Introduction
Osteosarcoma is the most common primary malignant
bone tumor in adolescents and young adults. Currently, the most
effective therapeutic option for the osteosarcoma is surgical
resection of all clinically detectable sites with systemic therapy
to control microscopic metastatic disease. Many studies have
reported significant advances in adjuvant chemotherapy for
osteosarcoma (1,2), however, in the last two decades,
treatment outcomes for patients with osteosarcoma have not improved
sufficiently, despite the implementation of several new therapeutic
interventions. In a comprehensive analysis of published phase I/II
clinical trials, researchers found that effective drugs tended to
have high toxicity (3). Therefore,
new therapeutic strategies for the treatment of osteosarcoma are
needed.
Mitochondria are cytoplasmic organelles that play
essential roles in cellular energy metabolism and programmed cell
death (4), and regulation of both
mitochondrial dysfunction and associated cellular biogenetics has
recently been identified as a promising target for anticancer
therapy (5). Moreover,
quantitative changes in mitochondrial numbers have been observed in
various cancers, with a decrease in hepatocellular carcinoma, renal
cell carcinoma, advanced gastric cancer and breast cancer (6–11)
and with an increase in head and neck cancers, ovarian cancer, and
esophageal squamous cell carcinoma (12–14).
Additionally, decreased mitochondrial numbers have been reported to
be associated with tumor progression and prognosis in patients with
hepatocellular carcinoma and breast cancer (6,7). We
previously reported that mitochondrial numbers were significantly
reduced in human sarcoma tissues, including osteosarcoma, compared
with that in normal muscles or benign musculoskeletal tumor
tissues, and that decreased mitochondrial numbers may be associated
with tumor progression in musculoskeletal malignancies (15). In mitochondrial biogenesis,
peroxisome proliferator-activated receptor-γ coactivator-1α
(PGC-1α) regulates the transcription of the gene coding
mitochondrial transcription factor A (TFAM), which reflects the
changing levels of mitochondrial DNA (mtDNA) in the cell, and plays
a crucial role in mtDNA maintenance (16). We have recently reported that
increasing mitochondrial numbers by forced expression of PGC-1α
could induce mitochondrial apoptosis in human sarcoma cell lines
(15), and the findings suggested
that mitochondrial biogenesis and/or numbers could be a therapeutic
target for human sarcomas. 5-Aminoimidazole-4-carboxiamide
ribonucleotide (AICAR) is widely used as a pharmacologic activator
of the energy-regulating enzyme AMP-activated protein kinase α
(AMPKα) (17,18). AICAR has been also reported to
stimulate mitochondrial biogenesis via PGC-1α activation in
skeletal muscles (19) and to be
an exercise mimetic (20).
Based on these previous studies, we hypothesized
that AICAR could increase PGC-1α expression through AMPK activation
and could induce mitochondrial apoptosis through the
PGC-1α/TFAM/mitochondrial pathway in tumor tissues similarly to
muscle tissues. Moreover, we expected that AICAR may show
anticancer effects in osteosarcoma cells. Therefore, in this study,
we evaluated the effects of AICAR on apoptotic activity through
AMPK phosphorylation in human osteosarcoma cells both in
vitro and in vivo.
Materials and methods
Reagent
AICAR was obtained from Abcam (Cambridge, UK)
(ab120358, Lot: APN12423-1-1;) for cell experiments, and LC
Laboratories (Woburn, MA, USA) (A-1098, Lot: ACR-101) for animal
experiments. AICAR was dissolved in distilled water and immediately
stored at −20°C. This stock solution was diluted in culture medium
for in vitro or saline for in vivo experiments,
immediately before use.
Cells
Two human osteosarcoma cell lines (MG63 and KHOS)
were used in this study. MG63 cells were obtained from the RIKEN
BRC through the National Bio-Resource Project of the MEXT (Ibaraki,
Japan), and KHOS cells were obtained from American Type Culture
Collection (ATCC, Manassas, VA, USA). Both cell lines were
routinely cultured in Dulbecco's modified Eagle's medium (DMEM)
containing 10% fetal bovine serum (FBS) and 100 U/ml
penicillin/streptomycin solution (all from Sigma-Aldrich, St.
Louis, MO, USA) at 37°C in a humidified 5% CO2
atmosphere. For all experiments, we used DMEM containing 10% FBS
without the antibiotic solution.
Animal studies
All animal experiments were approved by Kobe
University Animal Experimentation Regulations (permission no.
P-140504). Male BALB/c nude mice, aged 5 weeks, were purchased from
CLEA Japan Inc. (Tokyo, Japan) and were maintained in a facility
under specific pathogen-free conditions. Animals were fed
pathogen-free laboratory chow and allowed free access to autoclaved
water in an air-conditioned room with a 12-h light/dark cycle. For
in vivo experiments, both MG63 and KHOS osteosarcoma cells
were implanted into the dorsal, subcutaneous area of mice (n=12 for
each cell line) at a dose of 2.×106 cells in 500
µl phosphate-buffered saline (PBS), as previously described
(21), and mice were randomly
divided into two groups: the AICAR-treated group (n=6 for each cell
line) and the control group (n=6 for each cell line). Treatment
with 450 mg/kg AICAR or saline (as a control) by intraperitoneal
injection was started after 1 week of cell implantation and was
performed every day for 2 weeks. Tumor volume was calculated twice
a week, as previously described, according to the formula
V=π/6xa2xb, where a and b represent the shorter and
longer dimensions of the tumor, respectively (21). The body weight of each mouse was
also monitored throughout the experiment. At the end of the
experiment, all tumors were excised and immediately stored at
−80°C, and mitochondrial proliferation and apoptotic activity in
tumor tissues were evaluated by flow cytometry, immunoblotting, and
immunofluorescence staining.
Immunoblot analysis
Cell lysates were extracted from cells or implanted
tumors using whole cell lysis buffer (Mammalian Protein Extraction
reagent; Thermo Scientific, Rockford, IL, USA) and protein content
was quantified using BCA protein assay reagent (Bio-Rad, Hercules,
CA, USA). Samples containing equal amounts of protein were
electrophoresed by sodium dodecyl sulphate polyacrylamide gel
electrophoresis (SDS-PAGE) on 7.5–15% gradient gels and transferred
to polyvinylidene fluoride membranes. After blocking, membranes
were incubated overnight at 4°C with primary antibodies in CanGet
Signal Solution 1 (Toyobo Co., Ltd., Osaka, Japan). The following
primary antibodies were used in this study: anti-human
phospho-AMPKα (Thr172) antibody (1:1,000) (2531S, Lot: 12),
anti-human AMPKα antibody (1:1,000) (2532S, Lot: 19) (both from
Cell Signaling Technology, Danvers, MA, USA), anti-human PGC-1α
antibody (1:1,000) (H000110891-M12, Lot: 08162-3G11), anti-human
TFAM antibody (1:1,000) (H00007019-B01P, Lot: E1281) (both from
Abnova, Walnut, CA, USA), anti-human poly(ADP-ribose) polymerase
(PARP) antibody (1:1,000) (9542S, Lot: 13), anti-human cleaved PARP
antibody (1:1,000) (5625S, Lot: 8), anti-human caspase-3 antibody
(1:1,000) (9668S, Lot: 7), anti-human cleaved caspase-3 antibody
(1:1,000) (9664S, Lot: 20), anti-human caspase-9 antibody (1:1,000)
(9502S, Lot:8), anti-human cleaved caspase-9 antibody (1:500)
(7237S, Lot: 1) (all from Cell Signaling Technology), and
anti-human α-tubulin antibody (1:10,000) (T9026, Lot: 092M4792;
Sigma-Aldrich). After washing, membranes were incubated with the
appropriate secondary antibody conjugated to horseradish peroxidase
and were exposed with ECL Plus Western Blot Detection system
reagents (GE Healthcare Biosciences, Piscataway, NJ, USA), and
protein expression was detected using a Chemilumino Analyzer
LAS-3000 mini (Fujifilm, Tokyo, Japan). Membranes were reprobed
with anti-human α-tubulin antibody (Sigma-Aldrich) to confirm equal
protein loading. Positive bands in immunoblot analyses were
semiquantified by densitometrical analyses using the ImageJ
software (version 1.47; NIH, Bethesda, MD, USA) (http://rsbweb.nih.gov/ij/download.html).
Values were normalized against α-tubulin and presented as a
ratio.
Quantitative real-time polymerase chain
reaction (qPCR)
To evaluate mitochondrial numbers in AICAR-treated
osteosarcoma cell lines, we examined the relative amount of mtDNA
to nuclear DNA (nDNA). Genomic DNA was isolated from the cells
using a GenElute Mammalian Genomic DNA Miniprep kit
(Sigma-Aldrich). The sequences of the primers designed to amplify a
region corresponding to nn 16-408 of a D-loop of human mtDNA were
as follows: 5′-GCAGATTTGGGTACCACCCAAGTATTGACTCACCC-3′ (forward) and
5′-GCATGGAGAGCTCCCGTGAGTGGTTAATAGGGTGATAG-3′ (reverse). The PCR
conditions were as follows: 1 cycle at 95°C for 15 min, followed by
30 cycles at 95°C for 30 sec, 58°C for 30 sec, and 72°C for 90 sec
(22). The relative amount of
mtDNA to nDNA was then analysed using the ΔΔCt method.
Cell proliferation assays
To evaluate the effects of AICAR on in vitro
osteosarcoma cell proliferation, we performed WST-8 cell
proliferation assays using a cell counting kit-8 (CCK-8; Dojindo
Inc., Kumamoto, Japan). Cells were seeded in 96-well culture plates
at a density of 5,000 cells/well in 100 µl medium and
incubated with various concentrations of AICAR (0–2,000 µM).
At the indicated incubation times, 10 µl of the CCK-8
solution was added into each well and incubated for 1 h. Then, the
optical density was measured at a wavelength of 450 nm using a
Model 680 Microplate Reader (Bio-Rad), and the relative number of
viable cells in each well was calculated.
Immunofluorescence staining
Immunofluorescence staining was performed to verify
the relationship between mitochondrial proliferation and cellular
apoptosis in AICAR-treated osteosarcoma cells and implanted tumors
using an APO-Direct kit (BD Pharmingen, Franklin Lakes, NJ, USA)
and a MitoTracker Deep-Red FM (Invitrogen, Carlsbad, CA, USA)
according to the manufacturers' protocols. In vitro, cells
were incubated with or without AICAR and then fixed in 4%
paraformaldehyde for 30 min at room temperature. Cells were
incubated in the prepared DNA Labelling Solution (APO-Direct kit;
BD Pharmingen) for 1 h, and were then incubated in the MitoTracker
Deep-Red FM (Invitrogen) for 30 min. Nuclear staining was performed
using a propidium iodide, and stained cells were assessed using a
BZ-8000 confocal microscope (Keyence, Osaka, Japan). In
vivo, tumor tissue samples were embedded in an OCT compound
(Sakura Finetek Co., Tokyo, Japan), and 10-µm-thick sections
were prepared on a cryostat and stored frozen at −80°C. Sections
were incubated with anti-actin antibodies (Sigma-Aldrich) diluted
in PBS for 30 min at 37°C. After washing, sections were incubated
with the APO-Direct kit reagents (BD Pharmingen) and the
MitoTracker Deep-Red FM (Invitrogen) in PBS for 30 min in a dark
humid chamber at 37°C. The nucleus was stained with the
4′,6-diamidino-2-phenylindole (DAPI). Fluorescence images were
obtained using a BZ-8000 confocal microscope (Keyence), and
positive staining was semiquantified using ImageJ software.
Flow cytometric analysis
We also assessed apoptotic activity in AICAR-treated
cells or implanted tumors by flow cytometry. Briefly, cells were
collected from cultured cells or implanted tumors, suspended in 1%
paraformaldehyde in PBS, and resuspended in ice-cold ethanol at a
concentration of 1×106 cells/ml. Each cell pellet was
labelled using the APO-Direct kit (BD Pharmingen) according to the
manufacturers' protocols. Fluorescent intensity was analysed using
a BD FACSVerse (BD Biosciences, Franklin Lakes, NJ, USA).
Statistical analysis
Each experiment was performed independently at least
three times. All values are expressed as the mean ± standard error
of the mean (SEM). Comparisons between two continuous values were
made using one-way analysis of variance. Post-hoc analysis was
performed by Fisher's protected least significant difference test.
For distributed data, p-values <0.05 were considered
significant.
Results
AICAR increases AMPK phosphorylation and
induces mitochondrial proliferation through the PGC-1α/TFAM pathway
in osteosarcoma cells
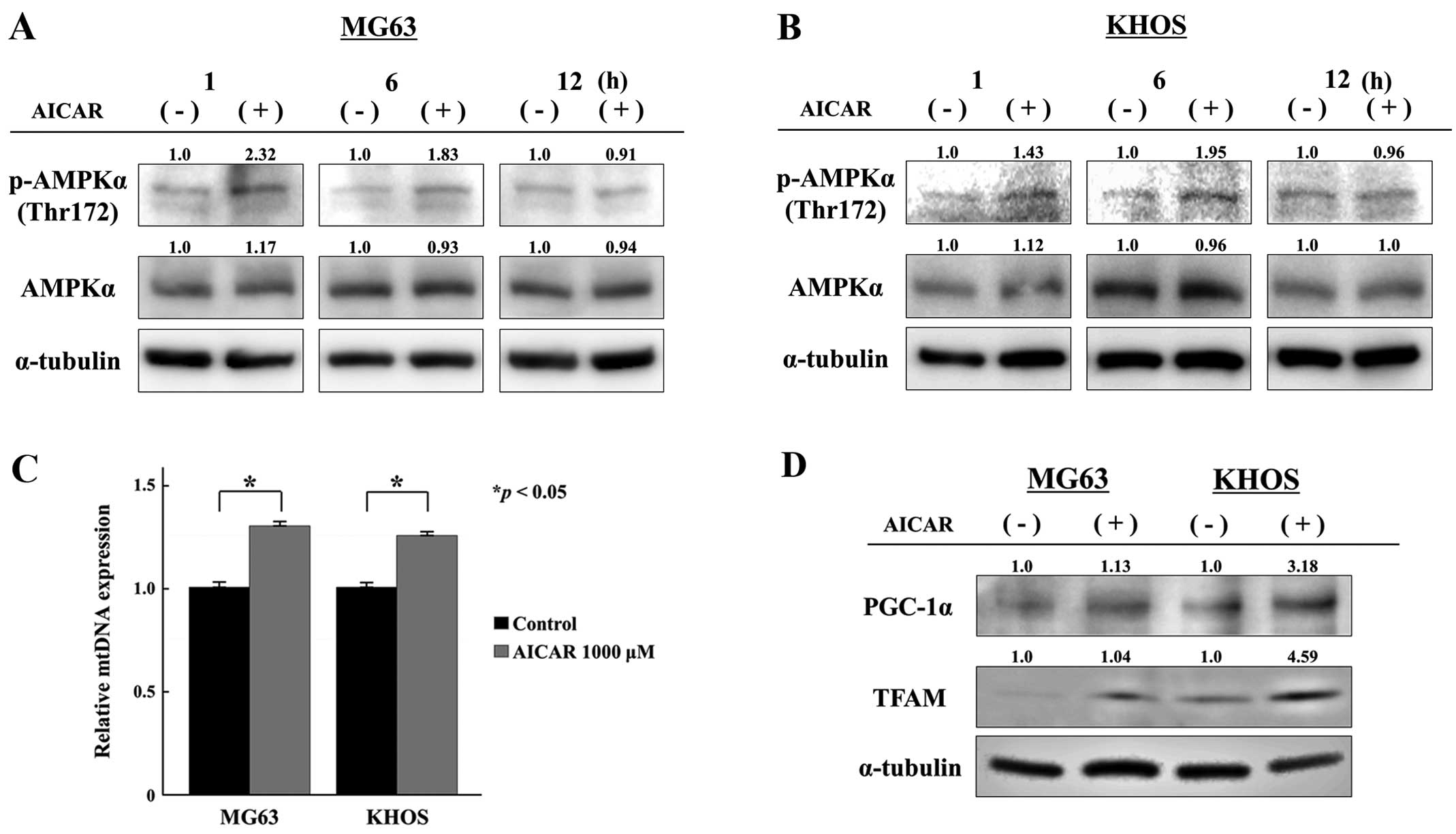
We first examined the effects of AICAR on AMPK
phosphorylation and mitochondrial biogenesis in osteosarcoma cells
in vitro. As shown in Fig.
1, the expression of phosphorylated AMPKα (Thr172) was strongly
increased immediately after AICAR treatment in both MG63 and KHOS
osteosarcoma cells (Fig. 1A and
B). Additionally, AICAR significantly increased the relative
number of mtDNA copies in both cell lines (Fig. 1C). Consistent with these results,
the expression levels of the mitochondria-related genes, PGC-1α and
TFAM, were increased in AICAR-treated osteosarcoma cells compared
with those in control cells (Fig.
1D). These findings strongly suggested that AICAR could induce
AMPK activation and increase mitochondrial biogenesis through the
PGC-1α/TFAM pathway in osteosarcoma cells.
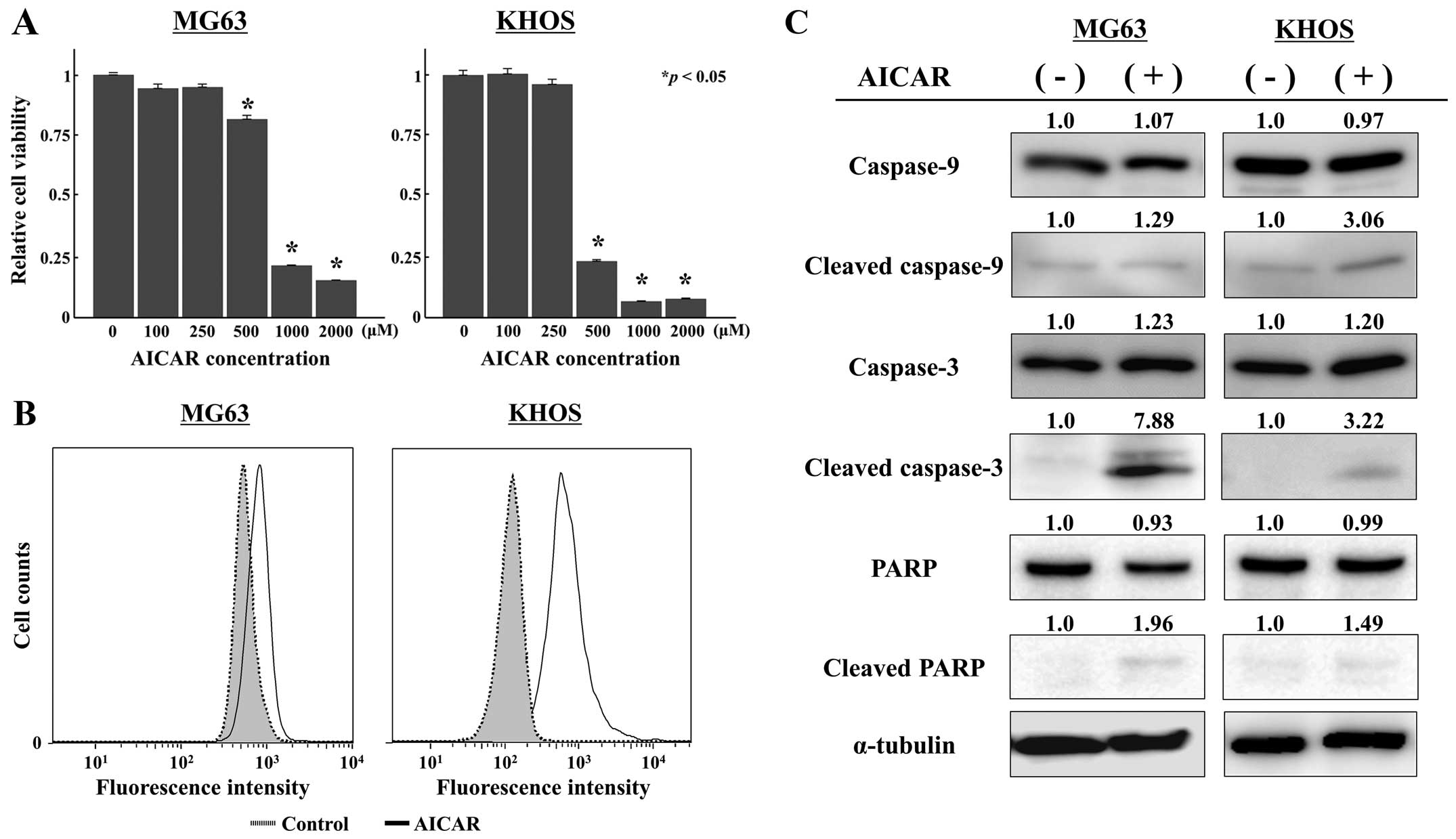
AICAR decreases osteosarcoma cell growth
and induces mitochondrial apoptosis and proliferation
To test the effects of mitochondrial proliferation
by AICAR on osteosarcoma cell growth and apoptosis, we assessed
cell viability and apoptotic activity in osteosarcoma cells after
AICAR treatment. AICAR showed concentration-dependent inhibitory
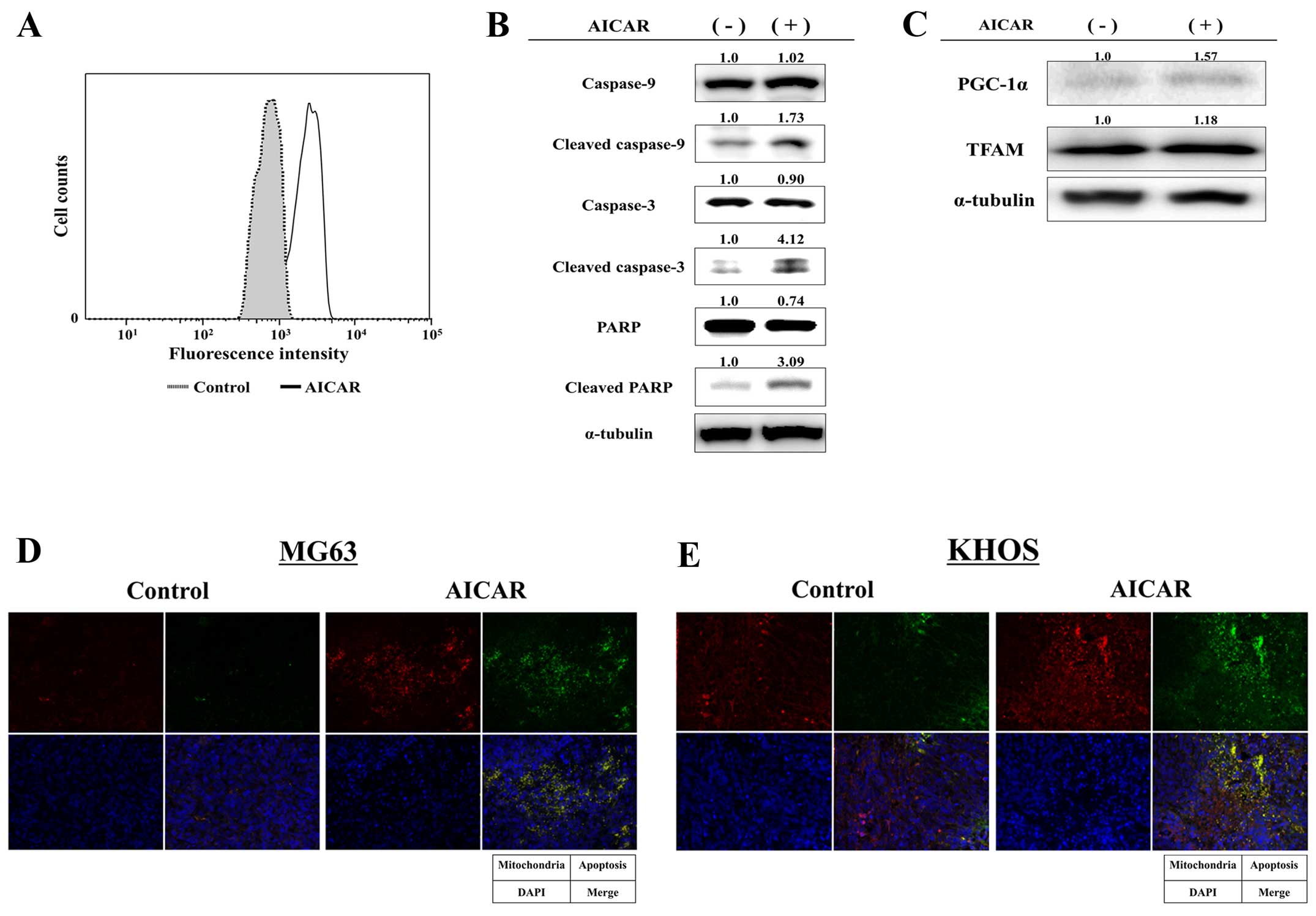
effects on cell viability in both osteosarcoma cell lines (Fig. 2A). Flow cytometric analyses
revealed that AICAR strongly increased the number of apoptotic
cells (Fig. 2B), and immunoblot
analyses demonstrated that cleaved forms of caspase-9, caspase-3
and PARP were strongly increased in osteosarcoma cells after 72 h
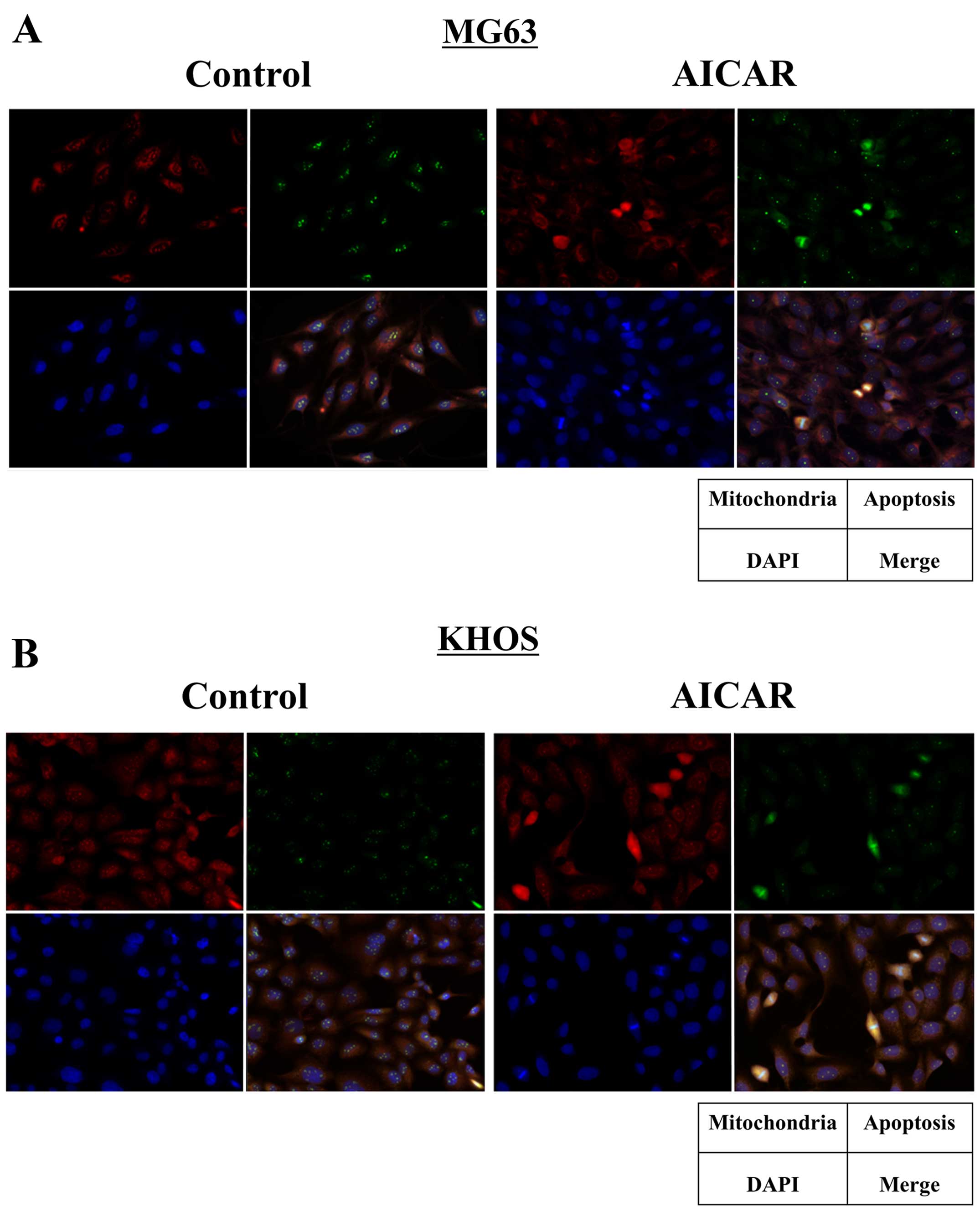
of treatment with 1,000 µM AICAR (Fig. 2C). Additionally, immunofluorescence
staining revealed that AICAR treatment increased the apoptotic cell
numbers along with increased mitochondrial proliferation in both
osteosarcoma cells (Fig. 3).
Semi-quantitative analyses of immunofluorescence positive staining
showed that apoptotic cells significantly increased in
AICAR-treated cells, and the relative positive staining to control
was a 6.87- and a 24.6-fold increase in MG63 and KHOS cells,
respectively (p<0.05). Positive staining of mitochondria also
significantly increased after AICAR treatment, and the relative
positive staining to control was a 3.43- and a 10.7-fold increase
in MG63 and KHOS cells, respectively (p<0.05). These
observations suggested that AICAR-dependent mitochondrial
proliferation induced mitochondrial apoptosis in human osteosarcoma
cells.
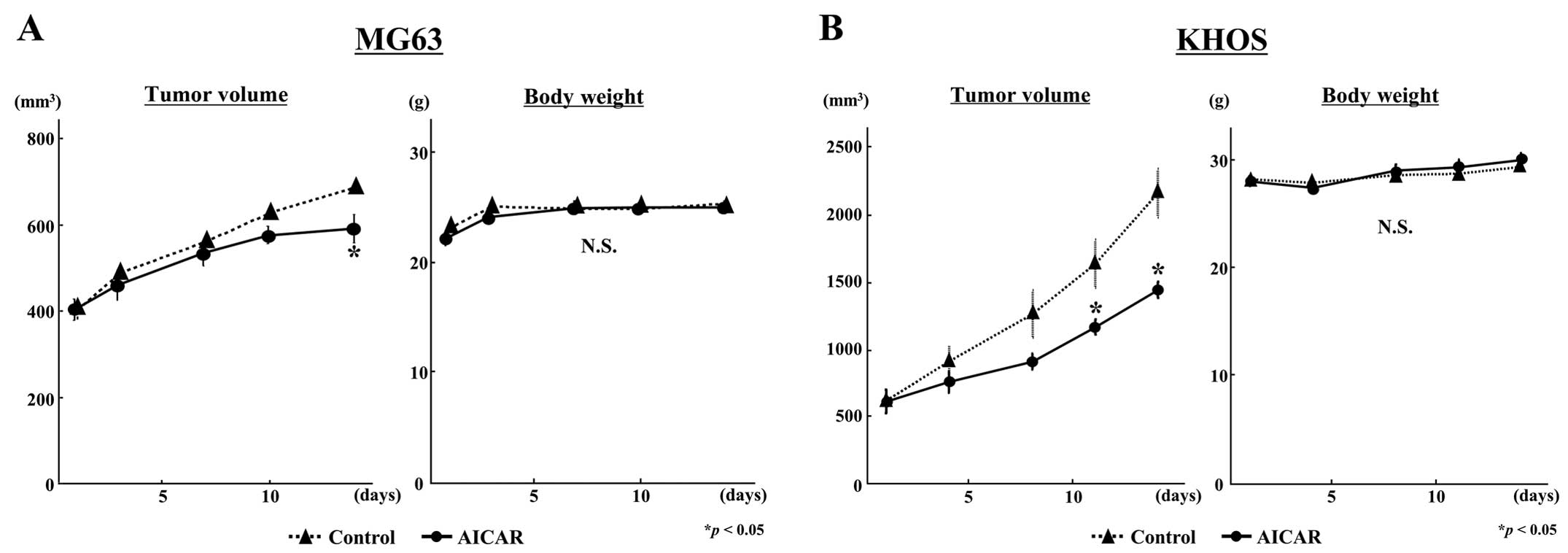
AICAR suppressed tumor growth in human
osteosarcoma xenografts with increased mitochondrial apoptosis
We evaluated the in vivo antitumor activity
of AICAR using human osteosarcoma xenografts. In both osteosarcoma
cell implanted models, AICAR significantly suppressed in
vivo osteosarcoma tumor growth compared with the growth of
control tumors (Fig. 4). At the
end of the experiment, tumor volume in the AICAR-treated group was
85.9% in MG63 (Fig. 4A) and 64.6%
in KHOS (Fig. 4B) of the volume in
each control group (p<0.05). No significant loss in body weight
was observed during the experimental period in either osteosarcoma
cell model (Fig. 4). In flow
cytometry (Fig. 5A) and immunoblot
analyses (Fig. 5B), increased
apoptotic activity was observed in AICAR-treated KHOS tumor
tissues, and the expression levels of the mitochondria-related
genes, PGC-1α and TFAM, were also increased in AICAR-treated KHOS
tumors (Fig. 5C). Additionally, in
immunofluorescence staining, apoptotic cells and mitochondrial
proliferation were observed in AICAR-treated both osteosarcoma
tumor tissues, consistent with the results from in vitro
experiments (Fig. 5D and E). These
findings suggested that AICAR could induce apoptosis via
mitochondrial proliferation in human osteosarcoma in
vivo.
Discussion
Osteosarcoma is the most common primary bone tumor
in childhood and adolescence and is a clinically aggressive
disease. Present therapeutic strategies, including a combination of
chemotherapy and surgery, have been shown to improve a long-term
survival in ~60–70% of cases; however, the prognosis of patients
with metastatic or recurrent disease is still poor because of the
lack of second-line chemotherapies (23,24).
Therefore, new therapeutic strategies against high-grade
osteosarcomas need to be established.
Mitochondria play essential roles in programmed cell
death (4), and in mitochondrial
biogenesis, the PGC-1α/TFAM pathway plays a crucial role in mtDNA
maintenance (16). In various
human malignancies, quantitative changes in mitochondrial numbers
have been observed (6–11), and decreased mitochondrial numbers
have been reported to be associated with tumor progression and
prognosis in patients with cancers (6,7).
Therefore, regulation of mitochondrial dysfunction has been
identified as a promising target for anticancer therapy (5). We have previously reported that
mitochondrial numbers are significantly reduced in human sarcoma
tissues, including osteosarcomas, and that increasing the
mitochondrial numbers by forced expression of PGC-1α induces
mitochondrial apoptosis in human sarcoma cell lines that exhibit
decreased mitochondrial numbers (15). Thus, because these findings suggest
that decreased mitochondrial numbers may influence tumor
progression in malignant musculoskeletal tumors, we hypothesized
that regulation of mitochondrial biogenesis could be a potent
therapeutic target for osteosarcoma.
Recent studies have highlighted the relationship
between cancer cell growth and cell metabolism, and the
energy-regulating enzyme AMPK is thought to be a key kinase that
mediates a metabolic cell cycle checkpoint (25). In human skeletal muscle, activation
of AMPK functions to maintain cellular energy stores and to switch
on catabolic pathways that produce ATP, mostly by enhancing
oxidative metabolism and mitochondrial biogenesis, while switching
off anabolic pathways that consume ATP; AMPK also directly
activates PGC-1α and regulates mitochondrial biogenesis (19). In tumor cells, the activation of
AMPK results in tumor suppression through cell cycle arrest or
apoptosis following phosphorylation of p53 and FOXO3a (26,27)
and induction of the cyclin-dependent kinase inhibitors
p21cip1 and p27kip1, leading to cell cycle
arrest (26,28). In this study, we examined the
therapeutic potential of mitochondrial proliferation via PGC-1α
activation by AMPK in human osteosarcoma cells.
In order to activate the AMPK/PGC-1α/TFAM axis in
mitochondrial biogenesis to fight cancer by interfering with energy
metabolism in cancer cells, AMPK activators, such as the guanidine
derivative metformin or AICAR, have been used (29–33).
AICAR is a nucleoside analog, which initially was developed as a
cardioprotective agent, but has a different mechanism of action
than standard nucleoside analogs such as fludarabine (34). In the 1980s and 1990s, AICAR was
under development as an agent that would reduce cardiac injury
associated with coronary artery bypass grafting. Although initial
trials clearly demonstrated success, later trials showed a minimal
efficacy as the morbidity and mortality rates associated with the
procedure decreased dramatically owing to better techniques and
increased familiarity with the procedures (35,36).
When added to cultured cells or administered into animals or
humans, AICAR is phosphorylated to become AICA-ribotide (ZMP), the
natural endogenous intermediate in de novo purine nucleotide
biosynthesis, which can function as an AMP mimic and can activate
AMPK. Previous studies have shown that AICAR has anticancer effects
in cancer cells (18,30,32,37).
For example, in HeLa cervical carcinoma cells, AMPK phosphorylation
by AICAR increases the levels of PGC-1α, NRF-1 and
TFAM mRNAs, resulting in upregulation of mtDNA replication
and transcription (37).
Additionally, other studies have revealed that AICAR inhibits cell
proliferation and induces apoptosis in various cancer cells,
including neuroblastoma (30),
colon cancer (32), breast cancer,
and prostate cancer cells (18).
However, in osteosarcoma, the potential role of AMPK and/or the
anticancer effects of AICAR through mitochondrial proliferation
have not been addressed. In the present study, we hypothesized that
AICAR could induce mitochondrial apoptosis in human osteosarcoma by
increasing PGC-1α expression through AMPK activation. In in
vitro studies, we proved that AICAR phosphorylated AMPK and
increased the expression of both PGC-1α and TFAM, resulting in
mitochondrial proliferation. Additionally, we demonstrated that the
apoptotic activity was increased along with mitochondrial
proliferation in AICAR-treated osteosarcoma cells. Furthermore, we
showed that AICAR suppressed in vivo osteosarcoma cell
growth by inducing mitochondrial apoptosis without apparent body
weight loss. Our results strongly suggested that decreased
mitochondrial numbers contributed to the progression of human
osteosarcoma, and that improvement of mitochondrial biogenesis by
AICAR through AMPK activation had anticancer effects on human
osteosarcoma via induction of mitochondrial proliferation and
apoptosis.
In the present study, we employed AICAR as an AMPK
activator for all experiments. Another AMPK activator, metformin,
which is typically the first drug used to treat patients with
diabetes mellitus type 2 (38),
activates AMPK by affecting the mitochondrial complex I of the
respiratory chain, leading to a drop in intracellular ATP levels
(39). Thus, metformin may inhibit
the function of isolated mitochondria (40–43),
while AICAR has been reported to activate mitochondrial function in
skeletal muscle via direct phosphorylation of PGC-1α (19). Additionally, metformin can induce
various side effects, including onset of lactic acidosis.
Therefore, in the clinical setting, metformin may not be feasible
to use in patients without diabetes mellitus. In contrast, AICAR is
relatively safe, as shown in clinical trials (36). Additionally, compared with standard
chemotherapies, AICAR exhibits preferential toxicity for tumor
cells and does not cause substantial damage to non-tumor cells
(44).
In conclusion, we demonstrated that AICAR induced
mitochondrial apoptosis via the PGC-1α/TFAM/mitochondrial pathway
by AMPK phosphorylation in human osteosarcoma. To the best of our
knowledge, this is the first study describing the apoptotic effects
of AICAR through the AMPK/PGC-1α/TFAM axis in the mitochondrial
pathway in human osteosarcoma. However, this study had several
limitations. We found that AICAR could decrease in vivo
osteosarcoma tumor growth but did not reduce tumor size;
unfortunately, we did not assess the antitumor effects of AICAR in
the context of AMPK inhibition by siRNA or AMPK inhibitors, such as
compound C (30,31). Although further studies are needed
to determine whether AICAR has sufficient effects in human
osteosarcoma and to elucidate the mechanisms mediating the
antitumor effects of AICAR, mitochondrial biogenesis via the
AMPK/PGC-1α/TFAM pathway should be an attractive target for
osteosarcoma, and AICAR may be a potent therapeutic agent for the
treatment of osteosarcoma.
Acknowledgments
We thank Minako Nagata, Maya Yasuda, and Kyoko
Tanaka for their expert technical assistance.
References
|
1
|
Hegyi M, Semsei AF, Jakab Z, Antal I, Kiss
J, Szendroi M, Csoka M and Kovacs G: Good prognosis of localized
osteosarcoma in young patients treated with limb-salvage surgery
and chemotherapy. Pediatr Blood Cancer. 57:415–422. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jaffe N: Osteosarcoma: Review of the past,
impact on the future. The American experience. Cancer Treat Res.
152:239–262. 2009. View Article : Google Scholar
|
|
3
|
van Maldegem AM, Bhosale A, Gelderblom HJ,
Hogendoorn PC and Hassan AB: Comprehensive analysis of published
phase I/II clinical trials between 1990–2010 in osteosarcoma and
Ewing sarcoma confirms limited outcomes and need for translational
investment. Clin Sarcoma Res. 2:52012. View Article : Google Scholar
|
|
4
|
Chan DC: Mitochondria: Dynamic organelles
in disease, aging, and development. Cell. 125:1241–1252. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Neuzil J, Dong LF, Rohlena J, Truksa J and
Ralph SJ: Classification of mitocans, anti-cancer drugs acting on
mitochondria. Mitochondrion. 13:199–208. 2013. View Article : Google Scholar
|
|
6
|
Yamada S, Nomoto S, Fujii T, Kaneko T,
Takeda S, Inoue S, Kanazumi N and Nakao A: Correlation between copy
number of mitochondrial DNA and clinico-pathologic parameters of
hepatocellular carcinoma. Eur J Surg Oncol. 32:303–307. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yu M, Zhou Y, Shi Y, Ning L, Yang Y, Wei
X, Zhang N, Hao X and Niu R: Reduced mitochondrial DNA copy number
is correlated with tumor progression and prognosis in Chinese
breast cancer patients. IUBMB Life. 59:450–457. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu CW, Yin PH, Hung WY, Li AF, Li SH, Chi
CW, Wei YH and Lee HC: Mitochondrial DNA mutations and
mitochondrial DNA depletion in gastric cancer. Genes Chromosomes
Cancer. 44:19–28. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee HC, Li SH, Lin JC, Wu CC, Yeh DC and
Wei YH: Somatic mutations in the D-loop and decrease in the copy
number of mitochondrial DNA in human hepatocellular carcinoma.
Mutat Res. 547:71–78. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xing J, Chen M, Wood CG, Lin J, Spitz MR,
Ma J, Amos CI, Shields PG, Benowitz NL, Gu J, et al: Mitochondrial
DNA content: Its genetic heritability and association with renal
cell carcinoma. J Natl Cancer Inst. 100:1104–1112. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tseng LM, Yin PH, Chi CW, Hsu CY, Wu CW,
Lee LM, Wei YH and Lee HC: Mitochondrial DNA mutations and
mitochondrial DNA depletion in breast cancer. Genes Chromosomes
Cancer. 45:629–638. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim MM, Clinger JD, Masayesva BG, Ha PK,
Zahurak ML, Westra WH and Califano JA: Mitochondrial DNA quantity
increases with histopathologic grade in premalignant and malignant
head and neck lesions. Clin Cancer Res. 10:8512–8515. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin CS, Chang SC, Wang LS, Chou TY, Hsu
WH, Wu YC and Wei YH: The role of mitochondrial DNA alterations in
esophageal squamous cell carcinomas. J Thorac Cardiovasc Surg.
139:189–197.e4. 2010. View Article : Google Scholar
|
|
14
|
Wang Y, Liu VW, Xue WC, Cheung AN and Ngan
HY: Association of decreased mitochondrial DNA content with ovarian
cancer progression. Br J Cancer. 95:1087–1091. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Onishi Y, Ueha T, Kawamoto T, Hara H, Toda
M, Harada R, Minoda M, Kurosaka M and Akisue T: Regulation of
mitochondrial proliferation by PGC-1α induces cellular apoptosis in
musculoskeletal malignancies. Sci Rep. 4:39162014. View Article : Google Scholar
|
|
16
|
Ekstrand MI, Falkenberg M, Rantanen A,
Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM and Larsson
NG: Mitochondrial transcription factor A regulates mtDNA copy
number in mammals. Hum Mol Genet. 13:935–944. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Corton JM, Gillespie JG, Hawley SA and
Hardie DG: 5-Aminoimidazole-4-carboxamide ribonucleoside. A
specific method for activating AMP-activated protein kinase in
intact cells? Eur J Biochem. 229:558–565. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Swinnen JV, Beckers A, Brusselmans K,
Organe S, Segers J, Timmermans L, Vanderhoydonc F, Deboel L, Derua
R, Waelkens E, et al: Mimicry of a cellular low energy status
blocks tumor cell anabolism and suppresses the malignant phenotype.
Cancer Res. 65:2441–2448. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jäger S, Handschin C, St-Pierre J and
Spiegelman BM: AMP-activated protein kinase (AMPK) action in
skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl
Acad Sci USA. 104:12017–12022. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Narkar VA, Downes M, Yu RT, Embler E, Wang
YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, et al:
AMPK and PPARdelta agonists are exercise mimetics. Cell.
134:405–415. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Okada Y, Akisue T, Hara H, Kishimoto K,
Kawamoto T, Imabori M, Kishimoto S, Fukase N, Onishi Y and Kurosaka
M: The effect of bevacizumab on tumour growth of malignant fibrous
histiocytoma in an animal model. Anticancer Res. 30:3391–3395.
2010.PubMed/NCBI
|
|
22
|
Yu M, Wan Y and Zou Q: Decreased copy
number of mitochondrial DNA in Ewing's sarcoma. Clin Chim Acta.
411:679–683. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Friebele JC, Peck J, Pan X, Abdel-Rasoul M
and Mayerson JL: Osteosarcoma: A Meta-Analysis and Review of the
Literature. Am J Orthop (Belle Mead NJ). 44:547–553. 2015.
|
|
24
|
Mirabello L, Troisi RJ and Savage SA:
Osteosarcoma incidence and survival rates from 1973 to 2004: Data
from the Surveillance, Epidemiology, and End Results Program.
Cancer. 115:1531–1543. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sanli T, Rashid A, Liu C, Harding S,
Bristow RG, Cutz JC, Singh G, Wright J and Tsakiridis T: Ionizing
radiation activates AMP-activated kinase (AMPK): A target for
radiosensitization of human cancer cells. Int J Radiat Oncol Biol
Phys. 78:221–229. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gwinn DM, Shackelford DB, Egan DF,
Mihaylova MM, Mery A, Vasquez DS, Turk BE and Shaw RJ: AMPK
phosphorylation of raptor mediates a metabolic checkpoint. Mol
Cell. 30:214–226. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shackelford DB and Shaw RJ: The LKB1-AMPK
pathway: Metabolism and growth control in tumour suppression. Nat
Rev Cancer. 9:563–575. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mihaylova MM and Shaw RJ: The AMPK
signalling pathway coordinates cell growth, autophagy and
metabolism. Nat Cell Biol. 13:1016–1023. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cheng J, Huang T, Li Y, Guo Y, Zhu Y, Wang
Q, Tan X, Chen W, Zhang Y, Cheng W, et al: AMP-activated protein
kinase suppresses the in vitro and in vivo proliferation of
hepatocellular carcinoma. PLoS One. 9:e932562014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guo D, Hildebrandt IJ, Prins RM, Soto H,
Mazzotta MM, Dang J, Czernin J, Shyy JY, Watson AD, Phelps M, et
al: The AMPK agonist AICAR inhibits the growth of
EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc
Natl Acad Sci USA. 106:12932–12937. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sauer H, Engel S, Milosevic N, Sharifpanah
F and Wartenberg M: Activation of AMP-kinase by AICAR induces
apoptosis of DU-145 prostate cancer cells through generation of
reactive oxygen species and activation of c-Jun N-terminal kinase.
Int J Oncol. 40:501–508. 2012.
|
|
32
|
Su RY, Chao Y, Chen TY, Huang DY and Lin
WW: 5-Aminoimidazole-4-carboxamide riboside sensitizes TRAIL-and
TNF{alpha}-induced cytotoxicity in colon cancer cells through
AMP-activated protein kinase signaling. Mol Cancer Ther.
6:1562–1571. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Woodard J and Platanias LC: AMP-activated
kinase (AMPK)-generated signals in malignant melanoma cell growth
and survival. Biochem Biophys Res Commun. 398:135–139. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Van Den Neste E, Cazin B, Janssens A,
González-Barca E, Terol MJ, Levy V, Pérez de Oteyza J, Zachee P,
Saunders A, de Frias M, et al: Acadesine for patients with
relapsed/refractory chronic lymphocytic leukemia (CLL): A
multicenter phase I/II study. Cancer Chemother Pharmacol.
71:581–591. 2013. View Article : Google Scholar :
|
|
35
|
Aronson S: An initial multicenter,
randomized controlled trial on the safety and efficacy of acadesine
in patients undergoing coronary artery bypass graft surgery. Anesth
Analg. 79:1021–1022. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Newman MF, Ferguson TB, White JA, Ambrosio
G, Koglin J, Nussmeier NA, Pearl RG, Pitt B, Wechsler AS, Weisel
RD, et al: RED-CABG Steering Committee and Investigators: Effect of
adenosine-regulating agent acadesine on morbidity and mortality
associated with coronary artery bypass grafting: The RED-CABG
randomized controlled trial. JAMA. 308:157–164. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fu X, Wan S, Lyu YL, Liu LF and Qi H:
Etoposide induces ATM-dependent mitochondrial biogenesis through
AMPK activation. PLoS One. 3:e20092008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhou G, Myers R, Li Y, Chen Y, Shen X,
Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al: Role of
AMP-activated protein kinase in mechanism of metformin action. J
Clin Invest. 108:1167–1174. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hawley SA, Ross FA, Chevtzoff C, Green KA,
Evans A, Fogarty S, Towler MC, Brown LJ, Ogunbayo OA, Evans AM, et
al: Use of cells expressing gamma subunit variants to identify
diverse mechanisms of AMPK activation. Cell Metab. 11:554–565.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
El-Mir MY, Nogueira V, Fontaine E, Avéret
N, Rigoulet M and Leverve X: Dimethylbiguanide inhibits cell
respiration via an indirect effect targeted on the respiratory
chain complex I. J Biol Chem. 275:223–228. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Owen MR, Doran E and Halestrap AP:
Evidence that metformin exerts its anti-diabetic effects through
inhibition of complex 1 of the mitochondrial respiratory chain.
Biochem J. 348:607–614. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Brunmair B, Staniek K, Gras F, Scharf N,
Althaym A, Clara R, Roden M, Gnaiger E, Nohl H, Waldhäusl W, et al:
Thiazolidinediones, like metformin, inhibit respiratory complex I:
A common mechanism contributing to their anti-diabetic actions?
Diabetes. 53:1052–1059. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Turner N, Li JY, Gosby A, To SW, Cheng Z,
Miyoshi H, Taketo MM, Cooney GJ, Kraegen EW, James DE, et al:
Berberine and its more biologically available derivative,
dihydroberberine, inhibit mitochondrial respiratory complex I: A
mechanism for the action of berberine to activate AMP-activated
protein kinase and improve insulin action. Diabetes. 57:1414–1418.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jose C, Hébert-Chatelain E, Bellance N,
Larendra A, Su M, Nouette-Gaulain K and Rossignol R: AICAR inhibits
cancer cell growth and triggers cell-type distinct effects on
OXPHOS biogenesis, oxidative stress and Akt activation. Biochim
Biophys Acta. 1807:707–718. 2011. View Article : Google Scholar : PubMed/NCBI
|