Introduction
Endometrial cancer is the most common malignancy of
the female genital system in developed countries. In Taiwan,
endometrial cancer ranks second among gynecological cancers in its
incidence. Risk factors for endometrial cancer are both
environmental and genetic. One major environmental risk factor is
endogenous or exogenous estrogen exposure: many sources of
exogenous estrogen exist, including oral contraceptives and hormone
replacement therapy (1). Some
genetic variants involved in the steroid hormone biosynthesis and
metabolism pathways may contribute to hyperestrogenic status, which
is associated with endometrial cancer risk (2,3). Use
of oral contraceptives provides significant long-term protection
against endometrial cancer (4).
Long-term sequential estrogen-plus-progestin therapy during
menopause increases the risk of endometrial cancer, whereas
short-term estrogen-plus-progestin use in menopause decreases the
risk of endometrial cancer (5).
Endometrial cancer patients who received extended adjuvant
tamoxifen do not appear to be at greater risk for endometrial
cancer (6).
Next-generation sequencing (NGS) will play an
important role in anticancer drug development (7) and targeted therapy (8). At present, there are multiple NGS
instruments in use, such as the Illumina HiSeq and MiSeq (Illumina,
Inc., San Diego, CA, USA), the Ion Torrent Proton and Personal
Genome Machine (Life Technologies, Carlsbad, CA, USA), as well as
the Roche 454 Sequencer and GS Junior (Roche Applied Biosystems,
Nutley, NJ, USA). The Cancer Genome Atlas (TCGA) project used NGS
techniques to identify hundreds of somatically altered genes
(9–12). This technical advance is rapidly
altering the routine practice of molecular pathology, from
single-gene tests (i.e., Sanger sequencing to assess KRAS
mutations in colorectal cancer) to multiplexed NGS assays. Several
NGS approaches have been commonly implemented clinically in
oncology, including hybrid capture-based panels, multiplexed
polymerase chain reaction (PCR)-based panels and comprehensive
genome/transcriptome/exome sequencing (13–16).
Whole genome sequencing offers high throughput, high accuracy
(<1 error per 100 kb) and affordable cost (<$5,000 USD in
reagents) (17). Recently, the
American College of Medical Genetics and Genomics (ACMG)
recommended reporting pathogenic findings in 56 genes with low
coverage. Low sequencing coverage may contribute to false negative
clinical exome results (18).
Based on histological subtype, endometrial cancer is
classified into two major types (I and II). Type I (endometrioid)
carcinoma is the most frequent type of endometrial cancer,
accounting for over 80% of cases. Type II (non-endometrioid)
carcinoma comprises a minority of endometrial cancer cases.
Whole-exome sequencing (WES) has led to the discovery of
ARID1A as a novel regulator of PI3K pathway activity in
endometrioid endometrial cancer (19). Through an integrated genomic,
transcriptomic and proteomic analysis of endome-trial cancer, TCGA
research has suggested a novel molecular classification for two
histological subtypes (12).
We performed WES on 14 patients with endometrial
cancers. Our results identify cancer driver and passenger genes,
and adhere to ACMG recommendations for the examination and
reporting of secondary genetic findings during clinical genomic
testing.
Materials and methods
Patients and samples
Fourteen patients with endometrial cancer were
recruited for this study. DNA was extracted using the QIAamp DNA
Micro kit (Qiagen, Heidelberg, Germany) following the
manufacturer's protocol. Extracted DNA samples were quantified
using NanoDrop 2000 spectrophotometer (Thermal Fisher Scientific,
Waltham, MA, USA) and a Qubit fluorometer (Invitrogen, Carlsbad,
CA, USA). This study was approved by the Institutional Review Board
of Kaohsiung Medical University Hospital (KMUH-IRB-970488).
Whole-exome sequencing
To generate standard exome capture libraries, we
used the Agilent SureSelect XT Reagent kit protocol for the
Illumina Hiseq paired-end sequencing library (cat. no. G9611A). In
all cases, the SureSelect XT Human All Exon Version 4 (51 Mb) probe
set was used. We used 50 ng genomic DNA for library construction
with the Agilent SureSelect XT Reagent kit. The adapter-ligated
sample was purified using Agencourt AMPure XP beads (Beckman
Coulter, Brea, CA, USA) and analyzed on a Bioanalyzer DNA 1000
chip. Part of the sample (750 ng) was prepared for hybridization
with the capture baits, and the sample was hybridized for 90 min at
65°C, captured with the Dynabeads MyOne Streptavidin T1 (Life
Technologies) and purified using Agencourt AMPure XP beads. The
Agilent protocol was used to add index tags by post-hybridization
amplification. Finally, all samples were sequenced on an Illumina
Hiseq system using 100 PE protocol. Metadata were deposited in the
NCBI Sequence Read Archive under the accession no. SRP099176.
Data analysis
To filter low-quality reads, we used the
FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit) to process
the raw read data files. There were two steps of sequence quality
processing. The command 'fastq_quality_filter -Q33 -q 30 -p 70'.
'-q 30' indicated that the minimum quality score was 30. '-p 70'
meant that bases must have a '-q' quality score of 70% or greater.
Sequences were retained if both forward and reverse sequencing
reads passed the first step.
An efficient sequence alignment tool, Bowtie2, was
used to align the retained reads with human genome (Grch38.p2)
(20). Based on the results of
sequence alignment, reads having only one chromosome location were
retained for further analysis. The Genome Analysis Tool kit, is a
widely used discovery tool to identify genetic variants based on
the results of sequence alignment (21).
Several databases and tools were used to annotate
identified genetic variants. dbSNP (b144) is an archive of genetic
variation within different species that provides information about
each genetic variant (22).
ClinVar is a database of clinically significant genetic variants
(23). COSMIC (v73) collects
somatic mutation information for human cancers (24). The TCGA project collects genomics,
methylomic and transcriptomic data across cancer types (25). Integrated mutation prediction
software (PolyPhen-2 and SIFT) were used for analyses of the
identified variants (26,27). DrGaP was used to identify driver
genes and driver signaling pathways (28).
Confirmation by Sanger sequencing
Potential mutations identified by whole-exome
sequencing were confirmed by PCR and Sanger sequencing. Specific
PCR primers were designed using Primer3 software (primers can be
provided on request). The products were sequenced directly with the
ABI PRISM terminator cycle sequencing kit v3.1 on an ABI 3130 DNA
sequencer (Applied Biosystems, Carlsbad, CA, USA).
Results
Whole-exome sequence analysis and
coverage
Using massive parallel sequencing on a HiSeq
platform, we generated ~45 billion bases of effective sequence data
with an average read length of 200 bases. After mapping to the
human reference genome (Grch38.p2) using the Bowtie 2 alignment
tool, we obtained an average depth of coverage for the target
regions of 20× for each sample (Table
I). The false positive and false negative rates were estimated
to be 2.75% (12/437) and 19.69% (76/386), respectively, after
confirmed by target sequencing of cancer-related genes (29). Table
II provides an overview of our approach to identify
variants.
 | Table ISummary of sequencing alignment and
coverage statistics in the exomes of 14 endometrial tumors. |
Table I
Summary of sequencing alignment and
coverage statistics in the exomes of 14 endometrial tumors.
| Patient ID | Total raw
reads | Total effective
reads | Reads mapped to
genome | Total effective
yield (Mb) | Average read length
(bp) | Covered ≥20X
(%) | Average sequencing
depth on target |
|---|
| F105T | 20,976,408 | 14,817,961 | 14,725,603 | 45974.96 | 200.01 | 43.06 | 19.43 |
| F139T | 18,071,410 | 12,999,809 | 12,909,672 | 43438.89 | 200.01 | 51.07 | 21.78 |
| F141T | 18,308,457 | 13,180,222 | 13,077,252 | 45838.97 | 200.10 | 44.58 | 19.06 |
| F61 | 21,965,842 | 15,337,823 | 15,247,340 | 47366.56 | 200.00 | 45.76 | 20.01 |
| F114T | 18,690,475 | 13,546,119 | 13,433,300 | 46105.01 | 200.16 | 48.37 | 20.01 |
| F123 | 20,115,993 | 14,018,792 | 13,919,598 | 46067.53 | 199.99 | 43.44 | 18.83 |
| F132 | 21,510,383 | 14,710,924 | 14,621,813 | 45507.75 | 199.83 | 45.5 | 20.56 |
| F134 | 20,126,153 | 14,005,289 | 13,909,284 | 45658.48 | 199.98 | 42.77 | 18.94 |
| F146 | 26,158,898 | 18,289,158 | 18,193,144 | 50310.04 | 200.03 | 46.31 | 21.53 |
| F147T | 19,394,632 | 14,099,623 | 13,962,761 | 45656.60 | 200.17 | 48.56 | 20.92 |
| F150T | 20,623,512 | 14,156,494 | 14,080,096 | 45931.50 | 199.85 | 48.22 | 20.58 |
| F152T | 19,092,410 | 14,153,725 | 14,048,858 | 46229.92 | 200.28 | 50.62 | 21.11 |
| F92T | 18,726,658 | 13,243,029 | 13,164,997 | 45153.23 | 200.09 | 43.91 | 18.60 |
| 03-3812T | 17,550,923 | 12,789,944 | 12,748,550 | 44627.55 | 200.18 | 52.99 | 21.17 |
| Average | 20,093,725 | 14,239,208 | 14,145,876 | 45990.50 | 200 | 47.03 | 20 |
| Table IIOverview of our approach to
identifying driver and passenger genes in endometrial cancer. |
Table II
Overview of our approach to
identifying driver and passenger genes in endometrial cancer.
| Type of
prioritization filter | Remaining variants
(n)
|
|---|
| Canonical
cancer-related genes | Non-canonical
cancer-related genes | Total |
|---|
| 1. All
variants | 2,936 variants in
586 genes | 60,259 variants in
13,214 genes | 63,185 variants in
13,800 genes |
| 2. Non-synonymous
variants | 726 variants in 352
genes | 14,970 variants in
6,974 genes | 15,716 variants in
7,326 genes |
| 3. The occurrence
of variants ≤50% | 603 variants in 322
genes | 11,293 variants in
6,110 genes | 12,561 variants in
6,432 genes |
| 4. Global minor
allele frequency <1% in dbSNP v144 or NA | 413 variants in 272
genes | 6,286 variants in
4,264 genes | 6,699 variants in
4,536 genes |
| 5. ClinVar or
PolyPhen-2 and SIFT predicted the variants to be pathogenic or
possibly/probably functionally impaired | 143 variants in 129
genes | 1,316 variants in
1,188 genes | 1,459 variants in
1,317 genes |
| 6. DrGaP identified
driver genes (P<0.01) | NA | 1,271 variants in
1,144 genes | 1,271 variants in
1,144 genes |
Mutational landscape in the 756 canonical
cancer-related genes
The 143 non-synonymous mutations identified in the
present study occurred in 129 genes, and included 141 missense
mutations, one nonsense mutation and one frame shift mutation
(details can be provided on request). The most frequently mutated
genes were PTEN (35.71%; 5/14), KRAS and
PIK3R1 (14.29%, 2/14).
Sixty-eight variants already existed in the dbSNP,
COSMIC, or TCGA databases and 75 variants in 71 genes did not. In
addition, we found 13 different mutations in the same codon of 12
canonical cancer-related genes compared with the TCGA database
(CDK8, DPYD, EPHA3, EPHA6, FLNA,
MAPK1, MAPK7, PLK1, PTEN,
RAPGEF2, RFC4 and ZNF521) (details can be
provided on request).
We ordered the 75 new variants according to
sequencing depth. Next, we selected 12 variants for Sanger
sequencing. Six variants had relatively high depth (208x, 156x,
131x, 105x, 97x and 89x). Several novel mutations were present only
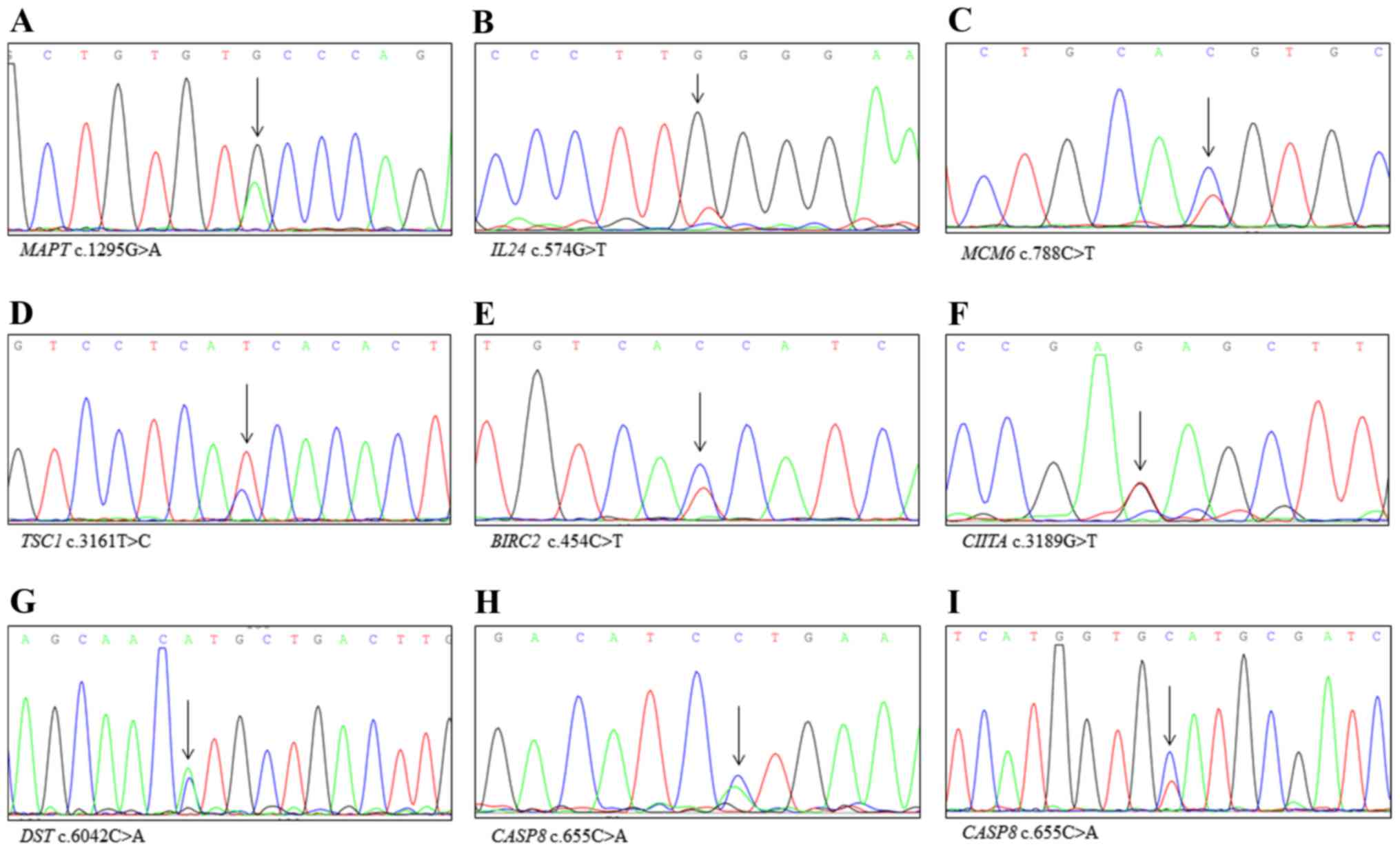
in cancer tissue, including MAPT p.C432Y, IL24
p.G192W, MCM6 p.R263H, TSC1 p.D1054G and BIRC2
p.P152S. However, KAT6B p.S1693N was also detected in paired
non-cancerous tissues. Six variants had low depth (22x, 21x, 21x,
20x, 20x and 20x). Novel mutations present only in cancer tissue
including CIITA p.E1063D, DST p.Q2014H, CASP8
p.L219M and NOTCH2 p.M2054I. FLNA p.V528L and
HSPA1L p.P93H were also detected in paired non-cancerous
tissues (Fig. 1).
Mutational landscape in the non-canonical
cancer-related genes
The 1,271 non-synonymous mutations identified in
this study, including 1,270 missense mutations and one nonsense
mutation, were located at 1,144 genes. In total, 723 variants had
previously been reported in the dbSNP, COSMIC or TCGA databases,
while 548 variants in 516 genes had not (details can be provided on
request).
However, nearly all mutations had been reported in
the 542 TCGA endometrial cancer samples. Only three mutations had
never been reported (ARMCX4, CUTA and SAP30L).
We selected these mutations for Sanger sequencing. CUTA
p.T187R and SAP30L p.H77Y were also detected in paired
non-cancerous tissues; ARMCX4 p.P2056H was the sole novel
mutation that was detected only in cancer tissue (Fig. 2). We also found 49 different
mutations in the same codon of 49 non-canonical cancer-related
genes compared with the TCGA database (details can be provided on
request).
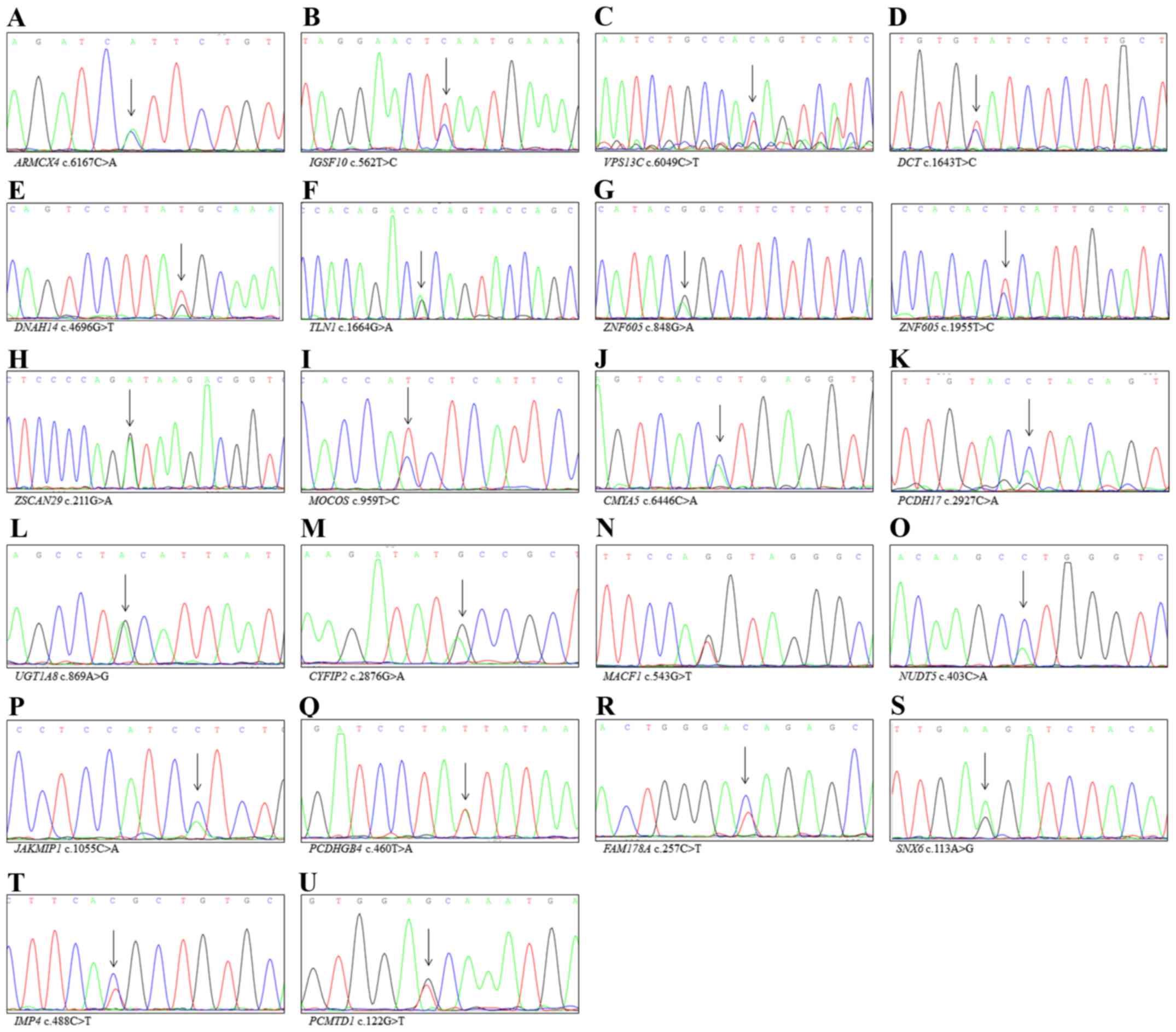 | Figure 2Confirmatory analysis by Sanger
sequencing of non-canonical cancer related genes detected via WES.
(A) ARMCX4, (B) IGSF10, (C) VPS13C, (D)
DCT, (E) DNAH14, (F) TLN1, (G) ZNF605,
(H) ZSCAN29, (I) MOCOS, (J) CMYA5, (K)
PCDH17, (L) UGT1A8, (M) CYFIP2, (N)
MACF1, (O) NUDT5, (P) JAKMIP1, (Q)
PCDHGB4, (R) FAM178A, (S) SNX6, (T)
IMP4 and (U) PCMTD1. |
The 1,144 genes were divided into five groups
according to mutation frequency. Fifteen non-canonical
cancer-related genes had high frequencies of mutation (35.71–50%;
5–7/14) and four variants in three genes (IGSF10,
FBXL13 and PRUNE2) were novel. We selected these new
genetic variants for Sanger sequencing. We detected IGSF10
p.R455W, FBXL13 p.G313A and PRUNE2 p.S2439I in paired
non-cancerous tissues, while IGSF10 p.K188Q was detected
only in cancer tissue (Fig.
2).
Six genes were mutated in 4 (28.57%) of the 14
patients. One gene (VPS13C) had a new genetic variant.
Sanger sequencing confirmed that VPS13C p.V2017M was a novel
mutation detected only in cancer tissue (Fig. 2).
Twenty-two genes were mutated in 3 (21.43%) of the
14 patients, and there were 12 new genetic variants in 8 genes. We
carried out Sanger sequencing on the new variants, and found that
ARAP2 p.Y1491C, DNAH14 p.D4225V, TLN1
p.D1325E, TREH p.M196T and ZSCAN29 p.G304V were also
present in paired non-cancerous tissues. Sanger sequencing also
confirmed that DCT p.Y548S, DNAH14 p.G1566C,
TLN1 p.A555V, ZNF605 p.P283Q, p.E652A and
ZSCAN29 p.P71S were novel mutations present only in cancer
tissue (Fig. 2). One variant
(TRANK1 p.W2445L) had an incorrect base call.
One hundred and nine genes were mutated in 2
(14.29%) of the 14 patients, and there were 73 new genetic variants
in 54 genes. We ordered the 73 new variants according to sequencing
depth. Next, we selected 10 variants for Sanger sequencing. Five
variants had high depth (163x, 108x, 104x, 99x and 92x). Several
novel mutations were detected only in cancer tissue, including
MOCOS p.I320T, CMYA5 p.P2149H and PCDH17
p.P976H (Fig. 2). MAGEE2
p.F249L and CRYBG3 p.Q632E were also detected in paired
non-cancerous tissues. Five variants had low depth (all 20x). Novel
mutations present only in cancer tissue including UGT1A8
p.Y290C and CYFIP2 p.C959Y (Fig. 2), while APOL4 p.A316V and
TRIM26 p.Q197H were also detected in paired non-cancerous
tissues. One variant (MYO3B p.G230W) had an incorrect base
call.
Among low-frequency mutations (7.14%; 1/14) in 992
non-canonical cancer-related genes, there were 458 new genetic
variants in 450 genes. We ordered these 458 variants according to
sequencing depth. Next, we selected 10 variants for Sanger
sequencing. Five variants had high depth (286x, 244x, 223x, 197x
and 187x). Several novel mutations were detected only in cancer
tissue, including MACF1 p.Q181H, NUDT5 p.G135S,
JAKMIP1 p.R352M and PCDHGB4 p.L154I (Fig. 2). MYH10 p.L1048X was also
detected in paired non-cancerous tissues. Five variants had low
depth (all 20x). Novel mutations detected only in cancer tissue
included FAM178A p.T86I, SNX6 p.L38R, IMP4
p.T163M and PCMTD1 p.A41D (Fig.
2). TNFAIP8L3 p.A38T was also detected in the paired
non-cancerous tissues.
ACMG gene
Of the 14 endometrial cancer samples, 28.57% (4/14)
harbored ACMG mutations, including RYR2, MSH6 and
TSC1, but did not have mutations in the remaining 53 genes.
Mutations in the RYR2 gene were detected in 14.29% (2/14) of
endometrial cancer specimens, with mutations at p.G1209R
(rs770286824) and p.F4960L (a novel mutation) identified in our
endometrial cancer samples. Both the MSH6 mutation at
p.Q572H (rs745772518) and the TSC1 mutation at p.D1054G were
detected in 7.14% (1/14) of endometrial cancer samples.
Altered pathways
Functional annotation of the 1,273 mutated genes was
performed using the DrGaP tool. Thirty-seven of the 1,273 mutated
genes were found in the Kyoto Encyclopedia of Genes and Genomics
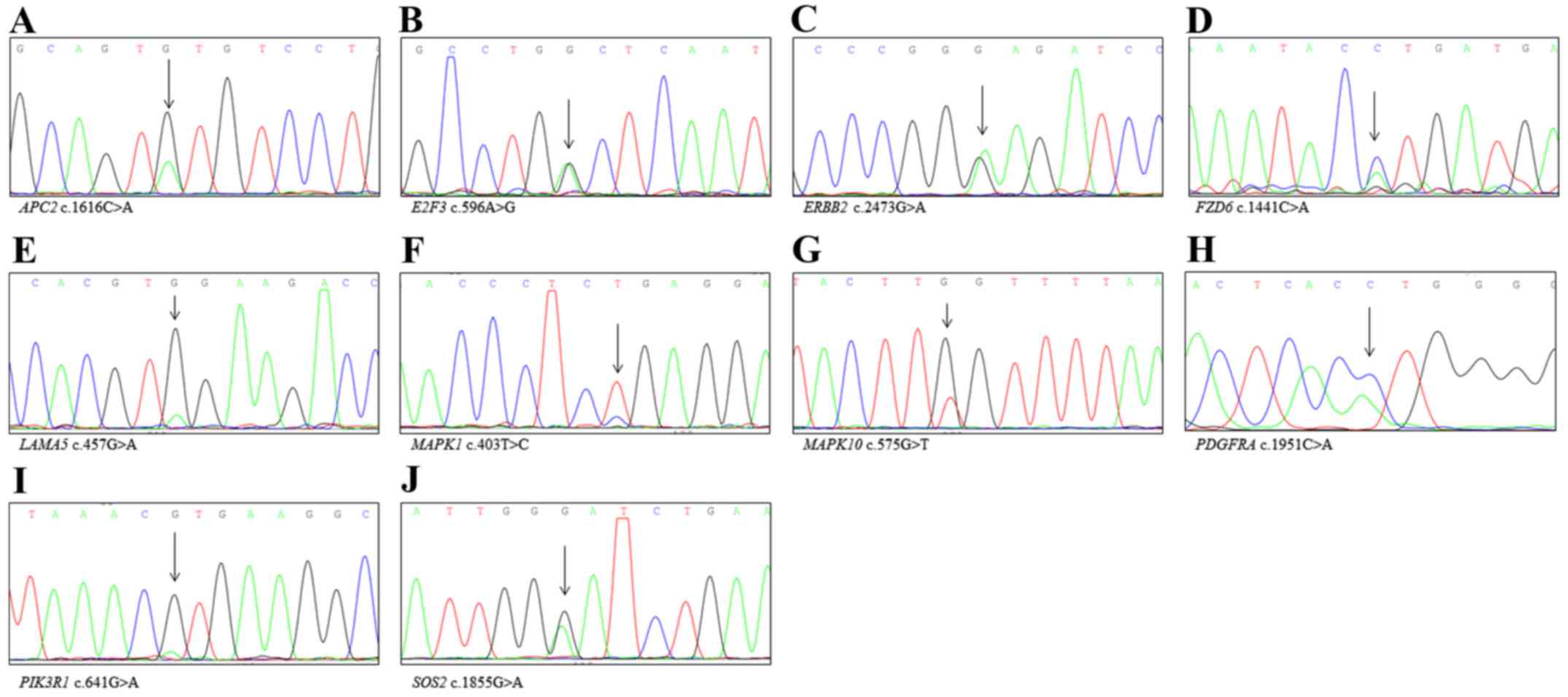
(KEGG) cancer pathways (hsa05200), including APC2,
BCR, BIRC2, CASP8, CCNA1,
CREBBP, CTNNA2, CTNNA3, CTNNB1,
E2F3, ERBB2, FGFR2, FOXO1, FZD6,
HHIP, ITGA2, ITGA3, KRAS, LAMA1,
LAMA2, LAMA3, LAMA5, LAMC1,
LAMC2, MAPK1, MAPK10, MSH6, MTOR,
PDGFRA, PIK3CA, PIK3R1, PLCG2, PTEN,
SOS2, STAT5A, TCF7 and TRAF5. We used
Sanger sequencing validation to confirm new variants Fig. 3). Moreover, we identified several
cellular pathways that were altered in endometrial cancer tissues
and we found that each sample had at least two pathways involved in
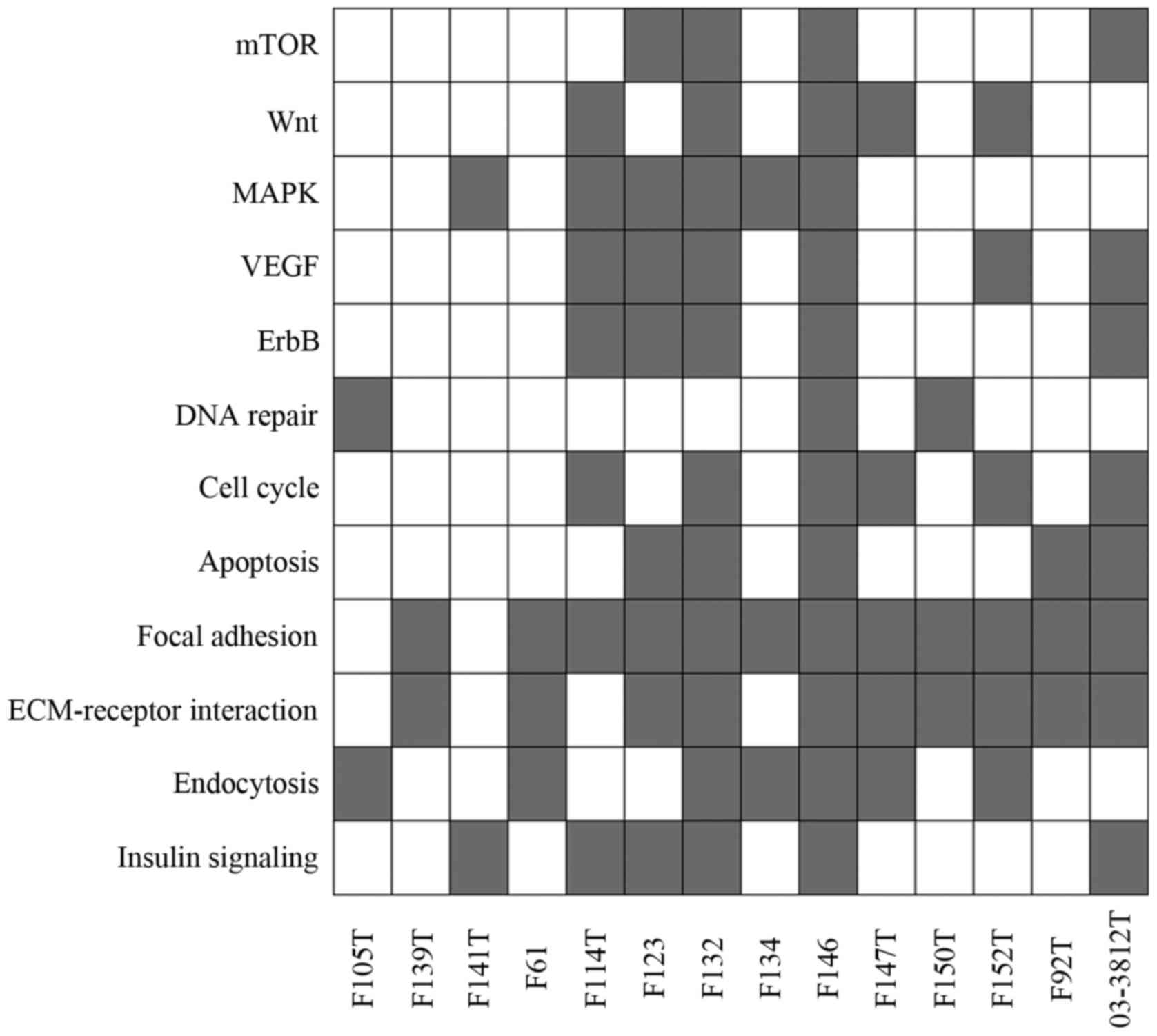
the carcinogenesis of endometrial cancer (Fig. 4).
Discussion
The present study described somatic mutation in the
whole endometrial cancer exome. We identified several cancer driver
and passengers genes from canonical and non-canonical
cancer-related genes. Overall, 35.71% of endometrial cancer cases
harbored PTEN mutations. Mutations were also found in other
canonical cancer-related genes, including KRAS and
PIK3R1 (14.29% each). To the best of our knowledge, several
sequencing variants have not been reported, including canonical
cancer-related genes (MAPT, IL24, MCM6,
TSC1, BIRC2, CIITA, DST, CASP8
and NOTCH2) and non-canonical cancer-related genes
(ARMCX4, IGSF10, VPS13C, DCT,
DNAH14, TLN1, ZNF605, ZSCAN29,
MOCOS, CMYA5, PCDH17, UGT1A8,
CYFIP2, MACF1, NUDT5, JAKMIP1,
PCDHGB4, FAM178A, SNX6, IMP4 and
PCMTD1).
Two canonical cancer-related genes (TSC1 and
BIRC2) were found in the Oncomine Cancer Research Panel,
which was used in the National Cancer Institute Match Trial
(30). TSC1 is a tumor
suppressor gene that encodes a growth inhibitory protein (hamartin)
thought to play a role in the stabilization of tuberin. TSC1
is a gene involved in the mTOR pathway. Mutations in TSC1,
TSC2 and MTOR have been associated with response to
rapalogs in patients with metastatic renal cell carcinoma (31). We identified a novel mutation,
p.D1054G, in one endometrial cancer patient.
BIRC2 is an oncogene that encodes c-IAP1,
which is a member of the apoptosis inhibitor family. Members of
this family inhibit apoptosis by binding to TRAF1 and TRAF2 and
likely interfering with activation of ICE-like proteases.
Previously, Choschzick et al (32) reported BIRC2 amplification
in uterine cervix cancer. In the present study, we identified a
novel mutation, p.P152S, in a patient with endometrial cancer.
The genetic alterations we found involved the mTOR,
Wnt, MAPK, VEGF and ErbB pathways, as well as aberrant DNA repair,
cell cycle control and apoptosis pathways. These pathways have
previously been shown to be involved in the multistep development
of endometrial cancer, and clinical trials of drugs for endometrial
cancer that target these pathways have been carried out (33). We also found genetic alterations in
the steroid hormone biosynthesis pathway (hsa00140), including
AKR1C3, CYP3A4, CYP3A43, HSD11B1,
SULT1E1 and UGT1A8.
NGS technology allows for the detection of
mutations and copy number variants. Although the hybrid capture
method was used, we failed to assess copy number alterations
because we sequenced only tumor samples. Sequencing coverage is
uneven across the genome owing to variability introduced by the
hybridization-capture step, and the development of a robust
algorithm is challenging (34). On
the other hand, multiplex PCR-based method fail to detection copy
number changes and/or gene fusions (13).
In summary, we performed WES of endometrial cancer
samples and identified several potential cancer driver and
passenger cancer genes (MAPT, IL24, MCM6,
TSC1, BIRC2, CIITA, DST, CASP8,
NOTCH2, ARMCX4, IGSF10, VPS13C,
DCT, DNAH14, TLN1, ZNF605,
ZSCAN29, MOCOS, CMYA5, PCDH17,
UGT1A8, CYFIP2, MACF1, NUDT5,
JAKMIP1, PCDHGB4, FAM178A, SNX6,
IMP4 and PCMTD1). The major limitation of this study
was the small sample size.
Acknowledgments
The authors would like to thank Wei-Chi Wang
(Health GeneTech Corporation, Taoyuan, Taiwan) for his invaluable
assistance in bioinformatics analysis. The present study was
supported by the National Health Research Institutes
(NHRI-EX104-10326BI), the Taiwan Ministry of Health and Welfare
Clinical Trial Center (MOHW106-TDU-B-212-113004) and the China
Medical University Hospital (DMR-106-105).
References
|
1
|
Sénéchal C, Cottereau E, de Pauw A, Elan
C, Dagousset I, Fourchotte V, Gauthier-Villars M, Lae M,
Stoppa-Lyonnet D and Buecher B: Environmental and genetic risk
factors for endometrial carcinoma. Bull Cancer. 102:256–269.
2015.In French. View Article : Google Scholar
|
|
2
|
Ashton KA, Proietto A, Otton G, Symonds I,
McEvoy M, Attia J, Gilbert M, Hamann U and Scott RJ: Polymorphisms
in genes of the steroid hormone biosynthesis and metabolism
pathways and endometrial cancer risk. Cancer Epidemiol. 34:328–337.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xu J, Lin X, Zhu H, Zhang Z and Yang B:
Genetic variation of the CYP17 and susceptibility to endometrial
cancer: A meta-analysis. Mol Biol Rep. 40:5085–5091. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Collaborative Group on Epidemiological
Studies on Endometrial Cancer: Endometrial cancer and oral
contraceptives: An individual participant meta-analysis of 27 276
women with endometrial cancer from 36 epidemiological studies.
Lancet Oncol. 16:1061–1070. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Trabert B, Wentzensen N, Yang HP, Sherman
ME, Hollenbeck AR, Park Y and Brinton LA: Is estrogen plus
progestin menopausal hormone therapy safe with respect to
endometrial cancer risk? Int J Cancer. 132:417–426. 2013.
View Article : Google Scholar
|
|
6
|
Brinton LA, Westhoff CL, Scoccia B, Lamb
EJ, Trabert B, Niwa S and Moghissi KS: Fertility drugs and
endometrial cancer risk: Results from an extended follow-up of a
large infertility cohort. Hum Reprod. 28:2813–2821. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ong M, Carreira S, Goodall J, Mateo J,
Figueiredo I, Rodrigues DN, Perkins G, Seed G, Yap TA, Attard G, et
al: Validation and utilisation of high-coverage next-generation
sequencing to deliver the pharmacological audit trail. Br J Cancer.
111:828–836. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
André F, Bachelot T, Commo F, Campone M,
Arnedos M, Dieras V, Lacroix-Triki M, Lacroix L, Cohen P, Gentien
D, et al: Comparative genomic hybridisation array and DNA
sequencing to direct treatment of metastatic breast cancer: A
multicentre, prospective trial (SAFIR01/UNICANCER). Lancet Oncol.
15:267–274. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cancer Genome Atlas Research Network:
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bell D, Berchuck A, Birrer M, Chien J,
Cramer DW, Dao F, Dhir R, DiSaia P, Gabra H, Glenn P, et al Cancer
Genome Atlas Research Network: Integrated genomic analyses of
ovarian carcinoma. Nature. 474:609–615. 2011. View Article : Google Scholar
|
|
11
|
Cancer Genome Atlas Network: Comprehensive
molecular characterization of human colon and rectal cancer.
Nature. 487:330–337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kandoth C, Schultz N, Cherniack AD, Akbani
R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, et al
Cancer Genome Atlas Research Network: Integrated genomic
characterization of endometrial carcinoma. Nature. 497:67–73. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Beadling C, Neff TL, Heinrich MC, Rhodes
K, Thornton M, Leamon J, Andersen M and Corless CL: Combining
highly multiplexed PCR with semiconductor-based sequencing for
rapid cancer genotyping. J Mol Diagn. 15:171–176. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Grasso C, Butler T, Rhodes K, Quist M,
Neff TL, Moore S, Tomlins SA, Reinig E, Beadling C, Andersen M, et
al: Assessing copy number alterations in targeted, amplicon-based
next-generation sequencing data. J Mol Diagn. 17:53–63. 2015.
View Article : Google Scholar
|
|
15
|
Zheng Z, Liebers M, Zhelyazkova B, Cao Y,
Panditi D, Lynch KD, Chen J, Robinson HE, Shim HS, Chmielecki J, et
al: Anchored multiplex PCR for targeted next-generation sequencing.
Nat Med. 20:1479–1484. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Samorodnitsky E, Datta J, Jewell BM,
Hagopian R, Miya J, Wing MR, Damodaran S, Lippus JM, Reeser JW,
Bhatt D, et al: Comparison of custom capture for targeted
next-generation DNA sequencing. J Mol Diagn. 17:64–75. 2015.
View Article : Google Scholar :
|
|
17
|
Drmanac R, Peters BA, Church GM, Reid CA
and Xu X: Accurate whole genome sequencing as the ultimate genetic
test. Clin Chem. 61:305–306. 2015. View Article : Google Scholar
|
|
18
|
Park JY, Clark P, Londin E, Sponziello M,
Kricka LJ and Fortina P: Clinical exome performance for reporting
secondary genetic findings. Clin Chem. 61:213–220. 2015. View Article : Google Scholar :
|
|
19
|
Liang H, Cheung LW, Li J, Ju Z, Yu S,
Stemke-Hale K, Dogruluk T, Lu Y, Liu X, Gu C, et al: Whole-exome
sequencing combined with functional genomics reveals novel
candidate driver cancer genes in endometrial cancer. Genome Res.
22:2120–2129. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Langmead B, Trapnell C, Pop M and Salzberg
SL: Ultrafast and memory-efficient alignment of short DNA sequences
to the human genome. Genome Biol. 10:R252009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M, et al: The Genome Analysis Toolkit: A MapReduce framework for
analyzing next-generation DNA sequencing data. Genome Res.
20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sherry ST, Ward MH, Kholodov M, Baker J,
Phan L, Smigielski EM and Sirotkin K: dbSNP: The NCBI database of
genetic variation. Nucleic Acids Res. 29:308–311. 2001. View Article : Google Scholar :
|
|
23
|
Landrum MJ, Lee JM, Riley GR, Jang W,
Rubinstein WS, Church DM and Maglott DR: ClinVar: Public archive of
relationships among sequence variation and human phenotype. Nucleic
Acids Res. 42(D1): D980–D985. 2014. View Article : Google Scholar :
|
|
24
|
Forbes SA, Beare D, Gunasekaran P, Leung
K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, et
al: COSMIC: Exploring the world's knowledge of somatic mutations in
human cancer. Nucleic Acids Res. 43(D1): D805–D811. 2015.
View Article : Google Scholar :
|
|
25
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R and Larsson E:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBio-Portal. Sci Signal. 6:pl12013. View Article : Google Scholar
|
|
26
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hua X, Xu H, Yang Y, Zhu J, Liu P and Lu
Y: DrGaP: A powerful tool for identifying driver genes and pathways
in cancer sequencing studies. Am J Hum Genet. 93:439–451. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chang YS, Huang HD, Yeh KT and Chang JG:
Genetic alterations in endometrial cancer by targeted
next-generation sequencing. Exp Mol Pathol. 100:8–12. 2016.
View Article : Google Scholar
|
|
30
|
Hovelson DH, McDaniel AS, Cani AK, Johnson
B, Rhodes K, Williams PD, Bandla S, Bien G, Choppa P, Hyland F, et
al: Development and validation of a scalable next-generation
sequencing system for assessing relevant somatic variants in solid
tumors. Neoplasia. 17:385–399. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kwiatkowski DJ, Choueiri TK, Fay AP, Rini
BI, Thorner AR, de Velasco G, Tyburczy ME, Hamieh L, Albiges L,
Agarwal N, et al: Mutations in TSC1, TSC2, and MTOR are associated
with response to rapalogs in patients with metastatic renal cell
carcinoma. Clin Cancer Res. 22:2445–2452. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Choschzick M, Tabibzada AM, Gieseking F,
Woelber L, Jaenicke F, Sauter G and Simon R: BIRC2 amplification in
squamous cell carcinomas of the uterine cervix. Virchows Arch.
461:123–128. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ma XY, Ma CX and Wang JH: Endometrial
carcinogenesis and molecular signaling pathways. Am J Mol Biol.
4:134–149. 2014. View Article : Google Scholar
|
|
34
|
Nam JY, Kim NK, Kim SC, Joung JG, Xi R,
Lee S, Park PJ and Park WY: Evaluation of somatic copy number
estimation tools for whole-exome sequencing data. Brief Bioinform.
17:185–192. 2016. View Article : Google Scholar
|