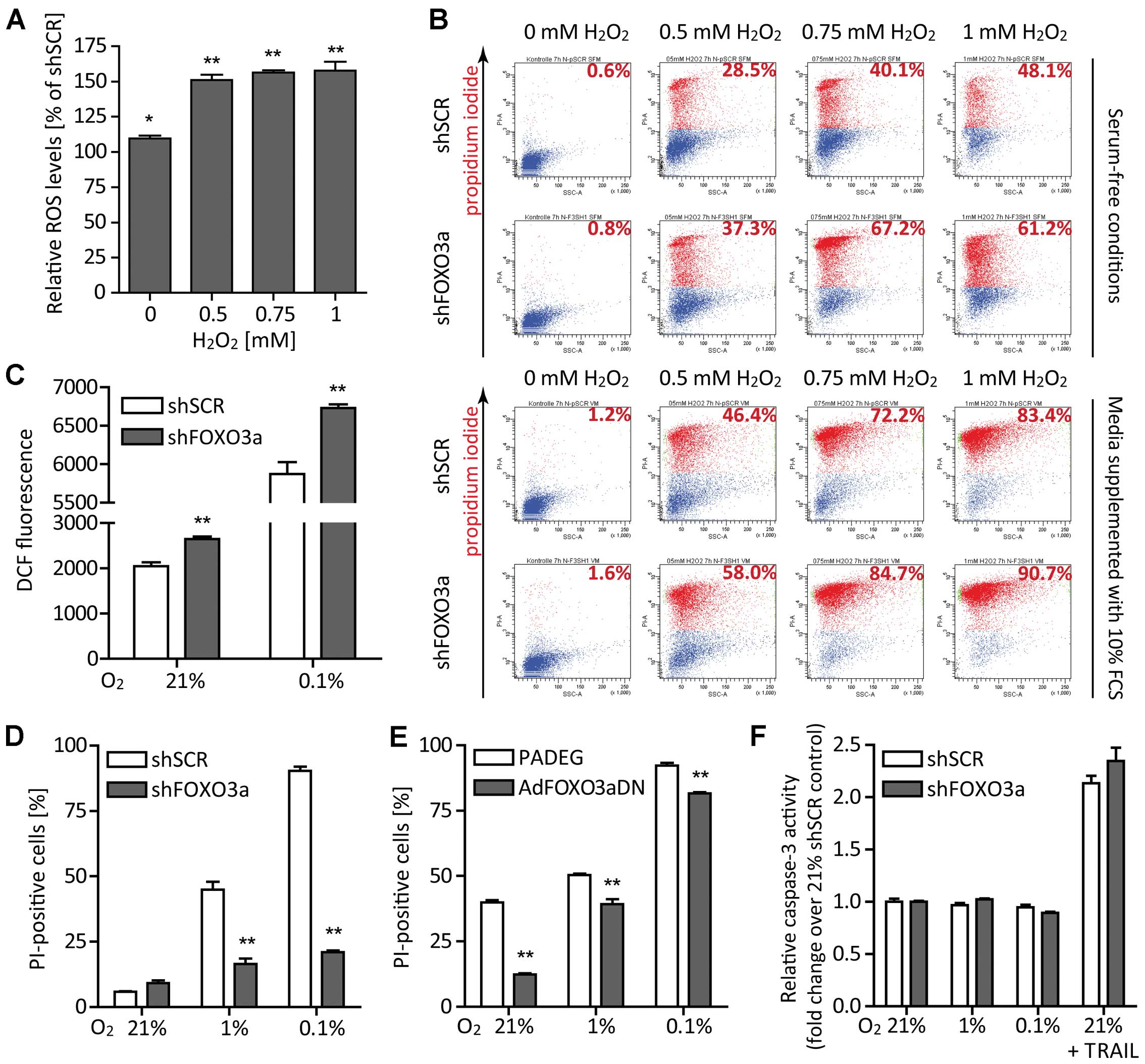 | Figure 3FOXO3a knockdown elevates ROS
levels, enhances cell death linked to oxidative stress and confers
protection from non-apoptotic cell death under starvation
conditions. (A) H2O2 was added to the medium
to give final concentrations of 0 mM (control), 0.5, 0.75 and 1 mM.
After a 6 h incubation period, DCF fluorescence was determined by
flow cytometry. Results are expressed as median fluorescence
intensities of LNT-229-shFOXO3a cells relative to the corresponding
LNT-229-shSCR (control) cells (mean ± SEM, *p<0.05
and **p<0.01). (B) In the absence (upper panel) or
presence (lower panel) of 10% FCS in medium supplemented with 5 mM
glucose, LNT-229-shFOXO3a and control cells were exposed to 0 mM
(control), 0.5, 0.75 and 1 mM H2O2. After 6
h, cell viability was assessed by PI staining and flow cytometry.
Results from one representative experiment are depicted. Numbers in
each upper right corner denote percentages of PI-positive (dead)
cells. (C) For 6 h, LNT-229-shFOXO3a and LNT-229-shSCR cells were
cultured under normoxic or hypoxic conditions in serum-free medium
containing 5 mM glucose. Thereafter, intracellular ROS generation
was estimated using H2DCFDA and flow cytometry. Data are
displayed as median fluorescence intensities and mean plus SEM of
three experiments, **p<0.01. (D) LNT-229-shFOXO3a
cells and their controls LNT-229-shSCR were cultured in serum-free
medium supplemented with 2 mM glucose. Following an incubation
period of 24 h under normoxic or hypoxic conditions, cell viability
was determined by PI staining and flow cytometry. Data are
presented as mean percentages of PI-positive cells and SEM
(**p<0.01). (E) The same experiment was performed
with LNT-229 cells adenovirally transduced with a dominant-negative
FOXO3a and its corresponding control. (F) Again applying the same
experimental design, we found that neither hypoxia (combined with
serum and glucose starvation) nor FOXO3a knockdown altered
DEVD-amc-cleaving caspase activity. Cells treated with TRAIL served
as positive controls. Results are displayed as fold-change over the
21% shSCR control plus SEM. |
FOXO3a promotes cell death in hypoxia in
a caspase-independent way
To simulate conditions of the ‘perinecrotic niche’,
we subjected LNT-229 cells to serum-free medium supplemented with 2
mM glucose and normoxia (21% O2), hypoxia (1%
O2) or severe hypoxia (0.1% O2).
Interestingly, FOXO3a gene suppression conferred robust
protection from cell death occurring under starvation conditions
(Fig. 3D). Experiments with three
different siRNAs targeting FOXO3a revealed the same
phenotype (data not shown). Additionally, we employed an adenoviral
dominant-negative FOXO3a construct (AdFOXO3aDN) and its control
(PADEG) (24). This approach also
confirmed the protective effect of FOXO3a inhibition under
starvation conditions (Fig. 3E).
We measured caspase-3 activity and found neither caspase-3
activation in hypoxia nor a difference between LNT-229-shFOXO3a and
control cells. Therefore, in this in vitro paradigm, cell
death under hypoxic conditions was non-apoptotic (47) and not affected by a pro-apoptotic
function of FOXO3a (Fig. 3F).
FOXO3a gene silencing saves glucose but
does not alter cell proliferation
To figure out the processes leading to cell death
under starvation conditions, we determined glucose concentrations
in culture supernatants of LNT-229-shFOXO3a and control cells.
Glucose consumption generally increased in hypoxia and decreased in
LNT-229-shFOXO3a cells (Fig. 4A).
Similar effects were observed in AdFOXO3aDN cells (Fig. 4B). This difference was not due to a
lower proliferation rate of LNT-229-shFOXO3a cells, as assessed by
BrdU-incorporation assays (Fig.
4C) and crystal violet staining (Fig. 4D). We also analyzed the cells’
glucose flux using the fluorescent D-glucose analog 2-NBDG
(48) and flow cytometry. Again,
LNT-229-shFOXO3a cells took up less 2-NBDG than the corresponding
control cells both in normoxia and in hypoxia (Fig. 4E).
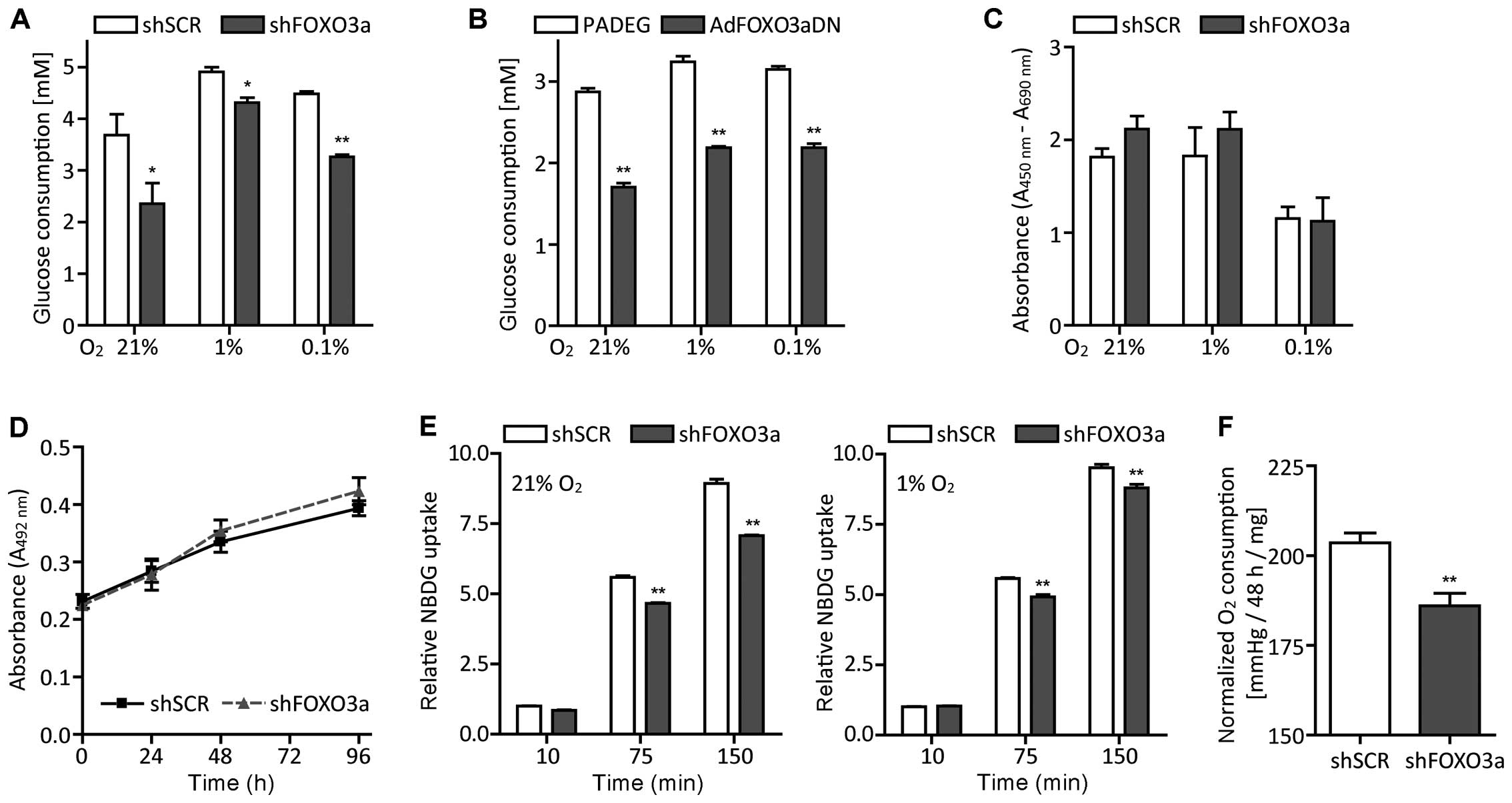 | Figure 4Attenuation of FOXO3a lowers glucose
and oxygen consumption whilst maintaining proliferative capacity.
(A) Twenty-four hours after seeding in medium supplemented with 10%
FCS and 25 mM glucose, LNT-229-shFOXO3a and control cells were
grown in serum-free medium containing 5 mM glucose for another 24
h. Then, residual glucose in supernatants was measured (mean ± SEM,
*p<0.05 and **p<0.01). (B) We also
observed this glucose-saving phenotype when using the adenoviral
dominant-negative FOXO3a construct (mean ± SEM,
*p<0.05 and **p<0.01). (C) BrdU
incorporation decreased in severe hypoxia, but did not differ
significantly between the two cell lines (mean ± SEM). (D) Glioma
cells were grown in medium containing 5 mM glucose and normoxic
cell density was assessed by crystal violet staining at day 1, 2, 3
and 4 (mean ± SEM). (E) LNT-229-shFOXO3a and control cells were
cultured under normoxic or hypoxic conditions for 24 h. Hereafter,
pre-incubated medium containing 2-NBDG, a fluorescent derivative of
D-glucose, was added. After 10, 75 and 150 min, cells were washed,
stained with PI and analyzed by flow cytometry. Data are presented
as median fluorescence intensities relative to the 10 min value of
LNT-229-shSCR (control) cells and SEM, **p<0.01. (F)
Oxygen consumption was calculated by subtracting oxygen
concentration after a 48 h incubation period from initial oxygen
concentration and to exclude any potential differences in
proliferation, normalized to protein content (mean ± SEM,
**p<0.01). |
FOXO3a knockdown attenuates oxygen
consumption
To investigate whether glioma cells compensate their
reduced glucose uptake by enhanced oxidative phosphorylation, we
determined oxygen consumption by ABL-80 FLEX blood gas analyzer.
Conversely, FOXO3a knockdown resulted in a decrease in oxygen
consumption (Fig. 4F). These
results were confirmed by experiments conducted with the XF96
extracellular flux analyzer (data not shown).
FOXO3a inhibition amplifies HIF1α
transcriptional activity in hypoxia
Analysis of primary tumor tissue revealed a
perinecrotic expression of FOXO3a matching the regions with higher
levels of HIF1α (Fig. 1B). We
therefore aimed to explore potential interactions between FOXO3a
and HIF1α. HIF-specific transcriptional activity, examined by
luciferase reporter assay (3HRE-pTK-luc construct), increased under
hypoxic conditions. FOXO3a knockdown augmented this effect
on HRE reporter gene activity (Fig.
5A). To confirm an inhibitory effect of FOXO3a on HIF1α, we
transiently overexpressed a constitutive nuclear FOXO3a-A3 where
all three PKB phosphorylation sites were mutated to alanine
(49). This resulted in a marked
reduction of HRE reporter gene activity (Fig. 5B). Additionally, we analyzed the
expression of several well-known HIF1 target genes using RT-qPCR
(50). The expression of vascular
endothelial growth factor (VEGF) and the monocarboxylate
transporter solute carrier family 16 member 3 (SLC16A3)
increased under hypoxic conditions and this was further enhanced by
FOXO3a knockdown, suggesting that FOXO3a inhibited HIF1
transcriptional activity in LNT-229 glioma cells (Fig. 5C). To study whether the protective
effect of FOXO3a knockdown on survival under hypoxia was
mediated by HIF1α, we applied siRNA targeting FOXO3a under
starvation conditions in LNT-229 cells stably transfected with an
shRNA targeting HIF1α or its control (Sima),
respectively (30). Loss of HIF1α
abolished the protection conferred by FOXO3a knockdown in
hypoxia (Fig. 5D).
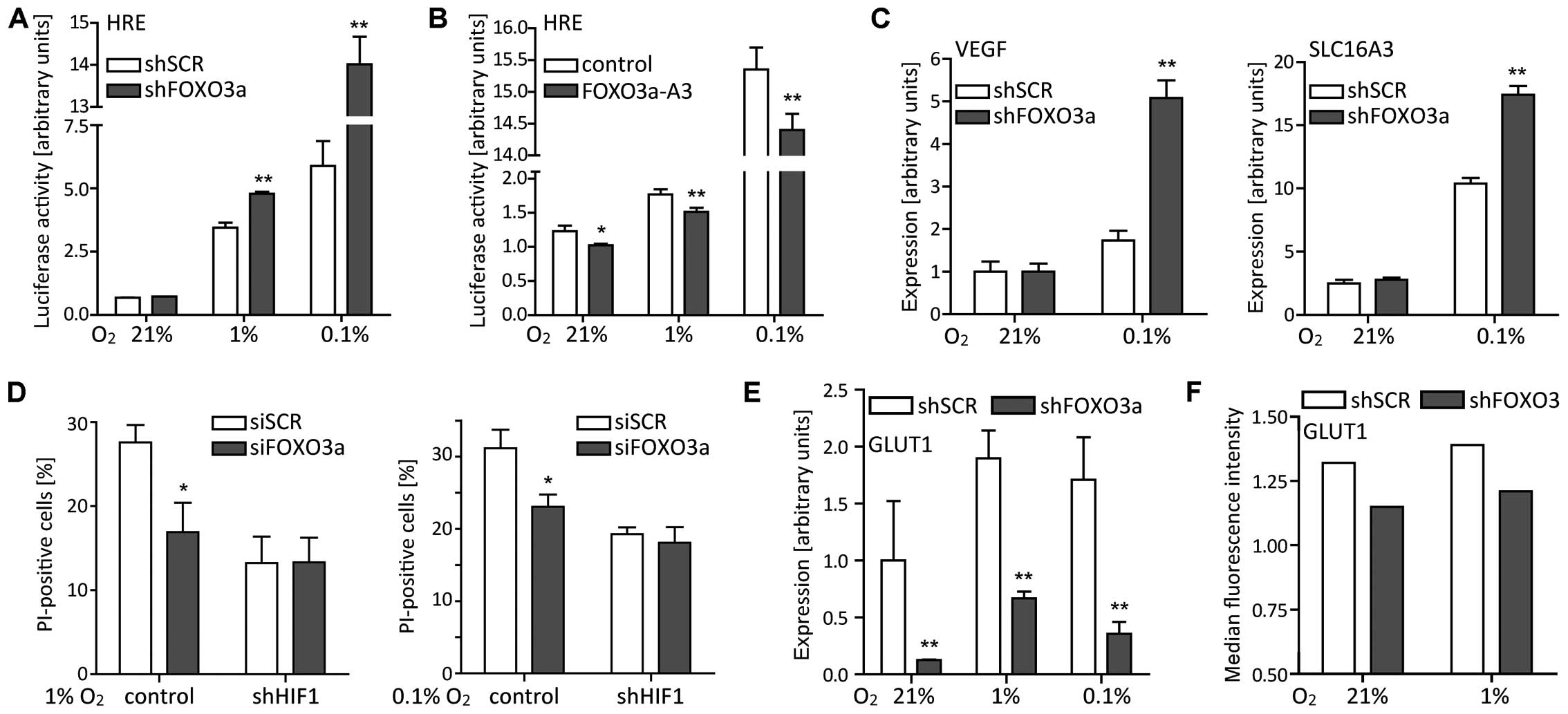 | Figure 5FOXO3a differentially affects
HRE-dependent transcriptional activity and target gene expression.
(A) LNT-229-shFOXO3a and control cells were cultured in serum-free
medium containing 5 mM glucose and under different oxygen
conditions. After 24 h, HIF-specific transcriptional activity was
examined by luciferase reporter assay (3HRE-pTK-luc construct, mean
± SEM, **p<0.01). (B) Inversely, transient
overexpression of constitutive nuclear FOXO3a-A3 reduced
HRE-dependent transcriptional activity (mean ± SEM,
*p<0.05 and **p<0.01). (C)
LNT-229-shFOXO3a and LNT-229-shSCR cells were grown in normoxia or
hypoxia for 24 h. Expression of VEGF and SLC16A3 was
analyzed by RT-qPCR (mean ± SEM, **p<0.01). (D) In
LNT-229 cells stably transfected with a shRNA construct targeting
HIF1α (shHIF1), additional FOXO3a knockdown failed to
protect cells under starvation conditions. Subsequent to 24 h of
serum, glucose and oxygen restriction (see Fig. 3), cell viability was determined by
PI staining and flow cytometry (mean ± SEM, *p<0.05).
(E) FOXO3a knockdown decreased GLUT1 expression (for
the experimental setup see above, mean ± SEM,
**p<0.01). (F) Following a 24 h incubation in
normoxia or hypoxia, cell-surface levels of GLUT1 were determined
by flow cytometry. PI-positive (dead) cells were excluded from the
analysis. Results from one representative experiment are shown. |
GLUT1 expression is reduced in the
absence of FOXO3a
The expression of GLUT1, another long-known
HIF1 target gene, decreased following FOXO3a knockdown.
Though induced under hypoxic conditions (51) both in LNT-229-shFOXO3a and control
cells, GLUT1 expression of control cells always exceeded
that of cells lacking FOXO3a (Fig. 5E). To verify the RT-qPCR data, we
analyzed surface GLUT1 levels by flow cytometry. This technique
also revealed higher GLUT1 amounts in hypoxia and lower amounts in
LNT-229-shFOXO3a cells (Fig. 5F).
The expression of HIF1 target genes was thus differentially
affected by FOXO3a knockdown in our glioma cell line.
The effect of FOXO3a knockdown is not
mediated by CITED2
As reported by Bakker et al, FOXO3a is
induced in hypoxia and activates the transcription of CITED2
(36). CITED2 in turn impairs HIF1
activity by competing with HIF1 for binding to the transcriptional
coactivator CBP/p300 (52). We
hypothesized that CITED2 also could be the mediator of the
phenotype observed in our cell line. Induction of CITED2 in
hypoxia was suppressed by FOXO3a knockdown (Fig. 6A). Nevertheless, a CITED2
knockdown did not display the protective phenotype of the
FOXO3a knockdown under hypoxic conditions (Fig. 6B). Regarding the glucose
consumption of cells transiently transfected with siRNA, a
CITED2 knockdown increased glucose consumption instead of
decreasing it (Fig. 6C).
Obviously, CITED2 was not the missing link between FOXO3a and HIF1
and did not account for either the diminished GLUT1
expression or finally the protection under starvation
conditions.
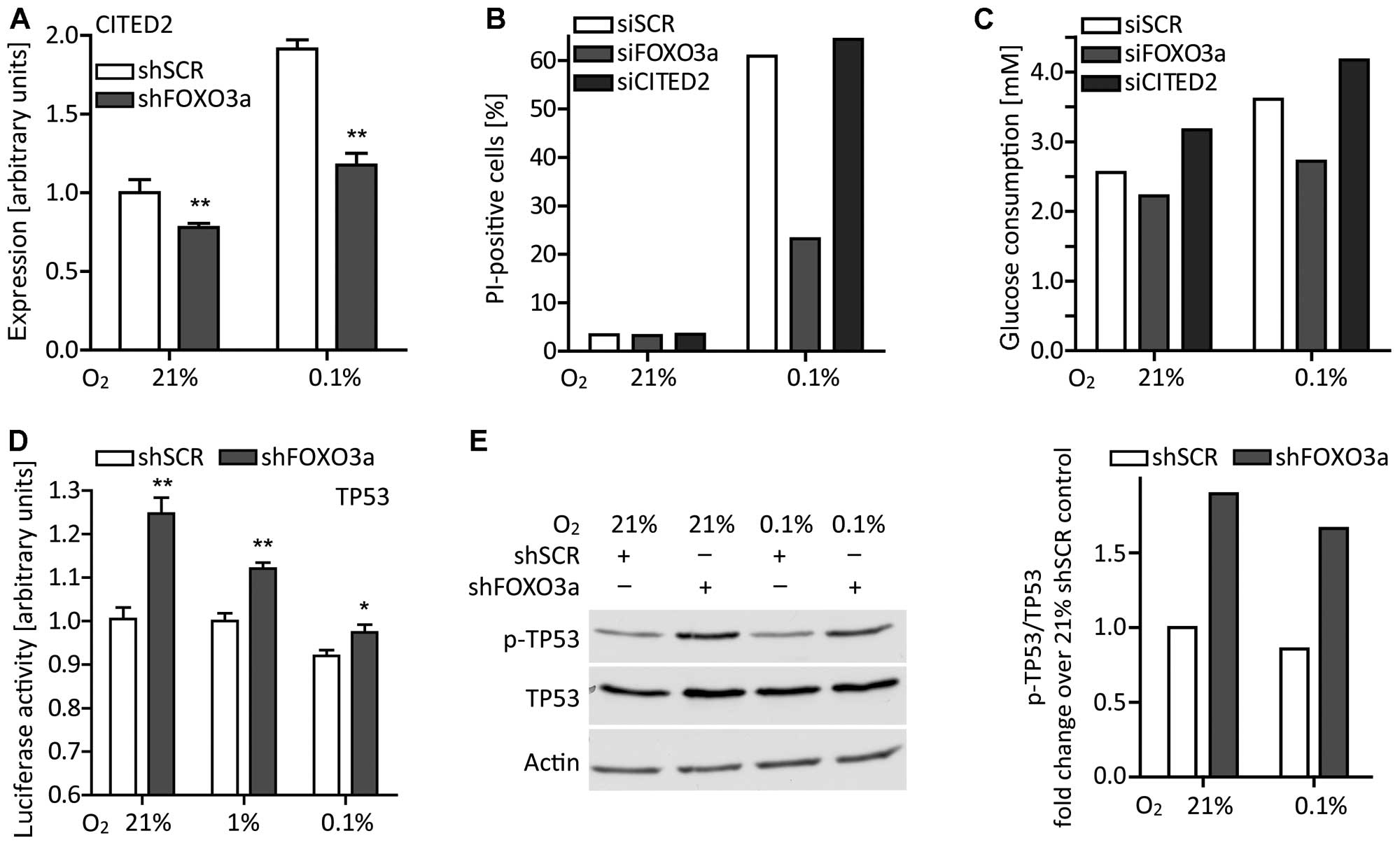 | Figure 6The effects of the FOXO3a
knockdown cannot be explained by the action of CITED2, but at least
partially by an interaction with TP53. (A) Under normoxic or
hypoxic conditions, LNT-229-shFOXO3a and control cells were grown
in serum-free medium supplemented with 5 mM glucose. After 24 h,
CITED2 expression was assessed by RT-qPCR (mean ± SEM,
**p<0.01). (B) LNT-229 cells transiently transfected with siRNA
targeting CITED2 were exposed to normoxia or hypoxia in
serum-free medium containing 2 mM glucose. After 24 h, cell death
was evaluated by PI staining and flow cytometry. Percentages of
PI-positive (dead) cells from one representative experiment are
shown. (C) Cells were grown in serum-free medium supplemented with
5 mM glucose and in normoxia or hypoxia. After 24 h, remaining
glucose concentrations were measured and glucose consumption was
calculated. Results from one representative experiment are shown.
(D) LNT-229-shFOXO3a and control cells were cultured in serum-free
medium containing 5 mM glucose and under different oxygen
conditions. After 24 h, TP53-mediated reporter gene expression was
examined (mean ± SEM, *p<0.05 and
**p<0.01). (E) In parallel, cell lysates were
analyzed for phosphorylated TP53 at serine 15 (p-TP53), total TP53
and, as a loading control, actin. Results of densitometric analysis
are expressed as p-TP53 relative to total TP53 and fold-change over
the 21% O2 condition of LNT-229-shSCR control cells. |
FOXO3a decreases TP53 transcriptional
activity
As GLUT1 expression is known to be
downregulated by TP53 (53) and
FOXO3a can promote the translocation of TP53 to the cytoplasm
(54), we reflected on TP53
potentially being involved in the reduced glucose consumption
associated with FOXO3a knockdown. Indeed, also in our glioma
cell line, FOXO3a knockdown increased TP53 transcriptional
activity (Fig. 6D) as well as its
phosphorylation at serine 15 (Fig.
6E) that is considered necessary for TP53 function (55). Considering our previous studies on
the role of TP53 in LNT-229 glioma cells (32), the interplay of FOXO3a and TP53 may
partially explain the phenotype observed under starvation
conditions because antagonizing TP53 enhances cell death when
availability of glucose and oxygen is restricted. Elevated TP53
activity in LNT-229-shFOXO3a cells could therefore improve cell
survival in unfavorable environments.
Discussion
Members of the FOXO family of transcription factors
are key players in the regulation of cell-fate decisions, such as
cell death, proliferation and metabolism. They are negatively
regulated by the serine/threonine kinase PKB that in turn is often
hyperactivated in glioblastoma, e.g., due to mutation and
inactivation of PTEN (56)
and upstream receptor tyrosine kinase signaling (12).
Our immunohistochemical studies on human glioma
tissue sections illustrated that FOXO3a expression increased with
WHO grade and was accentuated in perinecrotic tumor regions. This
finding disagrees with data published by Shi et al
suggesting lower FOXO3a levels in grade IV tumors (57). FOXO expression in areas of nutrient
deficiency may be comprehensible as a mechanism of survival in
nutrient-poor conditions when recalling the FOXO ortholog DAF-16 in
Caenorhabditis elegans and its facilitation of developmental
arrest at the dauer larval stage supporting survival until
environmental conditions improve (58). We analyzed 12 cell lines derived
from high grade gliomas and all of them were found to express
FOXO3a to a higher extent than normal brain. LNT-229 cells,
used for our further experiments and expressing moderate levels of
FOXO3a, were capable of modulating its expression, its
phosphorylation and its subcellular location and of regulating the
expression of its target genes. Hence, the FOXO3a system was
preserved and functional in our glioma cell line.
In the presence of enough glucose, a knockdown of
FOXO3a was detrimental to LNT-229 cells exposed to oxidative
stress: FOXO3a-dependent expression of MnSOD decreased,
intra-cellular ROS accumulated and cell death increased. Similarly
to our results, Bakker et al (36) and Jensen et al (21) observed an activation of FOXO3a in
hypoxia which promoted cell survival. However, in our glioma cell
line, upregulation of FOXO3a under hypoxic conditions was
not directly associated with HIF1 transcriptional activity.
In the absence of sufficient glucose, a knockdown of
FOXO3a turned out to be advantageous for glioma cell
survival by reducing the cells’ glucose and oxygen consumption. By
contrast, in neural stem/progenitor cells, FOXO3a knockout
led to increased respiration (59), and, in an immortalized retinal
pigment epithelial cell line, FOXO3a activation reduced oxygen
consumption rates (60). As
metabolism is known to be fine-tuned by numerous factors, those
differences may be ascribed to different experimental conditions or
cell-type specific characteristics.
Consistent with our observations, Emerling et
al described an increase in HIF1 transcriptional activity
following FOXO inactivation and the formation of a complex between
FOXO3a, HIF1 and p300 under hypoxic conditions resulting in a
suppression of HIF1 target genes (61). In our study, the impact of a
FOXO3a knockdown on GLUT1 was diametrically opposed to that
on other established HIF1 target genes. The downregulation of GLUT1
and the diminished glucose consumption could be attributed to a
strengthened activity of TP53 upon FOXO3a knockdown. In
previous studies, we observed that TP53 suppressed glycolysis and
induced cytochrome c oxidase assembly protein (SCO2) which
in turn promoted oxidative phosphorylation, reduced ROS levels and
thereby facilitated cell viability under starvation conditions
(32). TP53-induced glycolysis and
apoptosis regulator (TIGAR) activated the pentose phosphate pathway
(PPP), promoting NADPH production and protection against ROS
(62). TP53 and FOXO3a interrelate
with each other in various ways: Firstly, they share several target
genes, e.g. p53 upregulated modulator of apoptosis (PUMA),
cyclin-dependent kinase inhibitor 1A (CDKN1A) and growth
arrest and DNA damage-inducible 45 (GADD45) (63). Secondly, TP53 may promote FOXO3a
expression (64,65), but impair FOXO3a function
via serum- and glucocorticoid-inducible kinase 1 (SGK1) (66) as well as following oxidative stress
(67) and, via E3 ubiquitin
protein ligase (MDM2), endorse its degradation (68). Thirdly, FOXO3a can bind to TP53
(69) and compromise its
transcriptional activity (54).
This obviously complex regulatory system including feedback loops
allows to coordinate the cell’s response to environmental stress.
Known to interact with numerous transcription factors, CBP/p300 may
be the missing link between FOXO3a, HIF1 and TP53 (70). As its availability in the cell is
limited, multiple proteins compete with each other for binding to
CBP/p300 and thus modulate their action. Nevertheless, we did not
further address this topic and so it remains speculative.
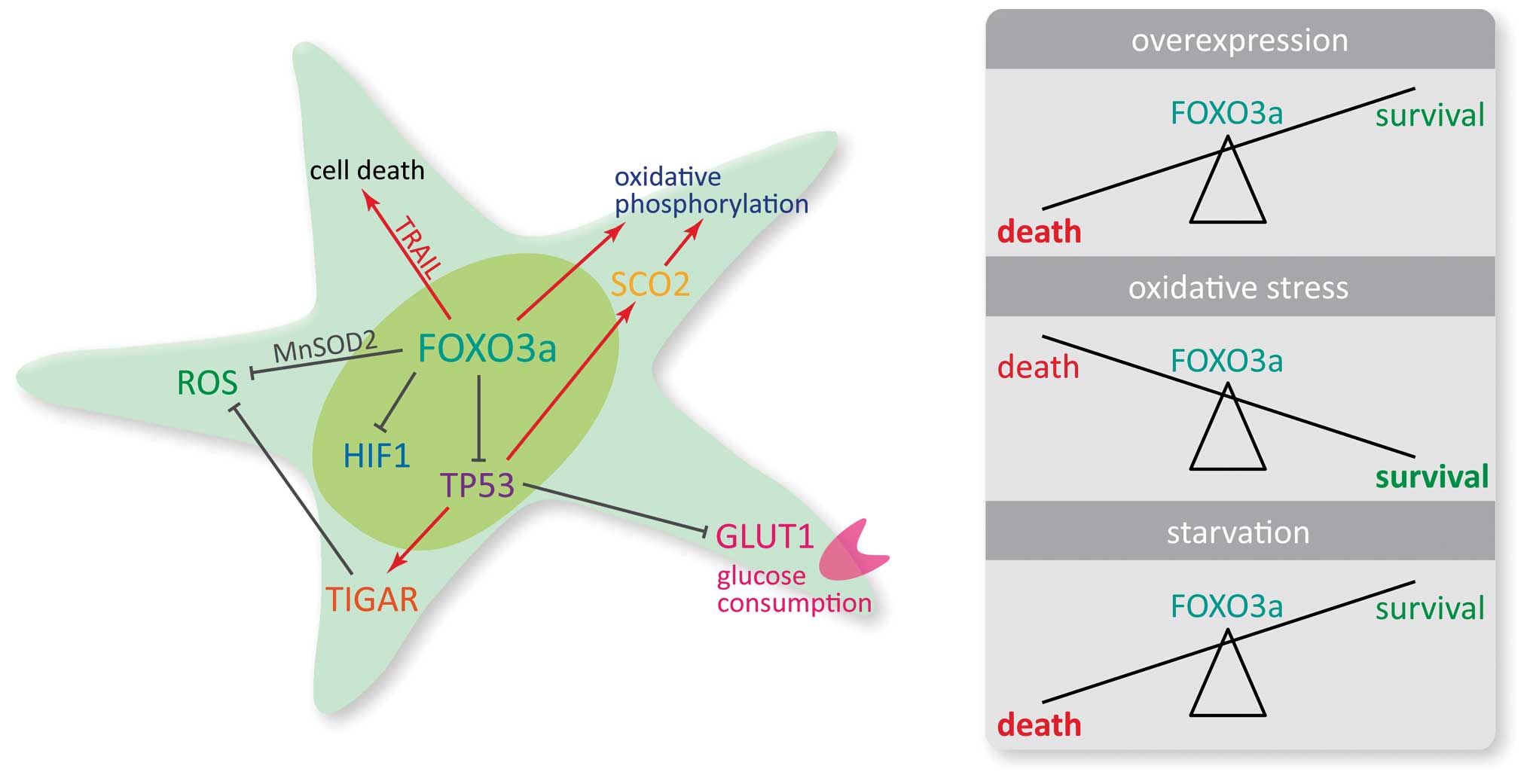
In conclusion, our results indicate three defined
effects of FOXO3a signaling depending on environmental conditions.
Firstly, overexpression of FOXO3a results in enhanced cell death
that is accompanied by increased expression of TRAIL.
Secondly, in the absence of other starvation signals, physiological
levels of FOXO3a prevent glioma cells from cell death following
oxidative stress, presumably by inducing MnSOD expression.
Thirdly, in situations of reduced oxygen and limited glucose
supply, FOXO3a sensitizes glioma cells to death by increasing
glucose and oxygen consumption. This phenotype may be attributable
to interactions with HIF1α and TP53 resulting in differential
regulation of target genes, in particular of GLUT1, which is
a key gene regulating uptake of glucose in cancer cells (71). The versatile role of FOXO3a in the
maintenance of metabolic homeostasis should therefore be taken into
consideration when targeting upstream regulators of FOXO3a
function, since they may profoundly alter the susceptibility of
glioma cells towards cell death. This could be of particular
importance in cells that inhabit the perinecrotic niche and may be
exposed to transient and recurrent episodes of hypoxia which have
been shown to promote tumor evolution and therapy resistance
(18,72–74).
Fig. 7 shows our findings on the
impact of FOXO3a on metabolic key parameters and cell
viability.
Acknowledgements
The Dr. Senckenberg Institute of Neurooncology was
supported by the Hertie foundation and the Dr. Senckenberg
foundation. JPS is ‘Hertie Professor for Neurooncology’.
Abbreviations:
|
2-NBDG
|
2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino)-2-deoxyglucose
|
|
BIM
|
Bcl-2-like protein 11
|
|
BrdU
|
5-bromo-2′-deoxyuridine
|
|
CBP
|
cAMP response element-binding
(CREB)-binding protein
|
|
CITED2
|
CBP/p300 interacting transactivator
with Glu/Asp rich carboxy-terminal domain 2
|
|
DAPI
|
4′,6-diamidino-2-phenylindole
|
|
GFP
|
green fluorescent protein
|
|
GLUT1
|
glucose transporter 1
|
|
HIF-1α
|
hypoxia-inducible factor-1α
|
|
IDH1
|
isocitrate dehydrogenase 1
|
|
MnSOD2
|
manganese superoxide dismutase
|
|
ROS
|
reactive oxygen species
|
|
SCO2
|
SCO2 cytochrome c oxidase
assembly protein
|
|
SDHA
|
succinate dehydrogenase complex,
subunit A
|
|
SEM
|
standard error of the mean
|
|
shRNA
|
short hairpin ribonucleic acid
|
|
siRNA
|
small interfering ribonucleic
acid
|
|
SLC16A3
|
solute carrier family 16, member 3
(monocarboxylic acid transporter 4)
|
|
TGF-β
|
transforming growth factor-β
|
|
TIGAR
|
TP53-induced glycolysis and apoptosis
regulator
|
|
TP53
|
tumor protein p53
|
|
TRAIL
|
tumor necrosis factor-related
apoptosis-inducing ligand
|
|
VEGF
|
vascular endothelial growth
factor
|
References
|
1
|
Coomans de Brachène A and Demoulin JB:
FOXO transcription factors in cancer development and therapy. Cell
Mol Life Sci. 73:1159–1172. 2016. View Article : Google Scholar
|
|
2
|
Eijkelenboom A and Burgering BMT: FOXOs:
Signalling integrators for homeostasis maintenance. Nat Rev Mol
Cell Biol. 14:83–97. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Charitou P, Rodriguez-Colman M, Gerrits J,
van Triest M, Groot Koerkamp M, Hornsveld M, Holstege F,
Verhoeven-Duif NM and Burgering BM: FOXOs support the metabolic
requirements of normal and tumor cells by promoting IDH1
expression. EMBO Rep. 16:456–466. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
van der Vos KE and Coffer PJ: The
extending network of FOXO transcriptional target genes. Antioxid
Redox Signal. 14:579–592. 2011. View Article : Google Scholar
|
|
5
|
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo
P, Hu LS, Anderson MJ, Arden KC, Blenis J and Greenberg ME: Akt
promotes cell survival by phosphorylating and inhibiting a Forkhead
transcription factor. Cell. 96:857–868. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ho KK, Myatt SS and Lam EW: Many forks in
the path: Cycling with FoxO. Oncogene. 27:2300–2311. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Naka K, Hoshii T, Muraguchi T, Tadokoro Y,
Ooshio T, Kondo Y, Nakao S, Motoyama N and Hirao A: TGF-beta-FOXO
signalling maintains leukaemia-initiating cells in chronic myeloid
leukaemia. Nature. 463:676–680. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tenbaum SP, Ordóñez-Morán P, Puig I,
Chicote I, Arqués O, Landolfi S, Fernández Y, Herance JR, Gispert
JD, Mendizabal L, et al: β-catenin confers resistance to PI3K and
AKT inhibitors and subverts FOXO3a to promote metastasis in colon
cancer. Nat Med. 18:892–901. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Calnan DR and Brunet A: The FoxO code.
Oncogene. 27:2276–2288. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
van der Horst A and Burgering BM:
Stressing the role of FoxO proteins in lifespan and disease. Nat
Rev Mol Cell Biol. 8:440–450. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Van Der Heide LP, Hoekman MF and Smidt MP:
The ins and outs of FoxO shuttling: Mechanisms of FoxO
translocation and transcriptional regulation. Biochem J.
380:297–309. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cancer Genome Atlas Research Network.
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sunayama J, Sato A, Matsuda K, Tachibana
K, Watanabe E, Seino S, Suzuki K, Narita Y, Shibui S, Sakurada K,
et al: FoxO3a functions as a key integrator of cellular signals
that control glioblastoma stem-like cell differentiation and
tumorigenicity. Stem Cells. 29:1327–1337. 2011.PubMed/NCBI
|
|
14
|
Lau CJ, Koty Z and Nalbantoglu J:
Differential response of glioma cells to FOXO1-directed therapy.
Cancer Res. 69:5433–5440. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Masui K, Tanaka K, Akhavan D, Babic I,
Gini B, Matsutani T, Iwanami A, Liu F, Villa GR, Gu Y, et al: mTOR
complex 2 controls glycolytic metabolism in glioblastoma through
FoxO acetylation and upregulation of c-Myc. Cell Metab. 18:726–739.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Firat E and Niedermann G: FoxO proteins or
loss of functional p53 maintain stemness of glioblastoma stem cells
and survival after ionizing radiation plus PI3K/mTOR inhibition.
Oncotarget. Jul 19–2016.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Clark PM, Mai WX, Cloughesy TF and
Nathanson DA: Emerging approaches for targeting metabolic
vulnerabilities in malignant glioma. Curr Neurol Neurosci Rep.
16:172016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liebelt BD, Shingu T, Zhou X, Ren J, Shin
SA and Hu J: Glioma stem cells: Signaling, microenvironment, and
therapy. Stem Cells Int. 2016:78498902016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Province P, Griguer CE, Han X, Nabors LB
and Shaykh HF: Hypoxia, angiogenesis and mechanisms for invasion of
malignant gliomas. Evolution of the Molecular Biology of Brain
Tumors and the Therapeutic Implications. Lichtor T: InTech; Rijeka:
2013, View Article : Google Scholar
|
|
21
|
Jensen KS, Binderup T, Jensen KT,
Therkelsen I, Borup R, Nilsson E, Multhaupt H, Bouchard C,
Quistorff B, Kjaer A, et al: FoxO3A promotes metabolic adaptation
to hypoxia by antagonizing Myc function. EMBO J. 30:4554–4570.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ronellenfitsch MW, Brucker DP, Burger MC,
Wolking S, Tritschler F, Rieger J, Wick W, Weller M and Steinbach
JP: Antagonism of the mammalian target of rapamycin selectively
mediates metabolic effects of epidermal growth factor receptor
inhibition and protects human malignant glioma cells from
hypoxia-induced cell death. Brain. 132:1509–1522. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Berra E, Benizri E, Ginouvès A, Volmat V,
Roux D and Pouysségur J: HIF prolyl-hydroxylase 2 is the key oxygen
sensor setting low steady-state levels of HIF-1alpha in normoxia.
EMBO J. 22:4082–4090. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Skurk C, Maatz H, Kim HS, Yang J, Abid MR,
Aird WC and Walsh K: The Akt-regulated forkhead transcription
factor FOXO3a controls endothelial cell viability through
modulation of the caspase-8 inhibitor FLIP. J Biol Chem.
279:1513–1525. 2004. View Article : Google Scholar
|
|
25
|
Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang
F, Yang JY, Zou Y, Bao S, Hanada N, Saso H, et al: IkappaB kinase
promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell.
117:225–237. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nakamura N, Ramaswamy S, Vazquez F,
Signoretti S, Loda M and Sellers WR: Forkhead transcription factors
are critical effectors of cell death and cell cycle arrest
downstream of PTEN. Mol Cell Biol. 20:8969–8982. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tran H, Brunet A, Grenier JM, Datta SR,
Fornace AJ Jr, DiStefano PS, Chiang LW and Greenberg ME: DNA repair
pathway stimulated by the forkhead transcription factor FOXO3a
through the Gadd45 protein. Science. 296:530–534. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Brummelkamp TR, Bernards R and Agami R: A
system for stable expression of short interfering RNAs in mammalian
cells. Science. 296:550–553. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Maurer GD, Tritschler I, Adams B,
Tabatabai G, Wick W, Stupp R and Weller M: Cilengitide modulates
attachment and viability of human glioma cells, but not sensitivity
to irradiation or temozolomide in vitro. Neuro Oncol. 11:747–756.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Henze AT, Riedel J, Diem T, Wenner J,
Flamme I, Pouyseggur J, Plate KH and Acker T: Prolyl hydroxylases 2
and 3 act in gliomas as protective negative feedback regulators of
hypoxia-inducible factors. Cancer Res. 70:357–366. 2010. View Article : Google Scholar
|
|
31
|
Wagenknecht B, Schulz JB, Gulbins E and
Weller M: Crm-A, bcl-2 and NDGA inhibit CD95L-induced apoptosis of
malignant glioma cells at the level of caspase 8 processing. Cell
Death Differ. 5:894–900. 1998. View Article : Google Scholar
|
|
32
|
Wanka C, Brucker DP, Bähr O,
Ronellenfitsch M, Weller M, Steinbach JP and Rieger J: Synthesis of
cytochrome c oxidase 2: A p53-dependent metabolic regulator that
promotes respiratory function and protects glioma and colon cancer
cells from hypoxia-induced cell death. Oncogene. 31:3764–3776.
2012. View Article : Google Scholar
|
|
33
|
Dyer BW, Ferrer FA, Klinedinst DK and
Rodriguez R: A noncommercial dual luciferase enzyme assay system
for reporter gene analysis. Anal Biochem. 282:158–161. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schneider CA, Rasband WS and Eliceiri KW:
NIH Image to ImageJ: 25 years of image analysis. Nat Methods.
9:671–675. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Obexer P, Geiger K, Ambros PF, Meister B
and Ausserlechner MJ: FKHRL1-mediated expression of Noxa and Bim
induces apoptosis via the mitochondria in neuroblastoma cells. Cell
Death Differ. 14:534–547. 2007. View Article : Google Scholar
|
|
36
|
Bakker WJ, Harris IS and Mak TW: FOXO3a is
activated in response to hypoxic stress and inhibits HIF1-induced
apoptosis via regulation of CITED2. Mol Cell. 28:941–953. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ullah MS, Davies AJ and Halestrap AP: The
plasma membrane lactate transporter MCT4, but not MCT1, is
up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J
Biol Chem. 281:9030–9037. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Harter PN, Jennewein L, Baumgarten P,
Ilina E, Burger MC, Thiepold AL, Tichy J, Zörnig M, Senft C,
Steinbach JP, et al: Immunohistochemical assessment of
phosphorylated mTORC1-pathway proteins in human brain tumors. PLoS
One. 10:e01271232015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rong Y, Durden DL, Van Meir EG and Brat
DJ: ‘Pseudopalisading’ necrosis in glioblastoma: A familiar
morphologic feature that links vascular pathology, hypoxia, and
angiogenesis. J Neuropathol Exp Neurol. 65:529–539. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ghaffari S, Jagani Z, Kitidis C, Lodish HF
and Khosravi-Far R: Cytokines and BCR-ABL mediate suppression of
TRAIL-induced apoptosis through inhibition of forkhead FOXO3a
transcription factor. Proc Natl Acad Sci USA. 100:6523–6528. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Modur V, Nagarajan R, Evers BM and
Milbrandt J: FOXO proteins regulate tumor necrosis factor-related
apoptosis inducing ligand expression. Implications for PTEN
mutation in prostate cancer. J Biol Chem. 277:47928–47937. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Warr MR, Binnewies M, Flach J, Reynaud D,
Garg T, Malhotra R, Debnath J and Passegué E: FOXO3A directs a
protective autophagy program in haematopoietic stem cells. Nature.
494:323–327. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Biggs WH III, Cavenee WK and Arden KC:
Identification and characterization of members of the FKHR (FOX O)
subclass of winged-helix transcription factors in the mouse. Mamm
Genome. 12:416–425. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kops GJ and Burgering BM: Forkhead
transcription factors: New insights into protein kinase B (c-akt)
signaling. J Mol Med (Berl). 77:656–665. 1999. View Article : Google Scholar
|
|
45
|
Chong ZZ, Li F and Maiese K: Activating
Akt and the brain’s resources to drive cellular survival and
prevent inflammatory injury. Histol Histopathol. 20:299–315.
2005.
|
|
46
|
Kops GJ, Dansen TB, Polderman PE, Saarloos
I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH and Burgering
BM: Forkhead transcription factor FOXO3a protects quiescent cells
from oxidative stress. Nature. 419:316–321. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Steinbach JP, Wolburg H, Klumpp A, Probst
H and Weller M: Hypoxia-induced cell death in human malignant
glioma cells: Energy deprivation promotes decoupling of
mitochondrial cytochrome c release from caspase processing and
necrotic cell death. Cell Death Differ. 10:823–832. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yoshioka K, Takahashi H, Homma T, Saito M,
Oh KB, Nemoto Y and Matsuoka H: A novel fluorescent derivative of
glucose applicable to the assessment of glucose uptake activity of
Escherichia coli. Biochim Biophys Acta. 1289:5–9. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ramaswamy S, Nakamura N, Sansal I,
Bergeron L and Sellers WR: A novel mechanism of gene regulation and
tumor suppression by the transcription factor FKHR. Cancer Cell.
2:81–91. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Semenza GL: HIF-1 mediates metabolic
responses to intra-tumoral hypoxia and oncogenic mutations. J Clin
Invest. 123:3664–3671. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Carruthers A, DeZutter J, Ganguly A and
Devaskar SU: Will the original glucose transporter isoform please
stand up! Am J Physiol Endocrinol Metab. 297:E836–E848. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bhattacharya S, Michels CL, Leung MK,
Arany ZP, Kung AL and Livingston DM: Functional role of p35srj, a
novel p300/CBP binding protein, during transactivation by HIF-1.
Genes Dev. 13:64–75. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Schwartzenberg-Bar-Yoseph F, Armoni M and
Karnieli E: The tumor suppressor p53 down-regulates glucose
transporters GLUT1 and GLUT4 gene expression. Cancer Res.
64:2627–2633. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
You H, Yamamoto K and Mak TW: Regulation
of transactivation-independent proapoptotic activity of p53 by
FOXO3a. Proc Natl Acad Sci USA. 103:9051–9056. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Loughery J, Cox M, Smith LM and Meek DW:
Critical role for p53-serine 15 phosphorylation in stimulating
transactivation at p53-responsive promoters. Nucleic Acids Res.
42:7666–7680. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Parsons DW, Jones S, Zhang X, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et
al: An integrated genomic analysis of human glioblastoma
multiforme. Science. 321:1807–1812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Shi J, Zhang L, Shen A, Zhang J, Wang Y,
Zhao Y, Zou L, Ke Q, He F, Wang P, et al: Clinical and biological
significance of forkhead class box O 3a expression in glioma:
Mediation of glioma malignancy by transcriptional regulation of
p27kip1. J Neurooncol. 98:57–69. 2010. View Article : Google Scholar
|
|
58
|
Hsu AL, Murphy CT and Kenyon C: Regulation
of aging and age-related disease by DAF-16 and heat-shock factor.
Science. 300:1142–1145. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yeo H, Lyssiotis CA, Zhang Y, Ying H,
Asara JM, Cantley LC and Paik JH: FoxO3 coordinates metabolic
pathways to maintain redox balance in neural stem cells. EMBO J.
32:2589–2602. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ferber EC, Peck B, Delpuech O, Bell GP,
East P and Schulze A: FOXO3a regulates reactive oxygen metabolism
by inhibiting mitochondrial gene expression. Cell Death Differ.
19:968–979. 2012. View Article : Google Scholar :
|
|
61
|
Emerling BM, Weinberg F, Liu JL, Mak TW
and Chandel NS: PTEN regulates p300-dependent hypoxia-inducible
factor 1 transcriptional activity through Forkhead transcription
factor 3a (FOXO3a). Proc Natl Acad Sci USA. 105:2622–2627. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wanka C, Steinbach JP and Rieger J:
Tp53-induced glycolysis and apoptosis regulator (TIGAR) protects
glioma cells from starvation-induced cell death by up-regulating
respiration and improving cellular redox homeostasis. J Biol Chem.
287:33436–33446. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Riley T, Sontag E, Chen P and Levine A:
Transcriptional control of human p53-regulated genes. Nat Rev Mol
Cell Biol. 9:402–412. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kurinna S, Stratton SA, Tsai WW, Akdemir
KC, Gu W, Singh P, Goode T, Darlington GJ and Barton MC: Direct
activation of forkhead box O3 by tumor suppressors p53 and p73 is
disrupted during liver regeneration in mice. Hepatology.
52:1023–1032. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Renault VM, Thekkat PU, Hoang KL, White
JL, Brady CA, Kenzelmann Broz D, Venturelli OS, Johnson TM, Oskoui
PR, Xuan Z, et al: The pro-longevity gene FoxO3 is a direct target
of the p53 tumor suppressor. Oncogene. 30:3207–3221. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
You H, Jang Y, You-Ten AI, Okada H, Liepa
J, Wakeham A, Zaugg K and Mak TW: p53-dependent inhibition of
FKHRL1 in response to DNA damage through protein kinase SGK1. Proc
Natl Acad Sci USA. 101:14057–14062. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Miyaguchi Y, Tsuchiya K and Sakamoto K:
p53 negatively regulates the transcriptional activity of FOXO3a
under oxidative stress. Cell Biol Int. 33:853–860. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Fu W, Ma Q, Chen L, Li P, Zhang M,
Ramamoorthy S, Nawaz Z, Shimojima T, Wang H, Yang Y, et al: MDM2
acts downstream of p53 as an E3 ligase to promote FOXO
ubiquitination and degradation. J Biol Chem. 284:13987–14000. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wang F, Marshall CB, Yamamoto K, Li GY,
Plevin MJ, You H, Mak TW and Ikura M: Biochemical and structural
characterization of an intramolecular interaction in FOXO3a and its
binding with p53. J Mol Biol. 384:590–603. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Wang F, Marshall CB and Ikura M:
Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis:
Structural and functional versatility in target recognition. Cell
Mol Life Sci. 70:3989–4008. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Chan DA, Sutphin PD, Nguyen P, Turcotte S,
Lai EW, Banh A, Reynolds GE, Chi JT, Wu J, Solow-Cordero DE, et al:
Targeting GLUT1 and the Warburg effect in renal cell carcinoma by
chemical synthetic lethality. Sci Transl Med. 3:94ra702011.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Horsman MR and Vaupel P:
Pathophysiological basis for the formation of the tumor
microenvironment. Front Oncol. 6:662016. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
LaGory EL and Giaccia AJ: The
ever-expanding role of HIF in tumour and stromal biology. Nat Cell
Biol. 18:356–365. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Weinmann M, Belka C, Güner D, Goecke B,
Müller I, Bamberg M and Jendrossek V: Array-based comparative gene
expression analysis of tumor cells with increased apoptosis
resistance after hypoxic selection. Oncogene. 24:5914–5922. 2005.
View Article : Google Scholar : PubMed/NCBI
|