Introduction
Ovarian cancer is the leading cause of death from
gynecologic malignancies in the United States (1). Most cases of ovarian cancer present
with advanced-stage disease and despite an initial high treatment
response rate, the majority of patients eventually develop
chemotherapy resistance (2).
High-grade serous ovarian cancer (HGSOC) accounts for 70–80% of
epithelial ovarian cancer cases. BRCA wild-type HGSOC
patients in particular have a worse survival compared to both
germline and somatic BRCA mutated HGSOC, and there remain
unmet therapeutic needs for them (3,4).
PARP inhibition has demonstrated clinical activity
against HGSOC, especially in deleterious germline BRCA
mutation carriers and platinum-sensitive diseases (5). Olaparib is the first US Food and Drug
Administration-approved PARP inhibitor (PARPi), licensed for use in
heavily pretreated germ-line BRCA mutation-associated
ovarian cancer (6). There are
several other PARPi currently in the late phase clinical trial
development, including niraparib, rucaparib, veliparib, and BMN 673
(talazoparib) (7). BMN 673 is a
potent oral PARPi with a greater DNA-PARP trapping activity
compared with other PARPi (8).
Thus far, PARPi monotherapy has shown limited activity against
BRCA wild-type HGSOC, highlighting the need for combination
strategies.
PARP1, the dominant isoform responsible for the
majority of PARP activity, is involved in various modes of cell
death including apoptosis, parthanatos, necroptosis, and autophagy,
in addition to DNA damage repair (9). PARP1 is cleaved and
poly(ADP-ribosyl)ation is inhibited during apoptosis (9). PARP1 activation and accumulation of
poly(ADP-ribose), by contrast, is essential for initiating or
progressing the other cell death pathways (9). Thus, drugs targeting apoptotic
pathways would be a suitable candidate for combination with
PARPi.
Bcl-2 family proteins play a critical role in
regulating the intrinsic apoptotic pathway. Major Bcl-2 family
proteins include the anti-apoptotic proteins (Bcl-2, Bcl-xL, Bcl-w,
and Mcl-1), pro-apoptotic effector proteins (Bax and Bak), and
pro-apoptotic BH3-only proteins (Bim, Bid, Bad, Puma, and Noxa)
(10). Bcl-xL is overexpressed in
the majority of recurrent, chemoresistant ovarian cancer, and its
expression in primary ovarian tumor is associated with a shorter
disease-free interval (11,12).
Preclinical studies showed inhibition of Bcl-xL increased
sensitivity of ovarian cancer cells to chemotherapeutic agents
(12,13). These data support anti-apoptotic
protein inhibition as a promising therapeutic direction for
recurrent ovarian cancer. ABT-263 is an orally available Bad-like
BH3 mimetic. ABT-263 competes with BH3-only proteins for binding to
anti-apoptotic proteins, thereby preventing anti-apoptotic proteins
from constraining pro-apoptotic proteins (10). ABT-263 has an inhibitory activity
against Bcl-2, Bcl-xL, and Bcl-w, but not against the other
anti-apoptotic protein Mcl-1 (10). This may partly explain the limited
monotherapy activity of ABT-263 against solid tumors (14), although ABT-263 treatment has
demonstrated favorable clinical outcome in hematological
malignancies as a mono-therapy (15).
We hypothesized that PARPi-induced cytotoxicity may
be augmented by inhibition of anti-apoptotic proteins. Our aim was
to evaluate the preclinical efficacy of the BH3-mimetic ABT-263 in
combination with the PARPi BMN 673 in HGSOC cells at clinically
achievable concentrations.
Materials and methods
Compounds
ABT-263 and BMN 673 were purchased from Selleck
Chemicals (Houston, TX, USA). Stock solutions of ABT-263 and BMN
673 were made in DMSO at 100 mM and 20 mM, respectively. Aliquots
were stored at −80°C and diluted in culture medium at least
1:10,000 immediately before use.
Cell culture
Three BRCA wild-type HGSOC cell lines were
used (16): OVCAR3 (purchased from
ATCC, Manassas, VA, USA), OVCAR8 (obtained from the NCI-Frederick
DCTD tumor/cell line repository, Frederick, MD, USA), and OV90
(generously gifted from Dr C.M. Annunziata, NCI/NIH, Bethesda, MD,
USA). All cell lines were cultured in RPMI-1640 medium with
L-glutamine, supplemented with 10% FBS and 1%
penicillin-streptomycin under a humidified atmosphere of 5%
CO2 at 37°C. Drug treatments were performed 24 h after
initial seeding. All cell lines were tested and authenticated in
March 2016 at NCI-Frederick Protein Expression Laboratory. Briefly,
DNA was extracted from each cell pellet and amplified by PCR using
the AmpFLSTR Identifiler PCR Amplification kit (Applied Biosystems,
Foster City, CA, USA). The Identifiler kit amplifies 15
tetranucleotide repeat loci and the Amelogenin gender determination
marker in a single PCR amplification. PCR-amplified fragments were
analyzed on the Applied Biosystems 3130xl genetic analyzer (Applied
Biosystems) in Hi-Di formamide with a size standard. Fragments were
then labeled and identified using GeneMapper software Version 4.0
(Applied Biosystems). Authenticity was confirmed against the ATCC
STR profiling database (www.atcc.org/en/STR_Database.aspx) and the NCI-60
published data (17).
Cell viability assay
Cells were seeded in 96-well plates at a density of
2,000 (OVCAR3 and OV90) or 1,000 (OVCAR8) cells per well, and
treated with ABT-263 and BMN 673 for the indicated time. Cell
viability was determined using the XTT Cell Proliferation Assay kit
(Trevigen, Gaithersburg, MD, USA) by comparing absorbance from the
drug-treated cells with that from untreated cells at the same
incubation period, unless otherwise specified. Absorbance at 450 nm
with a reference wavelength of 650 nm was measured by the
SpectraMax M5 reader (Molecular Devices, Sunnyvale, CA, USA). Each
drug concentration was examined in duplicate or triplicate, and
four independent experiments were performed. The IC50
values of ABT-263 and BMN 673 were calculated from
concentration-effect curves by applying a four-parameter non-linear
regression model using GraphPad Prism 7.0b software (GraphPad
Software Inc., La Jolla, CA, USA).
Drug combination analysis
Combination synergism was analyzed by two different
methods: the Chou-Talalay method using CompuSyn software (ComboSyn
Inc., Paramus, NJ, USA) (18) and
the Prichard-Shipman method using MacSynergy II software (Prichard
and Shipman, University of Michigan, Ann Arbor, MI, USA) (19). The resulting combination index (CI)
values define synergism (<0.9), additive (0.9–1.1), and
antagonism (>1.1) in drug combinations (18). Synergy/antagonism volumes were
statistically evaluated at the 95% confidence level and were
expressed in µM2%, which are used to categorize
the drug interactions: synergy (>25), additive (−25 to 25), and
antagonism (<−25) (20).
Confocal immunofluorescence microscopy of
γ-H2AX foci
Cells were seeded onto 12 mm poly L-lysine-coated
coverslips (Corning Inc., Oneonta, NY, USA) in 24-well plates, and
treated with 2 µM ABT-263 and 25 nM BMN 673 individually and
in combination for 24 and 48 h. DMSO (0.002%) treatment was used as
a vehicle control for this γ-H2AX immunofluorescence staining and
all the experiments described below. Cells were washed in
Dulbecco's PBS without calcium and magnesium upon completion of
incubation, then fixed in 4% paraformaldehyde in PBS for 10 min.
Permeabilization was performed with 0.25% Triton X-100 in PBS for
10 min, followed by blocking in 1% BSA in PBS for 1 h.
Immunostaining was performed using the anti-γ-H2AX antibody
(ab22551, 1:400 dilution, Abcam, Cambridge, MA, USA) for 1 h. Cells
were washed three times with PBS, then incubated with the secondary
antibody conjugated to Alexa Fluor 647 (A-21235, 1:200 dilution,
Thermo Fisher Scientific Inc., Grand Island, NY, USA) for 1 h in
the dark. Coverslips were mounted using Vectashield HardSet
Antifade Mounting Medium with DAPI (Vector Laboratories,
Burlingame, CA, USA), and slides were sealed and stored at 4°C
until imaging. The experiment was repeated at least four times on
independent occasions per cell line at each time point for this
γ-H2AX staining and all the experiments described below, unless
otherwise specified. Confocal immunofluorescence microscopy imaging
was performed using the LSM 780 laser-scanning confocal microscope
(Carl Zeiss, Thornwood, NY, USA) with a 63x/1.4 oil immersion
objective. A minimum of 120 cells were imaged per sample. Images
were analyzed by counting the number of γ-H2AX foci using Focinator
software (21), and noise
tolerance level was set to 80. Cells with >5 foci in the nucleus
were considered to be γ-H2AX foci-positive.
Cell cycle analysis
Cell cycle analysis was conducted using the APC BrdU
Flow kit (BD Biosciences, San Jose, CA, USA). Cells were treated as
described for the γ-H2AX immunofluorescence staining, then labeled
with 10 µM BrdU for 1 h at 37°C. Both floating and attached
cells were harvested and subjected to the flow cytometric analysis
according to the manufacturer's instructions. Stained cells were
acquired on the FACSCalibur flow cytometer operated by CellQuest
Pro software (BD Biosciences), and 25,000 events were collected per
sample. The cell cycle distribution was analyzed using Flowjo
10.0.8r1 software (Tree Star Inc., Ashland, OR, USA).
Annexin V binding assay
Cells were treated as above, then both floating and
attached cells were stained using the APC Annexin V Apoptosis
Detection kit with 7-AAD (Biolegend, San Diego, CA, USA). Stained
cells were measured using the FACSCalibur flow cytometer, and
25,000 events were collected per sample. The proportion of Annexin
V-positive cells were gated and calculated using Flowjo 10.0.8r1
software.
Caspase activity assay
Caspase activity was determined using the
Caspase-Glo 3/7 Assay kit (Promega, Madison, WI, USA). Cells were
treated as above, and both floating and attached cells were
harvested, pelleted, and subjected to the caspase activity assay.
Briefly, cell pellets were lysed in modified RIPA buffer (50 mM
Tris-HCl, pH 7.5, 150 mM NaCl, 10 µg/ml aprotinin, 1 mM
phenylmethylsulfonyl fluoride, 10 µg/ml leupeptin, 2 mM
Na3VO4, 4 mM EDTA, 10 mM NaF, 10 mM sodium
pyrophosphate, 1% Nonidet P-40, and 0.1% sodium deoxycholate),
supplemented with cOmplete Mini protease inhibitor cocktail tablets
and PhosSTOP phosphatase inhibitor cocktail tablets (Roche
Diagnostics, Indianapolis, IN, USA) and centrifuged at 14,000 rpm
for 20 min. Protein concentration was determined using the Pierce
BCA Protein Assay kit (Thermo Fisher Scientific Inc.). Ten
micrograms of protein in a 50 µl total volume was mixed with
50 µl of equilibrated Caspase-Glo 3/7 reagent. After 30 min
of incubation in the dark, luminescence was measured with the
SpectraMax M5 reader.
Western blot analysis
Whole cell lysates were prepared as described for
the caspase activity assay, and assessed by immunoblotting.
Briefly, equal amounts of protein between 12.8 and 32.0 µg
were separated by SDS-PAGE under reducing conditions on 14 or 16%
Novex Tris-Glycine gels (Thermo Fisher Scientific Inc.) and
electrophoretically transferred to 0.45 µm polyvinylidene
difluoride membranes (EMD Millipore, Billerica, MA, USA). The XCell
SureLock Mini-Cell and the XCell Blot Module (Thermo Fisher
Scientific Inc.) were used for gel electrophoresis and protein
transfer, according to the manufacturer's instructions. After
incubation with 5% non-fat dry milk in TBS containing 0.1% Tween-20
(TBST) for 1 h, membranes were washed once with TBST and probed
with the indicated primary antibodies overnight at 4°C. The
following primary antibodies were used: cleaved PARP (#5625),
β-tubulin (#2128), Bcl-2 (#4223), Bcl-xL (#2764), Mcl-1 (#5453),
Bax (#5023), Bak (#12105), Bim (#2933), Bid (#2002) (all from Cell
Signaling Technology, Danvers, MA, USA), Puma (ab33906), and Noxa
(ab13654) (both from Abcam). All the primary antibodies were
diluted 1:1,000 in TBST containing 5% BSA, except β-tubulin and
Bcl-xL, which were diluted 1:10,000. Membranes were washed three
times with TBST and incubated with 1:5,000 dilution of horseradish
peroxidase-conjugated anti-mouse or rabbit antibodies (Thermo
Fisher Scientific Inc.) for 1 h. After washing three times with
TBST, membranes were incubated with SuperSignal West Pico or Dura
Chemiluminescent Substrate (Thermo Fisher Scientific Inc.) for 5
min. Chemiluminescent signals were visualized by exposing the
membrane to X-ray film (HyBlot ES Autoradiography Film, Denville
Scientific Inc., Holliston, MA, USA). Immunoblot analyses were
performed at least three times for all the antibodies. Densitometry
was performed using ImageJ 1.50b software (NIH, Bethesda, MD, USA).
Densitometry values were normalized to the loading control
β-tubulin and expressed as relative fold changes compared to
DMSO-treated cells.
Statistical analysis
For multiple comparisons, the results were analyzed
by a two-way ANOVA and the Dunnett post hoc test to compare the
mean of each group with the combination treatment group using
GraphPad Prism 7.0b software. All differences were considered
statistically significant at p<0.05.
Results
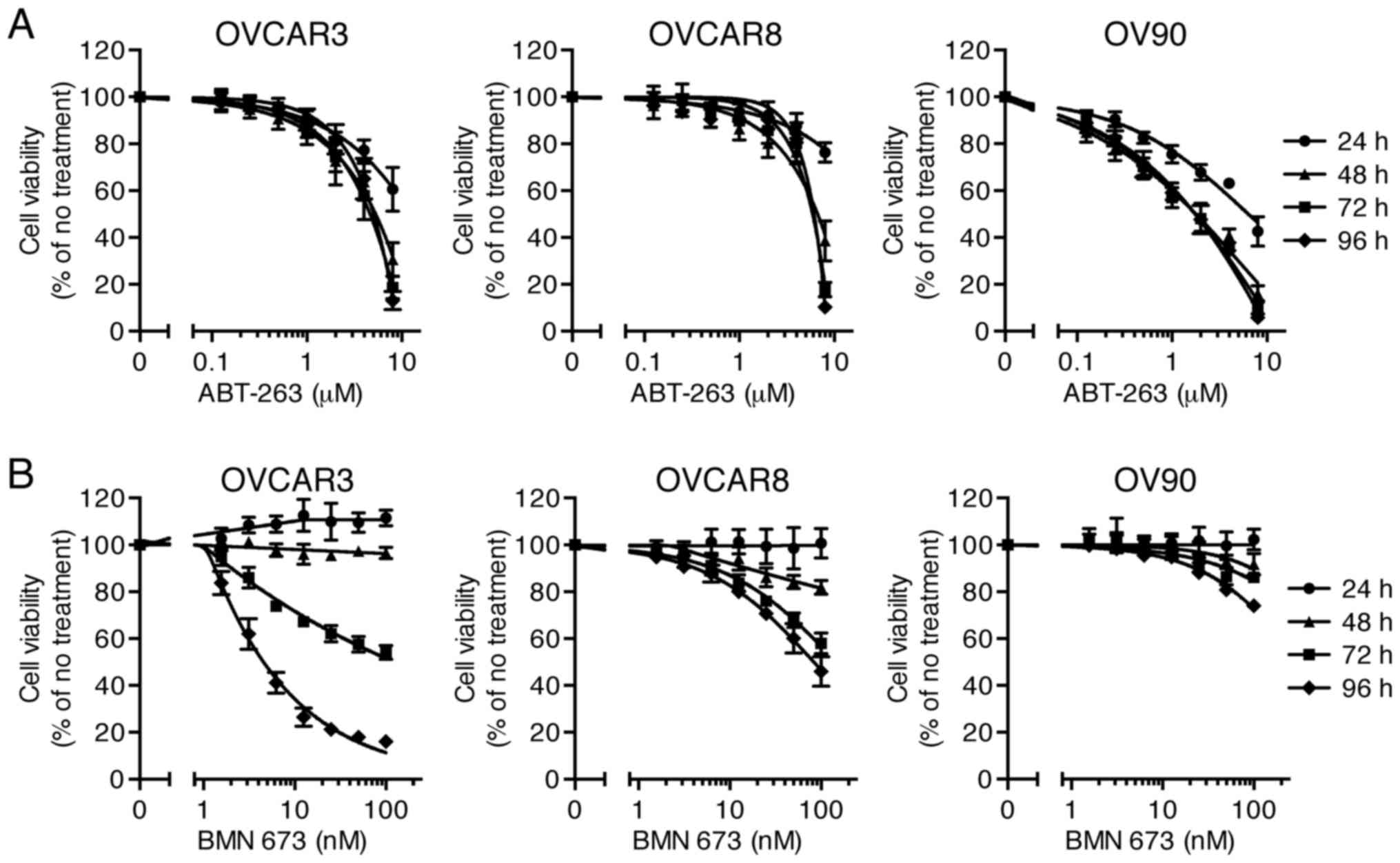
Sensitivity of HGSOC cells to single
agent treatment
Effects of ABT-263 (0.125–8 µM) and BMN 673
(1.56–100 nM) monotherapy on cell viability of three HGSOC cell
lines were evaluated at 24, 48, 72, and 96 h after initiation of
drug treatment. The concentration-effect curves are shown in
Fig. 1. Both ABT-263 and BMN 673
caused a concentration-dependent growth inhibition in all cell
lines. The effect of BMN 673 was deemed more treatment
time-dependent than that of ABT-263 in each cell line. BMN 673
treatment achieved modest cell growth inhibition during the first
48 h, followed by significant growth inhibition during the last 48
h. By contrast, ABT-263 treatment resulted in maximum cytotoxic
effects within 48 h, and almost no change in IC50 values
after 48 h (Table I).
 | Table ICytotoxic effects of ABT-263 and BMN
673. |
Table I
Cytotoxic effects of ABT-263 and BMN
673.
| ABT-263
IC50 (µM)
| BMN 673
IC50 (nM)
|
|---|
| 24 h | 48 h | 72 h | 96 h | <72 h | 96 h |
|---|
| OVCAR3 | ND | 5.00 (1.28) | 4.07 (0.87) | 4.50 (0.71) | ND | 4.66 (0.80) |
| OVCAR8 | ND | 7.04 (1.78) | 5.46 (0.58) | 5.42 (0.22) | ND | 88.2 (26.5) |
| OV90 | 6.70 (1.66) | 1.65 (0.39) | 1.51 (0.30) | 1.48 (0.24) | ND | ND |
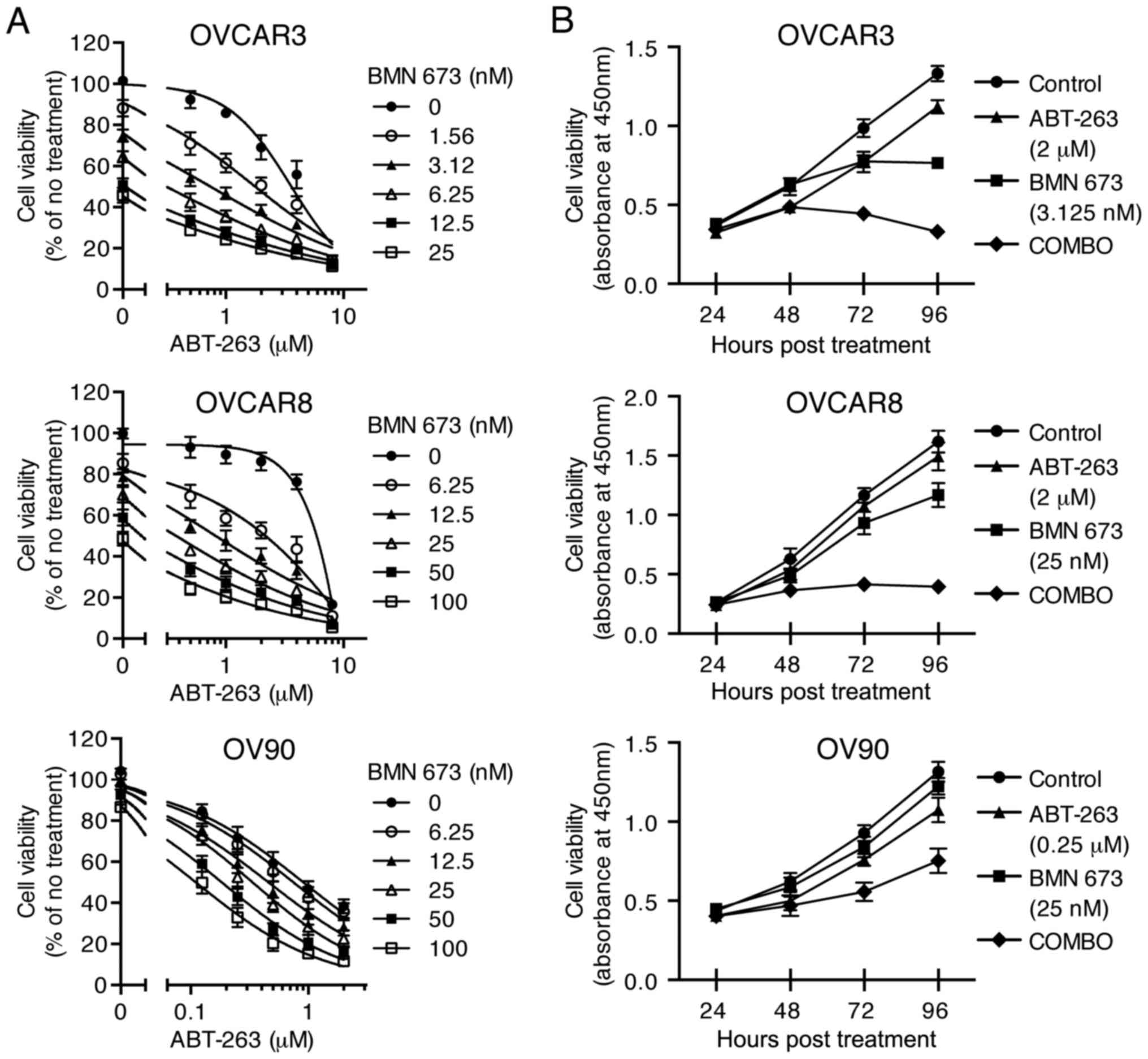
The combination of ABT-263 and BMN 673
synergistically inhibits HGSOC cell proliferation
To examine the cytotoxic effects of the combination
treatment, three HGSOC cell lines were treated with indicated
concentrations of ABT-263 alone and in combination with BMN 673 for
72 h (and 24–96 h for time course experiments). All the
concentration-effect curves for the combination treatment showed an
apparent leftward shift compared to the monotherapy curve in each
cell line (Fig. 2A), indicating
synergy (22). Table II shows the CI values and
synergy/antagonism volumes for each cell line with the combination
treatment for 72 h. Synergistic cytotoxicity was confirmed across
all cell lines with the CI values at 50, 75, and 90% inhibition all
<0.9 (18), and the
synergy/antagonism volumes at 95% confidence all >100 (20). Time course of cell viability was
measured as a function of the XTT assay absolute absorbance value.
The combination treatment completely inhibited growth of OVCAR3 and
OVCAR8 cells when each monotherapy showed a modest effect (Fig. 2B).
| Table IICombination index (CI) values and
synergy/antagonism volumes after 72 h of treatment. |
Table II
Combination index (CI) values and
synergy/antagonism volumes after 72 h of treatment.
| CI values at
inhibition of
| Synergy/antagonism
volumes (µM2%) |
|---|
| 50% | 75% | 90% |
|---|
| OVCAR3 | 0.52 (0.05) | 0.58 (0.09) | 0.71 (0.13) | 150/−5 |
| OVCAR8 | 0.34 (0.02) | 0.29 (0.06) | 0.29 (0.11) | 345/0 |
| OV90 | 0.57 (0.11) | 0.58 (0.25) | 0.89 (0.60) | 225/0 |
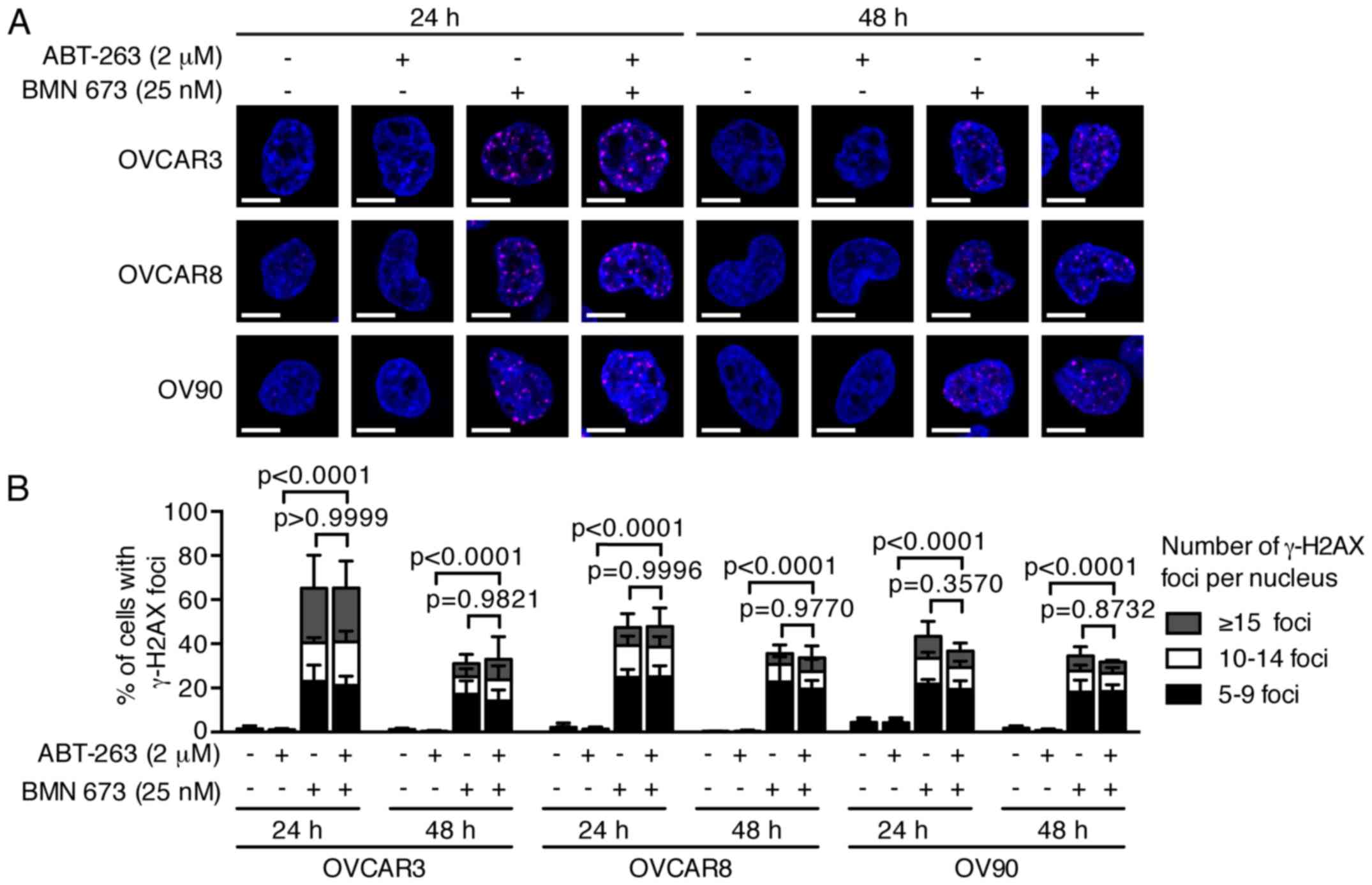
DNA fragmentation is significantly
increased by the combination treatment
PARPi cause DNA damage accumulation and G2/M cell
cycle arrest (23,24). Effects of ABT-263 and BMN 673 on
DNA double-strand break induction and cell cycle progression were
thus examined to dissect the potential mechanisms of cell growth
inhibition induced by the combination treatment. BMN 673
monotherapy induced γ-H2AX foci formation in all cell lines as
early as 24 h after treatment (Fig.
3). ABT-263 alone or in combination with BMN 673 did not
significantly induce γ-H2AX foci formation compared with the
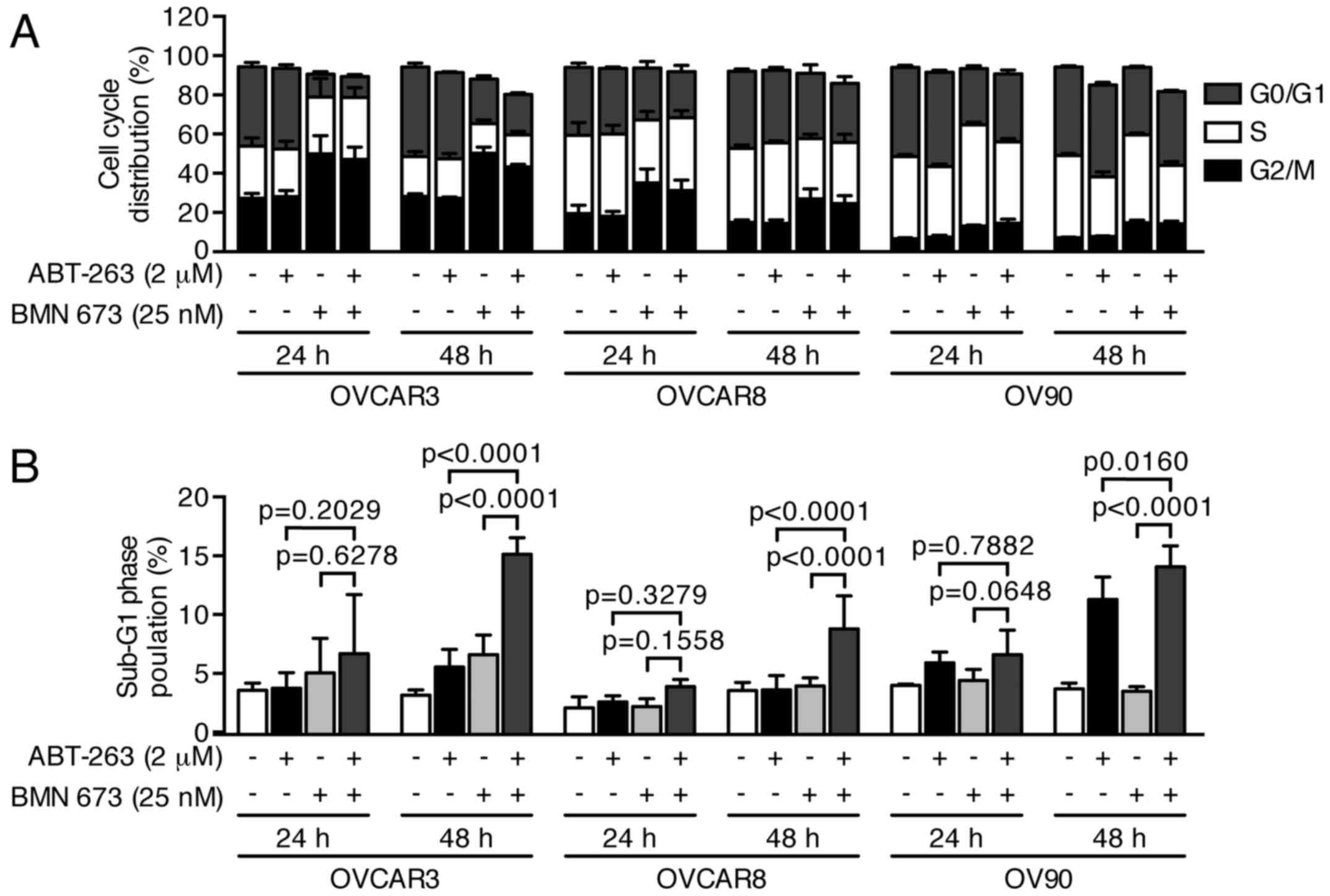
control or BMN 673 monotherapy, respectively. The cell cycle
analysis indicated that BMN 673 treatment induced G2/M phase
accumulation in all cell lines as early as 24 h after treatment, in
the presence or absence of ABT-263 (Figs. 4 and 5A). ABT-263 monotherapy did not
significantly change the cell cycle distribution compared with the
control. Notably, the combination treatment induced sub-G1 phase
accumulation compared with the control and each monotherapy after
48 h of treatment, indicating an increase in DNA fragmentation
(Figs. 4 and 5B).
Combination treatment significantly
induces apoptotic cell death via a caspase-dependent pathway
We next examined whether growth inhibition and DNA
fragmentation induced by the combination treatment were
attributable to apoptotic cell death. The combination treatment
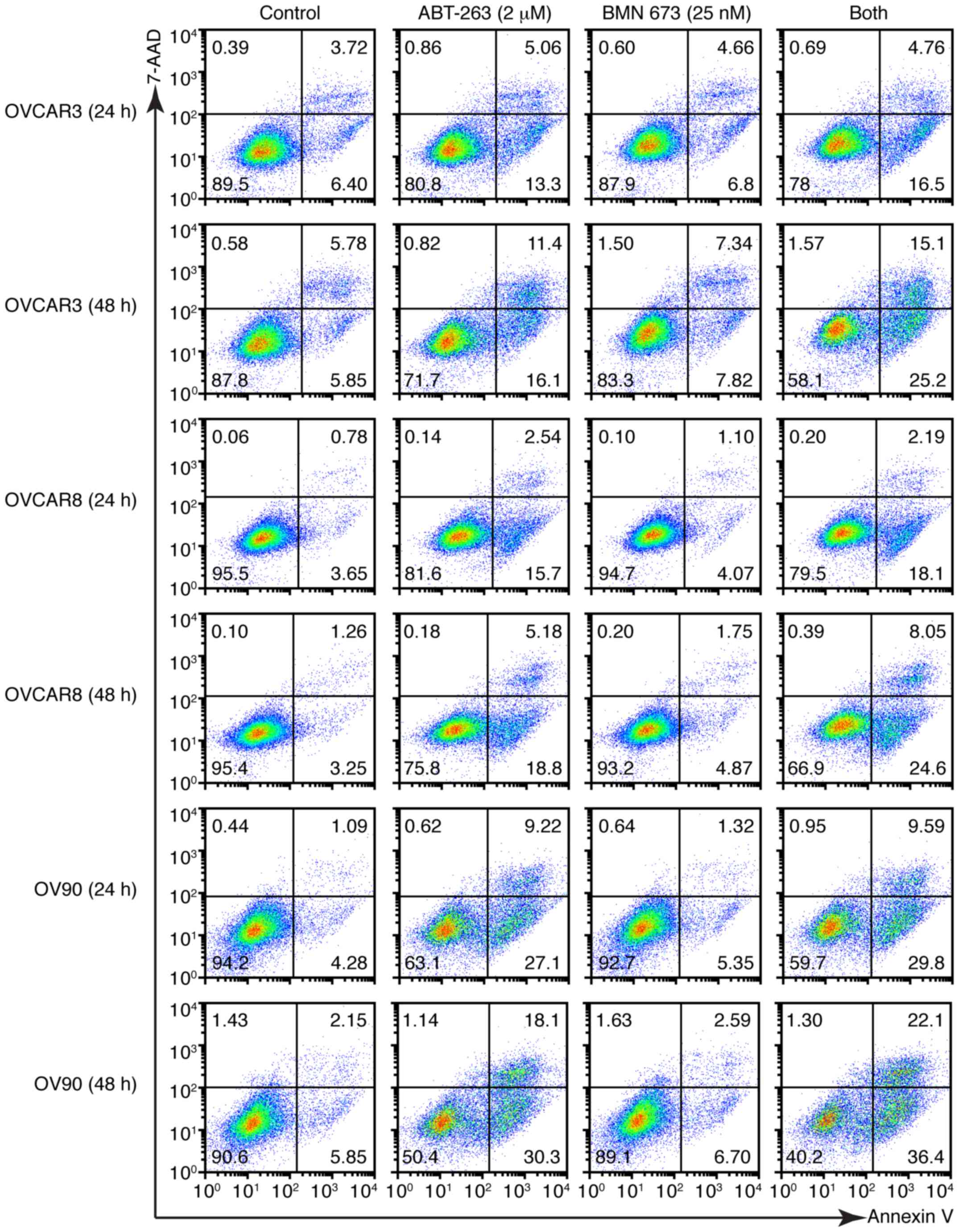
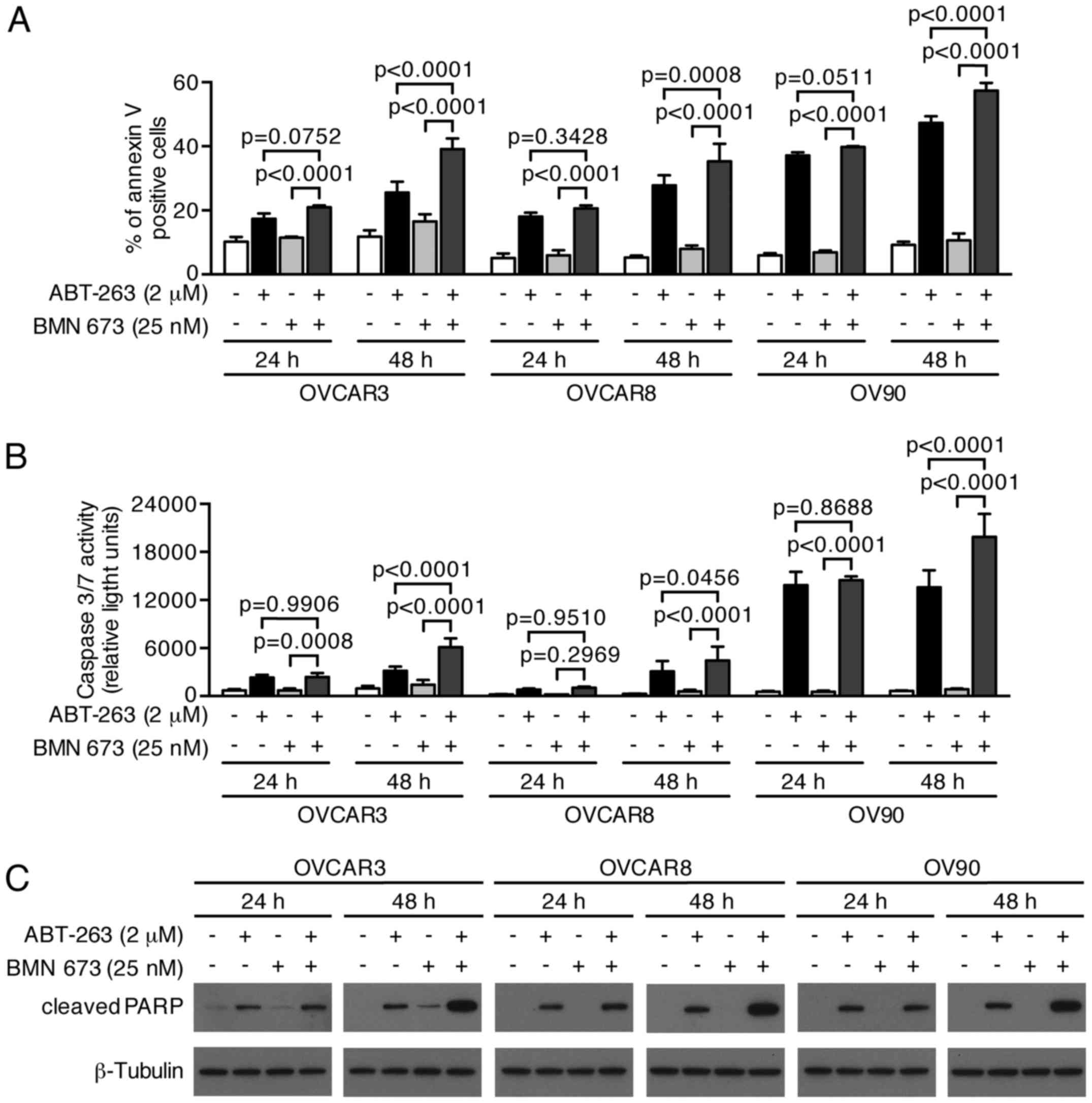
induced a higher percentage of Annexin V-positive cells compared
with the control and each monotherapy in all cell lines after 48 h
(Figs. 6 and 7A). We evaluated the caspase-3/7 activity
and PARP cleavage to further verify the apoptotic nature of cell
death induced by the combination treatment. Fig. 7B shows a significant increase of
caspase-3/7 activity after 48 h of the combination treatment
compared with the control and each monotherapy in all cell lines.
This was accompanied by an increased cleaved PARP expression after
48 h of the combination treatment, suggesting that a
caspase-dependent apoptotic pathway was activated in response to
the combination treatment (Fig.
7C).
ABT-263 alone and in combination with BMN
673 induces expression levels of Mcl-1, Bim, and Noxa
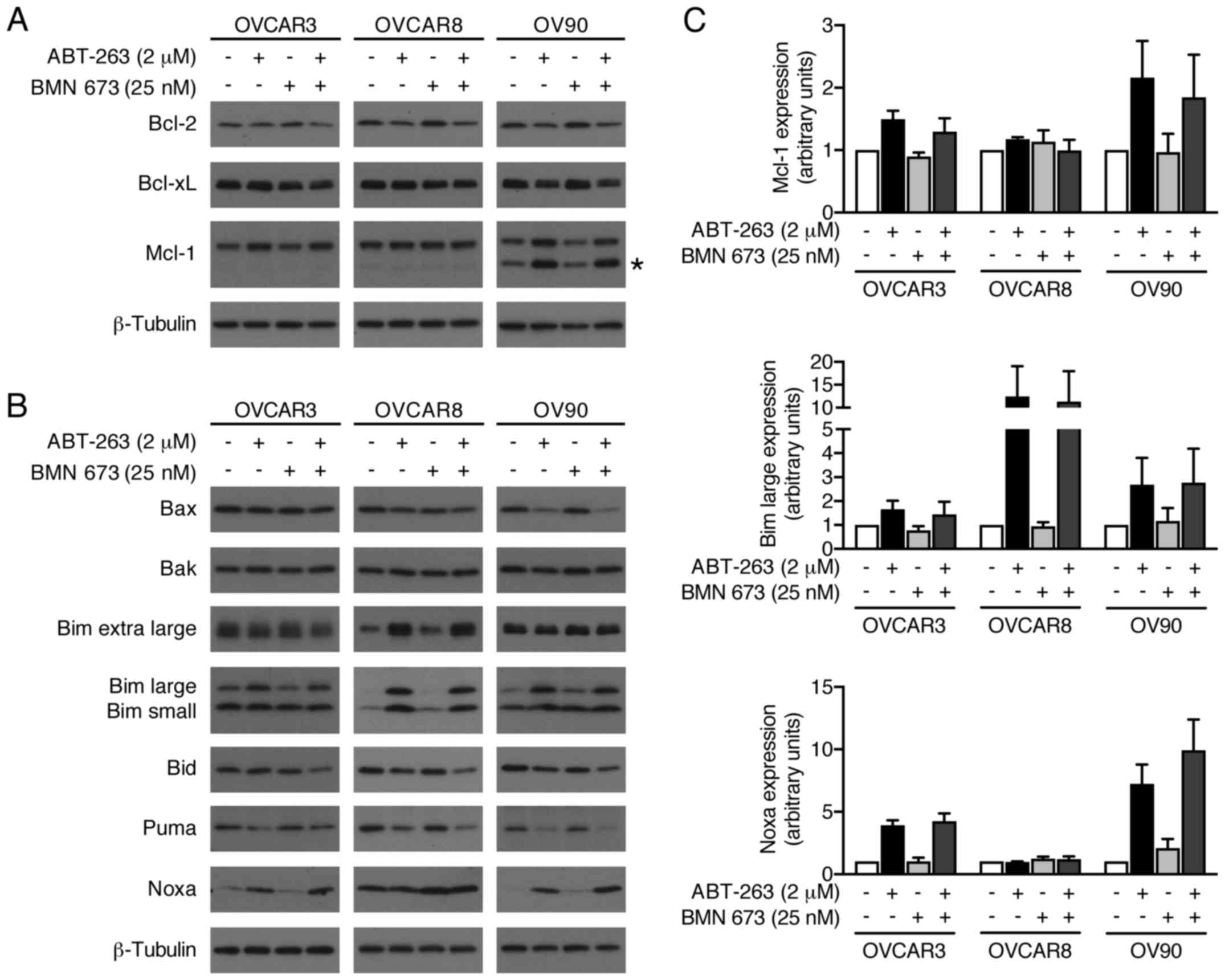
We evaluated possible alterations in the expression
levels of pro- and anti-apoptotic Bcl-2 family proteins to further
explore molecular mechanisms of increased apoptosis by the
combination treatment (Fig. 8).
BMN 673 monotherapy had little effect on most proteins examined
after 48 h of treatment. ABT-263 monotherapy and the combination
treatment increased Mcl-1, Bim, and Noxa protein expression.
Notably, Bid expression was decreased by the combination treatment,
but not by either monotherapy.
Discussion
Modulation of apoptotic pathways is a promising
therapeutic strategy in HGSOC. Here, we report a novel benefit of
the combination treatment with ABT-263 and BMN 673 against HGSOC
cells. The combination treatment showed synergistic cytotoxic
effects based on two different methods: the Chou-Talalay method
(18) and the Prichard-Shipman
method (19). Synergistic
cytotoxicity was further investigated by several assays, including
the Annexin V binding assay, the caspase-3/7 activity assay, and
cleaved PARP detection by immunoblotting. We observed significantly
increased apoptosis following the combination treatment compared
with either monotherapy.
The drug concentrations used in this study were
carefully selected to perform a clinically relevant in vitro
study (25). Synergistic cytotoxic
effects were induced by the combination treatment at clinically
achievable concentrations, which are below the peak plasma
concentrations of recommended phase 2 doses for each drug
(5.33–6.61 µM and 50 nM for ABT-263 and BMN 673,
respectively) (15,26,27).
We examined the drug effects on DNA damage and cell
cycle distribution to explore the potential mechanisms of the
observed synergistic cytotoxic effects. BMN 673 treatment induced
accumulation of γ-H2AX foci and the proportional increase of cells
in the G2/M phase, as previously reported (28). ABT-263 monotherapy did not induce
γ-H2AX foci formation or affect the cell cycle distribution,
consistent with previous reports (29,30).
The addition of ABT-263 did not affect either γ-H2AX foci formation
or G2/M cell cycle arrest induced by BMN 673 treatment, suggesting
the observed synergistic cytotoxicity was unlikely due to augmented
DNA damage accumulation.
Several chemotherapeutic agents have shown synergy
with ABT-263 based on two main mechanisms, either decreased
expression of Mcl-1 or increased expression of BH3-only proteins
(31). BMN 673 treatment neither
decreased the expression of Mcl-1 nor increased the expression of
BH3-only proteins. Our findings confirmed ABT-263 treatment
increased Mcl-1, Bim, and Noxa expression, as shown previously
(32–34). It has been reported that ABT-263
posttranscriptionally upregulates Mcl-1 expression through
ERK-mediated phosphorylation of Mcl-1 on Thr-163 (33,34).
Mcl-1 confers resistance to ABT-263 because it remains a potent
anti-apoptotic protein (31).
However, expression of Mcl-1 is not always sufficient to cause
resistance to ABT-263 given that occupancy of Mcl-1 by
pro-apoptotic proteins can effectively inactivate Mcl-1 (15,35,36).
The extent of pro-apoptotic Bcl-2 family proteins occupancy by
anti-apoptotic proteins correlates well with the observed clinical
response to chemotherapy, and could be modulated to enhance
chemosensitivity (37). Thus, we
speculated that ABT-263 treatment did not necessarily enhance the
effects of BMN 673 treatment, but might have lowered the apoptotic
threshold by increasing Bim and Noxa pro-apoptotic protein
expression, leading to greater susceptibility to addition of BMN
673 treatment (38). During
apoptosis, Bid can be cleaved and activated by caspase-3 as well as
caspase-8 (39). Although we did
not observe the occurrence of cleaved Bid, we found that the
expression of full length Bid was decreased by the combination
treatment, but not by either monotherapy, implying increased
caspase activity by the combination treatment.
ABT-263 monotherapy is currently under clinical
investigation in women with platinum resistant/refractory recurrent
ovarian cancer (NCT02591095). Platelet survival is dependent on
Bcl-xL and, as would be expected, clinically significant and
sometimes dose-limiting thrombocytopenia has been observed with
ABT-263 treatment (15,26). PARPi, as a class, also result in
hematotoxicity, including reduction in platelets with rare
significant thrombocytopenia, although this class of agents has
been documented to have activity at submaximal doses when used in
combination treatments (40,41).
Synergistic drug combinations carry the expectation of greater
therapeutic efficacy, although they may also increase the severity
of adverse effects. Careful dose escalation and close monitoring
would be needed for further clinical investigation.
In conclusion, combined treatment with ABT-263 and
BMN 673 synergistically inhibited HGSOC cell proliferation at
clinically achievable concentrations. The combination treatment
significantly increased apoptotic cell death through caspase
activation compared with either monotherapy. A pro-apoptotic
environment augmented by ABT-263 might be a possible mechanism of
the observed synergistic cytotoxic effects. Our results support
further evaluation of the therapeutic potential of the combination
treatment including BH3-mimetics and PARPi for HGSOC.
Acknowledgments
The authors would like to thank the Center for
Cancer Research Confocal Microscopy Core Facility (NCI/NIH,
Bethesda, MD, USA) for technical assistance. This study was
supported by the Intramural Program of the Center for Cancer
Research, National Cancer Institute, National Institutes of
Health.
Glossary
Abbreviations
Abbreviations:
|
HGSOC
|
high-grade serous ovarian cancer
|
|
PARPi
|
PARP inhibitor
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Coleman RL, Monk BJ, Sood AK and Herzog
TJ: Latest research and treatment of advanced-stage epithelial
ovarian cancer. Nat Rev Clin Oncol. 10:211–224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bowtell DD, Böhm S, Ahmed AA, Aspuria PJ,
Bast RC Jr, Beral V, Berek JS, Birrer MJ, Blagden S, Bookman MA, et
al: Rethinking ovarian cancer II: Reducing mortality from
high-grade serous ovarian cancer. Nat Rev Cancer. 15:668–679. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bell D, Berchuck A, Birrer M, Chien J,
Cramer DW, Dao F, Dhir R, DiSaia P, Gabra H, Glenn P, et al Cancer
Genome Atlas Research Network: Integrated genomic analyses of
ovarian carcinoma. Nature. 474:609–615. 2011. View Article : Google Scholar
|
|
5
|
Gelmon KA, Tischkowitz M, Mackay H,
Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M,
Gilks B, et al: Olaparib in patients with recurrent high-grade
serous or poorly differentiated ovarian carcinoma or
triple-negative breast cancer: A phase 2, multicentre, open-label,
non-randomised study. Lancet Oncol. 12:852–861. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim G, Ison G, McKee AE, Zhang H, Tang S,
Gwise T, Sridhara R, Lee E, Tzou A, Philip R, et al: FDA Approval
Summary: Olaparib monotherapy in patients with deleterious germline
BRCA-mutated advanced ovarian cancer treated with three or more
lines of chemotherapy. Clin Cancer Res. 21:4257–4261. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Parkes EE and Kennedy RD: Clinical
application of poly(ADP-ribose) polymerase inhibitors in high-grade
serous ovarian cancer. Oncologist. 21:586–593. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Murai J, Huang SY, Renaud A, Zhang Y, Ji
J, Takeda S, Morris J, Teicher B, Doroshow JH and Pommier Y:
Stereospecific PARP trapping by BMN 673 and comparison with
olaparib and rucaparib. Mol Cancer Ther. 13:433–443. 2014.
View Article : Google Scholar :
|
|
9
|
Aredia F and Scovassi AI:
Poly(ADP-ribose): A signaling molecule in different paradigms of
cell death. Biochem Pharmacol. 92:157–163. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Delbridge AR, Grabow S, Strasser A and
Vaux DL: Thirty years of BCL-2: Translating cell death discoveries
into novel cancer therapies. Nat Rev Cancer. 16:99–109. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Williams J, Lucas PC, Griffith KA, Choi M,
Fogoros S, Hu YY and Liu JR: Expression of Bcl-xL in ovarian
carcinoma is associated with chemoresistance and recurrent disease.
Gynecol Oncol. 96:287–295. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wong M, Tan N, Zha J, Peale FV, Yue P,
Fairbrother WJ and Belmont LD: Navitoclax (ABT-263) reduces
Bcl-x(L)-mediated chemoresistance in ovarian cancer models. Mol
Cancer Ther. 11:1026–1035. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Witham J, Valenti MR, De-Haven-Brandon AK,
Vidot S, Eccles SA, Kaye SB and Richardson A: The Bcl-2/Bcl-XL
family inhibitor ABT-737 sensitizes ovarian cancer cells to
carboplatin. Clin Cancer Res. 13:7191–7198. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rudin CM, Hann CL, Garon EB, Ribeiro de
Oliveira M, Bonomi PD, Camidge DR, Chu Q, Giaccone G, Khaira D,
Ramalingam SS, et al: Phase II study of single-agent navitoclax
(ABT-263) and biomarker correlates in patients with relapsed small
cell lung cancer. Clin Cancer Res. 18:3163–3169. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Roberts AW, Seymour JF, Brown JR, Wierda
WG, Kipps TJ, Khaw SL, Carney DA, He SZ, Huang DC, Xiong H, et al:
Substantial susceptibility of chronic lymphocytic leukemia to BCL2
inhibition: Results of a phase I study of navitoclax in patients
with relapsed or refractory disease. J Clin Oncol. 30:488–496.
2012. View Article : Google Scholar
|
|
16
|
Domcke S, Sinha R, Levine DA, Sander C and
Schultz N: Evaluating cell lines as tumour models by comparison of
genomic profiles. Nat Commun. 4:21262013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lorenzi PL, Reinhold WC, Varma S,
Hutchinson AA, Pommier Y, Chanock SJ and Weinstein JN: DNA
fingerprinting of the NCI-60 cell line panel. Mol Cancer Ther.
8:713–724. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Prichard MN and Shipman C Jr: A
three-dimensional model to analyze drug-drug interactions.
Antiviral Res. 14:181–205. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Prichard MK, Aseltine KR and Shipman C Jr:
MacSynergy II, version 1.0. User's manual. University of Michigan;
Ann Arbor, MI: 1993
|
|
21
|
Oeck S, Malewicz NM, Hurst S, Rudner J and
Jendrossek V: The Focinator - a new open-source tool for
high-throughput foci evaluation of DNA damage. Radiat Oncol.
10:1632015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao L, Wientjes MG and Au JL-S:
Evaluation of combination chemotherapy: Integration of nonlinear
regression, curve shift, isobologram, and combination index
analyses. Clin Cancer Res. 10:7994–8004. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Farmer H, McCabe N, Lord CJ, Tutt AN,
Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I,
Knights C, et al: Targeting the DNA repair defect in BRCA mutant
cells as a therapeutic strategy. Nature. 434:917–921. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Herriott A, Tudhope SJ, Junge G, Rodrigues
N, Patterson MJ, Woodhouse L, Lunec J, Hunter JE, Mulligan EA, Cole
M, et al: PARP1 expression, activity and ex vivo sensitivity to the
PARP inhibitor, talazoparib (BMN 673), in chronic lymphocytic
leukaemia. Oncotarget. 6:43978–43991. 2015.PubMed/NCBI
|
|
25
|
Smith MA and Houghton P: A proposal
regarding reporting of in vitro testing results. Clin Cancer Res.
19:2828–2833. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wilson WH, O'Connor OA, Czuczman MS,
LaCasce AS, Gerecitano JF, Leonard JP, Tulpule A, Dunleavy K, Xiong
H, Chiu YL, et al: Navitoclax, a targeted high-affinity inhibitor
of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study
of safety, pharmacokinetics, pharmacodynamics, and antitumour
activity. Lancet Oncol. 11:1149–1159. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
De Bono JSML, Gonzalez M, Curtin NJ, Wang
E, Henshaw JW, Chadha M, Sachdev JC, Matei D, Jameson GS, Ong M, et
al: First-in-human trial of novel oral PARP inhibitor BMN 673 in
patients with solid tumors. J Clin Oncol. 31:25802013.
|
|
28
|
Shen Y, Rehman FL, Feng Y, Boshuizen J,
Bajrami I, Elliott R, Wang B, Lord CJ, Post LE and Ashworth A: BMN
673, a novel and highly potent PARP1/2 inhibitor for the treatment
of human cancers with DNA repair deficiency. Clin Cancer Res.
19:5003–5015. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tan N, Wong M, Nannini MA, Hong R, Lee LB,
Price S, Williams K, Savy PP, Yue P, Sampath D, et al: Bcl-2/Bcl-xL
inhibition increases the efficacy of MEK inhibition alone and in
combination with PI3 kinase inhibition in lung and pancreatic tumor
models. Mol Cancer Ther. 12:853–864. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Green MM, Shekhar TM and Hawkins CJ: Data
on the DNA damaging and mutagenic potential of the BH3-mimetics
ABT-263/Navitoclax and TW-37. Data Brief. 6:710–714. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Stamelos VA, Redman CW and Richardson A:
Understanding sensitivity to BH3 mimetics: ABT-737 as a case study
to foresee the complexities of personalized medicine. J Mol Signal.
7:122012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang H, Yang YB, Shen HM, Gu J, Li T and
Li XM: ABT-737 induces Bim expression via JNK signaling pathway and
its effect on the radiation sensitivity of HeLa cells. PLoS One.
7:e524832012. View Article : Google Scholar
|
|
33
|
Wang B, Ni Z, Dai X, Qin L, Li X, Xu L,
Lian J and He F: The Bcl-2/xL inhibitor ABT-263 increases the
stability of Mcl-1 mRNA and protein in hepatocellular carcinoma
cells. Mol Cancer. 13:982014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hiraki M, Suzuki Y, Alam M, Hinohara K,
Hasegawa M, Jin C, Kharbanda S and Kufe D: MUC1-C stabilizes MCL-1
in the oxidative stress response of triple-negative breast cancer
cells to BCL-2 inhibitors. Sci Rep. 6:266432016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Morales AA, Kurtoglu M, Matulis SM, Liu J,
Siefker D, Gutman DM, Kaufman JL, Lee KP, Lonial S and Boise LH:
Distribution of Bim determines Mcl-1 dependence or codependence
with Bcl-xL/Bcl-2 in Mcl-1-expressing myeloma cells. Blood.
118:1329–1339. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yamaguchi R, Janssen E, Perkins G,
Ellisman M, Kitada S and Reed JC: Efficient elimination of cancer
cells by deoxyglucose-ABT-263/737 combination therapy. PLoS One.
6:e241022011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan
JA, Tammareddi A, Moore VG, Deng J, Anderson KC, Richardson P, Tai
YT, et al: Pretreatment mitochondrial priming correlates with
clinical response to cytotoxic chemotherapy. Science.
334:1129–1133. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lopez J and Tait SW: Mitochondrial
apoptosis: Killing cancer using the enemy within. Br J Cancer.
112:957–962. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Slee EA, Keogh SA and Martin SJ: Cleavage
of BID during cytotoxic drug and UV radiation-induced apoptosis
occurs downstream of the point of Bcl-2 action and is catalysed by
caspase-3: A potential feedback loop for amplification of
apop-tosis-associated mitochondrial cytochrome c release. Cell
Death Differ. 7:556–565. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee JM, Hays JL, Annunziata CM, Noonan AM,
Minasian L, Zujewski JA, Yu M, Gordon N, Ji J, Sissung TM, et al:
Phase I Ib study of olaparib and carboplatin in BRCA1 or BRCA2
mutation-associated breast or ovarian cancer with biomarker
analyses. J Natl Cancer Inst. 106:dju0892014. View Article : Google Scholar
|
|
41
|
Oza AM, Cibula D, Benzaquen AO, Poole C,
Mathijssen RH, Sonke GS, Colombo N, Špaček J, Vuylsteke P, Hirte H,
et al: Olaparib combined with chemotherapy for recurrent
platinum-sensitive ovarian cancer: A randomised phase 2 trial.
Lancet Oncol. 16:87–97. 2015. View Article : Google Scholar
|