Introduction
The colonisation of distant organs by disseminating
breast cancer cells is the main cause for cancer death in women.
Targeting neo-vascularisation with tailored drugs may prevent the
spreading of those cancer entities that expand through the blood
stream. In the early phases of breast cancer metastasis however,
cancer cells disperse through lymphatics. Before metastases
colonise organs the sentinel and post-sentinel lymph nodes fill up
with breast cancer cells and hence, the axillary lymph node status
is a major prognostic marker for clinical outcome (1). As blocking of intravasation would
improve prognosis, understanding of mechanistic details of breast
cancer intravasation into lymphatic vessels is crucial to develop
treatment strategies preventing lymph node metastasis. Although
prognostic markers exist, no predictive markers have been
identified to date, which could be targeted to combat
intravasation. Nevertheless, a few mechanisms supporting the
transmigration of cancer cells through the lymphatic barrier were
described (2–4). Intravasation is an interplay among
cancer cells and the endothelial barrier. Importantly, not only
cancer cells play an active role in this process but also lymph
endothelial cells (LECs) remarkably contribute to lymph node
metastasis i.e. by providing niches for cancer cell settlement
(5). To further study the role of
LECs at an early metastatic step, a quantitative assay based on a
three-dimensional (3D) cell model was developed consisting of
breast cancer spheroids placed on top of lymphendothelial
monolayers, which resembles the contact zone between the tumour and
lymphatics (6). This 3D in
vitro model is validated in scid mice and in tissue sections of
human patients (7) and was here
utilised to measure tumour cell-induced retraction of LECs, which
enables cancer cells to transmigrate the lymphatic barrier.
Focussing on the regulation and maintenance of the endothelial
barrier function several mechanisms of LECs, which are relevant to
withstand tumour intravasation, were found to become weakened by
signals of adjacent cancer cells (3,4).
Notably, LECs - upon activation of specific receptors - 'invite'
cancer cells to intravasate by reducing the resilience of the
endothelial wall (2,8,9)
through a process called endothelial-to-mesenchymal transition. In
turn, LECs acquire a migratory phenotype and open gates for cancer
cell transit as a rate limiting step of lymph node metastasis
(7). Hence, strengthening vascular
integrity and tone may improve resistance to intravasating cancer
cells.
As a pro-migratory mechanism ion channels modulate
adhesion and mobility of tumour cells and their dysregulation is
currently discussed to play a role in cancer metastasis (10). For example, the overexpression of
voltage gated K+ channels was observed in breast-,
colon- and prostate cancer and medullablastoma (11,12)
and their inhibition attenuates neuroblastoma and endothelial cell
migration (13,14). Consistently, the
Na+/K+-ATPase inhibitor ouabain (also known
as g-strophanthin) attenuates medullablastoma cell migration and
the localisation of Tyr397-phosphorylated FAK to lammelipodia
(15). Furthermore, the voltage
gated K+v1.2 channel induces directional migration of
mesenchymal bone marrow or CHO cells through phosphorylation of
Tyr397-FAK (16) as well as wound
healing in an animal model (17).
The antidyslipidemic drug fenofibrate closes K+ATP
channels (18,19) and voltage gated K+
channels (20), enhances
endothelial barrier function and integrity (5,21)
and is therefore, vasoprotective.
Furthermore, fenofibrate exhibits anticancer effects
(22). Independent of
K+ channel inhibition, fenofibrate binds to and
activates peroxisome proliferator-activated protein alpha (PPARα),
which directly binds to the IκBα promoter, induces IκBα protein
expression (23) and in turn
inhibits NF-κB (24,25). NF-κB plays a role in tumour
progression and intravasation through lymphendothelial barriers
(3,4). Hence, both activities of fenofibrate,
PPARα activation, which was shown to suppress cancer progression
(26,27), as well as K+ channel
inhibition (10,18,20)
may attenuate adhesion and cell mobility (28,29).
In the clinic fenofibrate is used to eliminate triglycerides from
the blood stream (30), thereby
reducing hypercholesterolemia and the risk of cardiovascular
diseases. Therefore, fenofibrate and a few more clinically used
drugs with reported ion channel inhibitory properties were here
investigated regarding their potential activities inhibiting
triple-negative (MDA-MB231) and estrogen receptor-positive (MCF-7)
breast cancer cell intravasation in vitro. Such drugs would
be directly available to patients.
Materials and methods
Antibodies and reagents
Polyclonal rabbit anti-focal adhesion kinase (FAK)
and polyclonal rabbit anti-phospho-Tyr397-FAK were from Cell
Signaling (Danvers, MA, USA). Polyclonal goat anti-CD54 (ICAM-1)
antibody was from R&D System (clone BBA-17, Minneapolis, MN,
USA) and monoclonal anti-β-actin from Sigma (Munich, Germany).
Monoclonal mouse anti-CD31 (JC70A), polyclonal rabbit anti-mouse,
anti-goat and anti-rabbit IgGs were from Dako (Glostrup,
Denmark).
The IκBα phosphorylation inhibitor
(E)-3-[(4-methylphenylsulfonyl]-2-propenenitrile (Bay11-7082) was
purchased from Biomol (Hamburg, Germany). Bepridil hydrochloride
(bepridil), niflumic acid, fenofibrate, cisapride monohydrate
(cisapride), primaquine, proadifen, digoxin, chlorotoxin and
ouabain octahydrate (ouabain) were from Sigma. Dyclonine was from
Abcam (Cambridge, UK).
Cell culture
Human MDA-MB231 and MCF-7 breast cancer cells were
purchased from the American Type Culture Collection (ATCC,
Rockville, MD, USA) and grown in MEM medium supplemented with 10%
foetal calf serum (FCS), 1% penicillin/streptomycin (PS) and 1%
non-essential amino acids (Gibco/Invitrogen, Karlsruhe, Germany).
Telomerase immortalized human lymph endothelial cells (LECs) were
grown in EGM2 MV (Clonetics CC-4147, Allendale, NJ, USA). The cells
were kept at 37°C in a humidified atmosphere containing 5%
CO2. For CCID formation assays, LECs were stained with
CellTracker™ green purchased from Invitrogen (Karlsruhe,
Germany).
Spheroid formation
MDA-MB231 cells (input of 6,000 cells per spheroid)
and MCF-7 cells (input of 3,000 cells per spheroid) were
transferred to 30 ml serum-free MEM medium containing 6 ml of a
1.6% methylcellulose solution (0.3% final concentration; cat. no.:
M-512, 4000 centipoises; Sigma-Aldrich, Munich, Germany). Cell
suspension (150 μl) were transferred to each well of a
96-well plate (Greiner Bio-one, Cellstar 650185, Kremsmünster,
Austria) to allow spheroid formation within 48 h.
CCID (circular chemorepellent induced
defect) assay
In this assay the sizes of the cell-free areas
(CCIDs), which are formed in the endothelial monolayer directly
underneath the tumour spheroids, were measured (7). MDA-MB231 spheroids were washed in PBS
and transferred to CellTracker (green)-stained LEC monolayers that
were seeded into 24-well plates (Costar 3524, Sigma-Aldrich) in 1
ml EGM2 MV medium. After 4 h of incubation, the CCID areas in the
LEC monolayers underneath the MDA-MB231 spheroids were photographed
using an Axiovert (Zeiss, Jena, Germany) fluorescence microscope to
visualise CellTracker-stained LECs underneath the spheroids. CCID
areas were calculated with the Zen Little 2012 (Zeiss). For each
condition the CCID size of at least 25 spheroids (unless otherwise
specified) was measured.
SDS gel electrophoresis and western
blotting
LECs were grown in T-25 tissue culture flasks (Nunc,
Roskilde, Denmark) to 80% confluence and then pre-treated with
indicated clinical drugs or inhibitors for 0.5, 1, 2 and 4 h. Then,
cells were processed for SDS gel electrophoresis and western
blotting as described before (4).
Chemo-luminescence was developed by Amersham ECL Prime kit (GE
Healthcare, Freiburg, Germany) and detected using a Lumi-Imager F1
Workstation (Roche, Basel, Switzerland). Densitometry of the
western blots was analysed with Image-J software (National
Institutes of Health, Bethesda, MD, USA).
Ethoxyresorufin-O-deethylase (EROD) assay
selective for CYP1A1 and CYP1A2 activity
MDA-MB231 cells were grown in phenol red-free
DMEM/F12 medium (Gibco, Karlsruhe, Germany) containing 10% FCS and
1% PS (Invitrogen, Karlsruhe, Germany). Before treatment, the cells
were transferred to DMEM/F12 medium supplemented with 10%
charcoal-stripped FCS (PAN Biotech, Aldenbach, Germany) and 1% PS.
After 24 h of treatment CYP1A1 activity was measured with minor
modifications as previously described (31). Briefly, ethoxyresorufin (final
concentration 5.0 μM, Sigma-Aldrich) was added and 0.4 ml
aliquots of the medium were sampled after 180 min and the formation
of resorufin was analysed by spectrofluorometry (PerkinElmer LS50B,
Waltham, MA, USA) with an excitation wavelength of 530 nm and an
emission wavelength of 585 nm. The pan-CYP inhibitor proadifen
(Sigma-Aldrich) was used as a positive control.
NF-κB transactivation assay
The transactivation of a NF-κB-driven luciferase
reporter was quantified in HEK293/NF-κB-luc cells (Panomics,
RC0014, Fremont, CA, USA) as previously described (32). In brief, cells were maintained at
37°C and 5% CO2, humidified atmosphere in Dulbecco's
modified Eagle's medium (DMEM; Lonza, Basel, Switzerland)
supplemented with 2 mM glutamine, 100 μg/ml hygromycin B,
100 U/ml benzylpenicillin, 100 μg/ml streptomycin, and 10%
FCS. One day before the experiments the cells were stained in
serum-free medium supplemented with 2 μM CellTracker Green
CMFDA (C2925; Invitrogen) for 1 h. Then, cells were reseeded in
96-well plates at a density of 4x104 cells/well in
phenol red-free and FCS-free DMEM overnight, and pre-treated with
the indicated compounds for 30 min prior to stimulation with 2
ng/ml TNFα for 4 h. The final concentration of DMSO in the
experiments was 0.1% or lower and an equal concentration of DMSO
was used as control. After cell lysis the luminescence of the
firefly luciferase and the fluorescence of the CellTracker Green
CMFDA were quantified on a GeniosPro plate reader (Tecan, Grodig,
Austria). The luciferase-derived signal from the NF-κB reporter was
normalized by the CellTracker Green CMFDA derived fluorescence to
account for differences in the cell number. The known NF-κB
inhibitor parthenolide (Sigma-Aldrich, Vienna, Austria) was used as
a positive control.
Adhesion assay
MDA-MB231 (40,000 cells/well) were seeded in
serum-free medium (DMEM containing 0.5% BSA, 2 mM CaCl2
and 2 mM MgCl2). Then 500X CellTracker (500X CellTracker
Solution from CytoSelect™ Tumor-endothelium Adhesion Assay from
Cell Biolabs, Inc., San Diego, CA, USA, CBA-215) was added to the
cell suspension (2 μl CellTracker to 1 ml suspension),
incubated for 1 h at 37°C and centrifuged at 1,000 rpm for 2 min.
Then the medium was aspirated, the cell pellet was washed ×3 with
serum-free medium (DMEM containing 0.5% BSA, 2 mM CaCl2
and 2 mM MgCl2) and then the cell pellet was
re-suspended in EGM2 MV medium. The CellTracker-stained MDA-MB231
cell suspension was treated either with Bay11-7082 as a positive
control, indicated compounds, or DMSO and incubated for 10 min at
room temperature. After incubation, the medium was aspirated from
the 48-well plate and 200 μl of the pre-treated MDA-MB231
cell suspension was added to LECs grown in monolayers and incubated
for 30 min at 37°C. Then the medium was aspirated and cells were
washed 3× with 250 μl 1X Wash Buffer (10X Wash Buffer was
from CytoSelect Tumor-endothelium Adhesion Assay). Before the third
wash, cells were inspected for morphological changes under the
microscope. After the final wash the plate was tapped on a
flint-free paper towel and 150 μl of 1X Lysis Buffer (4X
Lysis Buffer were from CytoSelect Tumor-endothelium Adhesion Assay)
was added to each well and cells were lysed by the shearing forces
through a pipette tip. Lysate (100 μl) was transferred to
96-well black-wall clear bottom plates (Nunc, Thermo Scientific,
Rochester, NY, USA) and the fluorescence was measured with a
fluorescence plate reader at 485/530 nm.
Statistical analysis
For statistical analyses Excel 2013 software and
Prism 6 software package (GraphPad, San Diego, CA, USA) were used.
The values were expressed as mean ± SEM and the Student's t-test
and ANOVA with Tukey's post-test was used to compare differences
between control samples and treatment groups as well as difference
among treatment groups. Statistical significance level was set to
P<0.05.
Results
Fenofibrate inhibits NF-κB, ICAM-1, FAK,
adhesion and CCID formation in LECs
Fenofibrate inhibited TNFα-induced NF-κB activation
(Fig. 1A) and transiently
downregulated expression of the adhesion molecules ICAM-1 and CD31
in LECs (Fig. 1B). In addition,
Bay11-7802 (a specific NF-κB inhibitor) suppressed ICAM-1 (Fig. 1C) and both compounds inhibited the
adhesion of MDA-MB231 cells to LECs (Fig. 1D), which is a prerequisite for
intravasation and CCID formation (3). Accordingly, fenofibrate attenuated
CCID formation in the MDA-MB231/LEC model (Fig. 1E) and in the MCF-7/LEC model
(Fig. 1F). The inhibition of CCID
formation by fenofibrate was achieved at concentrations as low as 5
μM. However, the inhibition of NF-κB and of cell adhesion
was observed at higher concentrations indicating that also another
mechanism must have been involved in the CCID- inhibitory effect of
fenofibrate. It was shown that feno-fibrate inhibits cell migration
and stabilises HUVEC barrier function, which is accompanied by the
downregulation of FAK activity (5). FAK activity is required for
cell-matrix adhesion and directional migration (33,34)
and adhesion and migration is also necessary for CCID formation
(7). In LEC fenofibrate inhibited
the phosphorylation of Tyr397-FAK (Fig. 1G), which is indicative for its
activity.
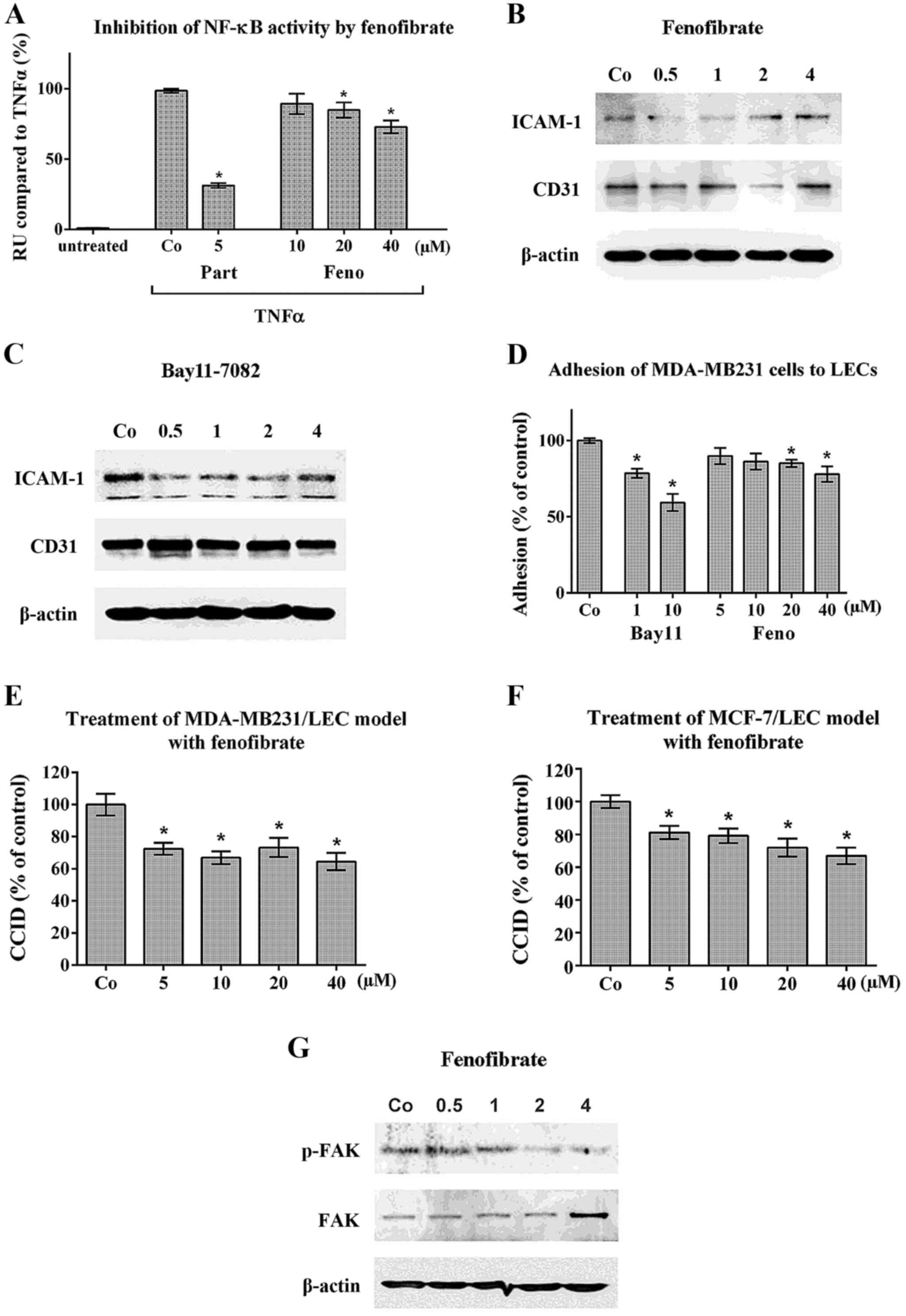 | Figure 1Inhibition of NF-κB activity,
adhesion and CCID formation upon fenofibrate treatment. (A)
HEK293-NFκB-Luc cells were pretreated with 5 μM parthenolide
(Part) as a specific inhibitor of NF-κB or with increasing
concentrations of fenofibrate (Feno) or solvent (Co-DMSO) for 1 h.
Then, cells were stimulated with 2 ng/ml human recombinant TNFα for
an additional 4 h when NF-κB activity was measured. Three
independent experiments with 4 replicates were analysed. (B) LECs
were grown to ~80% confluence and then pretreated with solvent (Co)
or 20 μM fenofibrate or (C) 5 μM Bay11-7082 as a
specific inhibitor of NF-κB for 0.5, 1, 2 and 4 h. Then, cells were
lysed, proteins separated by SDS gel electrophoresis and subjected
to western blotting using the indicated antibodies. Staining with
Ponceau S and immunoblotting with anti-β-actin antibody controlled
equal sample loading. (D) MDA-MB231 cells were pre-treated with
Bay11-7082 (Bay11) or with increasing concentrations of
fenofibrate, or solvent (Co) and then placed on confluent LEC
monolayers, which were also pre-treated with respective compounds.
After 30 min the adhesion of MDA-MB231 cells to LECs was measured
as described in Materials and methods. Experiments were performed
in triplicate. (E) MDA-MB231 spheroids or (F) MCF-7 spheroids were
pre-treated for 30 min with solvent (Co) or the indicated
fenofibrate concentrations. Then, spheroids were placed on top of
LEC monolayers and co-cultivated for 4 h when CCIDs were analysed.
Three independent experiments with at least 8 replicates were
analysed. (G) LECs were grown to ~80% confluence and then
pretreated with solvent (Co) or fenofibrate for 0.5, 1, 2 and 4 h
when cells were lysed, proteins separated by SDS gel
electrophoresis and subjected to western blotting using the
indicated antibodies. Error bars indicate means ± SEM and asterisks
significance (P<0.05; ANOVA together with Tukey's
post-test). |
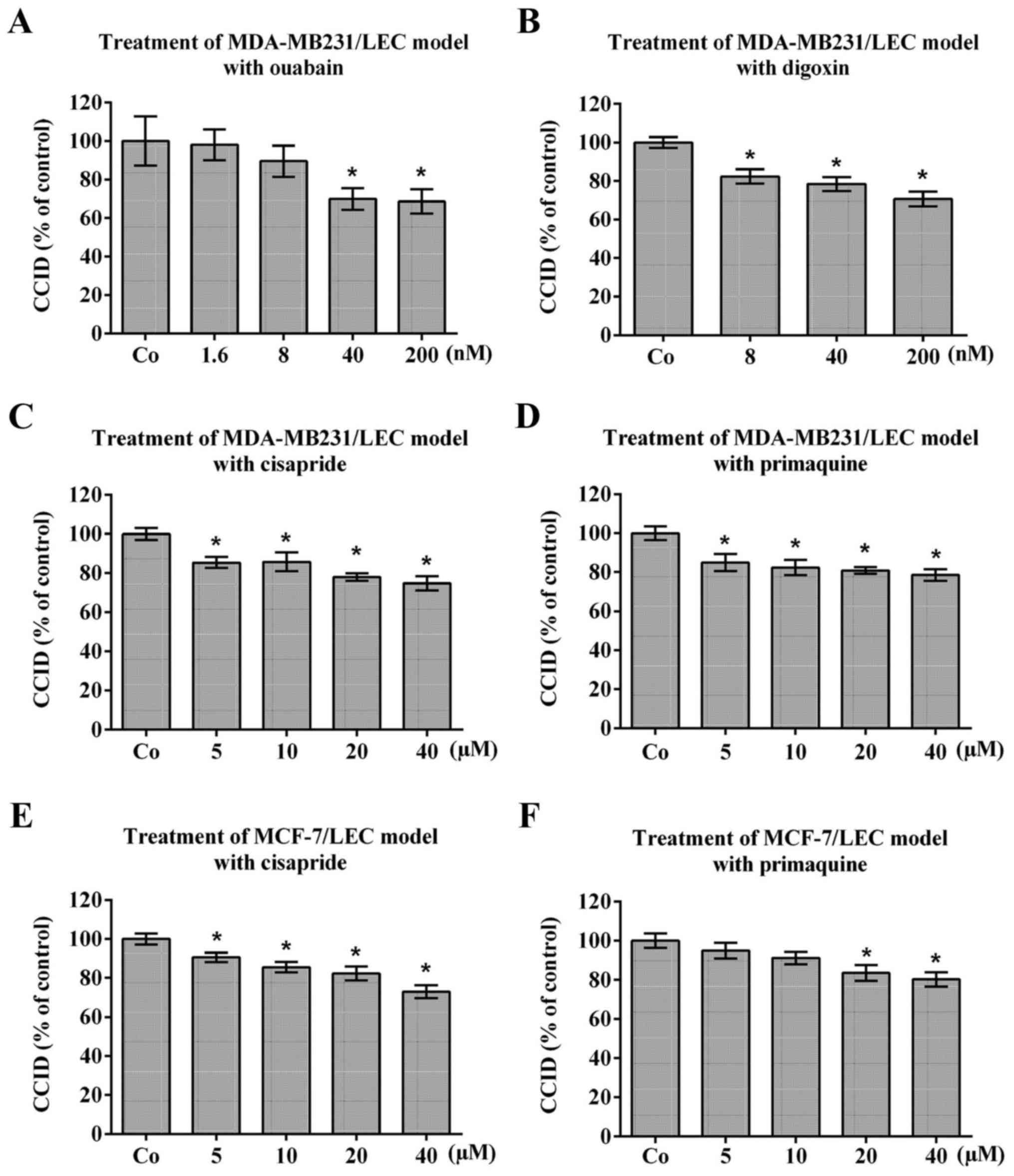
Bona fide K+channel inhibitors
attenuate CCID formation
To investigate whether potassium channels contribute
to CCID formation, the MDA-MB231/LEC model was treated with ouabain
and digoxin, which are used to specifically inhibit
Na+/K+-ATPase (35,36).
Ouabain and digoxin dose-dependently attenuated MDA-MB231-triggered
CCID formation (Fig. 2A and B;
respectively). Ouabain inhibits cell migration through inactivation
of FAK (37) and also digoxin
inhibits cell migration (38).
Similarly, cisapride (a serotonin 5-HT 4 receptor agonist used to
treat gastroesophageal reflux disease) and primaquine (used to
treat malaria through binding to Plasmodium DNA), which were both
reported to inhibit hERG-K+ channel (39,40),
inhibited MDA-MB231-triggered CCID formation (Fig. 2C and D; respectively) and
MCF-7-trigerred CCID formation (Fig.
2E and F; respectively). As the treatment with known
K+ ATPase- and hERG-K+ channel inhibitors
resulted in significant inhibition of CCID formation, it was
concluded that the activity of K+ channels contributes
to breast cancer intravasation in vitro. Based on these
results fenofibrate seems to inhibit CCID formation through
inhibition of NF-κB activity as well as of K+ channels.
Reportedly, the delayed rectifier K+v2.1 channel induces
the phosphorylation of Tyr397-FAK (16,17).
Therefore, fenofibrate may have inhibited FAK phosphorylation
through inhibition of K+v1.2 channel. Furthermore,
primaquine, but not cisapride inhibited the phosphorylation of
Tyr397-FAK (data not shown).
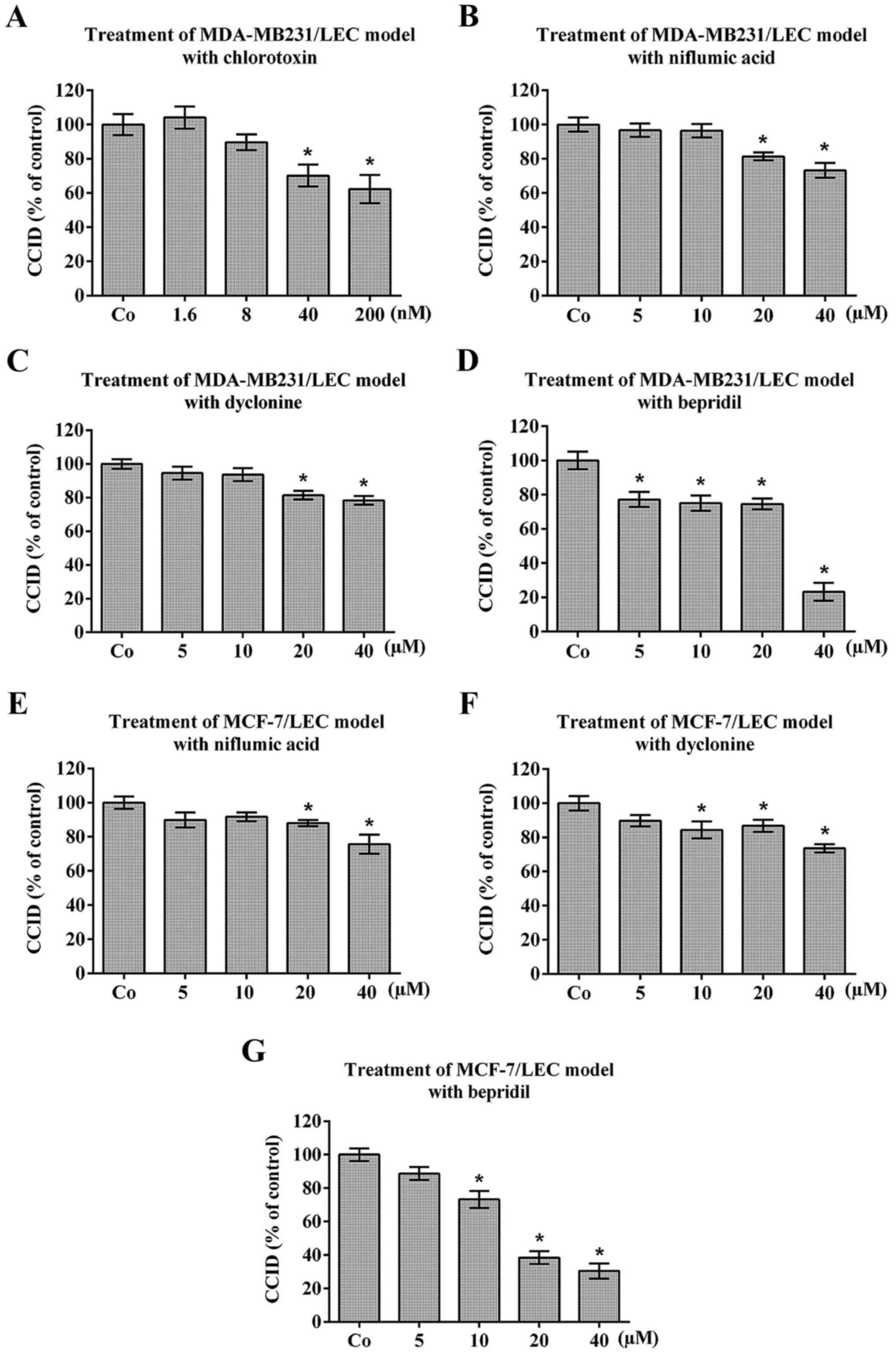
Niflumic acid, dyclonine and bepridil
hydrochloride inhibit CCID formation
As K+-channels, also Cl−-,
Na+-, and Ca2+-channels are dis-regulated
during carcinogenesis (10).
Therefore, the 3D-models were treated with the reported
Cl−-channel inhibitors chlorotoxin and niflumic acid
(41,42) (Fig.
3A, B and E, respectively), the Na+-channel
inhibitor dyclonine (43)
(Fig. 3C and F) and the
Ca2+-channel inhibitor bepridil hydrochloride (44) (Fig. 3D
and G), which inhibited MDA-MB231- and MCF-7- triggered CCID
formation in LEC barriers.
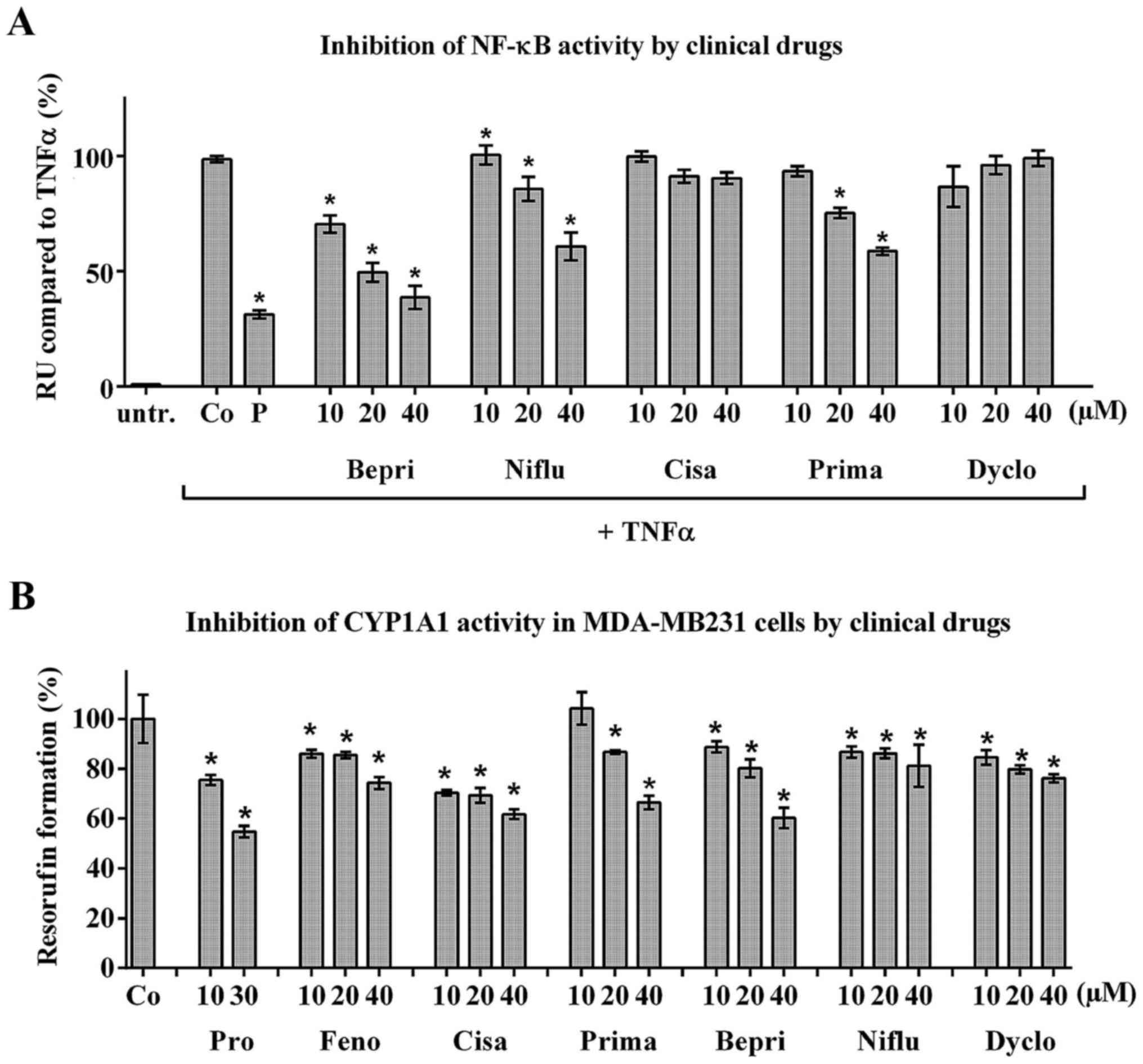
Fenofibrate, primaquine, cisapride,
dyclonine, bepridil and niflumic acid inhibit CYP1A1 in MDA-MB231
cells
Intravasation is an interplay between tumour emboli
and the vessel endothelium beneath. In the former experiments the
effect of fenofibrate was investigated in LECs. Herein, the effect
of fenofibrate and the other clinical drugs was studied in
MDA-MB231 breast cancer cells. Recently, it was shown that
MDA-MB231 cell intravasation depends on NF-κB- and CYP1A1-activity
of the cancer cells (4,45). Niflumic acid, bepridil and
primaquine, but not dyclonine or cisapride, inhibited TNFα-induced
NF-κB activity in HEK293/NF-κB-luc cells (Fig. 4A). Furthermore, the treatment of
MDA-MB231 cells with fenofibrate, primaquine, dyclonine, bepridil,
niflumic acid and cisapride significantly inhibited the formation
of resorufin indicating the inhibition of CYP1A1 activity (tested
by EROD assay; Fig. 4B). Hence,
the here tested drugs also exerted their anti-intravasative
properties by attenuating CYP1A1 activity in the tumour cells and
some, additionally, by inhibiting NF-κB.
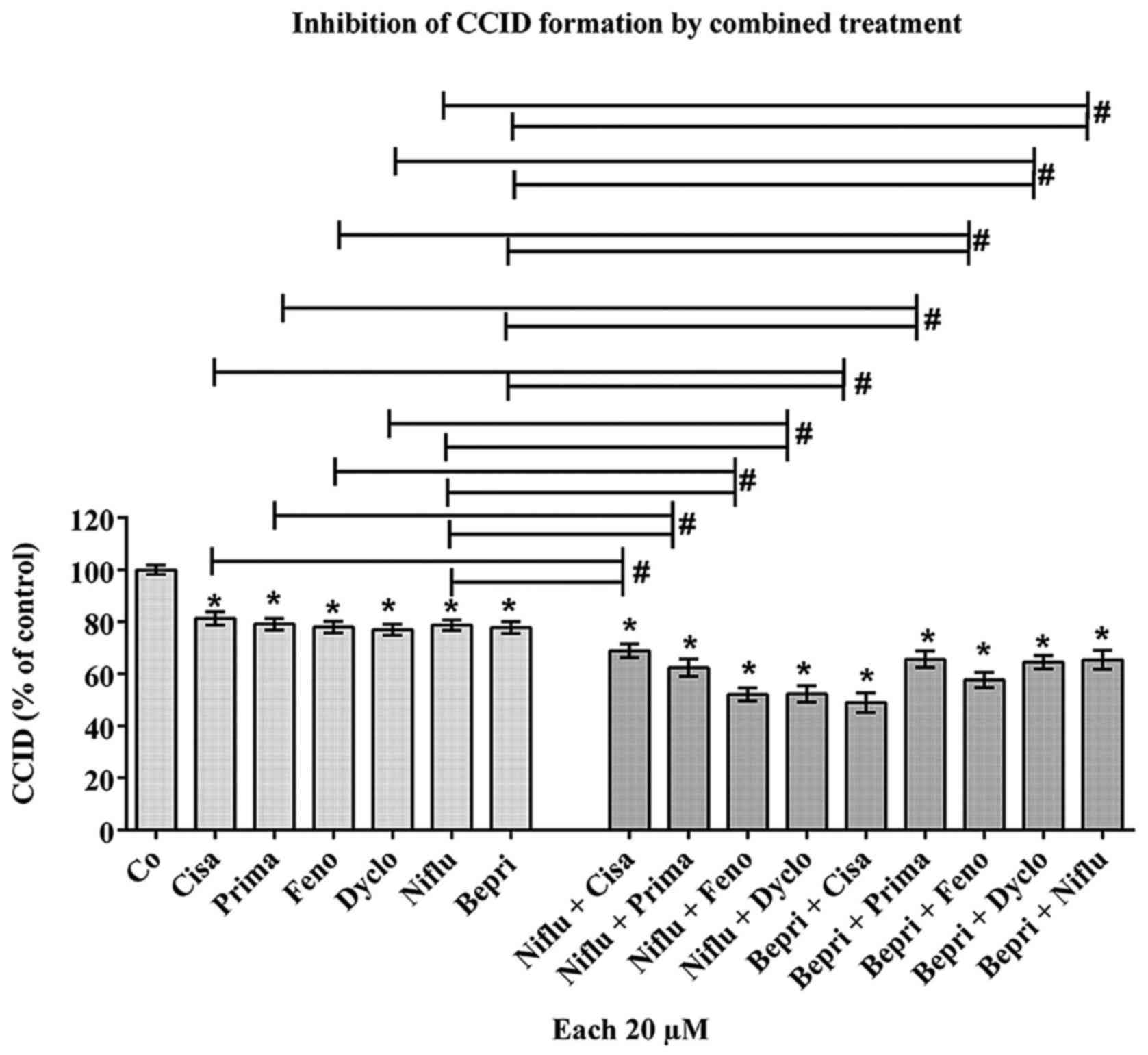
Drug combinations further reduce
intravasation
The drugs at 20 μM of fenofibrate, cisapride,
primaquine, niflumic acid, bepridil and dyclonine acid inhibited
CCID formation by ~20%. Therefore, we investigated whether drug
combinations may improve the CCID-inhibitory effect. Combining only
bona fide K+ channel inhibitors or K+ channel
inhibitors together with the Na+ channel inhibitor
dyclonine did not exhibit improved effects (Table I). In contrast, the combinations of
bepridil hydrochloride and primaquine, or bepridil hydrochloride
and dyclonine, or bepridil hydrochloride and niflumic acid, or
niflumic acid and cisapride, or niflumic acid and primaquine
inhibited CCID formation additively. Synergistic effects were
achieved when combining bepridil hydrochloride and cisapride, or
bepridil hydrochloride and fenofibrate, or niflumic acid and
fenofibrate, or niflumic acid and dyclonine (Fig. 5). Since different drug combinations
resulted in additive, synergistic, or unchanged inhibition of CCID
formation, this implicated that the treatment with these drugs
affected intravasative mechanisms non-randomly.
 | Table IAnalysis of drug combinations on CCID
formation. |
Table I
Analysis of drug combinations on CCID
formation.
| Drugs | Concentration
(μM) | CCID formation (%
of control ± SD) | P-value compared to
control |
|---|
| Cisa | 20 | 79.97±10.60 | <0.0001 |
| Prima | 20 | 81.04±9.70 | <0.0001 |
| Feno | 20 | 79.21±21.85 | 0.0089 |
| Dyclo | 20 | 81.50±13.75 | <0.0001 |
| Cisa + Prima | 20/20 | 84.43±21.20 | 0.0305 |
| Cisa + Feno | 20/20 | 85.51±19.31 | 0.0309 |
| Cisa + Dyclo | 20/20 | 84.85±13.23 | 0.0006 |
| Prima + Feno | 20/20 | 90.97±17.96 | 0.0305 |
| Prima +Dyclo | 20/20 | 84.75±12.32 | 0.0009 |
| Feno + Dyclo | 20/20 | 82.32±16.17 | <0.0001 |
We searched for potential drug-drug interactions
(DDIs) such as between fenofibrate, cisapride, and niflumic acid,
which are recorded in the data base of the -Austria-Codex
Fachinformation (Oesterreichische Apotheker-Verlagsgesellschaft
m.b.H., date of release: 01/09/2016) regarding known cross
reactions. Moreover, DDIs between these drugs and standard
chemotherapeutic drugs used in breast cancer were screened, yet no
dangerous DDIs were retrieved. Bepridil, dyclonine, and primaquine
were not listed as these drugs are not licensed in Austria and
Germany.
Discussion
Adhesion of breast cancer cells to the LEC wall is
obligatory to enable subsequent intravasation into the vessel lumen
(3). It was shown that cancer cell
adhesion (10) and adhesion of
endothelial cells to monocytes depends on K+ channel
activity (46). Reportedly, the
antidislypidemic and therefore, vasoprotective drug fenofibrate
inhibits K+-ATPase and voltage gated K+
channels (19,20) and tumour invasivity (5). As dysregulated ion channels assist
tumour progression (10), this
tempted us to investigate whether fenofibrate and other ion channel
inhibitors that are in clinical use attenuate breast cancer cell
intravasation. Fenofibrate modulates LEC adhesion (5) by activating PPARα which in turn
inhibits NF-κB (24,25) and by inhibiting K+
channel activity (20). Herein, we
demonstrate a novel property of fenofibrate as an inhibitor of CCID
formation within the LEC barrier, which correlated with the
inhibition of NF-κB, the downregulation of ICAM-1 in LECs and
subsequent attenuation of adhesion of cancer cells.
Almost all phosphorylation-regulated ion channels,
or K+ channels containing RGD sequences, may contribute
to signalling networks (47)
thereby modulating FAK activity. The activity of FAK is necessary
for loose adhesion to the matrix and rapid directional migration
(33,34) as observed in CCID formation
(7) and in a current study
establishing the role of FAK in this process we find that knockdown
of FAK substantially inhibits CCIDs (Hong et al, unpublished
data). The inhibition of FAK-phosphorylation in LECs by the
Na+ channel inhibitor carbamazepine (48) correlates with the inhibition of
CCID formation (31) and this
supports the notion that also other ion channels contribute to
endothelial barrier disintegration and cancer progression. To this
end, it was shown that voltage gated Na+ channels are
differently expressed in asterocytoma and breast cancer cells
(49,50). Therefore, the inactivation of FAK,
which was most likely achieved through the inhibition of
K+-ATPase (37) was
another, and NF-κB-independent mechanism of fenofibrate
compromising metastatic outspread.
Furthermore, the central role of Ca2+
signalling and the relevance of Ca2+ channels as central
mediators and amplifiers of migration and CIDD formation in LECs
and cancer associates fibroblasts (CAFs) was reported (8,51).
Therefore, the Ca2+ channel inhibitor bepridil
hydrochloride was tested in combination with other bona fide ion
channel inhibitors. Some drug combinations improved the CCID
inhibitory effect in an additive and even synergistic manner.
Whereas fenofibrate inhibited FAK activity and
ICAM-1 expression in LECs, the activity of CYP1A1 was inhibited in
MDA-MB231 cancer cells. CYP1A1 was demonstrated to significantly
contribute to breast cancer intravasation in vitro (45). CYP1A1 resides in heterogenous
membrane regions of the endoplasmatic reticulum (52–54)
and interactions between P450 molecules and juxtaposed proteins
within the lipid layer (such as ion channels) (55) are discussed to have profound
effects on CYP function (56). In
fact, some studies put CYP function upstream of Na+- and
K+ channels demonstrating that CYP epoxigenase activity
mediates arachidonic acid-triggered sodium and potassium channel
inhibition (57,58). Furthermore, epoxyeicosatrienoic
acids, which are endothelium-derived CYP metabolites of arachidonic
acid, relax vascular smooth muscle by calcium-activated potassium
channel activation (59) and
CYP-omega-hydroxylation-dependent metabolites inhibit 10 pS
chloride channel (60).
Directionally migrating cancer cells regulate their cell volume by
Cl− channels at their leading edge and inhibiting ClC-3
channel by shRNA affects glioma cell migration (61).
Nevertheless, fenofibrate-mediated inhibition of
K+ channels and inhibition of CYP1A1 in MDA-MB231 cells
could have been entirely separate events. The comparison of MCF-7
(estrogen-dependent cell line)- and MDA-MB231 (triple-negative cell
line)-based 3D models revealed differences in the drug activities
on the inhibition of CCID formation, which can only be explained by
their impact on the respective breast cancer cells but not on LECs.
The mechanisms behind these differences await to be elucidated.
Besides the usage of a few anti-angiogenic
therapeutics, no anti-intravasative drugs are currently available
and none of the here tested drugs have been designed to inhibit
malignant tumour progression. However, pharmaceutically used drugs
exhibit side effects and these unintended activities might inhibit
steps of the metastatic cascade. Importantly, all tested drugs are
directly available to patients and the used concentrations can be
reached in humans. Our new concept may lead to an improved therapy
of metastasis in cancer patients.
Acknowledgments
We wish to thank Toni Jäger for preparing the
figures. C.H.N was supported by technology grant (TSA Doktorat)
financed by the Austria Federal Ministry of Science and Research
(BMFW) in frame of Asea Uninet, A.F. was supported by DIKTI-OeAD
fellowship, and J.H. by State Scholarship Fund of China Scholarship
Council, the National Natural Science Foundation of China (no.
81202853) and the Natural Science Foundation of Jiangsu Province
(no. BK2012444). The work was further supported by a grant of the
Austrian Science Fund (FWF: S10704-B13) to V.M.D.
References
|
1
|
Sobin LH, Gospodarowicz MK and Wittekind
C: TNM Classification of Malignant Tumours (UICC International
Union Against Cancer). 7th edition. Wiley-Blackwell; New York:
2009
|
|
2
|
Vonach C, Viola K, Giessrigl B, Huttary N,
Raab I, Kalt R, Krieger S, Vo TP, Madlener S, Bauer S, et al: NF-κB
mediates the 12(S)-HETE-induced endothelial to mesenchymal
transition of lymphendothelial cells during the intravasation of
breast carcinoma cells. Br J Cancer. 105:263–271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Viola K, Kopf S, Huttary N, Vonach C,
Kretschy N, Teichmann M, Giessrigl B, Raab I, Stary S, Krieger S,
et al: Bay11-7082 inhibits the disintegration of the
lymphendothelial barrier triggered by MCF-7 breast cancer
spheroids; the role of ICAM-1 and adhesion. Br J Cancer.
108:564–569. 2013. View Article : Google Scholar :
|
|
4
|
Nguyen CH, Senfter D, Basilio J, Holzner
S, Stadler S, Krieger S, Huttary N, Milovanovic D, Viola K,
Simonitsch-Klupp I, et al: NF-κB contributes to MMP1 expression in
breast cancer spheroids causing paracrine PAR1 activation and
disintegrations in the lymph endothelial barrier in vitro.
Oncotarget. 6:39262–39275. 2015.PubMed/NCBI
|
|
5
|
Piwowarczyk K, Wybieralska E, Baran J,
Borowczyk J, Rybak P, Kosińska M, Włodarczyk AJ, Michalik M,
Siedlar M, Madeja Z, et al: Fenofibrate enhances barrier function
of endothelial continuum within the metastatic niche of prostate
cancer cells. Expert Opin Ther Targets. 19:163–176. 2015.
View Article : Google Scholar
|
|
6
|
Madlener S, Saiko P, Vonach C, Viola K,
Huttary N, Stark N, Popescu R, Gridling M, Vo NT, Herbacek I, et
al: Multifactorial anticancer effects of digalloyl-resveratrol
encompass apoptosis, cell-cycle arrest, and inhibition of
lymphendothelial gap formation in vitro. Br J Cancer.
102:1361–1370. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kerjaschki D, Bago-Horvath Z, Rudas M,
Sexl V, Schneckenleithner C, Wolbank S, Bartel G, Krieger S, Kalt
R, Hantusch B, et al: Lipoxygenase mediates invasion of
intrametastatic lymphatic vessels and propagates lymph node
metastasis of human mammary carcinoma xenografts in mouse. J Clin
Invest. 121:2000–2012. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nguyen CH, Brenner S, Huttary N, Li Y,
Atanasov AG, Dirsch VM, Holzner S, Stadler S, Riha J, Krieger S, et
al: 12(S)-HETE increases intracellular Ca(2+) in lymph-endothelial
cells disrupting their barrier function in vitro; stabilization by
clinical drugs impairing calcium supply. Cancer Lett. 380:174–183.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nguyen CH, Stadler S, Brenner S, Huttary
N, Krieger S, Jäger W, Dolznig H and Krupitza G: Cancer
cell-derived 12(S)-HETE signals via 12-HETE receptor, RHO, ROCK and
MLC2 to induce lymph endothelial barrier breaching. Br J Cancer.
115:364–370. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Litan A and Langhans SA: Cancer as a
channelopathy: Ion channels and pumps in tumor development and
progression. Front Cell Neurosci. 9:862015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Comes N, Serrano-Albarrás A, Capera J,
Serrano-Novillo C, Condom E, Ramón Y, Cajal S, Ferreres JC and
Felipe A: Involvement of potassium channels in the progression of
cancer to a more malignant phenotype. Biochim Biophys Acta.
1848.2477–2492. 2015.
|
|
12
|
Northcott PA, Dubuc AM, Pfister S and
Taylor MD: Molecular subgroups of medulloblastoma. Expert Rev
Neurother. 12:871–884. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Su C, Shi A, Cao G, Tao T, Chen R, Hu Z,
Shen Z, Tao H, Cao B, Hu D, et al: Fenofibrate suppressed
proliferation and migration of human neuroblastoma cells via
oxidative stress dependent of TXNIP upregulation. Biochem Biophys
Res Commun. 460:983–988. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Goetze S, Eilers F, Bungenstock A,
Kintscher U, Stawowy P, Blaschke F, Graf K, Law RE, Fleck E and
Gräfe M: PPAR activators inhibit endothelial cell migration by
targeting Akt. Biochem Biophys Res Commun. 293:1431–1437. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wolle D, Lee SJ, Li Z, Litan A, Barwe SP
and Langhans SA: Inhibition of epidermal growth factor signaling by
the cardiac glycoside ouabain in medulloblastoma. Cancer Med.
3:1146–1158. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hu X, Wei L, Taylor TM, Wei J, Zhou X,
Wang JA and Yu SP: Hypoxic preconditioning enhances bone marrow
mesenchymal stem cell migration via Kv2.1 channel and FAK
activation. Am J Physiol Cell Physiol. 301:C362–C372. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wei JF, Wei L, Zhou X, Lu ZY, Francis K,
Hu XY, Liu Y, Xiong WC, Zhang X, Banik NL, et al: Formation of
Kv2.1-FAK complex as a mechanism of FAK activation, cell
polarization and enhanced motility. J Cell Physiol. 217:544–557.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bajwa PJ, Alioua A, Lee JW, Straus DS,
Toro L and Lytle C: Fenofibrate inhibits intestinal Cl- secretion
by blocking baso-lateral KCNQ1 K+ channels. Am J Physiol
Gastrointest Liver Physiol. 293:G1288–G1299. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shimomura K, Shimizu H, Ikeda M, Okada S,
Kakei M, Matsumoto S and Mori M: Fenofibrate, troglitazone, and
15-deoxy-Delta12,14-prostaglandin J2 close KATP channels and induce
insulin secretion. J Pharmacol Exp Ther. 310:1273–1280. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shimomura K, Ikeda M, Ariyama Y, Proks P,
Shimomura Y, Mori M and Matsumoto S: Effect of peroxisome
proliferator-activated receptor alpha ligand fenofibrate on K(v)
channels in the insulin-secreting cell line HIT-T15. Gen Physiol
Biophys. 25:455–460. 2006.
|
|
21
|
De Ciuceis C, Amiri F, Iglarz M, Cohn JS,
Touyz RM and Schiffrin EL: Synergistic vascular protective effects
of combined low doses of PPARalpha and PPARgamma activators in
angiotensin II-induced hypertension in rats. Br J Pharmacol.
151:45–53. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Grabacka M, Plonka PM, Urbanska K and
Reiss K: Peroxisome proliferator-activated receptor alpha
activation decreases metastatic potential of melanoma cells in
vitro via down-regulation of Akt. Clin Cancer Res. 12:3028–3036.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang N, Chu ES, Zhang J, Li X, Liang Q,
Chen J, Chen M, Teoh N, Farrell G, Sung JJ, et al: Peroxisome
proliferator activated receptor alpha inhibits hepatocarcinogenesis
through mediating NF-κB signaling pathway. Oncotarget. 5:8330–8340.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rival Y, Benéteau N, Taillandier T, Pezet
M, Dupont-Passelaigue E, Patoiseau JF, Junquéro D, Colpaert FC and
Delhon A: PPARalpha and PPARdelta activators inhibit
cytokine-induced nuclear translocation of NF-kappaB and expression
of VCAM-1 in EAhy926 endothelial cells. Eur J Pharmacol.
435:143–151. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Han D, Wei W, Chen X, Zhang Y, Wang Y,
Zhang J, Wang X, Yu T, Hu Q, Liu N, et al: NF-κB/RelA-PKM2 mediates
inhibition of glycolysis by fenofibrate in glioblastoma cells.
Oncotarget. 6:26119–26128. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chandran K, Goswami S and Sharma-Walia N:
Implications of a peroxisome proliferator-activated receptor alpha
(PPARα) ligand clofibrate in breast cancer. Oncotarget.
7:15577–15599. 2016.
|
|
27
|
Abu Aboud O, Wettersten HI and Weiss RH:
Inhibition of PPARα induces cell cycle arrest and apoptosis, and
synergizes with glycolysis inhibition in kidney cancer cells. PLoS
One. 8:e711152013. View Article : Google Scholar
|
|
28
|
Huang WP, Yin WH, Chen JW, Jen HL, Young
MS and Lin SJ: Fenofibrate attenuates endothelial monocyte adhesion
in chronic heart failure: An in vitro study. Eur J Clin Invest.
39:775–783. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tsai SC, Tsai MH, Chiu CF, Lu CC, Kuo SC,
Chang NW and Yang JS: AMPK-dependent signaling modulates the
suppression of invasion and migration by fenofibrate in CAL 27 oral
cancer cells through NF-κB pathway. Environ Toxicol. 31:866–876.
2016. View Article : Google Scholar
|
|
30
|
Koh KK, Oh PC, Sakuma I, Lee Y, Han SH and
Shin EK: Vascular and metabolic effects of omega-3 fatty acids
combined with fenofibrate in patients with hypertriglyceridemia.
Int J Cardiol. 221:342–346. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Teichmann M, Kretschy N, Kopf S,
Jarukamjorn K, Atanasov AG, Viola K, Giessrigl B, Saiko P, Szekeres
T, Mikulits W, et al: Inhibition of tumour spheroid-induced
prometastatic intravasation gates in the lymph endothelial cell
barrier by carbamazepine: Drug testing in a 3D model. Arch Toxicol.
88:691–699. 2014.
|
|
32
|
Rozema E, Atanasov AG, Fakhrudin N,
Singhuber J, Namduang U, Heiss EH, Reznicek G, Huck CW, Bonn GK,
Dirsch VM, et al: Selected extracts of Chinese herbal medicines:
Their effect on NF-κB, PPARα and PPARγ and the respective bioactive
compounds. Evid Based Complement Alternat Med. 2012:9830232012.
View Article : Google Scholar
|
|
33
|
Gu J, Tamura M, Pankov R, Danen EH, Takino
T, Matsumoto K and Yamada KM: Shc and FAK differentially regulate
cell motility and directionality modulated by PTEN. J Cell Biol.
146:389–403. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang HB, Dembo M, Hanks SK and Wang Y:
Focal adhesion kinase is involved in mechanosensing during
fibroblast migration. Proc Natl Acad Sci USA. 98:11295–11300. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cherniavsky Lev M, Karlish SJ and Garty H:
Cardiac glycosides induced toxicity in human cells expressing α1-,
α2-, or α3-isoforms of Na-K-ATPase. Am J Physiol Cell Physiol.
309:C126–C135. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rodrigues-Mascarenhas S, Da Silva de
Oliveira A, Amoedo ND, Affonso-Mitidieri OR, Rumjanek FD and
Rumjanek VM: Modulation of the immune system by ouabain. Ann NY
Acad Sci. 1153:153–163. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pongrakhananon V, Chunhacha P and
Chanvorachote P: Ouabain suppresses the migratory behavior of lung
cancer cells. PLoS One. 8:e686232013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lin SY, Chang HH, Lai YH, Lin CH, Chen MH,
Chang GC, Tsai MF and Chen JJ: Digoxin suppresses tumor malignancy
through inhibiting multiple Src-related signaling pathways in
non-small cell lung cancer. PLoS One. 10:e01233052015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Walker BD, Singleton CB, Bursill JA, Wyse
KR, Valenzuela SM, Qiu MR, Breit SN and Campbell TJ: Inhibition of
the human ether-a-go-go-related gene (HERG) potassium channel by
cisapride: Affinity for open and inactivated states. Br J
Pharmacol. 128:444–450. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kim KS, Lee HA, Cha SW, Kwon MS and Kim
EJ: Blockade of hERG K(+) channel by antimalarial drug, primaquine.
Arch Pharm Res. 33:769–773. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
McFerrin MB and Sontheimer H: A role for
ion channels in glioma cell invasion. Neuron Glia Biol. 2:39–49.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cruickshank SF, Baxter LM and Drummond RM:
The Cl(−) channel blocker niflumic acid releases Ca(2+) from an
intracellular store in rat pulmonary artery smooth muscle cells. Br
J Pharmacol. 140:1442–1450. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sahdeo S, Scott BD, McMackin MZ, Jasoliya
M, Brown B, Wulff H, Perlman SL, Pook MA and Cortopassi GA:
Dyclonine rescues frataxin deficiency in animal models and buccal
cells of patients with Friedreich's ataxia. Hum Mol Genet.
23:6848–6862. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Flaim SF, Ratz PH, Swigart SC and Gleason
MM: Bepridil hydrochloride alters potential-dependent and
receptor-operated calcium channels in vascular smooth muscle of
rabbit aorta. J Pharmacol Exp Ther. 234:63–71. 1985.PubMed/NCBI
|
|
45
|
Nguyen CH, Brenner S, Huttary N, Atanasov
AG, Dirsch VM, Chatuphonprasert W, Holzner S, Stadler S, Riha J,
Krieger S, et al: AHR/CYP1A1 interplay triggers lymphatic barrier
breaching in breast cancer spheroids by inducing 12(S)-HETE
synthesis. Hum Mol Genet. Sep 27–2016.Epub ahead of print.
View Article : Google Scholar
|
|
46
|
Burgazli KM, Venker CJ, Mericliler M,
Atmaca N, Parahuleva M and Erdogan A: Importance of large
conductance calcium-activated potassium channels (BKCa) in
interleukin-1b-induced adhesion of monocytes to endothelial cells.
Eur Rev Med Pharmacol Sci. 18:646–656. 2014.PubMed/NCBI
|
|
47
|
McPhee JC, Dang YL, Davidson N and Lester
HA: Evidence for a functional interaction between integrins and G
protein-activated inward rectifier K+ channels. J Biol Chem.
273:34696–34702. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Camerino DC, Tricarico D and Desaphy JF:
Ion channel pharmacology. Neurotherapeutics. 4:184–198. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Driffort V, Gillet L, Bon E,
Marionneau-Lambot S, Oullier T, Joulin V, Collin C, Pagès JC,
Jourdan ML, Chevalier S, et al: Ranolazine inhibits NaV1.5-mediated
breast cancer cell invasiveness and lung colonization. Mol Cancer.
13:2642014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Brisson L, Driffort V, Benoist L, Poet M,
Counillon L, Antelmi E, Rubino R, Besson P, Labbal F, Chevalier S,
et al: NaV1.5 Na+ channels allosterically regulate the
NHE-1 exchanger and promote the activity of breast cancer cell
invadopodia. J Cell Sci. 126:4835–4842. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Stadler S, Nguyen CH, Schachner H,
Milovanovic D, Holzner S, Brenner S, Eichsteininger J, Stadler M,
Senfter D, Krenn L, et al: Colon cancer cell-derived 12(S)-HETE
induces the retraction of cancer-associated fibroblast via MLC2,
RHO/ROCK and Ca2+ signalling. Cell Mol Life Sci. Dec
24–2016.Epub ahead of print.
|
|
52
|
Park JW, Reed JR, Brignac-Huber LM and
Backes WL: Cytochrome P450 system proteins reside in different
regions of the endoplasmic reticulum. Biochem J. 464:241–249. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Park JW, Reed JR and Backes WL: The
Localization of cytochrome P450s CYP1A1 and CYP1A2 into different
lipid microdomains is governed by their N-terminal and internal
protein regions. J Biol Chem. 290:29449–29460. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Brignac-Huber L, Reed JR and Backes WL:
Organization of NADPH-cytochrome P450 reductase and CYP1A2 in the
endoplasmic reticulum - microdomain localization affects
mono-oxygenase function. Mol Pharmacol. 79:549–557. 2011.
View Article : Google Scholar :
|
|
55
|
Berka K, Paloncýová M, Anzenbacher P and
Otyepka M: Behavior of human cytochromes P450 on lipid membranes. J
Phys Chem B. 117:11556–11564. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Scott EE, Wolf CR, Otyepka M, Humphreys
SC, Reed JR, Henderson CJ, McLaughlin LA, Paloncýová M, Navrátilová
V, Berka K, et al: The role of protein-protein and protein-membrane
interactions on P450 function. Drug Metab Dispos. 44:576–590. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wei Y, Lin DH, Kemp R, Yaddanapudi GS,
Nasjletti A, Falck JR and Wang WH: Arachidonic acid inhibits
epithelial Na channel via cytochrome P450 (CYP)
epoxygenase-dependent metabolic pathways. J Gen Physiol.
124:719–727. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang Z, Wei Y, Falck JR, Atcha KR and Wang
WH: Arachidonic acid inhibits basolateral K channels in the
cortical collecting duct via cytochrome P-450 epoxygenase-dependent
metabolic pathways. Am J Physiol Renal Physiol. 294:F1441–F1447.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Campbell WB, Holmes BB, Falck JR,
Capdevila JH and Gauthier KM: Regulation of potassium channels in
coronary smooth muscle by adenoviral expression of cytochrome P-450
epoxygenase. Am J Physiol Heart Circ Physiol. 290:H64–H71. 2006.
View Article : Google Scholar
|
|
60
|
Gu RM, Yang L, Zhang Y, Wang L, Kong S,
Zhang C, Zhai Y, Wang M, Wu P, Liu L, et al:
CYP-omega-hydroxylation-dependent metabolites of arachidonic acid
inhibit the basolateral 10 pS chloride channel in the rat thick
ascending limb. Kidney Int. 76:849–856. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Cuddapah VA and Sontheimer H: Molecular
interaction and functional regulation of ClC-3 by
Ca2+/calmodulin-dependent protein kinase II (CaMKII) in
human malignant glioma. J Biol Chem. 285:11188–11196. 2010.
View Article : Google Scholar : PubMed/NCBI
|