Introduction
B-cell acute lymphoblastic leukemia (B-ALL) is an
aggressive hematological malignancy characterized by uncontrolled
and rapid proliferation of B-lymphoid precursor cells within the
bone marrow (1,2). B-ALL represents the most common type
of ALL and accounts for ~75 and 85% of adult and childhood ALL
cases, respectively (3,4). It is estimated that nearly 4,000
de novo cases are diagnosed each year in the US (5). Although advances in risk-adapted
intensive chemotherapy have dramatically improved the disease
outcomes of childhood patients with B-ALL, this approach is less
effective in adults, with a disease-related mortality of ~60%
(6,7). Relapse occur frequently following
remission induction chemotherapy, and the expected survival of
relapsed patients was less than 6 months (8,9).
Especially, the 5-year survival rate for adult patients is a dismal
7% at time of relapse (10).
Chemotherapy-refractory and relapsed B-ALL remains the primary
cause of cancer-related mortality in children and young adults
(5,11,12).
Therefore, there is an urgent need to discover new therapeutic
targets for B-ALL. Understanding the critical factors and molecular
mechanisms associated with B-ALL initiation and progression is
essential for further advances in therapy. Currently, many
cytogenetic and genomic lesions have been shown to play important
roles in the pathogenesis of B-ALL, which include hyperdiploidy
(11,13,14),
hypodiploidy (6,13,14),
rearrangements of TEL, TCF3, MLL, BCR/ABL and IgH@ gene
(6,12,14,15),
mutations of PAX5, CRLF2, IKZF1 and JAK gene (6,11,15),
deletion of CDKN2A/B (12). These
aberrations are strongly correlated with disease characteristics,
patient prognosis and the response to chemotherapy, however,
understanding of the molecular mechanisms of this disease remains
limited. In particular, the regulatory networks of B-ALL genes
expression are still unclear.
miRNAs are small noncoding RNAs of ~18 to 25
nucleotides that inhibit gene expression at the
post-transcriptional level through binding to the 3′-UTR of target
mRNAs (16). A growing body of
research evidence has confirmed the crucial roles of miRNA in
various types of human cancers, including B-ALL (17). It has been reported that
miR-125b-5p was upreg-ulated in patients with B-ALL carrying the
t(11;14)(q24;q32) translocation. Ectopic expression of miR-125b-5p
would induce hematopoietic fetal liver cells progresses to B-ALL,
T-cell acute lymphoblastic leukemia (T-ALL) or a myeloproliferative
neoplasm in xenograft mouse models (18). Furthermore, elevated levels of
miR-125b-5p, miR-99a and miR-100 were correlated with resistance to
daunorubicin and vincristine, while decreased expression of miR-708
was related to resistance to glucocorticoids in pediatric B-ALL
(16,17). miR-17-92 was significantly
downregulated in the BCR-ABL-positive human B-ALL samples and its
overexpression induced apoptosis of B-ALL cells in a
BCR-ABL-dependent manner by targeting Bcl-2 (19).
Transcription factors (TFs) are protein factors that
regulate gene transcription by specifically binding to
cis-regulatory elements located in the promoter regions of
target genes, which we now call transcription factor binding site
(TFBS) (20). As key regulators of
gene expression in organisms, TFs and miRNAs are able to mutually
regulate one another in forms of feedback loops (FBLs) or
cooperatively regulate the same target genes in a combinatorial
manner to form feed-forward loops (FFLs) hence ensuring the precise
and accurate control of gene expression (20,21).
Recent investigations concerning the gene expression regulation by
miRNA and TF in leukemia have provided new insights into disease
pathogenesis (21–23). miR-223 and two TFs (C/EBPα and
NFI-A) formed a regulatory circuitry to control the differentiation
of acute promyelocytic leukemia cells (21). miR-196b can exert its effects by
targeting the TF MYC and induce cell apoptosis in B-ALL (22). miR-19 was overexpressed in T-ALL
cell lines and patients. Ectopic expression of miR-19 sustained
activation of NF-κB pathway in T-ALL involving a feed-forward
circuitry in miR-19, CYLD and TF NF-κB (23). However, global regulation of miRNA-
and TF-mediated control of gene expression in adult B-ALL is still
poorly understood. Thus, an integrated analysis of miRNAs and TFs,
as their cooperative interaction relates to B-ALL pathogenesis,
requires examination.
In this study, we applied Illumina deep-sequencing
and TF array technology to analyze the miRNA and TF expression
profiles, respectively, in bone marrow samples of adult B-ALL
patients and their controls. Based on a combined bioinformatics
approach for prediction of miRNA and TF targets, we integrated
differentially expressed miRNAs and TFs as well as B-ALL candidate
genes to construct a B-ALL-related miRNA and TF regulatory network.
After analyzing the network function and topology, we found some
B-ALL-related pathways and hub regulators from the network, and
further investigated their regulation properties in the subnetwork.
This study will be helpful to decipher the complex molecular
mechanisms underlying B-ALL, as well as to provide potential
therapeutic targets for B-ALL in the future.
Materials and methods
Patient samples
A total of 25 bone marrow samples, including 15
patients derived from newly diagnosed and untreated adult B-ALL (7
males and 8 females; 18–71 years old; average age, 36.5 years) and
10 controls (6 males and 4 females; 10-73 years old; average age,
39.0 years), were used in this study. All samples were collected at
the Affiliated Hospital of Guangdong Medical University (Zhanjiang,
China). Diagnosis of B-ALL was determined according to the accepted
WHO criteria (24–26). Control samples were obtained from
10 patients with unexplained fever or anemia whose bone marrow
aspirates was demonstrated to be normal bone marrow morphology. All
patients gave informed consent for participation in this study
according to the Helsinki convention criteria, and the study was
approved by the Ethics Committee of Affiliated Hospital of
Guangdong Medical University.
Total RNA isolation
Total RNA was isolated using RNeasy kit (Qiagen,
Hilden, Germany) and TRIzol reagent (Invitrogen, Carlsbad, CA, USA)
including a DNase digestion step, in accordance with the
manufacturer's instructions. RNA yield and purity was assessed by
determining the absorbance at 230, 260 and 280 nm in a ND-1000
spectrophotometer (NanoDrop, Wilmington, DE, USA).
Illumina deep-sequencing and analysis of
miRNAs
Small RNA libraries were generated as follows: i)
3′-RNA adapters were ligated to total RNA; ii) 5′-RNA adapters were
ligated to total RNA; iii) ligation products were reverse
transcribed to cDNA and amplified by PCR; iv) 130-155 bp PCR
amplified products (correspond to small RNA fragments ranging from
15-35 nucleotides) were isolated and purified from polyacryl-amide
gels. After quantified with 2100 Bioanalyzer (Agilent, Palo Alto,
CA, USA), the entire libraries were denatured with 0.1 M NaOH and
subsequently loaded on the cBot instrument for amplification in
situ using TruSeq Rapid SR cluster kit (both from Illumina, San
Diego, CA, USA), as recommended by the manufacturer. Finally, small
RNA libraries were quantified and sequenced for 36 cycles on
Illumina HiSeq 2000 analyzer using TruSeq Rapid SBS kit (Illumina)
following the manufacturer's standard workflow.
After sequencing was complete, image analysis and
base calling were performed by utilizing Off-Line Basecaller
software (version 1.8.0; Illumina). Clean reads which passed Solexa
Chastity quality filter, were trimmed to remove the 3′-adapter
sequence, and the reads shorter than 15 nucleotides were discarded.
To identify known miRNAs, trimmed reads were compared with the
human miRNA precursors in miRBase database (version 19.0,
http://www.mirbase.org/) by using Novoalign
software (release v2.07.11, http://www.novocraft.com/). Subsequently, raw counts
of miRNA reads were normalized to transcripts per million (TPM)
values. Differentially expressed miRNAs between samples may be
identified by the fold-change filtering using a threshold of at
least 2-fold-changes according to the normalized most abundant tag
counts.
Transcription factor array and data
analysis
Transcription factor array analysis was performed
using TranSignal Combo Protein/DNA arrays (Panomics, Redwood City,
CA, USA) according to the manufacturer's directions, which enables
simultaneous detection of 345 major transcription factors. In
general, nuclear extracts were prepared using the nuclear extract
kit (Panomics) and the protein concentrations were measured with
BCA protein assay kit (KangChen, Shanghai, China) as per the
manufacturer's instructions. Ten micrograms of nuclear proteins
were incubated for 30 min at 15°C with 10 µl of probe mixture
containing 345 biotin-labeled TF-specific DNA oligonucleotides
(Panomics) to allow the formation of TF/DNA complexes. Such
complexes were separated from the free probes using spin column
separation system (Panomics). The TF-specific probes were then
extracted from the TF/DNA complexes and hybridized to the
TranSignal array membrane at 42°C overnight. After three washings
and addition of horseradish peroxidase (HRP)-conjugated
streptavidin, hybridization signals were generated by standard
chemiluminescence procedures with ECL-Hyperfilm (Amersham Pharmacia
Biotech, Uppsala, Sweden). The signals were detected with a GBox
Imaging system (Syngene, Cambridge, UK) and spot intensities were
analyzed using ScanAlyze software (version 1.0.3, http://graphics.stanford.edu/software/scanalyze/).
The data points showing at least 2-fold signal difference between
B-ALL samples and their controls are considered significant.
Quantitative PCR of miRNA and TF
Total RNA samples were reverse transcribed (RT) to
generate cDNA with Stem-loop RT primer (for miRNAs) and oligo(dT)
primer (for TFs) using the SuperScript III Reverse Transcriptase
kit (Invitrogen) following the instructions of the manufacturer.
QPCR was carried out in triplicate on a Prism 7500 Real-time PCR
system (Applied Biosystems, Foster City, CA, USA). The reaction
mixtures consisted of 5 µl of 2X SYBR-Green PCR master mix (Applied
Biosystems), 2 µl of template cDNA, 0.5 µl of 10 µM PCR forward
primer, 0.5 µl of 10 µM PCR reverse primer and 2 µl of
double-distilled water. The PCR reaction was conducted at 95°C for
10 min, followed by 40 cycles of 95°C for 10 sec and 60°C for 60
sec. To evaluate the reaction specificity, a melting curve analysis
was performed immediately after PCR amplification. The expression
levels of miRNAs and IFs were normalized to those of the human U6
snRNA and 18S rRNA, respectively. Relative expression was analyzed
using the comparative 2−ΔΔCt method (27). Differences between the two groups
were compared using the Student's t-test and considered significant
at P<0.05. All primers used in this study are listed in Tables ITable II–III.
 | Table IPrimers used for cDNA synthesis of
miRNAs. |
Table I
Primers used for cDNA synthesis of
miRNAs.
| miRNAs | Primer for cDNA
synthesis (5′-3′) |
|---|
| miR-200c-3p |
CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTCCAT |
| miR-200b-3p |
CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTCATCATT |
| miR-194-5p |
CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTCCACATG |
| miR-26b-5p |
GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGACACCTAT |
| miR-16-5p |
GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGACCGCCAAT |
| miR-424-5p |
GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGACTTCAAA |
| U6 snRNA |
CGCTTCACGAATTTGCGTGTCAT |
| Table IIPrimers used for quantitative PCR of
miRNAs. |
Table II
Primers used for quantitative PCR of
miRNAs.
| miRNAs | Sense primer
(5′-3′) | Antisense primer
(5′-3′) | Product (bp) |
|---|
| miR-200c-3p |
ACACTCCAGCTGGGTAATACTGCCGGGTAATG |
CTCAACTGGTGTCGTGGA | 73 |
| miR-200b-3p |
ACACTCCAGCTGGGTAATACTGCCTGGTAATG |
CTCAACTGGTGTCGTGGA | 73 |
| miR-194-5p |
ACACTCCAGCTGGGTGTAACAGCAACTCCATG |
CTCAACTGGTGTCGTGGA | 73 |
| miR-26b-5p |
GGGGTTCAAGTAATTCAGG |
TGCGTGTCGTGGAGTC | 65 |
| miR-16-5p |
GGGTAGCAGCACGTAAATA |
CAGTGCGTGTCGTGGAGT | 65 |
| miR-424-5p |
GGGCAGCAGCAATTCATGT |
GTGCGTGTCGTGGAGTCG | 63 |
| U6 snRNA |
GCTTCGGCAGCACATATACTAAAAT |
CGCTTCACGAATTTGCGTGTCAT | 89 |
| Table IIIPrimers used for quantitative PCR of
TFs. |
Table III
Primers used for quantitative PCR of
TFs.
| TFs | Sense primer
(5′-3′) | Antisense primer
(5′-3′) | Product (bp) |
|---|
| STAT5A |
GGCCATCCTAGGTTTTGTG |
CCCGATTTCTGAGTCACTAAAG | 100 |
| FOXD1 |
CTTTTCTCGTCTTGGTGGTT |
CAGTTTTGTCCGAGAATTCG | 104 |
| MEF2A |
AGTTTCCAACAGCTGTCCTT |
GATTGGGACCACCTGTGTAC | 90 |
| FOXF2 |
CACTCCAGCATGTCCTCCTA |
CTAGCTGAGGGATGGAAAGA | 170 |
| HLF |
CGATCACCTTCTGCTCCTA |
GATTCGGATAAGGCTTTGC | 110 |
| FOXO4 |
ATAGCACCACCTCCAGTCA |
CATGTCACACTCCAGGTTCTC | 150 |
| 18S rRNA |
CCTGGATACCGCAGCTAGGA |
GCGGCGCAATACGAATGCCCC | 112 |
Association analysis of miRNA and TF
expression profiles
Identification of miRNA-gene/TF
regulatory relation- ships
To acquire regulatory relationships of miRNA-gene
and miRNA-TF, target prediction for differentially expressed miRNAs
(fold-change, ≥2.0) was carried out using three different online
databases, including PicTar (release 2007, http://pictar.mdc-berlin.de/cgi-bin/PicTar_vertebrate.cgi),
TargetScan (release 6.2, http://www.targetscan.org/) and miRanda (version v5,
http://www.ebi.ac.uk/enright-srv/microcosm/htdocs/targets/v5/)
(28,29). To reduce false-positive results,
only the targets predicted by at least two of the aforementioned
databases were retained in this study (28,29).
The identified targets were then incorporated into the
experimentally verified targets which were obtained from miRTarBase
(release 4.5, http://mirtarbase.mbc.nctu.edu.tw/) (30). After compiling B-ALL candidate
genes/TFs from the MalaCards database (version 1.08.564, http://www.malacards.org/) (31), these results were overlapped with
B-ALL candidate genes/TFs to avoid the redundancy and form
miRNA→gene and miRNA→TF regulatory relationships. The extracted
miRNA and target gene/TF pairs were subsequently subjected to TFBS
analysis.
Identification of TF-gene/miRNA
regulatory relationships
TF-gene and TF-miRNA regulatory relations were
analyzed using the predicted TFBS information obtained from the
TFBS Conserved Track (http://genome.ucsc.edu/cgi-bin/hgTables?hgsid=350051003&hgta_doSchemaDb=hg19&hgta_doSchemaTable=tfbsConsFactors)
at UCSC Genome Browser database (32), and TFBSs were required to be
conserved among human/mouse/rat. To reduce false-positive
predictions, a P-value of 0.05 was selected as a cut-off. Next, the
5,000 bp upstream to 1,000 bp downstream of the transcription start
site (TSS) was defined as putative promoter region for each gene
(33,34). A gene is considered to be the
target of a TF when at least one TFBS is located within the gene's
promoter region and its P<0.05. As a part of the common
transcription unit, miRNAs usually co-expressed with known host
genes (protein coding genes/lncRNAs). Therefore, the putative
promoter region (spans from 5,000 bp upstream to 1,000 bp
downstream of the TSS) for host genes is used as the miRNA promoter
(34,35). miRNAs having a TFBS that completely
falls within the promoter region of their host gene and its
P<0.05 were considered as TF targets. To further increase the
prediction accuracy, the predicted TFs were then overlapped with
differentially expressed TFs (fold-change, ≥2.0) obtained from the
TF profiling data. After extraction of the TF-miRNA pairs and
TF-miRNA target gene pairs, the predicted TF→miRNA and TF→gene
relations were retrieved, respectively.
miRNA-TF regulatory network generation
and network node analysis
By merging the regulatory relationships between TFs,
miRNAs and target genes (TF→miRNA, TF→gene, miRNA→gene and
miRNA→TF), we constructed a comprehensive miRNA-TF regulatory
network for B-ALL using Gephi software (release 0.8.1-β, http://gephi.github.io/). GO database (www.geneontology.org) (36) and Kyoto Encydopedia of Gene and
Genomes (KEGG) database (http://www.genome.jp/kegg/) (37) were then used to analyze the
functional categories and pathways of the network nodes, which were
considered to be significant when P<0.05. For further evaluation
of network characteristics, the node degree is determined by
calculating the number of direct links between nodes within the
network. To identify network hubs, we classified the nodes by
descending based on their degrees. The nodes having a total degree
(sum of the in-degree and out-degree) ≥10 were considered as the
hub components. Any neighboring node connected to hubs was
extracted from the network to construct the sub-network.
Results
Alterations in miRNA and TF expression in
adult B-ALL
To acquire B-ALL-related miRNAs and TFs, we analyzed
the global expression change of miRNAs and TFs in adult B-ALL
samples compared with their controls, using Illumina
deep-sequencing and TF array technology, respectively. The miRNA
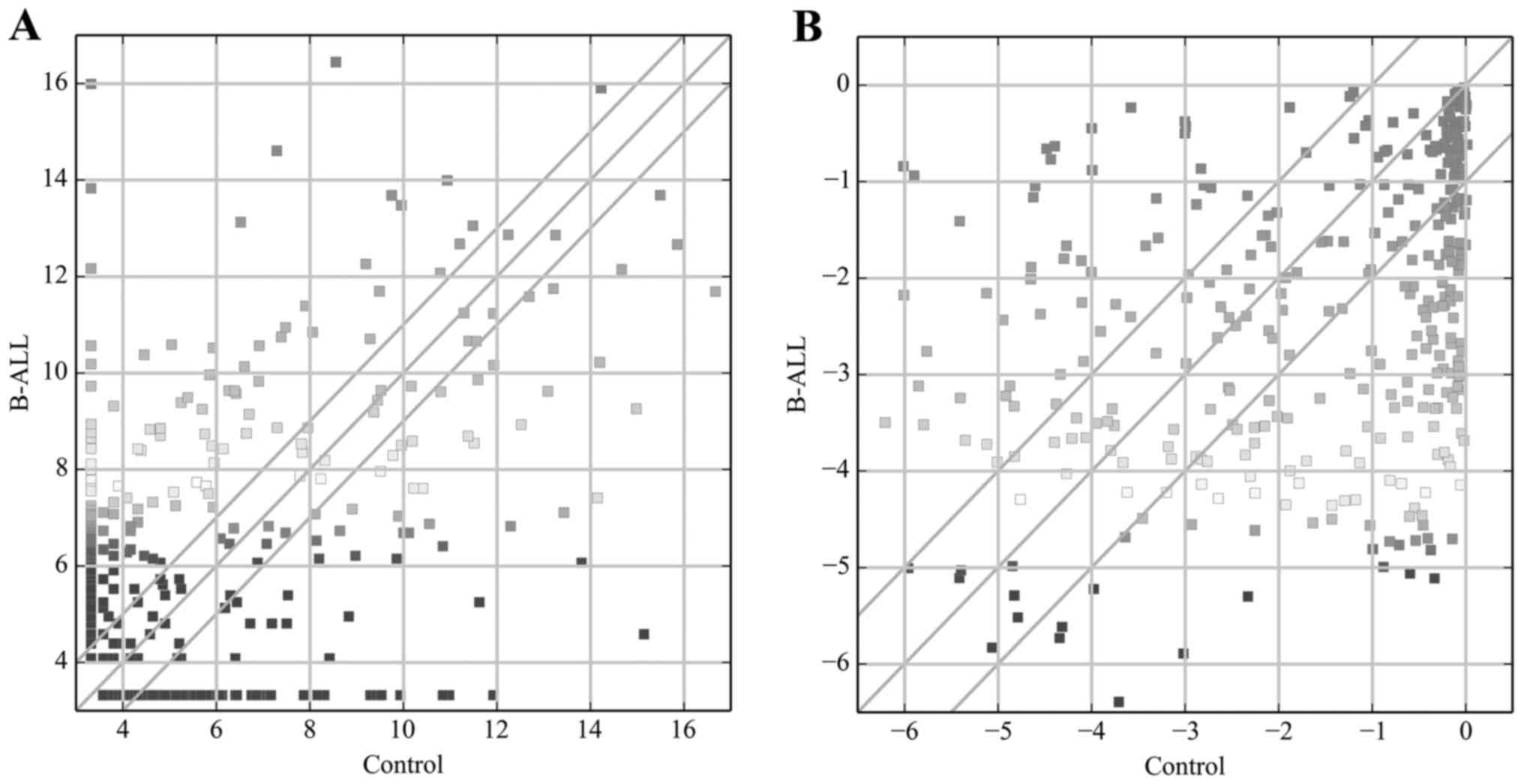
expression profiling data showed 291 miRNAs were differentially
expressed (fold-change, ≥2.0) among groups (Fig. 1A), including 168 that were
upregulated and 123 that were downregulated. miR-1246 (fold-change,
6516.2) was the most upregulated miRNA, while miR-106b-5p
(fold-change, 1509.5) was the most downregulated miRNA.
Analysis of the TF array results revealed 201 TFs
(57 upregulated and 144 downregulated) that were differentially
expressed (fold-change ≥2.0) between the control and adult B-ALL
samples (Fig. 1B). Among these
TFs, MYC (fold-change, 36.0) exhibited the highest degree of
upregulation, while MYOG (fold-change, 91.4) was the most
downregulated TF.
Verification of Illumina sequencing and
TF array results by qPCR
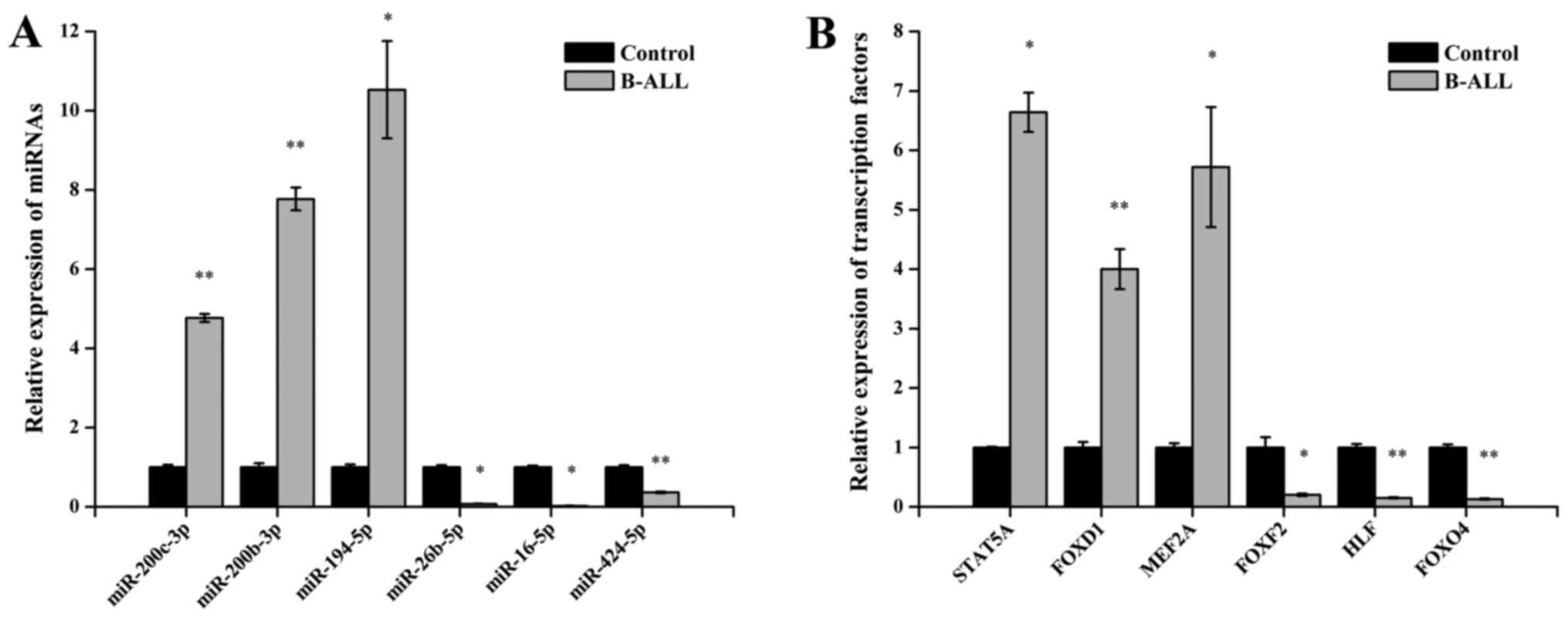
To demonstrate the reliability of the Illumina
sequencing and TF array data, 6 miRNAs and 6 TFs that are
differentially expressed were chosen for further verification by
qPCR. As shown in Fig. 2A,
miR-200c-3p, miR-200b-3p and miR-194-5p were upregulated and that
miR-26b-5p, miR-16-5p and miR-424-5p were downregulated in adult
B-ALL samples relative to their controls (all P<0.05).
Furthermore, qPCR analysis also confirmed statistically significant
differences between groups in signal transducer and activator of
transcription 5A (STAT5A), FOXD1, MEF2A, FOXF2, HLF and FOXO4
(P<0.05 for each TFs) (Fig.
2B). These results were consistent with the Illumina sequencing
and TF array data.
miRNA and TF regulatory network in adult
B-ALL
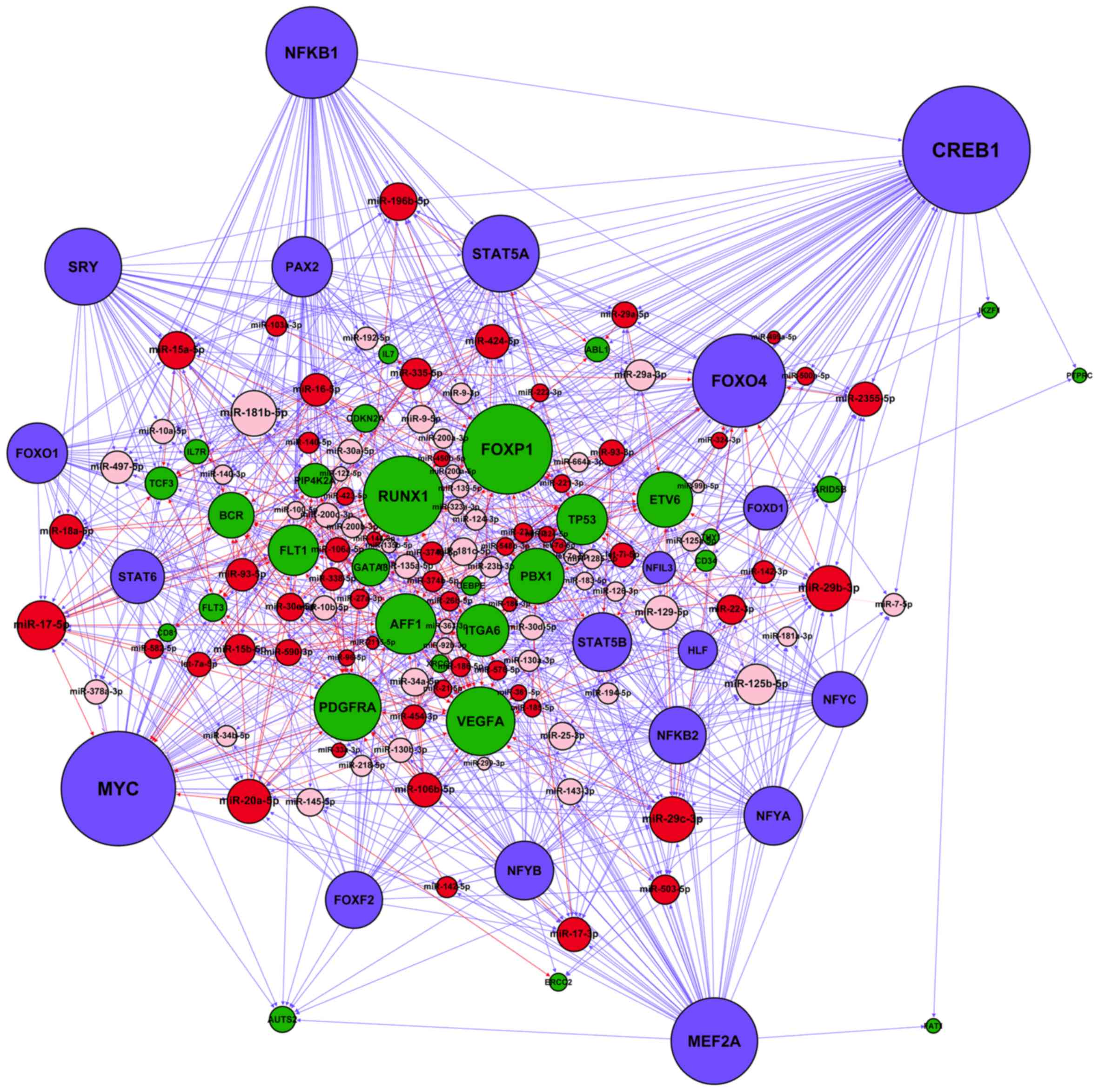
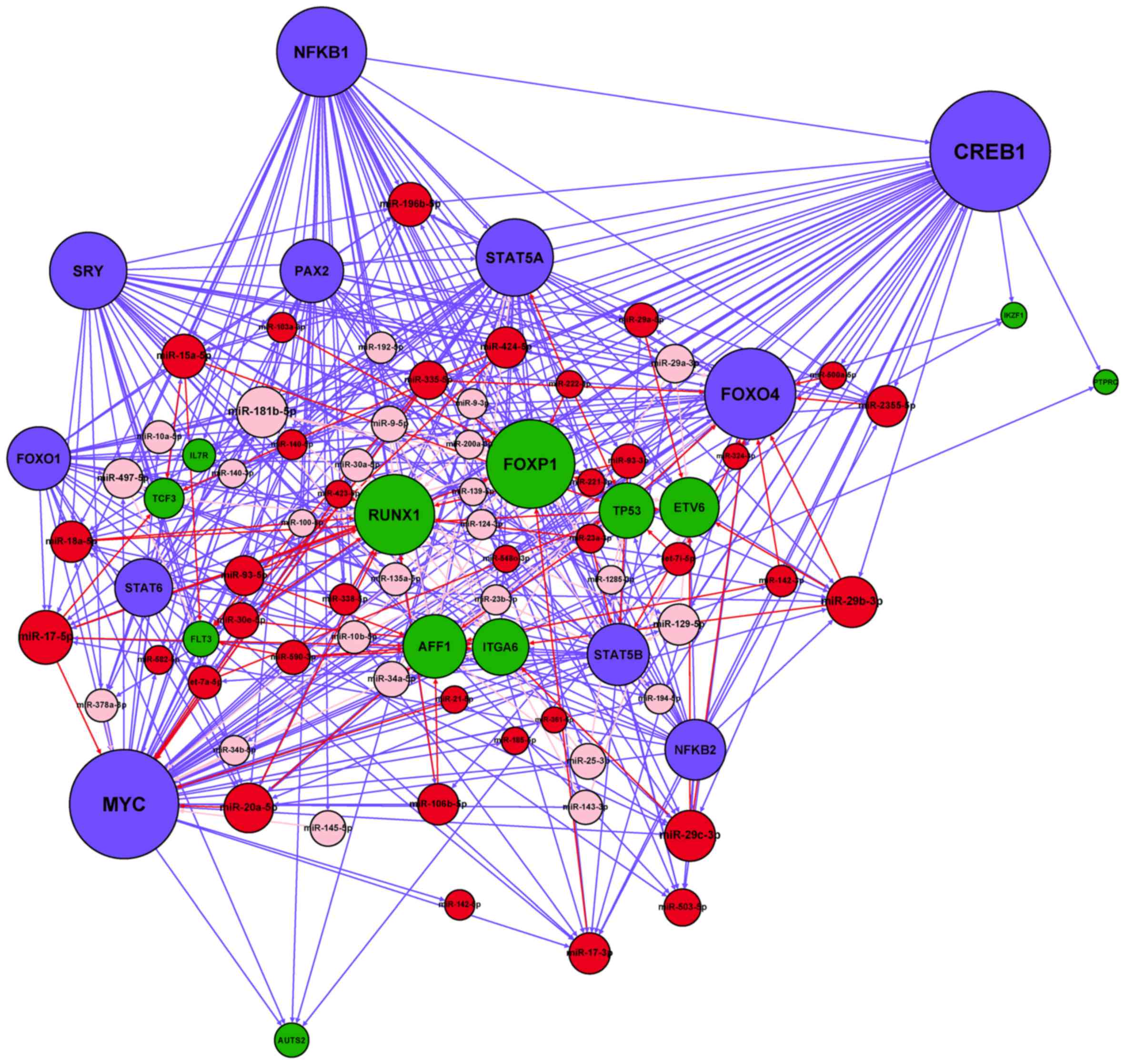
After merging the four types of regulatory
interactions (miRNA→gene, miRNA→TF, TF→gene and TF→miRNA)
identified in the Materials and methods section, we constructed a
miRNA-TF regulatory network specific for adult B-ALL (Fig. 3). A total of 41 miRNA-regulated TFs
and 19 TFs regulated miRNAs in the network. On average, each miRNA
was regulated by 0.24 TFs, whereas each TF was regulated by 10.25
miRNAs. The numbers of nodes and regulatory relationships of the
resultant network are listed in Table
IV. FFLs and FBLs are motifs known to play key roles in gene
regulatory networks (21).
Typically, FFLs can be categorized into three different types based
on the master regulators: miRNA-FFL, TF-FFL and composite FFL
(21,38). In Table V, we summarized the FFLs and FBLs
in the B-ALL network and found ~98% of motifs were FFL. Among the
526 merged FFLs, 492 (93.5%) were TF-FFLs, in which TF was the
master regulator, 30 (5.7%) belonged to miRNA-FFLs, and 4 (0.8%)
belonged to composite-FFLs. These results indicate that TF-FFL was
the dominant motif in the network.
| Table IVSummary of relationships in the
B-ALL-related miRNA and transcription factor (TF) regulatory
network. |
Table IV
Summary of relationships in the
B-ALL-related miRNA and transcription factor (TF) regulatory
network.
| Relationship | No. of pairs | No. of miRNAs | No. of genes | No. of TFs |
|---|
| miRNA-genea | 431 | 131 | 27 | - |
| miRNA-TFb | 47 | 41 | - | 4 |
| TF-genec | 176 | - | 35 | 19 |
| TF-miRNAd | 429 | 80 | - | 19 |
| Table VSummary of feed-forward loops (FFLs)
and feedback loops (FBLs) based on B-ALL-related network data. |
Table V
Summary of feed-forward loops (FFLs)
and feedback loops (FBLs) based on B-ALL-related network data.
| Motif types | No. of motifs | No. of nodes
| No. of links
|
|---|
| Genes | miRNAs | TFs | miRNA-gene | miRNA-TF | TF-gene | TF-miRNA |
|---|
| miRNA-FFL | 30 | 9 | 15 | 4 | 27 | 19 | 16 | - |
| TF-FFL |
492 | 20 | 70 | 19 |
189 | - |
104 |
280 |
| Composite-FFL | 4 | 4 | 2 | 2 | 4 | 2 | 4 | 2 |
| FBL | 9 | - | 9 | 3 | - | 9 | - | 9 |
Functional analysis of the miRNA and TF
mediated regulatory network
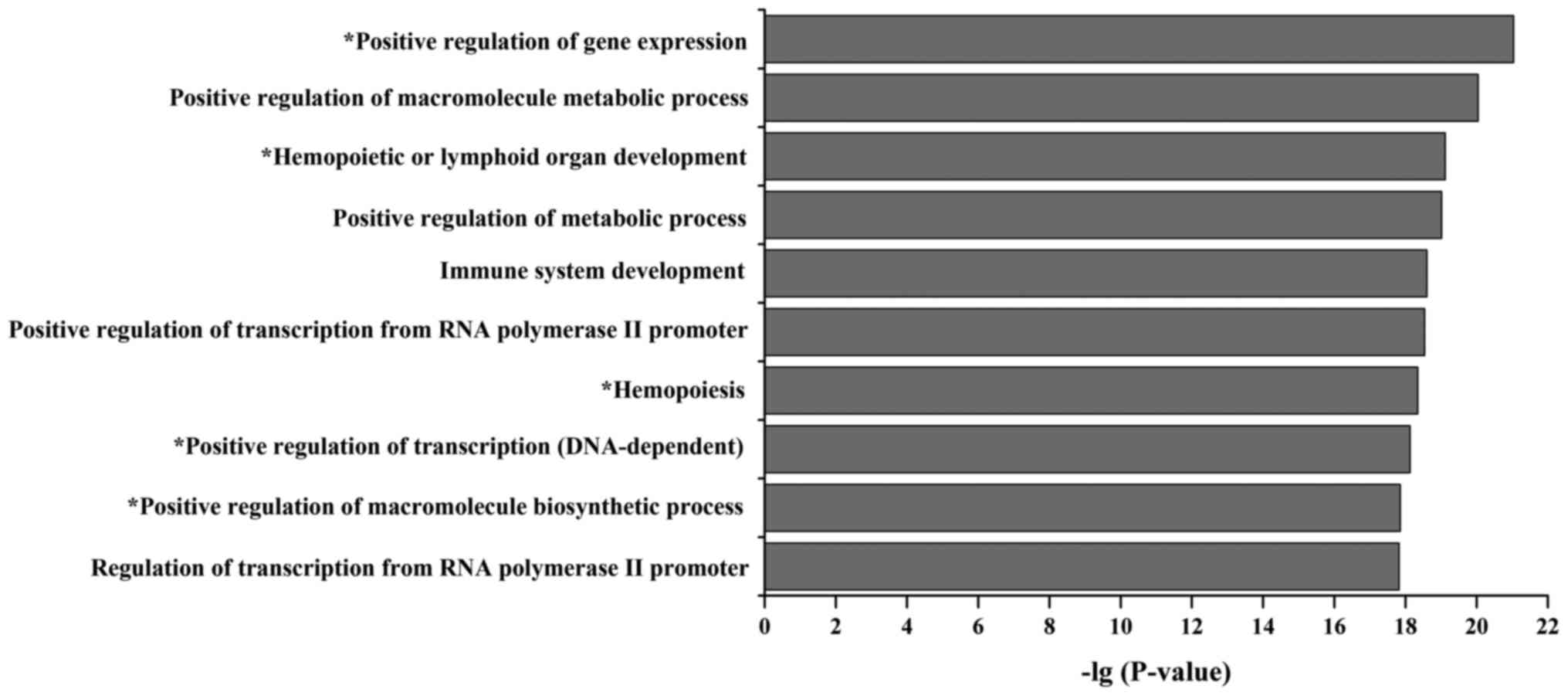
Enrichment analysis is an effective method to
understand functional nodes and network motifs. To investigate the
functional categories of the B-ALL miRNA-TF regulatory network, we
performed the GO enrichment analysis for all network nodes and
identified 10 highly-enriched biological process terms (Fig. 4). Among these GO terms, 5 terms
were related to the initiation and progression of B-ALL, such as
positive regulation of gene expression, hemopoietic or lymphoid
organ development, hemopoiesis, positive regulation of
transcription (DNA-dependent) and positive regulation of
macromolecule biosynthetic process (6,7,11,39).
Moreover, significantly-enriched biological pathways for the
network nodes were also examined by applying KEGG pathway
enrichment analysis. The nodes were significantly enriched in 25
different pathways, of which 13 pathways were directly related to
leukemia or hematopoiesis (Table
VI). Among these pathways, PI3K-Akt signaling pathway, Jak-STAT
signaling pathway, Ras signaling pathway and cell cycle pathway
were well-known to be involved in B-ALL (6,9,11,13,39).
| Table VIPathway analysis for nodes in
B-ALL-related miRNA and transcription factor regulatory
network. |
Table VI
Pathway analysis for nodes in
B-ALL-related miRNA and transcription factor regulatory
network.
| Pathway ID | Definition | Gene count | % | Fisher-P-value |
|---|
| hsa05200 | Pathways in
cancera | 15 | 9.43 | 7.67E-11 |
| hsa05202 | Transcriptional
misregulation in cancera | 12 | 7.55 | 1.23E-10 |
| hsa05220 | Chronic myeloid
leukemiaa | 9 | 5.66 | 1.54E-10 |
| hsa05166 | HTLV-I
infectiona | 11 | 6.92 | 1.10E-07 |
| hsa05221 | Acute myeloid
leukemiaa | 6 | 3.77 | 6.18E-07 |
| hsa05161 | Hepatitis B | 7 | 4.40 | 1.37E-05 |
| hsa04151 | PI3K-Akt signaling
pathwaya | 10 | 6.29 | 1.42E-05 |
| hsa05219 | Bladder cancer | 4 | 2.52 | 5.48E-05 |
| hsa04640 | Hematopoietic cell
lineagea | 5 | 3.14 | 0.000120 |
| hsa05203 | Viral
carcinogenesisa | 7 | 4.40 | 0.000125 |
| hsa05215 | Prostate
cancer | 5 | 3.14 | 0.000126 |
| hsa05206 | MicroRNAs in
cancer | 8 | 5.03 | 0.000191 |
| hsa04630 | Jak-STAT signaling
pathwaya | 6 | 3.77 | 0.000206 |
| hsa05212 | Pancreatic
cancer | 4 | 2.52 | 0.000478 |
| hsa04612 | Antigen processing
and presentation | 4 | 2.52 | 0.000945 |
| hsa05222 | Small cell lung
cancer | 4 | 2.52 | 0.001299 |
| hsa04012 | ErbB signaling
pathway | 4 | 2.52 | 0.001356 |
| hsa04014 | Ras signaling
pathwaya | 6 | 3.77 | 0.001509 |
| hsa05152 | Tuberculosis | 5 | 3.14 | 0.003049 |
| hsa04060 | Cytokine-cytokine
receptor interaction | 6 | 3.77 | 0.003687 |
| hsa04110 | Cell cyclea | 4 | 2.52 | 0.004927 |
| hsa05162 | Measles | 4 | 2.52 | 0.006479 |
| hsa04010 | MAPK signaling
pathwaya | 5 | 3.14 | 0.013771 |
| hsa05169 | Epstein-Barr virus
infectiona | 4 | 2.52 | 0.026308 |
| hsa04510 | Focal adhesion | 4 | 2.52 | 0.027579 |
Network topological analysis and
sub-network construction
To assess the global properties of this network, we
calculated degrees and analyzed degree distributions for all nodes,
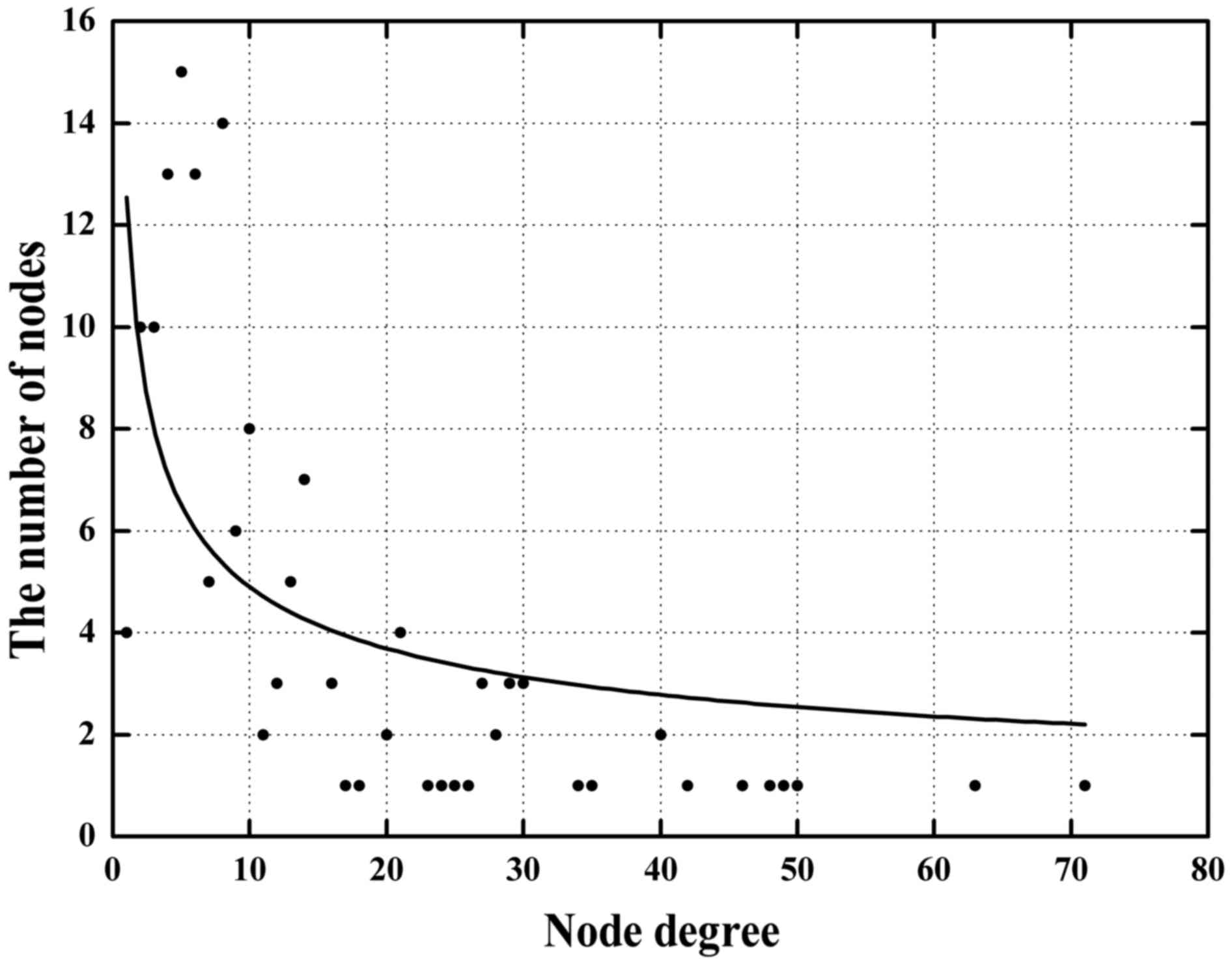
which are primary topological network measures. In the network, the
average node degree of miRNAs, TFs and genes was 7.6 (range from 1
to 23), 35 (range from 13 to 71) and 15.7 (range from 2 to 48),
respectively. These results indicate that TFs are located at higher
degrees within the network. Only a few nodes displayed a high node
degree (hubs) while most nodes had a low degree, which reflected a
scale-free feature of this network and further demonstrate that hub
nodes may play vital roles in the network stability and robustness
(Fig. 5). We categorized the nodes
in descending order according to their total degrees and identified
hub nodes (total degrees ≥10) inside the network. Twenty-eight hub
miRNAs and 19 hub TFs were obtained from the network, respectively
(Tables VII and VIII). Notably, 6 of the 28 hub miRNAs
belonged to members of the miR-15 family, namely
miR-15a-5p/15b-5p/16-5p/424-5p/497-5p/503-5p (Table VII), indicating an important role
of miR-15 family in B-ALL-related regulatory network. Moreover,
more than half of the 19 hub TFs were either well-known leukemia
regulators, such as MYC (40),
STAT5A (41,42) and STAT5B (41,42),
or associated with leukemia development and progression, such as
CREB1 (43), FOXO1 (44), FOXO4 (45), NFKB1 (46), NFKB2 (47), STAT6 (48) and HLF (11,49),
which preliminarily reflected the robustness of our network. Among
the 47 hub regulators, 4 hub miRNAs [miR-17-5p (19), miR-125b-5p (50), miR-18a-5p (19) and miR-34a-5p (4)] and 5 hub TFs [MYC (43,51,52),
STAT5A (41,53), STAT5B (41,54),
FOXO1 (44) and HLF (11)] have been previously implicated in
B-ALL. Of these regulators, 3 hub TFs (MYC, STAT5A and STAT5B)
participated in Jak-STAT signaling pathway which was directly
related to B-ALL pathogenesis. Therefore, we constructed a
sub-network from these 3 TFs and all their direct linked nodes in
the network, and defined it as the B-ALL-related Jak-STAT signaling
sub-network, which included 436 edges, 12 genes, 61 miRNAs and 11
TFs (Fig. 6).
| Table VIIHub miRNAs in B-ALL-related miRNA and
transcription factor regulatory network. |
Table VII
Hub miRNAs in B-ALL-related miRNA and
transcription factor regulatory network.
| Top | miRNA | In-degree | Out-degree | Total degree |
|---|
| 1 | miR-17-5p | 12 | 11 | 23 |
| 2 | miR-181b-5p | 12 | 9 | 21 |
| 3 | miR-29b-3p | 17 | 4 | 21 |
| 4 | miR-29c-3p | 17 | 4 | 21 |
| 5 | miR-20a-5p | 12 | 8 | 20 |
| 6 | miR-125b-5p | 11 | 7 | 18 |
| 7 | miR-196b-5p | 13 | 3 | 16 |
| 8 | miR-15a-5p | 11 | 5 | 16 |
| 9 | miR-129-5p | 8 | 6 | 14 |
| 10 | miR-18a-5p | 12 | 2 | 14 |
| 11 | miR-17-3p | 12 | 2 | 14 |
| 12 | miR-2355-5p | 13 | 1 | 14 |
| 13 | miR-424-5p | 10 | 4 | 14 |
| 14 | miR-106b-5p | 6 | 8 | 14 |
| 15 | miR-497-5p | 9 | 4 | 13 |
| 16 | miR-93-5p | 6 | 7 | 13 |
| 17 | miR-16-5p | 7 | 6 | 13 |
| 18 | miR-29a-3p | 7 | 5 | 12 |
| 19 | miR-15b-5p | 7 | 5 | 12 |
| 20 | miR-335-5p | 5 | 7 | 12 |
| 21 | miR-22-3p | 9 | 2 | 11 |
| 22 | miR-503-5p | 10 | 1 | 11 |
| 23 | miR-25-3p | 6 | 4 | 10 |
| 24 | miR-9-5p | 4 | 6 | 10 |
| 25 | miR-145-5p | 6 | 4 | 10 |
| 26 | miR-34a-5p | 2 | 8 | 10 |
| 27 | miR-181c-5p | 1 | 9 | 10 |
| 28 | miR-30e-5p | 6 | 4 | 10 |
| Table VIIIHub transcription factors in
B-ALL-related miRNA and transcription factor regulatory
network. |
Table VIII
Hub transcription factors in
B-ALL-related miRNA and transcription factor regulatory
network.
| Top | Transcription
factor | In-degree | Out-degree | Total degree |
|---|
| 1 | CREB1 | 8 | 63 | 71 |
| 2 | MYC | 14 | 49 | 63 |
| 3 | FOXO4 | 18 | 32 | 50 |
| 4 | NFKB1 | 0 | 49 | 49 |
| 5 | MEF2A | 0 | 46 | 46 |
| 6 | SRY | 0 | 40 | 40 |
| 7 | STAT5A | 8 | 32 | 40 |
| 8 | PAX2 | 0 | 30 | 30 |
| 9 | FOXO1 | 0 | 30 | 30 |
| 10 | NFYB | 0 | 29 | 29 |
| 11 | NFYA | 0 | 29 | 29 |
| 12 | STAT5B | 12 | 17 | 29 |
| 13 | NFKB2 | 0 | 28 | 28 |
| 14 | FOXF2 | 0 | 28 | 28 |
| 15 | NFYC | 0 | 27 | 27 |
| 16 | STAT6 | 0 | 26 | 26 |
| 17 | FOXD1 | 0 | 20 | 20 |
| 18 | HLF | 0 | 17 | 17 |
| 19 | NFIL3 | 0 | 13 | 13 |
Discussion
B-ALL is a genetically heterogeneous disease and
many different types of cytogenetic abnormalities and molecular
genetic mutations are important initiating events in the
leukemogenesis of B-ALL (6,11,39).
However, these genetic alterations are insufficient to fully
explain the biology and heterogeneity of this disease (11). The pathogenesis of B-ALL is still
not entirely clear to date. In particular, the heterogeneity and
complexity makes it difficult to discover hub regulators and
regulatory motifs involved in the pathogenesis of B-ALL. By
integrating high-throughput expression profiling technology and
bioinformatics approaches, systems biology focuses on exploring the
interactions between biological molecules and explaining the
biological phenomenon on the network level, which may accelerate
the discovery of hub regulators and pathways that control various
aspects of the disease pathogenesis (55,56).
To improve our understanding of the molecular
mechanism of B-ALL, we applied systems biology approach to obtain
and integrate expression changes of miRNAs and TFs between bone
marrow samples from adult B-ALL patients and their controls, and
constructed a miRNA-TF mediated regulatory network associated with
this disease (Fig. 3).
Subsequently, we identified the miRNA and TF mediated regulatory
motifs from the network, including FFLs and FBLs (Table V), which are significant and
prevalent motifs in gene regulatory networks (21). It has been reported that FFLs may
work as the core of the global gene regulatory network and play
critical roles in disease pathogenesis (23,57).
The majority of FFLs in our network are TF-FFLs, in which TF as the
main regulator controls the expression of both miRNA and their
common target gene. This finding is consistent with a recent study
that TF-FFL was the dominant motif in the development of B
lymphocytes (57), revealing an
important role of TF-FFL in B cell development and B-ALL
pathogenesis.
For evaluating the functional properties of this
network, GO and KEGG pathway annotation were used to analyze the
enrichment of the network nodes (Fig.
4 and Table VI). Among 10
highly-enriched biological process terms, one half of these terms
were related to the occurrence and development of B-ALL. Moreover,
more than half of significantly-enriched pathways were directly
associated with leukemia or hematopoiesis. Among them, four
pathways have been reported to be implicated in B-ALL pathogenesis,
such as PI3K-Akt signaling pathway, Jak-STAT signaling pathway, Ras
signaling pathway and cell cycle pathway. Further network
topological analysis identified 28 hub miRNAs and 19 hub TFs within
the network (Tables VII and
VIII). Of these, 4 hub miRNAs
(miR-17-5p, miR-125b-5p, miR-18a-5p and miR-34a-5p) and 5 hub TFs
(MYC, STAT5A, STAT5B, FOXO1 and HLF) were functionally associated
with B-ALL, which have been confirmed in previous studies.
Collectively, these results preliminarily verified the reliability
of our network. Among these hub TFs, MYC, STAT5A and STAT5B
participated in Jak-STAT signaling pathway which is the well-known
pathway involved in B-ALL. As a member of the
helix-loop-helix/leucine-zipper transcription factor family, MYC is
a master regulator of important biological processes such as cell
survival, proliferation, differentiation, metabolism and
pluripotency maintenance (58,59).
Constitutive activation of MYC exhibited in a variety of B-ALL cell
lines (60). Double trans-genesis
of MYC and Bcl-2 caused hyperproliferation of pre-B and B cells and
promoted initiation of B-ALL in mice (51). Inhibition of MYC expression and
STAT5 phosphorylation by JQ1 significantly prolonged survival in
mice xenografted with primary human CRLF2-rearranged B-ALL
(52). STAT5 is a cytoplasmic
transcription factor that is usually transiently activated by
phosphorylation in response to a wide variety of growth factors and
cytokines (61). STAT5 consists of
two separate genes, STAT5A and STAT5B, which collectively are key
regulators of cell proliferation and survival in early B
lymphopoiesis as well as plays an important role in ordering
immunoglobulin recombination during B cell development (41). Constitutively high expression of
STAT5A induced by retrovirus integration in mice perturbed
signaling in early pre-B cells leading to spontaneous
pre-B-ALL/pre-B lymphomas (62).
Overexpression of STAT5A in mice cooperate with the loss of p53
function also promoted faster initiation of B-ALL/B-cell lymphoma
(55). Furthermore, transgenic
expression of STAT5B strongly synergized with adapter protein BLNK
deficiency to induce pre-B-ALL in mice by inhibiting expression of
p27 (54). To further investigate
the regulation of MYC, STAT5A and STAT5B on highly complex
molecular network in B-ALL, we subsequently constructed a
B-ALL-related Jak-STAT signaling sub-network (Fig. 6) by extracting MYC, STAT5A, STAT5B
and all their directly linking nodes from the network.
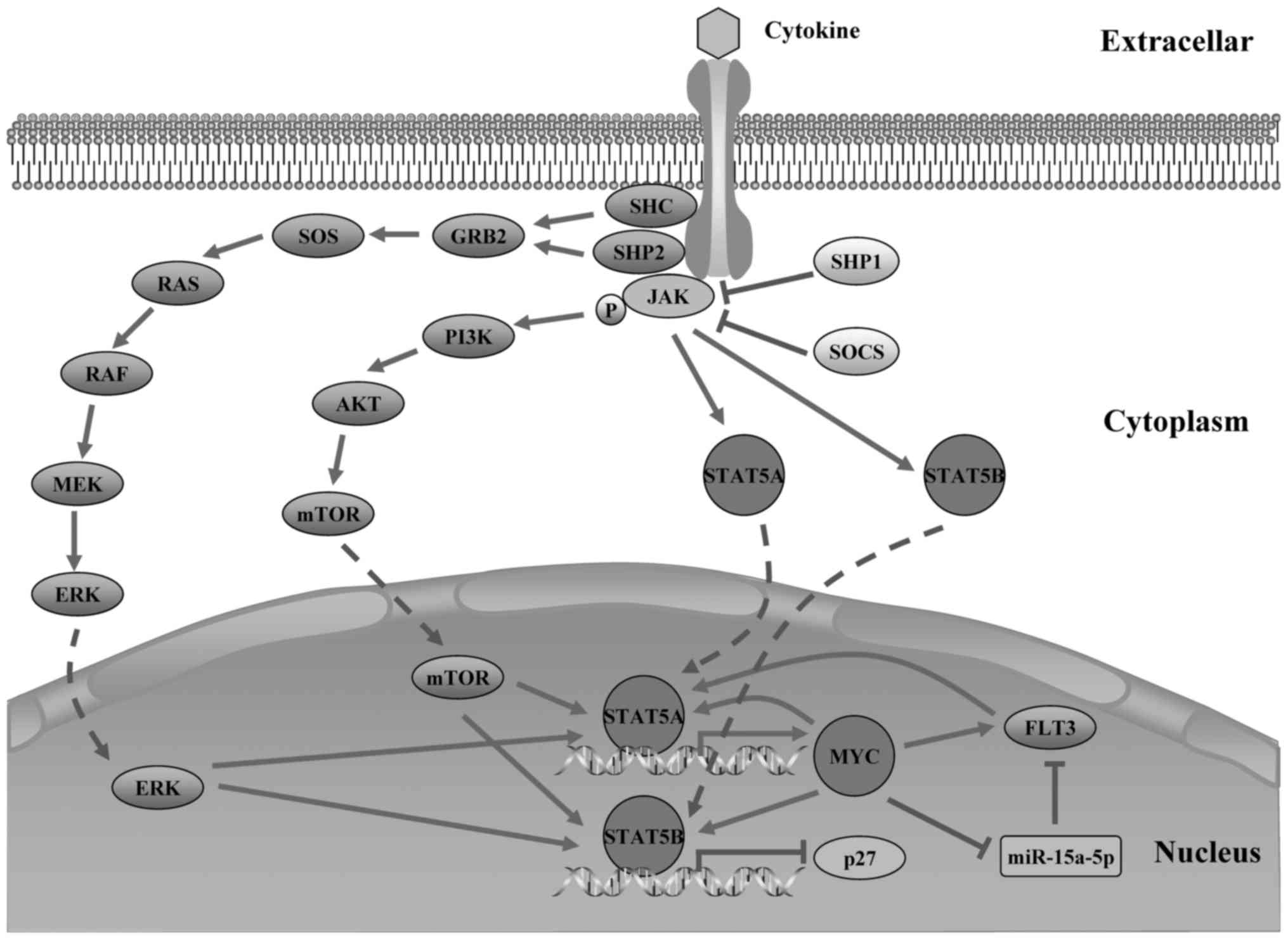
In the sub-network, MYC stood out as a key regulator
due to its direct linking to STAT5A and STAT5B, which may establish
cross-talk between MYC and all sub-network nodes. Notably, MYC and
miR-15a-5p were predicted to cooperatively regulate their common
target genes FMS-like tyrosine kinase 3 (FLT3), which constitute a
MYC/miR-15a-5p/FLT3 TF-FFL. As a member of the miR-15 family as
well as a hub miRNA in our network, miR-15a-5p is a potential tumor
suppressor and is downregulated in glioma, bladder cancer,
hepatoma, prostate cancer, chronic lymphocytic leukemia and
different kinds of lymphoma, exerting its function by inducing
apoptosis, inhibiting cell proliferation or suppressing
tumorigenicity (63,64). In accord with the above-mentioned
reports, decreased expression of miR-15a-5p was also observed in
our study, indicating a potential relationship between miR-15a-5p
and B-ALL. However, the exact effect of miR-15a-5p on B-ALL
occurrence and development is still unknown. It has been reported
that MYC transcriptionally repressed miR-15a-5p expression by
binding to its host gene (DLEU2) promoter (58). Inhibition of miR-15a-5p expression
by MYC may promote expression of their common target gene, the
FLT3. Therefore, we speculate that MYC may synergize with
miR-15a-5p to induce FLT3 expression in MYC/miR-15a-5p/FLT3 TF-FFL.
FLT3 is a type III receptor tyrosine kinase, which is primarily
expressed in lymphoid progenitors and early myeloid and plays a
crucial role in their differentiation and proliferation (65). Highly expressed FLT3 was observed
in nearly all pre-B-ALL and most acute myeloid leukemia (AML)
patient samples as well as the majority of pre-B-ALL and AML cell
lines (65–67). In B-ALL patients with an MLL-AF4
gene rearrangement, a high level of FLT3 expression was
significantly correlated with a high risk of therapy failure
(39). It has been reported that
FLT3 may collaborate with MLL gene fusions to promote
leukemogenesis of B-ALL (65).
IMC-EB10, an anti-FLT3 monoclonal antibody, can inhibit FLT3
signaling as well as significantly prolongs survival and decrease
the engraftment of NOD/SCID mice injected with human B-ALL cells,
indicating an important role of FLT3 in the pathogenesis of B-ALL
(67). FLT3 was also demonstrated
to potently activate the STAT5A in both B-ALL and AML cell lines
(65). Recently, STAT5A has been
shown to bind to the 3′ super-enhancer of MYC and recruited BRD2
proteins to induce MYC gene expression (59), which establish a regulatory circuit
that FLT3 may regulate MYC expression via activating the STAT5A. In
our sub-network, upregulation of MYC expression may further promote
the expression of FLT3 by the MYC/miR-15a-5p/FLT3 FFL, implying a
positive feedback between MYC and FLT3 which will be involved in
the pathogenesis of B-ALL. In the discussion above, we present a
model of MYC/miR-15a-5p/FLT3 FFL involving in Jak-STAT signaling
pathway and regulatory network in B-ALL (Fig. 7). Based on the literature review
and our sub-network analysis, we propose that MYC/miR-15a-5p/FLT3
FFL is a potential key motif in the B-ALL miRNA-TF regulatory
network, which may be associated with the occurrence and
development of B-ALL.
In conclusion, the miRNA and TF-based network
analysis allows us to acquire an overall view of the molecular
regulatory network involved in B-ALL. It also identified some key
regulators and regulatory motifs for B-ALL, as well as provided new
clues for deciphering the B-ALL pathogenesis at the transcriptional
and post-transcriptional levels.
Abbreviations:
|
B-ALL
|
B-cell acute lymphoblastic
leukemia
|
|
ALL
|
acute lymphoblastic leukemia
|
|
T-ALL
|
T-cell acute lymphoblastic
leukemia
|
|
miRNA
|
microRNA
|
|
TF
|
transcription factor
|
|
TFBS
|
transcription factor binding site
|
|
FBL
|
feedback loop
|
|
FFL
|
feed-forward loop
|
|
WHO
|
World health Organization
|
|
HRP
|
horseradish peroxidase
|
|
qPCR
|
quantitative PCR
|
|
TSS
|
transcription start site
|
|
GO
|
Gene Oncology
|
|
KEGG
|
Kyoto Encydopedia of Gene and
Genomes
|
|
STAT5
|
signal transducer and activator of
transcription 5
|
|
FLT3
|
FMS-like tyrosine kinase 3
|
|
AML
|
acute myeloid leukemia
|
Acknowledgments
This study was financially supported by the
Guangdong Provincial Natural Science Foundation of China (no.
2016A030313677), the Medical Science and Technology Research Fund
of Guangdong Province (no. A2016193) and the Open Project Program
of Guangdong Provincial Key Laboratory of Medical Molecular
Diagnostics (no. FZZD201604). We sincerely thank all donors for
taking part in this investigation.
References
|
1
|
Purizaca J, Meza I and Pelayo R: Early
lymphoid development and microenvironmental cues in B-cell acute
lymphoblastic leukemia. Arch Med Res. 43:89–101. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sanjuan-Pla A, Bueno C, Prieto C, Acha P,
Stam RW, Marschalek R and Menéndez P: Revisiting the biology of
infant t(4;11)/MLL-AF4+ B-cell acute lymphoblastic
leukemia. Blood. 126:2676–2685. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Korfi K, Smith M, Swan J, Somervaille TC,
Dhomen N and Marais R: BIM mediates synergistic killing of B-cell
acute lymphoblastic leukemia cells by BCL-2 and MEK inhibitors.
Cell Death Dis. 7:e21772016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Luna-Aguirre CM, de la Luz Martinez-Fierro
M, Mar-Aguilar F, Garza-Veloz I, Treviño-Alvarado V, Rojas-Martinez
A, Jaime-Perez JC, Malagon-Santiago GI, Gutierrez-Aguirre CH,
Gonzalez-Llano O, et al: Circulating microRNA expression profile in
B-cell acute lymphoblastic leukemia. Cancer Biomark. 15:299–310.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tasian SK and Gardner RA: CD19-redirected
chimeric antigen receptor-modified T cells: A promising
immunotherapy for children and adults with B-cell acute
lymphoblastic leukemia (ALL). Ther Adv Hematol. 6:228–241. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Woo JS, Alberti MO and Tirado CA:
Childhood B-acute lymphoblastic leukemia: A genetic update. Exp
Hematol Oncol. 3:162014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou Y, You MJ, Young KH, Lin P, Lu G,
Medeiros LJ and Bueso-Ramos CE: Advances in the molecular
pathobiology of B-lymphoblastic leukemia. Hum Pathol. 43:1347–1362.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pegram HJ, Smith EL, Rafiq S and Brentjens
RJ: CAR therapy for hematological cancers: Can success seen in the
treatment of B-cell acute lymphoblastic leukemia be applied to
other hematological malignancies? Immunotherapy. 7:545–561. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang XH, Wang CC, Jiang Q, Yang SM, Jiang
H, Lu J, Wang QM, Feng FE, Zhu XL, Zhao T, et al: ADAM28
overex-pression regulated via the PI3K/Akt pathway is associated
with relapse in de novo adult B-cell acute lymphoblastic leukemia.
Leuk Res. 39:1229–1238. 2015. View Article : Google Scholar
|
|
10
|
Sikaria S, Aldoss I and Akhtari M:
Monoclonal antibodies and immune therapies for adult precursor
B-acute lymphoblastic leukemia. Immunol Lett. 172:113–123. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mullighan CG: Molecular genetics of
B-precursor acute lymphoblastic leukemia. J Clin Invest.
122:3407–3415. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Othman MA, Grygalewicz B, Pienkowska-Grela
B, Rygier J, Ejduk A, Rincic M, Melo JB, Carreira IM, Meyer B and
Liehr T: A novel IGH@ gene rearrangement associated with
CDKN2A/B deletion in young adult B-cell acute lymphoblastic
leukemia. Oncol Lett. 11:2117–2122. 2016.PubMed/NCBI
|
|
13
|
Iacobucci I, Iraci N, Messina M, Lonetti
A, Chiaretti S, Valli E, Ferrari A, Papayannidis C, Paoloni F,
Vitale A, et al: IKAROS deletions dictate a unique gene expression
signature in patients with adult B-cell acute lymphoblastic
leukemia. PLoS One. 7:e409342012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Moorman AV: The clinical relevance of
chromosomal and genomic abnormalities in B-cell precursor acute
lymphoblastic leukaemia. Blood Rev. 26:123–135. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yoda A, Yoda Y, Chiaretti S, Bar-Natan M,
Mani K, Rodig SJ, West N, Xiao Y, Brown JR, Mitsiades C, et al:
Functional screening identifies CRLF2 in precursor B-cell acute
lympho-blastic leukemia. Proc Natl Acad Sci USA. 107:252–257. 2010.
View Article : Google Scholar
|
|
16
|
Luan C, Yang Z and Chen B: The functional
role of microRNA in acute lymphoblastic leukemia: Relevance for
diagnosis, differential diagnosis, prognosis, and therapy. Onco
Targets Ther. 8:2903–2914. 2015.PubMed/NCBI
|
|
17
|
Cocco C and Airoldi I: Cytokines and
microRNA in pediatric B-acute lymphoblastic leukemia. Cytokine
Growth Factor Rev. 22:149–156. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bousquet M, Harris MH, Zhou B and Lodish
HF: MicroRNA miR-125b causes leukemia. Proc Natl Acad Sci USA.
107:21558–21563. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Scherr M, Elder A, Battmer K, Barzan D,
Bomken S, Ricke-Hoch M, Schröder A, Venturini L, Blair HJ, Vormoor
J, et al: Differential expression of miR-17-92 identifies BCL2 as a
therapeutic target in BCR-ABL-positive B-lineage acute
lymphoblastic leukemia. Leukemia. 28:554–565. 2014. View Article : Google Scholar
|
|
20
|
Piriyapongsa J, Jordan IK, Conley AB,
Ronan T and Smalheiser NR: Transcription factor binding sites are
highly enriched within microRNA precursor sequences. Biol Direct.
6:612011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang HM, Kuang S, Xiong X, Gao T, Liu C
and Guo AY: Transcription factor and microRNA co-regulatory loops:
Important regulatory motifs in biological processes and diseases.
Brief Bioinform. 16:45–58. 2015. View Article : Google Scholar
|
|
22
|
Bhatia S, Kaul D and Varma N: Potential
tumor suppressive function of miR-196b in B-cell lineage acute
lymphoblastic leukemia. Mol Cell Biochem. 340:97–106. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ye H, Liu X, Lv M, Wu Y, Kuang S, Gong J,
Yuan P, Zhong Z, Li Q, Jia H, et al: MicroRNA and transcription
factor co-regulatory network analysis reveals miR-19 inhibits CYLD
in T-cell acute lymphoblastic leukemia. Nucleic Acids Res.
40:5201–5214. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vardiman JW, Thiele J, Arber DA, Brunning
RD, Borowitz MJ, Porwit A, Harris NL, Le Beau MM,
Hellström-Lindberg E, Tefferi A, et al: The 2008 revision of the
World Health Organization (WHO) classification of myeloid neoplasms
and acute leukemia: Rationale and important changes. Blood. 114.
pp. 937–951. 2009, View Article : Google Scholar
|
|
25
|
Alvarnas JC, Brown PA, Aoun P, Ballen KK,
Bellam N, Blum W, Boyer MW, Carraway HE, Coccia PF, Coutre SE, et
al National Comprehensive Cancer Network: Acute lymphoblastic
leukemia. J Natl Compr Canc Netw. 10:858–914. 2012.PubMed/NCBI
|
|
26
|
Allen A, Gill K, Hoehn D, Sulis M, Bhagat
G and Alobeid B: C-myc protein expression in B-cell acute
lymphoblastic leukemia, prognostic significance? Leuk Res.
38:1061–1066. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pfaffl MW: A new mathematical model for
relative quantification in real-time RT-PCR. Nucleic Acids Res.
29:e452001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sui W, Lin H, Peng W, Huang Y, Chen J,
Zhang Y and Dai Y: Molecular dysfunctions in acute rejection after
renal transplantation revealed by integrated analysis of
transcription factor, microRNA and long noncoding RNA. Genomics.
102:310–322. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dong M, Wang X, Zhao HL, Chen XL, Yuan JH,
Guo JY, Li KQ and Li G: Integrated analysis of transcription
factor, microRNA and LncRNA in an animal model of obliterative
bronchiolitis. Int J Clin Exp Pathol. 8:7050–7058. 2015.PubMed/NCBI
|
|
30
|
Lin Y, Wu J, Chen H, Mao Y, Liu Y, Mao Q,
Yang K, Zheng X and Xie L: Cyclin-dependent kinase 4 is a novel
target in micoRNA-195-mediated cell cycle arrest in bladder cancer
cells. FEBS Lett. 586:442–447. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rappaport N, Nativ N, Stelzer G, Twik M,
Guan-Golan Y, Stein TI, Bahir I, Belinky F, Morrey CP, Safran M, et
al: MalaCards: An integrated compendium for diseases and their
annotation. Database (Oxford). 2013:bat0182013. View Article : Google Scholar
|
|
32
|
Felice B, Cattoglio C, Cittaro D, Testa A,
Miccio A, Ferrari G, Luzi L, Recchia A and Mavilio F: Transcription
factor binding sites are genetic determinants of retroviral
integration in the human genome. PLoS One. 4:e45712009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bang SY, Kim JH, Lee PY, Chi SW, Cho S, Yi
GS, Myung PK, Park BC, Bae KH and Park SG: Candidate target genes
for the Saccharomyces cerevisiae transcription factor, Yap2. Folia
Microbiol (Praha). 58:403–408. 2013. View Article : Google Scholar
|
|
34
|
Lin XC, Xu Y, Sun GP, Wen JL, Li N, Zhang
YM, Yang ZG, Zhang HT and Dai Y: Molecular dysfunctions in acute
myeloid leukemia revealed by integrated analysis of microRNA and
transcription factor. Int J Oncol. 48:2367–2380. 2016.PubMed/NCBI
|
|
35
|
Alexiou P, Vergoulis T, Gleditzsch M,
Prekas G, Dalamagas T, Megraw M, Grosse I, Sellis T and
Hatzigeorgiou AG: miRGen 2.0: A database of microRNA genomic
information and regulation. Nucleic Acids Res. 38:D137–D141. 2010.
View Article : Google Scholar :
|
|
36
|
Harris MA, Clark J, Ireland A, Lomax J,
Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C,
et al Gene Ontology Consortium: The Gene Ontology (GO) database and
informatics resource. Nucleic Acids Res. 32:D258–D261. 2004.
View Article : Google Scholar
|
|
37
|
Kanehisa M, Araki M, Goto S, Hattori M,
Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T,
et al: KEGG for linking genomes to life and the environment.
Nucleic Acids Res. 36:D480–D484. 2008. View Article : Google Scholar :
|
|
38
|
Arora S, Rana R, Chhabra A, Jaiswal A and
Rani V: miRNA-transcription factor interactions: A combinatorial
regulation of gene expression. Mol Genet Genomics. 288:77–87. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhou Y, Kanagal-Shamanna R, Zuo Z, Tang G,
Medeiros LJ and Bueso-Ramos CE: Advances in B-lymphoblastic
leukemia: Cytogenetic and genomic lesions. Ann Diagn Pathol.
23:43–50. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Brondfield S, Umesh S, Corella A, Zuber J,
Rappaport AR, Gaillard C, Lowe SW, Goga A and Kogan SC: Direct and
indirect targeting of MYC to treat acute myeloid leukemia. Cancer
Chemother Pharmacol. 76:35–46. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Malin S, McManus S and Busslinger M: STAT5
in B cell development and leukemia. Curr Opin Immunol. 22:168–176.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Berger A, Sexl V, Valent P and Moriggl R:
Inhibition of STAT5: A therapeutic option in BCR-ABL1-driven
leukemia. Oncotarget. 5:9564–9576. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chae HD, Mitton B, Lacayo NJ and Sakamoto
KM: Replication factor C3 is a CREB target gene that regulates cell
cycle progression through the modulation of chromatin loading of
PCNA. Leukemia. 29:1379–1389. 2015. View Article : Google Scholar
|
|
44
|
Köhrer S, Havranek O, Seyfried F, Hurtz C,
Coffey GP, Kim E, Ten Hacken E, Jäger U, Vanura K, O'Brien S, et
al: Pre-BCR signaling in precursor B-cell acute lymphoblastic
leukemia regulates PI3K/AKT, FOXO1 and MYC, and can be targeted by
SYK inhibition. Leukemia. 30:1246–1254. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Coomans de Brachène A and Demoulin JB:
FOXO transcription factors in cancer development and therapy. Cell
Mol Life Sci. 73:1159–1172. 2016. View Article : Google Scholar
|
|
46
|
Grosjean-Raillard J, Adès L, Boehrer S,
Tailler M, Fabre C, Braun T, De Botton S, Israel A, Fenaux P and
Kroemer G: Flt3 receptor inhibition reduces constitutive NFkappaB
activation in high-risk myelodysplastic syndrome and acute myeloid
leukemia. Apoptosis. 13:1148–1161. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Vacca A, Felli MP, Palermo R, Di Mario G,
Calce A, Di Giovine M, Frati L, Gulino A and Screpanti I: Notch3
and pre-TCR interaction unveils distinct NF-kappaB pathways in
T-cell development and leukemia. EMBO J. 25:1000–1008. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bruns HA and Kaplan MH: The role of
constitutively active Stat6 in leukemia and lymphoma. Crit Rev
Oncol Hematol. 57:245–253. 2006. View Article : Google Scholar
|
|
49
|
LeBrun DP: E2A basic helix-loop-helix
transcription factors in human leukemia. Front Biosci. 8:s206–s222.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Puissegur MP, Eichner R, Quelen C, Coyaud
E, Mari B, Lebrigand K, Broccardo C, Nguyen-Khac F, Bousquet M and
Brousset P: B-cell regulator of immunoglobulin heavy-chain
transcription (Bright)/ARID3a is a direct target of the oncomir
microRNA-125b in progenitor B-cells. Leukemia. 26:2224–2232. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Strasser A, Harris AW, Bath ML and Cory S:
Novel primitive lymphoid tumours induced in transgenic mice by
cooperation between myc and bcl-2. Nature. 348:331–333. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ott CJ, Kopp N, Bird L, Paranal RM, Qi J,
Bowman T, Rodig SJ, Kung AL, Bradner JE and Weinstock DM: BET
bromodomain inhibition targets both c-Myc and IL7R in high-risk
acute lymphoblastic leukemia. Blood. 120:2843–2852. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Joliot V, Cormier F, Medyouf H, Alcalde H
and Ghysdael J: Constitutive STAT5 activation specifically
cooperates with the loss of p53 function in B-cell lymphomagenesis.
Oncogene. 25:4573–4584. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Nakayama J, Yamamoto M, Hayashi K, Satoh
H, Bundo K, Kubo M, Goitsuka R, Farrar MA and Kitamura D: BLNK
suppresses pre-B-cell leukemogenesis through inhibition of JAK3.
Blood. 113:1483–1492. 2009. View Article : Google Scholar :
|
|
55
|
Cho JH, Gelinas R, Wang K, Etheridge A,
Piper MG, Batte K, Dakhallah D, Price J, Bornman D, Zhang S, et al:
Systems biology of interstitial lung diseases: Integration of mRNA
and microRNA expression changes. BMC Med Genomics. 4:82011.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Li H: Systems genetics in '-omics' era:
Current and future development. Theory Biosci. 132:1–16. 2013.
View Article : Google Scholar
|
|
57
|
Lin Y, Zhang Q, Zhang HM, Liu W, Liu CJ,
Li Q and Guo AY: Transcription factor and miRNA co-regulatory
network reveals shared and specific regulators in the development
of B cell and T cell. Sci Rep. 5:152152015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Jackstadt R and Hermeking H: MicroRNAs as
regulators and mediators of c-MYC function. Biochim Biophys Acta.
1849:544–553. 2015. View Article : Google Scholar
|
|
59
|
Pinz S, Unser S and Rascle A: Signal
transducer and activator of transcription STAT5 is recruited to
c-Myc super-enhancer. BMC Mol Biol. 17:102016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Han SS, Han S and Kamberos NL:
Piperlongumine inhibits the proliferation and survival of B-cell
acute lymphoblastic leukemia cell lines irrespective of
glucocorticoid resistance. Biochem Biophys Res Commun. 452:669–675.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Schepers H, Wierenga AT, Vellenga E and
Schuringa JJ: STAT5-mediated self-renewal of normal hematopoietic
and leukemic stem cells. JAK-STAT. 1:13–22. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Tsuruyama T, Nakamura T, Jin G, Ozeki M,
Yamada Y and Hiai H: Constitutive activation of Stat5a by
retrovirus integration in early pre-B lymphomas of SL/Kh strain
mice. Proc Natl Acad Sci USA. 99:8253–8258. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Gao SM, Yang J, Chen C, Zhang S, Xing CY,
Li H, Wu J and Jiang L: miR-15a/16–1 enhances retinoic
acid-mediated differentiation of leukemic cells and is upregulated
by retinoic acid. Leuk Lymphoma. 52:2365–2371. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Huang E, Liu R and Chu Y: miRNA-15a/16: As
tumor suppressors and more. Future Oncol. 11:2351–2363. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Gu TL, Nardone J, Wang Y, Loriaux M,
Villén J, Beausoleil S, Tucker M, Kornhauser J, Ren J, MacNeill J,
et al: Survey of activated FLT3 signaling in leukemia. PLoS One.
6:e191692011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Annesley CE and Brown P: The biology and
targeting of FLT3 in pediatric leukemia. Front Oncol. 4:2632014.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Small D: Targeting FLT3 for the treatment
of leukemia. Semin Hematol. 45(Suppl 2): S17–S21. 2008. View Article : Google Scholar : PubMed/NCBI
|