Introduction
Fibrosarcoma is a malignant and highly metastatic
mesenchymal tumor derived from fibrous connective tissue. The tumor
typically grows as a solitary, longstanding mass or swelling in the
subcutaneous tissue, which can be found in both children and adults
(1). The poor prognosis of
fibrosarcoma can be attributed to both aggressive characteristic of
this cancer and the lack of efficacy in current therapies to
prevent, counteract or slow tumor progression, especially as a
result of drug resistance development. To overcome the
chemoresistance to current therapies and improve patient outcome,
novel treatment agents are urgently needed to target mechanisms
where fibrosarcoma cell grow and survive.
Microtubules, composed by α- and β-tubulin
heterodimers, are pivotal elements of cytoskeleton of eukaryotic
cells and involved in many fundamental processes including
regulation of cell motility, maintenance of cell shape,
localization of organelle, transportation of intracellular material
and coordination of cell division (2–4).
Since specific inhibition of microtubule dynamic can disrupt
chromosome segregation and consequently block mitosis, more and
more investigators have focused on microtubule target for new
anticancer drug development (5,6).
There are some well-characterized binding sites for ligands in
tubulin, including taxanes, laulimalide, peloruside A, vinca
alkaloids, and colchicine sites (3,5).
These agents can be divided into two groups:
microtubule-destabilizing agents and microtubule-stabilizing agents
(3,5,7). In
addition, there are also some compounds targeting the actin,
another critical element of cytoskeleton in eukaryotic cells
(8,9). Although many compounds targeting
microtubules or actin have been used successfully in the therapy of
many cancers, the effectiveness might be weakened by multidrug
resistance, tubulin isotype variation, tubulin mutations, and
microtubule regulatory protein alteration (3,10).
As a result, development of new microtubule inhibitors is still
needed to increase the efficacy and overcome drug resistance.
Combretastatin A-4 (CA-4), a phenolic cis-stilbene
natural product, has been reported as one of the most potent
antitumor agents of combretastatins (11,12).
However, the application of CA-4 as an anticancer drug is limited
by its low bioavailability, as it tends to isomerize to the
thermodynamically more stable and inactive trans-isomer, which
decreases its half-life and reduces its activity. Despite the
limited bioavailability, the relatively simple structure and the
high affinity to the tubulin binding site render CA-4 as an
attractive lead compound for the development of new anticancer
agents by our group and many other groups (13–17).
Our group previously reported that
5-(3-amino-4-methoxyphenyl)-4-(3,4,5-trimethoxyphenyl)-3H-1,2-dithiole-3-one
(4-d), a tubulin inhibitor designed through the replacement of the
double bond with a heterocyclic moiety to improve stability of
CA-4, showed potent anti-proliferative activity (18). Based on these results, the purpose
of our current study was to synthesize a series of analogues of 4-d
to further elucidate the structure activity relationship (SAR) and
discover promising compounds. Among these new synthetic compounds,
5-(furan-2-yl)-4-(3,4,5-trimethoxyphenyl)-3H-1,2-dithiol-3-one
oxime (6f) exhibited significant anticancer activity against many
human cancer cell lines. As a result, the cytotoxic efficacy and
related molecular mechanisms underlying the anticancer activity of
6f were further evaluated in fibrosarcoma HT-1080 cells, a human
tumorigenic cell line commonly used to study the effects of new
anticancer drugs. Our study proved that 6f destroyed microtubule
polymerisation, induced G2/M arrest and subsequent
caspase-dependent apoptosis in HT-1080 cells.
Materials and methods
Synthysis of target compounds
The synthetic methods for the target compounds are
summarized as follows (Fig. 1).
The synthetic route outlined in Fig.
1 allowed rapid SAR development to explore the possibility of
replacing the carboxylic acid through parallel synthesis in the
first stage of the synthetic sequence. Acylation of methyl
2-(3,4,5-trimethoxyphenyl) acetate (1) by carboxylic acids 2a-f under basic
conditions in DMF provided intermediates 3a-f. The ring closure of
3a-f using Lawesson's reagent in refluxing toluene led to
4,5-diaryl-1,2-dithiole-3-thiones 4a-f. These products were
synthesized using potassium permanganate to yield the
4,5-diaryl-1,2-dithiole-3-ones 5a-f. In addition, 6a-f were
prepared by the treatment of 4a-f with hydroxylamine hydrochloride
in refluxing ethanol.
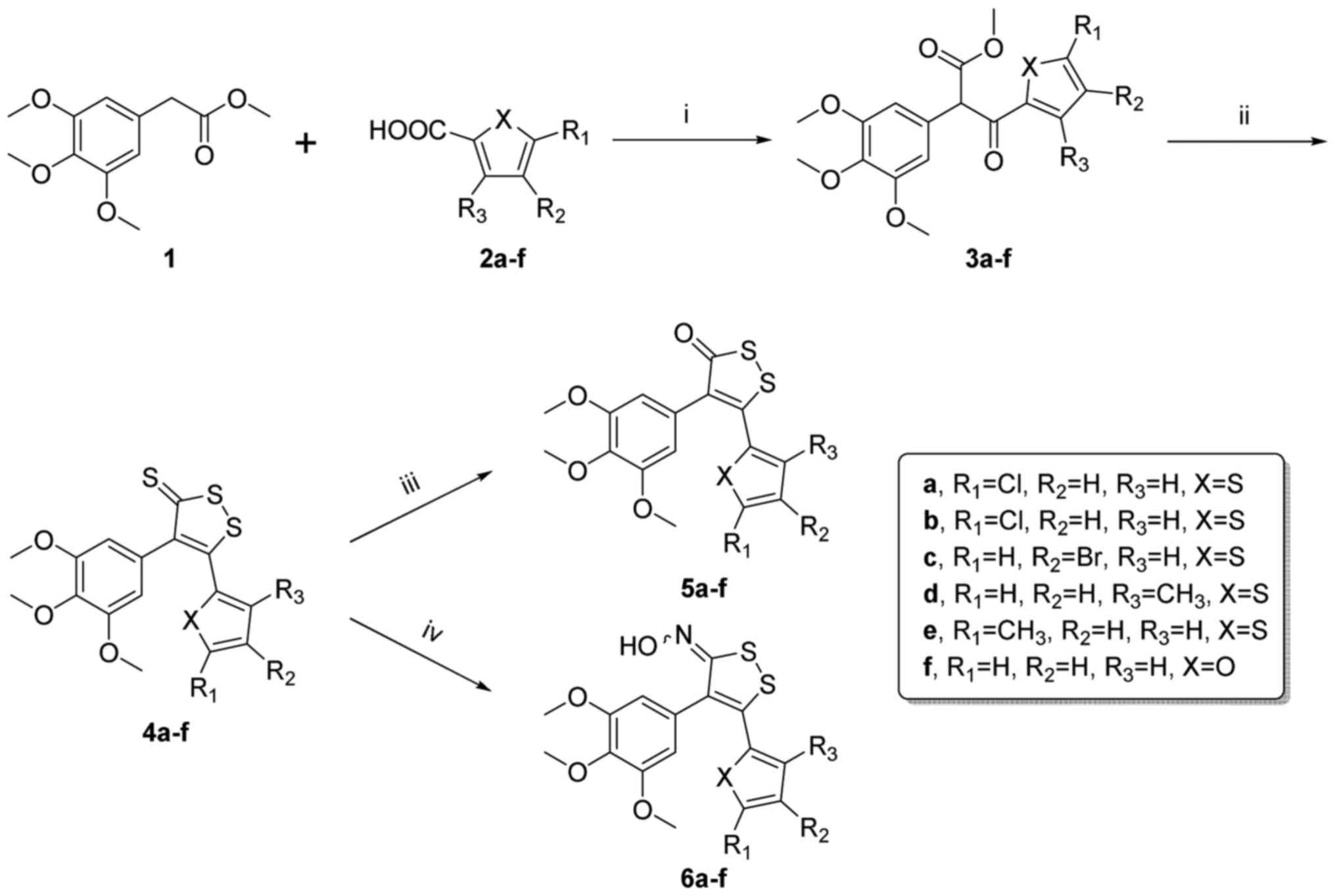 | Figure 1The synthesis process of the new
compounds. Reagents and conditions: (i) NaH, CDI, DMF, 0°C, 4 h;
(ii) Lawesson's reagent, toluene, reflux, 4 h; (iii) KMno4,
acetone, rt, 12 h; (iv) NH2OH·HCl, NaOAc, EtOH, reflux,
12 h. |
Cell cultures
Human cervical cancer cell line HeLa, human breast
cancer cell line MCF-7, human non-small cell lung cancer cell line
A549, human liver carcinoma cell line HepG-2, human oral squamous
cell carcinoma cell line KB, human gastric adenocarcinoma cell line
SGC-7901 and human fibrosarcoma cell line HT-1080 were obtained
from American Type Culture Collection (ATCC). Human normal liver
cell line HL-7702 was obtained from Boster (Wuhan, China). These
cells were cultured in RPMI-1640 (Gibco, Grand Island, NY, USA)
containing 10% fetal bovine serum (FBS) (TBD Biotechnology,
Tianjin, China), 100 U/ml streptomycin and 100 U/ml penicillin at
37°C in humidified atmosphere with 5% CO2.
MTT assay
The in vitro antiproliferative activities
were determined by MTT assay. Briefly, cells were seeded into
96-well plates at a density of 3–5×103/well. At 24 h,
triplicate wells were treated with media, CA-4 or different
concentrations of new designed compounds for 24, 48 or 72 h at 37°C
in 5% CO2. Then, the drug containing medium was removed
and replaced by 100 µl fresh medium with 5 mg/ml MTT
solution (Ameresco, Inc., Framingham, MA, USA). After 3 h of
incubation, the OD490 was tested by a microplate reader
(MK3, Thermo fisher Scientific, inc., Hanau, Germany). The
percentage of cell growth inhibition was calculated according to a
previous study (15).
Observation of cellular morphology
HT-1080 cells were seeded onto 24-well plates at a
density of 1×105 cells per well. After incubation with
6f for 24 h at 37°C, cellular morphology was photographed by
inverted microscope (Motic AE2000).
Acridine orange (AO) staining
The nuclear morphology was studied by the
fluorescent DNA-binding dye AO. After treatment with 6f or CA-4 for
24 h, the cells were stained with AO (10 µg/ml) (Sigma, St.
Louis, MO, USA) for 10 min, then the nuclear morphology was
photographed under a fluorescence microscope (Olympus, Tokyo,
Japan).
Molecular modeling studies
The molecular modelling studies were performed by
Accelrys Discovery Studio 3.0. The crystal structure of tubulin
complexed with DAMA-colchicine (PDB: 1SA0) was from the RCSB
Protein Data Bank (http://www.rcsb.org/pdb). The protein protocol was
prepared via several operations, including the standardization of
atom names, insertion of missing atoms in residues and removal of
alternate conformations, insertion of missing loop regions based on
SEQRES data, optimization of short and medium sized loop regions
with the Looper Algorithm, minimization of remaining loop regions,
calculation of pK, and protonation of the structure. The receptor
model was then typed with the CHARMm force field, and a binding
sphere with radius of 9.0 Å was defined with the original ligand
(DAMA-colchicine) as the binding site. The CA-4 (1a) and 6f were
drawn with Chemdraw and fully minimized using the CHARMm force
field. finally, they were docked into the binding site using the
CDOCKER protocol with the default settings.
Tubulin polymerisation assay
The effect of 6f on microtubule polymerisation was
tested using a tubulin polymerisation assay kit (Cytoskeleton cat.
# BK011P). Briefly, tubulin was re-suspended in ice cold G-PEM
buffer (80 mM PIPES, 2 mM MgCl2, 0.5 mM EGTA, 1 mM GTP,
15% (v/v) glycerol) and added to a 96-well plate containing
different concentration of 6f, CA-4, paclitaxel or vehicle. Samples
were mixed well and tubulin assembly was monitored (emission
wavelength is 420 nm; excitation wavelength is 360 nm) at 1 min
intervals for 90 min at 37°C using a plate reader (FASCalibur, BD
Biosciences, San Diego, CA, USA). IC50 values were
calculated at 20 min by SPSS 19.0.
Immunofluorescence staining
Immunostaining was carried out according to previous
method (15). Briefly, HT-1080
cells were seeded at 1.5×104 per well on a 24-well plate
and treated with media, CA-4 or 6f for 24 h. The primary α-tubulin
antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA)
diluted (1:100) with 2% BSA in PBS was incubated with cells
overnight at 4°C. Then cells were incubated with FITC-conjugated
anti-mouse secondary antibody, diluted (1:100) with 2% BSA in PBS,
for 2 h at 37°C. The nucleus was stained with
4,6-diamino-2-phenolindol dihydrochloride (DAPI) (Beyotime, Haimen,
China) and then, fluorescence microscope (Olympus) was used to
detect the immunofluorescence.
Cell cycle analysis
HT-1080 cells (4×105 cells) were
incubated with media, CA-4 or various concentration of 6f,
respectively, for indicated time. Then, the cells were collected
and fixed in ice-cold 70% ethanol overnight. After that, the cells
were incubated in 500 µl of PBS containing 20 µg/ml
RNase for 30 min at 37°C. Then, the cells were stained with 50
µg/ml propidium iodide (PI) (Sigma) at 4°C in the dark for
30 min. The samples were analyzed by FACScan flow cytometry (Becton
Dickinson, Franklin Lakes, NJ, USA).
Measurement of mitochondrial membrane
potential (MMP)
MMP was tested using fluorescent probe Rhodamine 123
(Sigma) as previously described (19). Briefly, HT-1080 cells were seeded
onto 6-well plates at a density of 4×105 cells per well.
After incubation with 6f for 24, 36 and 48 h, HT-1080 cells were
collected and suspended in 1 ml PBS containing 10 µg/ml
Rh123 and incubated at 37°C for 30 min. The fluorescent intensity
of the cells was analyzed by FACScan flow cytometry (excitation
wavelength is 480 nm and emission wavelength is 525 nm).
Western blot analysis
Protein preparation and immunoblot analyses were
carried out according to previous procedures (15). In brief, the protein content of the
supernatant was determined using a protein assay reagent (Bio-Rad
Laboratories, Hercules, CA, USA). The protein lysates were
separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred to a nitrocellulose
membrane (Millipore Corp., Bedford, MA, USA). The membranes were
probed with primary antibodies (1:300–1:1000) (Santa Cruz
Biotechnology, Inc.) overnight at 4°C and then incubated with a
horseradish peroxidase (HRP)-conjugated secondary antibodies (1:
800) (Santa Cruz Biotechnology, Inc.) for 2 h at 37°C. Proteins
were visualized using enhanced chemiluminescence (Amersham
Biosciences, Amersham, UK). Densitometry analysis was done using
ImageJ 1.44 software.
Statistical analysis
All data were expressed as the mean ± standard
deviation (SD) of three independent experiments unless stated
otherwise. Statistical differences between groups were assessed by
the one-way analysis of variance (ANOVA) followed by LSD t-test or
Dunnett's T3 using SPSS software 16.0 and p-values <0.05 were
considered to indicate statistically significant differences.
Results
In vitro anti-proliferative activity of
target compounds
In order to evaluate the anti-proliferative activity
of various
5-aryl-4-(3,4,5-trimethoxyphenyl)-3H-1,2-dithiol-3-ones and
oximes to cancer cells, the target compounds 4a-f, 5a-f, 6a-f and
reference drug adriamycin (ADM) were screened against three human
cancer cell lines (SGC-7901, A549 and HT-1080 cells) using MTT
assay (Table I). Among these
compounds, 6f exhibited better activity than other compounds. As a
result, we explored its anticancer activity and the underlying
mechanisms in the following experiments.
 | Table IIn vitro anti-proliferative
activities of the target compounds as measured with MTT test in
three human cancer cell lines (mean ± SD, n=3). |
Table I
In vitro anti-proliferative
activities of the target compounds as measured with MTT test in
three human cancer cell lines (mean ± SD, n=3).
| Compound | IC50
(µM) ± SD
|
|---|
| SGC-7901 | A549 | HT-1080 |
|---|
| 4a | >50 | >50 | >50 |
| 4b | 16.72±1.21 | 30.73±1.98 | 10.06±0.86 |
| 4c | >50 | >50 | >50 |
| 4d | 15.25±1.42 | 22.82±1.63 | 18.54±1.55 |
| 4e | 7.87±0.97 | 16.13±1.27 | 5.69±0.73 |
| 4f | 45.35±2.51 | 16.07±1.35 | 41.20±2.79 |
| 5a | >50 | >50 | >50 |
| 5b | >50 | >50 | >50 |
| 5c | >50 | >50 | >50 |
| 5d | >50 | >50 | >50 |
| 5e | 9.57±1.13 | >50 | 1.86±0.08 |
| 5f | 31.00±2.25 | 10.96±1.57 | 8.7±1.24 |
| 6a | >50 | >50 | >50 |
| 6b | >50 | 7.43 | >50 |
| 6c | >50 | >50 | >50 |
| 6d | >50 | >50 | >50 |
| 6e | 6.41±1.03 | >50 | 2.87±0.86 |
| 6f |
2.33±0.54 |
2.72±0.50 |
1.96±0.14 |
| ADMa | 0.37±0.05 | 0.55±0.07 | 0.11±0.01 |
6f attenuates the proliferation of
different cancer cells
To clarify the anti-proliferative sensitivity of 6f
to different cell lines including HeLa, MCf-7, A549, HepG-2, KB,
SGC-7901, HT-1080 and HL-7702 cells, the cell viability was
measured by MTT assay after the treatment of cells with various
concentrations of 6f for 72 h. Table
II shows the IC50 of 6f for eight cell lines. All
the tested cancer cell lines except HL-7702 showed susceptibility
to 6f with IC50 values ranging from 1.84±0.52 to
7.98±2.52 µM. Among these cells, HeLa and HT-1080 cells were
the most sensitive cells to 6f treatment. As a result, HT-1080
cells were selected for the following investigations. In addition,
the higher IC50 values of 6f to human original normal
liver cell line HL-7702 indicated its low toxicity to normal
cells.
| Table IIIC50 of 6f in various
human cell lines (mean ± SD, n=3). |
Table II
IC50 of 6f in various
human cell lines (mean ± SD, n=3).
| Cell line | Cell type | IC50
(µM) |
|---|
| HeLa | Human cervical
cancer | 1.84±0.52 |
| MCF-7 | Human breast
carcinoma | 7.98±2.52 |
| A549 | Human non-small
cell lung cancer | 2.72±0.50 |
| HepG-2 | Human liver
carcinoma | 2.24±0.32 |
| KB | Human oral squamous
cell carcinoma | 2.22±0.21 |
| SGC-7901 | Human gastric
adenocarcinoma | 2.33±0.54 |
| HT-1080 | Human
fibrosarcoma | 1.96±0.14 |
| HL-7702 | Human normal liver
cells | 94.32±7.85 |
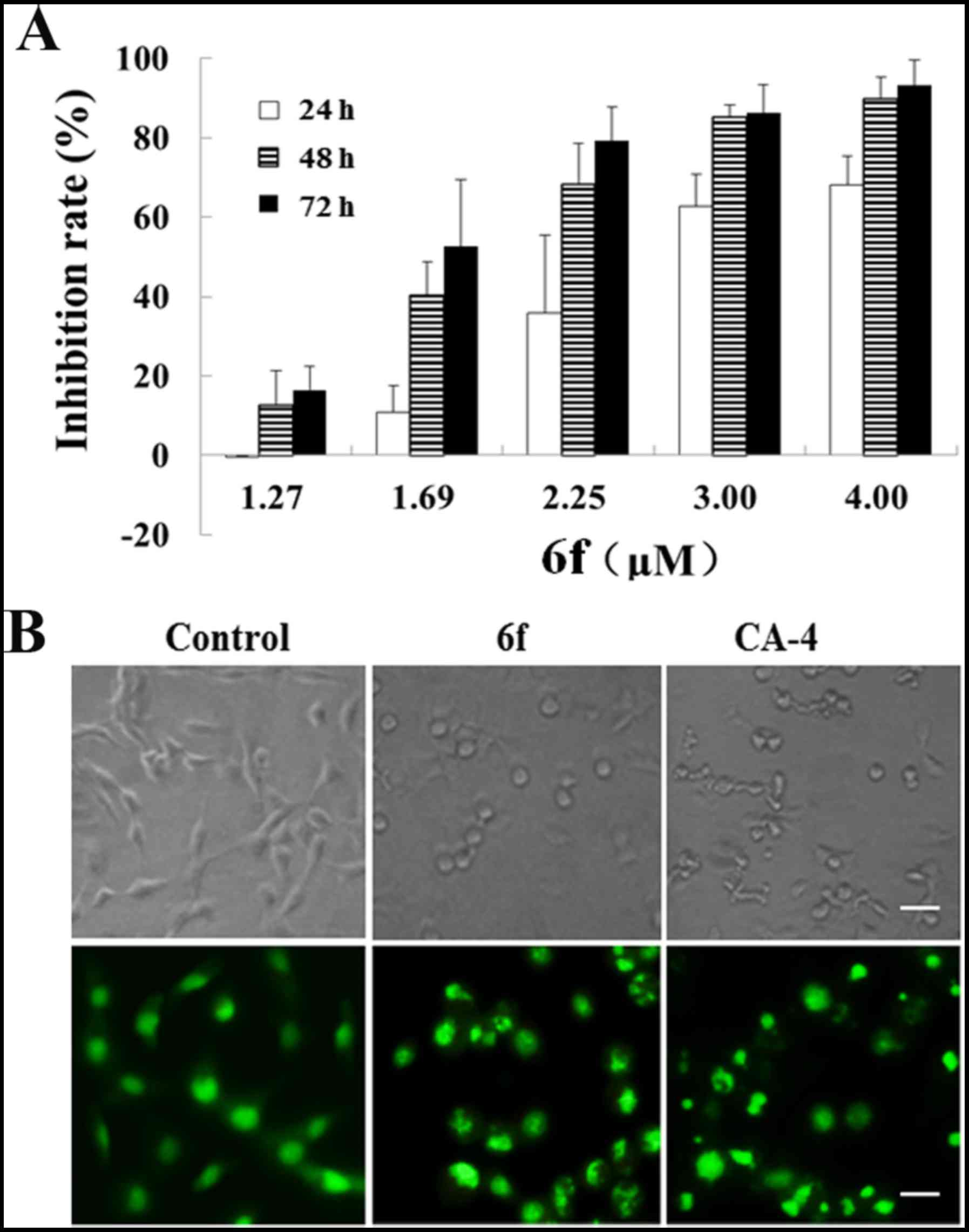
6f inhibits HT-1080 cell proliferation in
a dose- and time-dependent manner
MTT assay was used to further evaluate the
anti-proliferative characteristics of 6f against HT-1080 cells. As
shown in Fig. 2, 6f treatment exhibited potent cytotoxicity
against HT-1080 cells (Fig. 2A)
dose- and time-dependently. Moreover, cellular morphology of
HT-1080 cells was observed by inverted microscope and AO staining.
As shown in the upper part of Fig.
2B, cell adherence and growth in the control group were well
within normal shape, with smooth surface and clear boundary. After
6f treatment for 24 h, the cell morphology became round, cell size
was reduced, cell membrane of most cells was not complete, and
disintegrated fragments were visible. Nuclear morphology with AO
staining in the lower part of Fig.
2B showed that the nuclei in the control group were stained
homogeneously with AO, whereas exposure to 6f caused marked
chromatin condensation and nuclear fragmentation, an indication of
apoptosis.
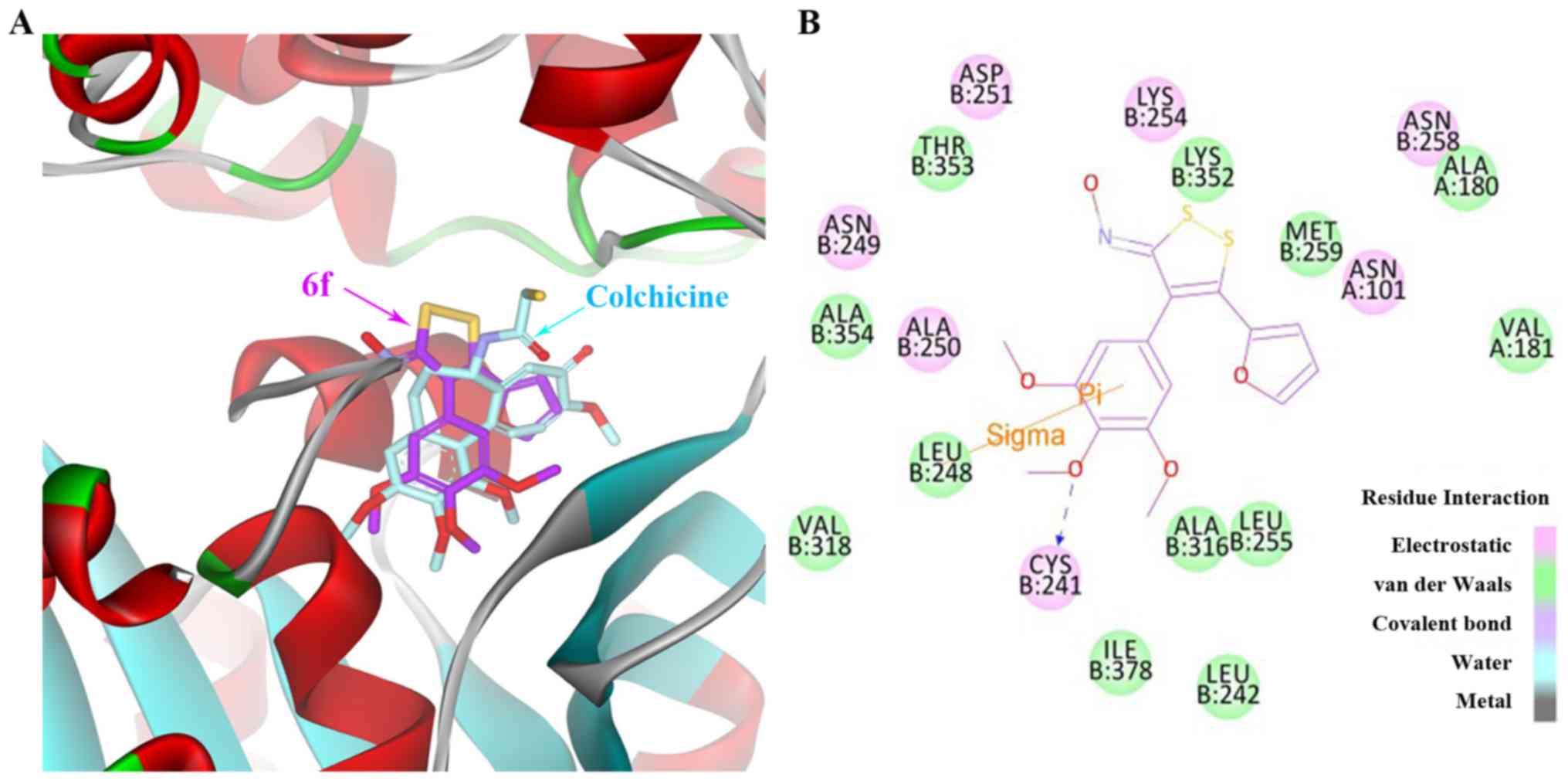
Molecular modelling
To explore the interactions between the newly
synthesized compounds and tubulin, the potential binding mode of
compound 6f at the colchicine site in the tubulin dimer was
investigated (Fig. 3). The X-ray
crystal structure of the DAMA-colchicine-tubulin complex (PDB code
1SA0) was used as the tubulin protein template. In the binding
models shown in Fig. 3A, the
binding orientations of compound 6f (purple) superimposed well with
X-ray crystal structure of the DAMA-colchicine (cyan). In the
binding mode for 6f presented in Fig.
3B, the trimethoxyphenyl ring of the compound fostered σ-π
interactions with Leu-248. Furthermore, there is a potential
hydrogen bond between the trimethoxyphenyl moiety and Cys-241, an
interaction observed with other colchicine site agents. The furan
moiety is locked by Van-der-Waals interactions with Met-259,
Ala-180 and Val-181 (Fig. 3B). In
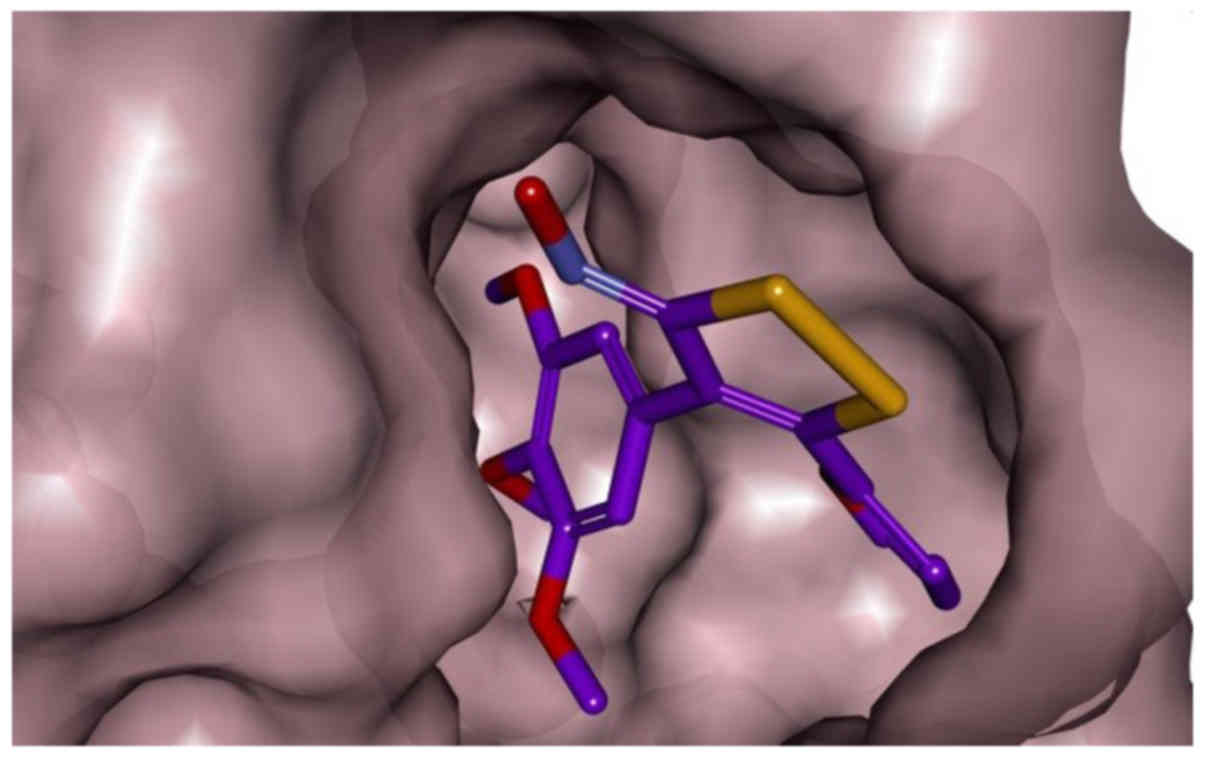
addition, as shown in 3D representation of docked ligand (in red)
into colchicine-binding site of tubulin (Fig. 4), 6f shown as stick model (carbon
colored purple) fitted well with the colchicine-binding pocket of
tubulin in surface (pink) representation.
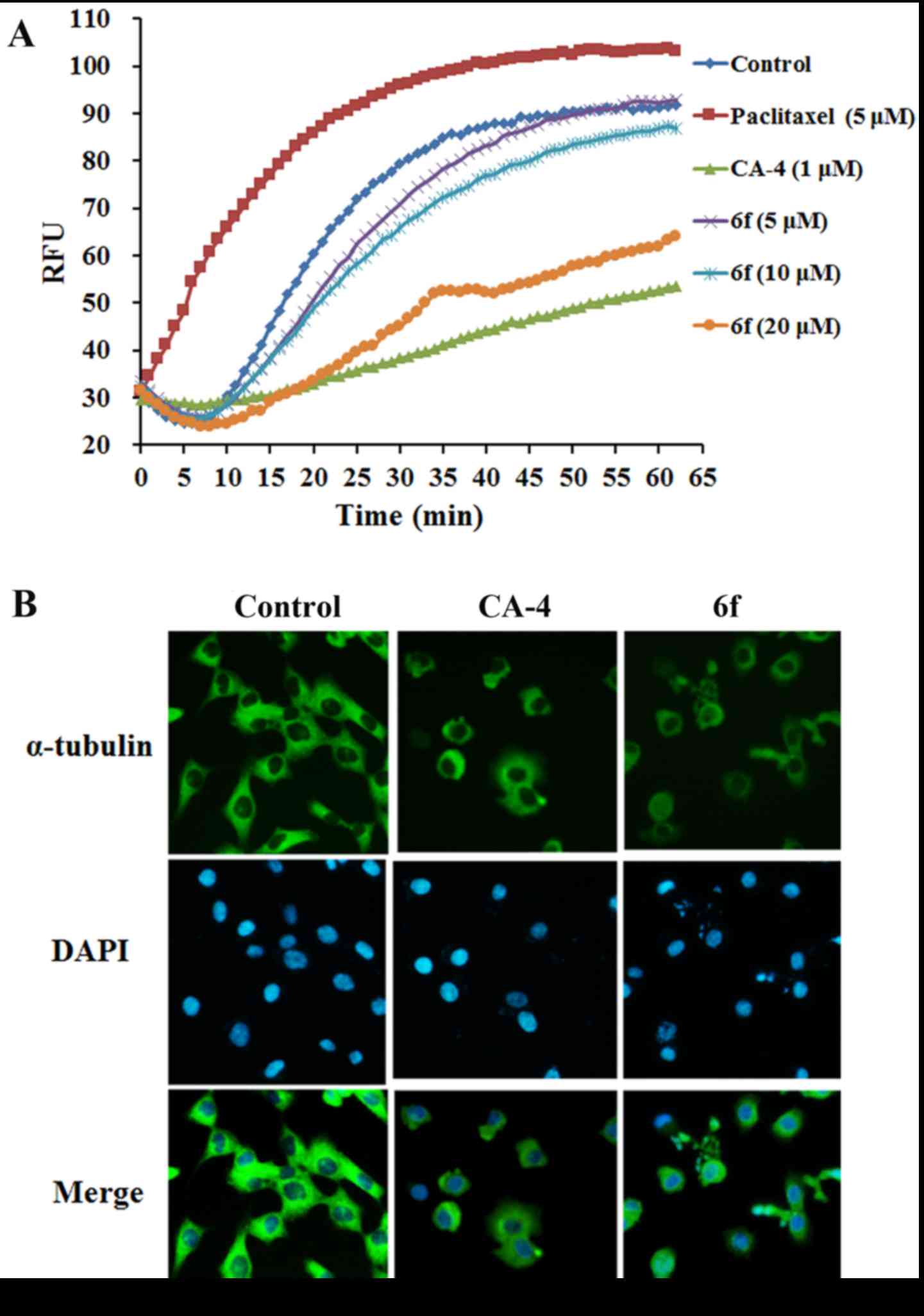
6f disturbs microtubule
polymerization
To further confirm that the anti-proliferative
activity of 6f was related to microtubule, the tubulin
polymerisation was monitored kinetically by a fluorescent plate
reader. As shown in Fig. 5A,
6f (5–20 µM)
dose-dependently inhibited microtubule assembly, which suggested
that the anti-proliferative activity of 6f was associated with
microtubule depolymerisation. Furthermore, the immunofluorescence
analysis using specific antibodies to α-tubulin was applied to
characterize the change of microtubule after 6f treatment. Our data
revealed that the microtubule arrangement in the control group was
intact. However, 6f (2 µM) and CA-4 (8 nM) treated cells
showed cellular microtubule depolymerisation with scattered
microtubule fragments in the cytoplasm of HT-1080 cells (Fig. 5B). These data further proved the
potential tubulin-targeting activity of 6f.
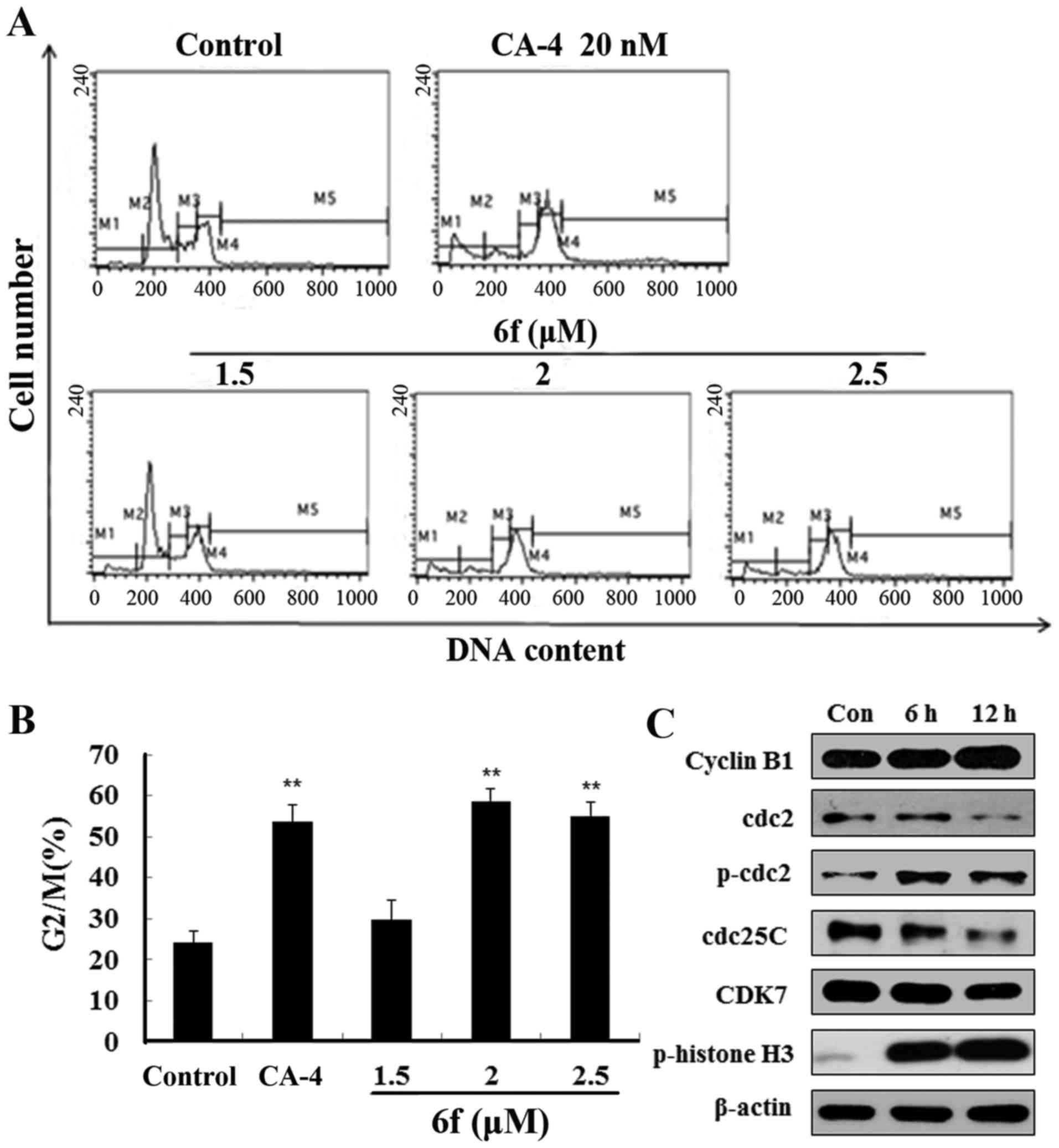
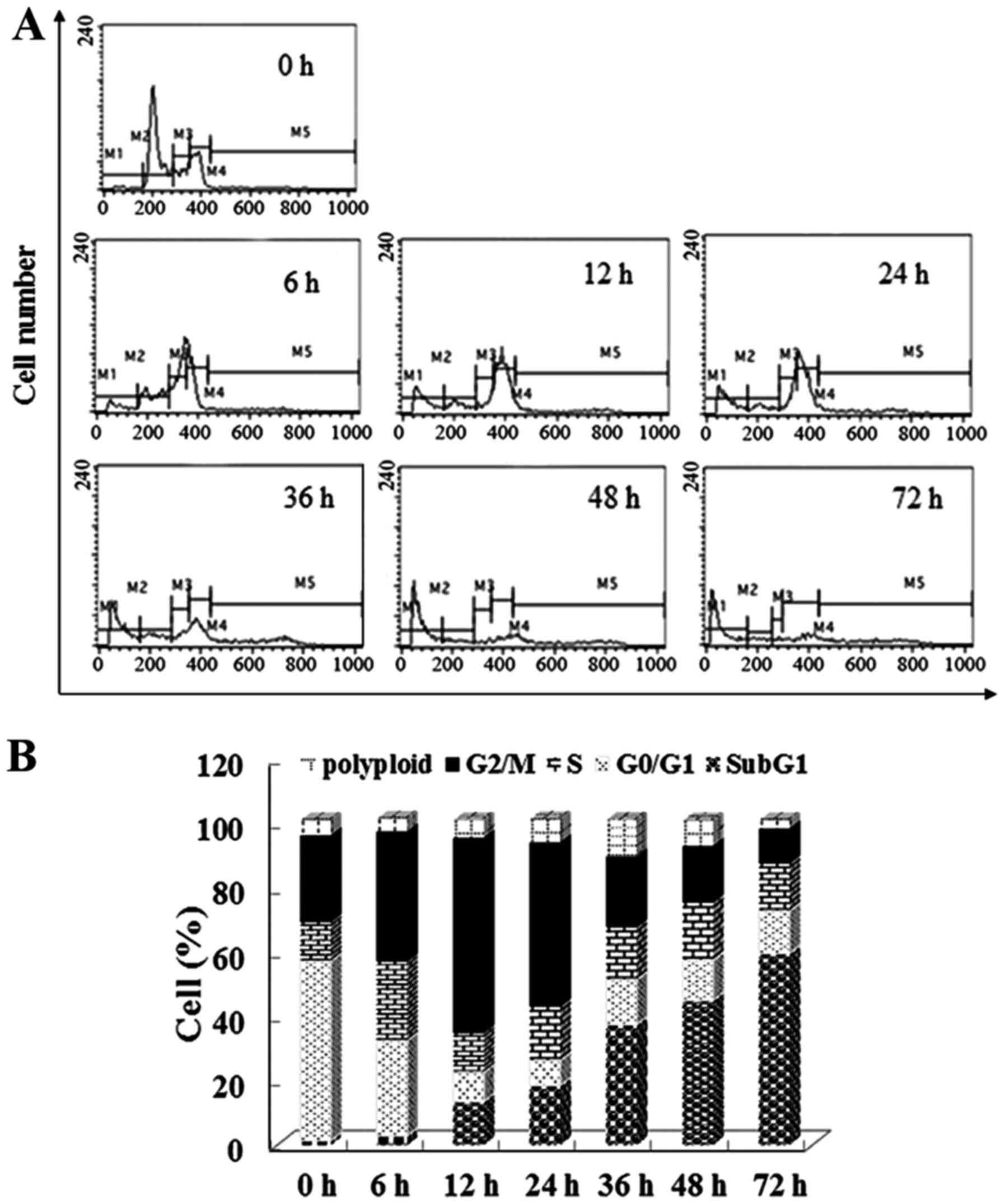
6f induces G2/M phase arrest and changes
cell cycle regulatory protein expression
Since most tubulin-targeting agents induce cell
cycle arrest, cell cycle distribution of 6f-treated cells was next
evaluated by flow cytometry. The results in Fig. 6A and B show that 6f (1.5–2.5
µM) treatment for 12 h dose-dependently increased HT-1080
cell accumulation in the G2/M phase. Moreover, the increased ratio
of G2/M phase was concomitant with the decreased ratio of G0/G1
phase and S phase, while the ratio of polyploidy did not change
significantly. These results suggested that 6f induced HT-1080 cell
arrest in G2/M phase after treatment for 12 h.
To better explore the mechanisms of 6f-induced G2/M
arrest, we examined the changes of G2/M phase related proteins. As
shown in Fig. 6C, 6f (2 µM) significantly upregulated
the expression of p-cdc2, cyclin B1, p-histone H3 and downregulated
cdc2, cdc25c and CDK7 at 6 and 12 h after 6f treatment in HT-1080
cells.
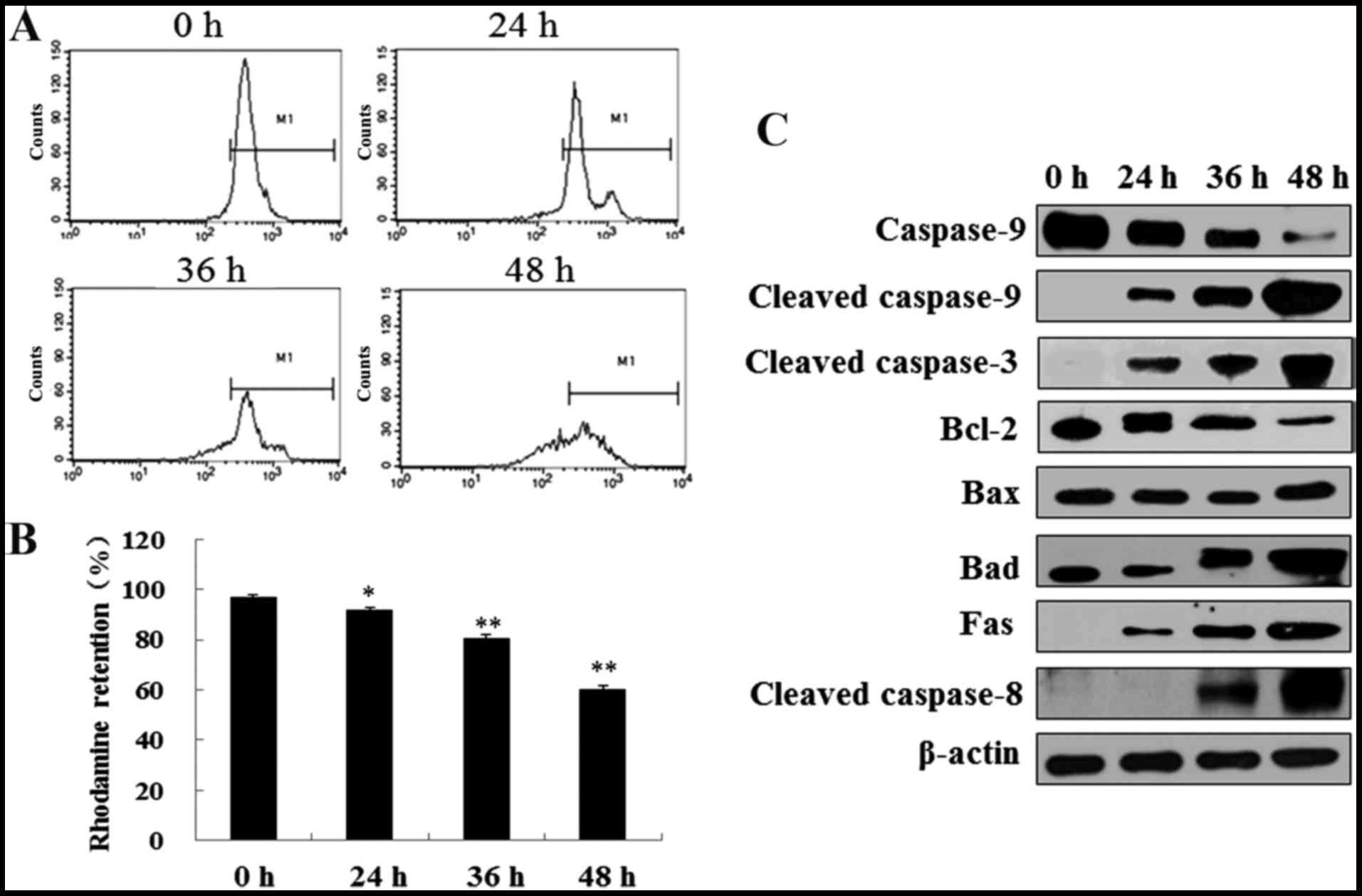
6f induces MMP decrease and apoptosis
through caspase activation
The above data proved that 6f induced profound G2/M
cell cycle arrest after treatment for 12 h in HT-1080 cells. Since
the cell cycle is closely regulated and controlled, a prolonged
mitotic arrest will most likely result in apoptosis induction
(20). We next tested that if 6f
could induce apoptosis after prolonged 6f treatment by flow
cytometry. As demonstrated in Fig. 7A
and B, 6f (2 µM) treatment for 12 h induced a dramatic
increase of cells in G2/M phase. After that, cells in G2/M phase
were gradually decreased and apoptosis characterized by sub-G1
phase were gradually increased from 24 to 72 h.
Mitochondria is involved in cell apoptosis and MMP
reflects the function of mitochondria (21). To determine whether 6f treated
cells were truly undergoing apoptotic death, we assessed the
changes of MMP in HT-1080 cells using Rhodamine staining and flow
cytometry. As showed in Fig. 8A and
B, MMP represented by Rhodamine retention was decreased
time-dependently in HT-1080 cells after 6f (2 µM) treatment
for 24, 36 and 48 h.
To gain insight into the mechanism of 6f-induced
apoptosis in HT-1080 cells, the effects of 6f on the expression of
apoptosis-related proteins were examined by western blotting. As
shown in Fig. 8C, 6f (2 µM) time-dependently
upregulated Fas, cleaved caspase-3, cleaved caspase-8, cleaved
caspase-9, Bax and Bad levels and downregulated caspase-9 and Bcl-2
levels in HT-1080 cells.
Discussion
A major feature distinguishing cancer cells from
non-malignant cells is their ability to grow and divide
uncontrollably. Microtubule-targeted agents interfere with the
dynamics of the spindle microtubules to inhibit mitosis in cancer
cells (22). Therefore,
microtubule-targeted agents are an important constituent of several
well-established therapies for the treatment or management of
fibrosarcoma.
The present study designed and synthesized a series
of new compounds which were analogues of CA-4 and expected them to
be microtubule-targeted agents. The MTT assay was used to confirm
the anti-proliferative effects of these new compounds. Among these
compounds, 6f exhibited better anticancer activity in three cell
lines including SGC-7901, A549 and HT-1080 than other compounds.
Further study proved that 6f inhibited cell proliferation in seven
cancer cell lines. We selected HT-1080, one of sensitive cell
lines, to further study the cellular and molecular mechanisms
underlying 6f-induced cytotoxicity for the first time.
Molecular docking studies suggested that 6f
interacts very closely with the colchicine docking pose through
hydrogen bonds at the colchicine binding site of tubulin, which
indicates that 6f might destabilize microtubule polymerisation like
colchicine. To further prove this hypothesis, tubulin
polymerisation assay was applied. Our results proved that 6f
dose-dependently inhibits microtubule polymerisation in
vitro. Furthermore, immunofluorescent staining of α-tubulin
proved that 6f treatment disturbed microtubule network and
organization of HT-1080 cells. These data indicate that 6f is a
novel promising microtubule-depolymerizing agent.
Several microtubule-targeted agents, including vinca
alkaloids, taxanes and colchicine, bind to tubulin and alter the
polymerisation dynamics of microtubules, in turn disrupting cell
cycle and inducing apoptosis (23–25).
Therefore, in the present study, the effect of 6f on cell cycle
progression was tested by flow cytometry. Similar with other
microtubule inhibitors (15,26),
our data also proved that 6f treatment caused cell cycle arrest at
the G2/M phase. Cell cycle checkpoints are pivotal in controlling
cell cycle progression (27). It
is well-known that different types of cyclins and their
cyclin-dependent kinases control cell cycle progression (28). Progression from G2 to M phase is
controlled via a series of the cyclin family members, particularly
cyclin B1 and cdc2. CyclinB1 can be accumulated from prophase to
metaphase, and degraded as cells progressing into anaphase
(29). Cdc2 is a cell-cycle kinase
responsible for the regulation of G2 progression and G2/M
transition in all eukaryotic cells. The conversion of cdc2 from
inactive to active form is controlled by the specific phosphatase
cdc25c. Phosphorylation of cdc25c self-activates its phosphatase
function and subsequent activates cdc2/cyclin B1 kinase to
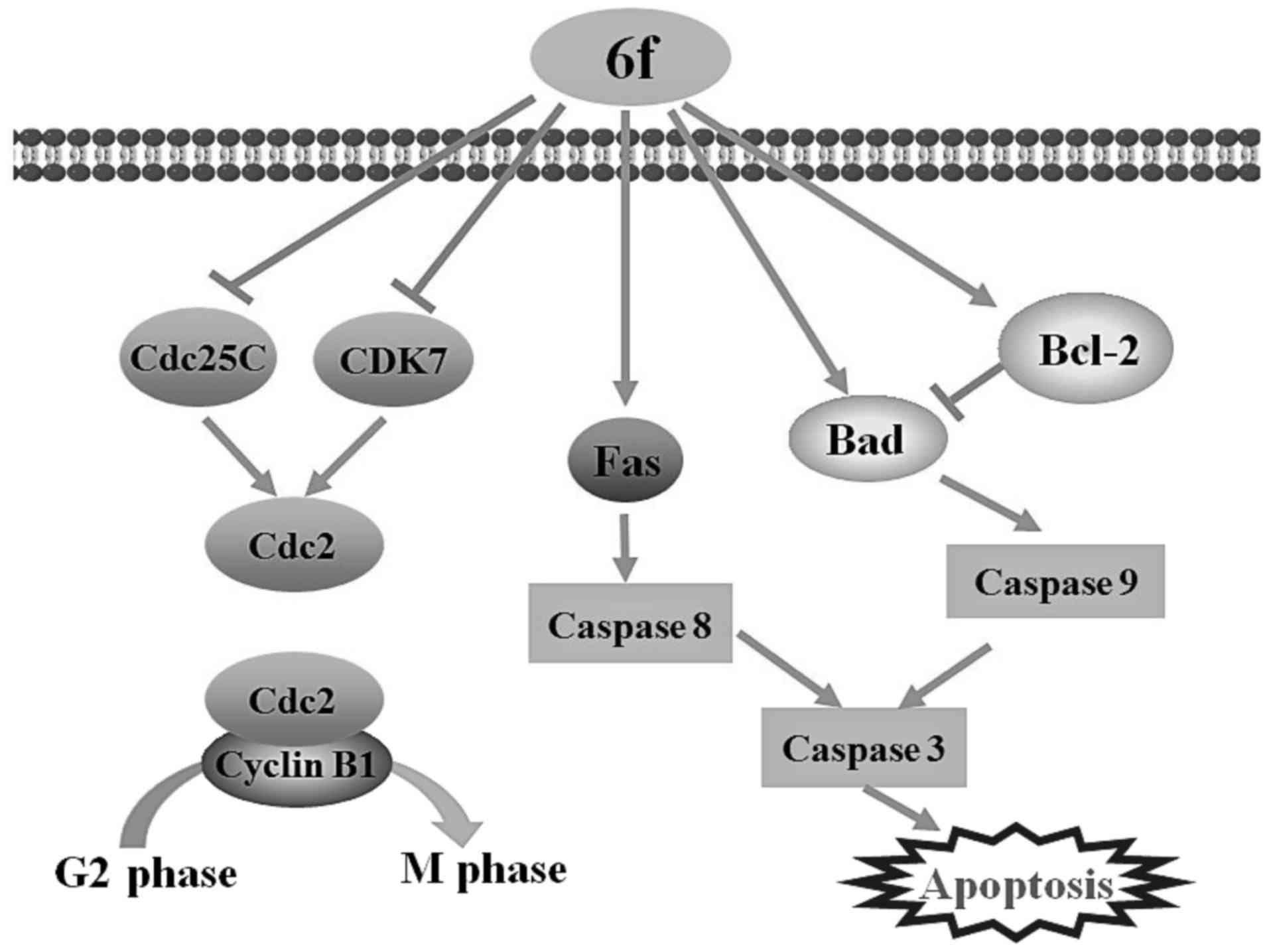
facilitate cell cycle entry into the M phase (26). In this study, we demonstrated that
cells treated with 6f led to upregulation of p-cdc2, cyclin B1,
p-histone H3 and downregulation of cdc2, cdc25c and CDK7, which
might decrease the activity of cdc2/cyclin B1, result in G2/M
arrest and eventually lead to cell apoptosis in HT-1080 cells
(fig. 9).
Apoptosis, characterized by cytoplasmic shrinkage,
chromatin condensation and DNA fragmentation, is an active form of
cell death that occurs in response to different factors, including
anticancer drugs (30,31). Compounds that induce apoptosis in
tumor cells are considered as promising agents against cancer
(31,32). It is widely accepted that
microtubule-interfering agents may activate apoptotic signaling
pathways and then induce apoptosis after initiating cell cycle
arrest (33–35). Similar with other agents, our
results also proved that 6f induced apoptosis characterized by
sub-G1 phase increase by flow cytometry after G2/M arrest.
Therefore, the mechanism of apoptotic cell death was also
investigated in 6f treated cells.
There are two main pathways leading to cellular
apoptosis: the mitochondria-dependent intrinsic pathway and the
death-receptor-mediated extrinsic pathway (36,37).
In the extrinsic pathway, membrane-bound death receptors on the
cell surface receive stimuli of pro-apoptotic ligands and transmit
signals by activating downstream initiators (38). The interaction between death
receptor and its ligand triggers the formation of a death-inducing
signaling complex, which in turn recruits pro-caspase-8.
Pro-caspase-8 undergoes autoproteolytic cleavage to form active
caspase-8, an initiator caspase of the extrinsic pathway (37,39).
The intrinsic pathway is initiated with loss of MMP and then Bcl-2
protein family-induced cytochrome c release following activation of
caspase-9, an initiator caspase of the intrinsic pathway. In both
pathways, the activated initiator caspase leads to their own
autoactivation and further activates caspase-3 and -7, the effector
caspase, and then leads to the cleavage of PARP, one of its
downstream substrates (37,40,41).
In our studies, 6f induced cell apoptosis by upregulating Fas,
caspase-3, -8, -9 and pro-apoptotic protein Bax and Bad levels and
downregulating pro-caspase-9 and anti-apoptotic protein Bcl-2
levels in HT-1080 cells. Moreover, MMP of HT-1080 cells was
significantly decreased after 6f treatment. These results indicate
that 6f-induced cell apoptosis was associated with both the
mitochondria-dependent intrinsic pathway and the
death-receptor-mediated extrinsic pathway (Fig. 9).
In conclusion, we synthesized a series of new
compounds and proved for the first time that the novel compound 6f
showed strong anticancer activity against HT-1080 cells through
disrupting microtubule polymerisation and inducing G2/M arrest, and
then causing cell apoptosis. Therefore, 6f is a potential
microtubule depolymerising agent for the therapy of different
cancers especially fibrosarcoma. Furthermore, 6f-induced cell
apoptosis was associated with both the mitochondria-dependent
intrinsic pathway and the death-receptor-mediated extrinsic
pathway. The present mechanistic studies also provide a good
foundation for further research and development of novel compounds
as potential therapeutic anticancer agents.
Acknowledgments
This work was supported by grants from the National
Natural Science Foundation (81602969 and 81673293), Young and
middle age backbone personnel training program of Shenyang
Pharmaceutical University (ZQN2015003) and Liaoning BaiQianWan
Talents Program.
References
|
1
|
Lombardi R, Jovine E, Zanini N, Salone MC,
Gambarotti M, Righi A, Balladelli A, Colangeli M and Rocca M: A
case of lung metastasis in myxoinflammatory fibroblastic sarcoma:
Analytical review of one hundred and thirty eight cases. Int
Orthop. 37:2429–2436. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Amos LA: What tubulin drugs tell us about
microtubule structure and dynamics. Semin Cell Dev Biol.
22:916–926. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Perez EA: Microtubule inhibitors:
Differentiating tubulin-inhibiting agents based on mechanisms of
action, clinical activity, and resistance. Mol Cancer Ther.
8:2086–2095. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
de Forges H, Bouissou A and Perez F:
Interplay between microtubule dynamics and intracellular
organization. Int J Biochem Cell Biol. 44:266–274. 2012. View Article : Google Scholar
|
|
5
|
Dumontet C and Jordan MA:
Microtubule-binding agents: A dynamic field of cancer therapeutics.
Nat Rev Drug Discov. 9:790–803. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim SN, Kim NH, Park YS, Kim H, Lee S,
Wang Q and Kim YK: 7-Diethylamino-3(2′-benzoxazolyl)-coumarin is a
novel microtubule inhibitor with antimitotic activity in multidrug
resistant cancer cells. Biochem Pharmacol. 77:1773–1779. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chang LC, Yu YL, Hsieh MT, Wang SH, Chou
RH, Huang WC, Lin HY, Hung HY, Huang LJ and Kuo SC: A novel
microtubule inhibitor, MT3-037, causes cancer cell apoptosis by
inducing mitotic arrest and interfering with microtubule dynamics.
Am J Cancer Res. 6:747–763. 2016.PubMed/NCBI
|
|
8
|
Velasco-Velázquez MA, Agramonte-Hevia J,
Barrera D, Jiménez-Orozco A, García-Mondragón MJ, Mendoza-Patiño N,
Landa A and Mandoki J: 4-Hydroxycoumarin disorganizes the actin
cytoskeleton in B16-F10 melanoma cells but not in B82 fibroblasts,
decreasing their adhesion to extracellular matrix proteins and
motility. Cancer Lett. 198:179–186. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gismondi A, Nanni V, Reina G, Orlanducci
S, Terranova ML and Canini A: Nanodiamonds coupled with
5,7-dimethoxycoumarin, a plant bioactive metabolite, interfere with
the mitotic process in B16F10 cells altering the actin
organization. Int J Nanomed. 11:557–574. 2016. View Article : Google Scholar
|
|
10
|
Kavallaris M, Annereau JP and Barret JM:
Potential mechanisms of resistance to microtubule inhibitors. Semin
Oncol. 35(Suppl 3): S22–S27. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pettit GR, Singh SB, Hamel E, Lin CM,
Alberts DS and Garcia-Kendall D: Isolation and structure of the
strong cell growth and tubulin inhibitor combretastatin A-4.
Experientia. 45:209–211. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Aziz G, Odlo K, Hansen TV, Paulsen RE and
Mathisen GH: Combretastatin A-4 and structurally related triazole
analogues induce caspase-3 and reactive oxygen species-dependent
cell death in PC12 cells. Eur J Pharmacol. 703:25–32. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu Q, Qi H, Sun M, Zuo D, Jiang X, Wen Z,
Wang Z, Wu Y and Zhang W: Synthesis and biological evaluation of
3-alkyl-1,5-diaryl-1h-pyrazoles as rigid analogues of
combretastatin a-4 with potent antiproliferative activity. PLoS
One. 10:e01287102015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mahal K, Biersack B, Caysa H, Schobert R
and Mueller T: Combretastatin A-4 derived imidazoles show
cytotoxic, antivascular, and antimetastatic effects based on
cytoskeletal reorganisation. Invest New Drugs. 33:541–554. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zuo D, Guo D, Jiang X, Guan Q, Qi H, Xu J,
Li Z, Yang F, Zhang W and Wu Y:
3-(3-Hydroxy-4-methoxyphenyl)-4-(3,4,5-trimethoxyphenyl)-1,2,5-selenadiazole
(G-1103), a novel combretastatin A-4 analog, induces G2/M arrest
and apoptosis by disrupting tubulin polymerization in human
cervical HeLa cells and fibrosarcoma HT-1080 cells. Chem Biol
interact. 227:7–17. 2015. View Article : Google Scholar
|
|
16
|
Wen Z, Li X, Zuo D, Lang B, Wu Y, Jiang M,
Ma H, Bao K, Wu Y and Zhang W: Ultrasound-promoted two-step
synthesis of 3-arylselenylindoles and 3-arylthioindoles as novel
combretastatin A-4 analogues. Sci Rep. 6:239862016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Duan YT, Man RJ, Tang DJ, Yao YF, Tao XX,
Yu C, Liang XY, Makawana JA, Zou MJ, Wang ZC, et al: Design,
synthesis and antitumor activity of novel link-bridge and b-ring
modified combretastatin a-4 (ca-4) analogues as potent antitubulin
agents. Sci Rep. 6:253872016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Z, Qi H, Shen Q, Lu G, Li M, Bao K,
Wu Y and Zhang W: 4,5-Diaryl-3H-1,2-dithiole-3-thiones and related
compounds as combretastatin A-4/oltipraz hybrids: Synthesis,
molecular modelling and evaluation as antiproliferative agents and
inhibitors of tubulin. Eur J Med Chem. 122:520–529. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Qiao F, Zuo D, Shen X, Qi H, Wang H, Zhang
W and Wu Y: DAT-230, a novel microtubule inhibitor, exhibits potent
antitumor activity by inducing G2/M phase arrest, apoptosis in
vitro and perfusion decrease in vivo to HT-1080. Cancer Chemother
Pharmacol. 70:259–270. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Eisenlöffel C, Schmöle AC, Pews-Davtyan A,
Brennführer A, Kuznetsov SA, Hübner R, Frech S, Schult C, Junghanss
C, Beller M, et al: Interference of a novel indolylmaleimide with
microtubules induces mitotic arrest and apoptosis in human
progenitor and cancer cells. Biochem Pharmacol. 85:763–771. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu G, Chen X, Chen S, Ye W, Hou K and
Liang M: Arsenic trioxide reduces chemo-resistance to
5-fluorouracil and cisplatin in HBx-HepG2 cells via complex
mechanisms. Cancer Cell int. 15:1162015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Islam MN and Iskander MN: Microtubulin
binding sites as target for developing anticancer agents. Mini Rev
Med Chem. 4:1077–1104. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang Z, Xu Y and Peng W: Colchicine
induces apoptosis in HT-29 human colon cancer cells via the AKT and
c-Jun N-terminal kinase signaling pathways. Mol Med Rep.
12:5939–5944. 2015.PubMed/NCBI
|
|
24
|
Chiu WH, Luo SJ, Chen CL, Cheng JH, Hsieh
CY, Wang CY, Huang WC, Su WC and Lin CF: Vinca alkaloids cause
aberrant ROS-mediated JNK activation, Mcl-1 downregulation, DNA
damage, mitochondrial dysfunction, and apoptosis in lung
adenocarcinoma cells. Biochem Pharmacol. 83:1159–1171. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ganansia-Leymarie V, Bischoff P, Bergerat
JP and Holl V: Signal transduction pathways of taxanes-induced
apoptosis. Curr Med Chem Anticancer Agents. 3:291–306. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hsieh CC, Kuo YH, Kuo CC, Chen LT, Cheung
CH, Chao TY, Lin CH, Pan WY, Chang CY, Chien SC, et al:
Chamaecypanone C, a novel skeleton microtubule inhibitor, with
anticancer activity by trigger caspase 8-Fas/FasL dependent
apoptotic pathway in human cancer cells. Biochem Pharmacol.
79:1261–1271. 2010. View Article : Google Scholar
|
|
27
|
Chen CH, Liao CH, Chang YL, Guh JH, Pan SL
and Teng CM: Protopine, a novel microtubule-stabilizing agent,
causes mitotic arrest and apoptotic cell death in human
hormone-refractory prostate cancer cell lines. Cancer Lett.
315:1–11. 2012. View Article : Google Scholar
|
|
28
|
Vermeulen K, Van Bockstaele DR and
Berneman ZN: The cell cycle: A review of regulation, deregulation
and therapeutic targets in cancer. Cell Prolif. 36:131–149. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu H, Zhang J, Xue N, Hu Y, Yang B and He
Q: Novel combretastatin A-4 derivative XN0502 induces cell cycle
arrest and apoptosis in A549 cells. Invest New Drugs. 28:493–501.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tao L, Fu R, Wang X, Yao J, Zhou Y, Dai Q,
Li Z, Lu N and Wang W: LL-202, a newly synthesized flavonoid,
inhibits tumor growth via inducing G(2)/M phase arrest and cell
apoptosis in MCF-7 human breast cancer cells in vitro and in vivo.
Toxicol Lett. 228:1–12. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT,
Liu B and Bao JK: Programmed cell death pathways in cancer: A
review of apoptosis, autophagy and programmed necrosis. Cell
Prolif. 45:487–498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mollinedo F and Gajate C: Microtubules,
microtubule-interfering agents and apoptosis. Apoptosis. 8:413–450.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jeung HC, Che XF, Haraguchi M, Furukawa T,
Zheng CL, Sumizawa T, Rha SY, Roh JK and Akiyama S: Thymidine
phosphorylase suppresses apoptosis induced by
microtubule-interfering agents. Biochem Pharmacol. 70:13–21. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhou L, Cai X, Han X, Xu N and Chang DC:
CDK1 switches mitotic arrest to apoptosis by phosphorylating
Bcl-2/Bax family proteins during treatment with microtubule
interfering agents. Cell Biol Int. 38:737–746. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Khan KH, Blanco-Codesido M and Molife LR:
Cancer therapeutics: Targeting the apoptotic pathway. Crit Rev
Oncol Hematol. 90:200–219. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ola MS, Nawaz M and Ahsan H: Role of Bcl-2
family proteins and caspases in the regulation of apoptosis. Mol
Cell Biochem. 351:41–58. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Parrish AB, Freel CD and Kornbluth S:
Cellular mechanisms controlling caspase activation and function.
Cold Spring Harb Perspect Biol. 5:52013. View Article : Google Scholar
|
|
39
|
Golks A, Brenner D, Fritsch C, Krammer PH
and Lavrik IN: c-FLIPR, a new regulator of death receptor-induced
apoptosis. J Biol Chem. 280:14507–14513. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Saelens X, Festjens N, Vande Walle L, van
Gurp M, van Loo G and Vandenabeele P: Toxic proteins released from
mitochondria in cell death. Oncogene. 23:2861–2874. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang CC, Liu HE, Lee YL, Huang YW, Chen
YJ, Liou JP and Huang HM: Mpt0b169, a novel tubulin inhibitor,
induces apoptosis in taxol-resistant acute myeloid leukemia cells
through mitochondrial dysfunction and mcl-1 downregulation. Tumour
Biol. 37:6065–6072. 2016. View Article : Google Scholar
|