Introduction
Lung cancer has become one of the major causes for
cancer-related deaths in the last few years, most cases of which
are non-small cell lung cancer (NSCLC). Although, in the past
decade, target therapy such as epidermal growth factor receptor
(EGFR) antagonist has been applied in NSCLC treatment, mutations of
target genes and drug resistance damaged the curative effect of
target therapy (1). As a novel
therapeutic strategy, epigenetics approach impairs the drug
resistance resulted from cancer heterogeneity by regulating the
cluster of tumor suppression genes. Among these epigenetics
approaches, hypomethylating agents and histone deacetylase (HDAC)
inhibitors have been widely used in clinical treatment of various
tumors including lung cancer (2).
However, the tumor-related genes and signal pathways targeted by
epigenetic therapy are largely unknown.
Histone deacetylases (HDACs) are the critical
enzymes that catalyze the removal of acetyl groups from the lysine
residues within histones. Therefore, HDAC family displays a
determinant role in transcriptional regulation by controlling
chromatin condensation. Based on their structure and homology,
human HDACs have been classified into four classes (classes I, II,
III and IV) (3). HDAC5 belongs to
class II HDACs (4–10) which play a crucial role in
tumorigenesis (4,5). In the present study, overexpression
of HDAC5 was found to be closely associated with activation of
tumor drivers and inhibition of tumor suppressors in NSCLC cells.
Consistently, numerous studies showed that HDACs are aberrantly
expressed in various cancer types and then change the epigenetic
status which blocks the expressions of tumor suppressors (6). Consequently, abnormal expressions of
HDACs promote cell proliferation, invasion and even resistance to
multi-drugs which contribute to a poor prognosis in different
cancers (7,8). Although some members of HDACs have
been well investigated in several cancer types, the molecular
mechanisms for the functions and deregulation of HDAC5 in human
NSCLC remain unclear.
Earlier research found that overexpression of
miR-589 partially reversed the TGF-β1 induced
epithelial-mesenchymal transition (EMT) change of human peritoneal
mesothelial cells (9). Although,
as an important microRNA, miR-589 were rarely studied in tumor
progression until recently. Li et al reported that miR-589
was significantly downregulated in cisplatin-resistant A549, and
transfection with miR-589 resulted in an increased sensitivity to
cisplatin (10). A later study
showed that miR-589-5p could downregulate the stemness
characteristics of CD90+ cancer stem cells (CSCs) of HCC
in part by silencing MAP3K8 (11).
Our findings revealed that the loss of miR-589-5p by
hypermethylation accelerated NSCLC development through increasing
the expression of HDAC5.
In the present study, higher expression of HDAC5
accompanied with decreased miR-589-5p was observed in NSCLC
compared with normal tissues. Furthermore, miR-589-5p significantly
reduced HDAC5 expression by targeting the 3′UTR of HDAC5 mRNA.
Promoter hypermethylation of the miR-589 gene was also observed in
NSCLC cells, but not in lung epithelial cells, which caused the
loss of miR-589-5p. Consequently, aberrant regulation of
miR-589-5p/HDAC5 pathway promoted the cell cycle and EMT-related
gene clusters and then conferred the aggressiveness of NSCLC
cells.
Materials and methods
Cell culture and drug treatment
Human NSCLC cell lines A549, H1299, and human lung
epithelial cell line BEAS-2B were purchased from the American Type
Culture Collection (ATCC). A549 and H1299 cells were cultured in
Dulbecco's modified Eagle's medium (DMEM, Corning) supplemented
with 10% fetal bovine serum (FBS, Gibco), 100 U/ml penicillin, and
100 U/ml streptomycin. BEAS-2B cells were cultured in RPMI-1640
(Corning) supplemented with same additive. All cells were grown at
37°C in 5% CO2. A549 and H1299 cell lines were treated
with 5-Aza-dC (Sigma) at a final concentration of 4 µM.
Ethics approval
All procedures performed in studies involving human
participants were in accordance with the ethical standards of the
institutional and/or national research committee and with the 1964
Helsinki declaration and its later amendments or comparable ethical
standards.
mRNA and miRNA quantification
TRIzol reagent was used to extract total RNA. miRNA
was amplified using the TaqMan MicroRNA Reverse Transcription kit
(Applied Biosystems). Mature miR-589-5p transcript levels were
measured with TaqMan MicroRNA assay primers (Applied Biosystems)
and the mRNA levels of indicated genes were measured using SYBR
Premix Ex Taq™ (Takara). miRNA or mRNA levels were quantified with
the ABI PRISM 7500 real-time PCR system (Applied Biosystems).
DNA sodium bisulfite conversion
Genomic DNA was extracted from indicated cells by
phenol-chloroform technique. Bisulfite conversion was performed as
described (12). Specific primers
for converted promoter region were used to generate PCR product.
Forward, TTTTGGAGTTTTTTTTGGTTTTT; reverse, CAAAAACAAAACCAAAATCA.
PCR products were cloned in pUCm-T followed by sequencing with T7
primer.
QPCR array
The high-throughput profiling of gene clusters were
analyzed by The ExProfile™ Gene qPCR array (GeneCopoeia) according
to the manufacturer's instructions. In 96-well plate, there are 50
pairs of qPCR primers and 12-wells of controls which are used to
monitor the efficiency of the entire experimental process (from
reverse transcription to qPCR reaction). A cDNA pool, containing
reverse transcript products from total RNA of the indicated cells,
was used as the qPCR validation template.
CCK-8 assay
Cells in logarithmic growth were plated and
transfected. After indicated time of culture, CCK-8 (Cell Counting
Kit-8) (Dojindo Molecular Technologies, MD, USA) was added, and the
OD450 was measured using an automatic plate reader.
Cell cycle analysis
Cells were treated with the indicated reagents and
fixed in 70% ethanol overnight. Fixed cells were treated with 100
mg/ml RNaseA (Roche) before addition of 50 mg/ml PI (Sigma) and
analyzed by FACSCalibur flow cytometer.
EdU incorporation assay
The indicated cells were seeded in 96-well plates.
DNA synthesis was assessed using the Cell-Light EdU
(5-ethynyl-2′-deoxyuridine) DNA Cell Proliferation kit (RiboBio)
according to the manufacturer's instructions. Images of the cells
were cap tured with a fluorescence microscope. ImageJ software was
used to count the fluorescent points.
Wound healing assay
Transfected cells were grown to 80% confluence in
6-well tissue culture plates and wounded with a sterile tip to
remove cells by perpendicular linear scrapes. After that, the cells
were cultured for 24 h. The wound was photographed and quantified
immediately after wounding and again 24 h later.
Cell invasion assays
Cell invasion were assessed in Boyden chambers with
Matrigel according to the manufacturer's protocol (Invitrogen).
First, an 8-mm-porosity polycarbonate membrane was covered with 200
µl of serum-free medium containing 1×105 cells
per well. The plates were then incubated with 10% FBS medium for 48
h at 37°C in a 5% CO2 incubator. The invasion cells on
the bottom surface of the filter were fixed, stained, and counted
using optical microscopy.
Colony formation in soft agar
For colony formation experiment, cells were
suspended in 0.3% agar and seeded into 6-well plate pre-coated with
1.0 ml of 0.6% agar. Medium was changed every 5 days for three
weeks. The number and size of colonies were examined and data were
obtained by analyzing with ImageJ software.
Nude mouse xenograft assay
Twenty-five female BALB/c nude mice were injected
with A549 cells with stable overexpression of miR-589-5p or HDAC5.
All of the mice were randomly divided into five groups (n=5 of each
group), and each received a subcutaneous injection of a viable cell
suspension mixture (5×106) containing 90% of A549 cells
with indicated transfection. When the tumors could be palpated,
tumor size was measured using calipers every three days. All of the
mice were sacrificed on the fifth week after injection, and the
individual tumors were weighed.
Immunohistochemistry
The study was performed with the approval of the
Ethics Committee of The First Affiliated Hospital of Xi'an Jiaotong
University and the Second of Dalian Medical University. NSCLC
tissues were collected by needle biopsy or surgery from 190
patients at The First Affiliated Hospital of Xi'an Jiaotong
University (Xi'an, China) and The Second of Dalian Medical
University (Dalian, China) from 2008 to 2010. All specimens were
fixed in 10% neutral formalin, embedded in paraffin and cut into
4-µm sections for immunohistochemical staining. The
EnVision™ two-step method was used (Dako, Hamburg, Germany), as
well as the following antibody: antibody against HDAC5. To estimate
the score for each slide, at least 10 individual fields at 200X
were chosen, and 100 cancer cells were counted in each field. The
immunostaining intensity was divided into four grades: 0, no
expression; 1, mildly positive; 2, moderately positive; and 3,
markedly positive. The proportion of positive-staining cells was
divided into five grades: 0, <10%; 1, 11–25%; 2, 26–50%; 3,
51–75%; and 4, >75%. The staining results were assessed and
confirmed by two independent investigators blinded to the clinical
data. The percentage of positivity of the tumor cells and the
staining intensities were then multiplied in order to generate the
IHC score, and graded as 0–6, low expression; 7–12, high
expression. Cases with a discrepancy in scores were discussed to
obtain a consensus.
Statistical analysis
All statistical analyses were carried out using the
SPSS 17.0 statistical software. Experimental data were presented as
mean ± SD from at least three independent experiments. The data
were analyzed by Student's t-test. Statistical analysis to evaluate
correlation was performed using Pearson's correlation analysis. A
p<0.05 was considered statistically significant.
Results
A significant negative correlation
between miR-589-5p and HDAC5 was found in NSCLC specimens
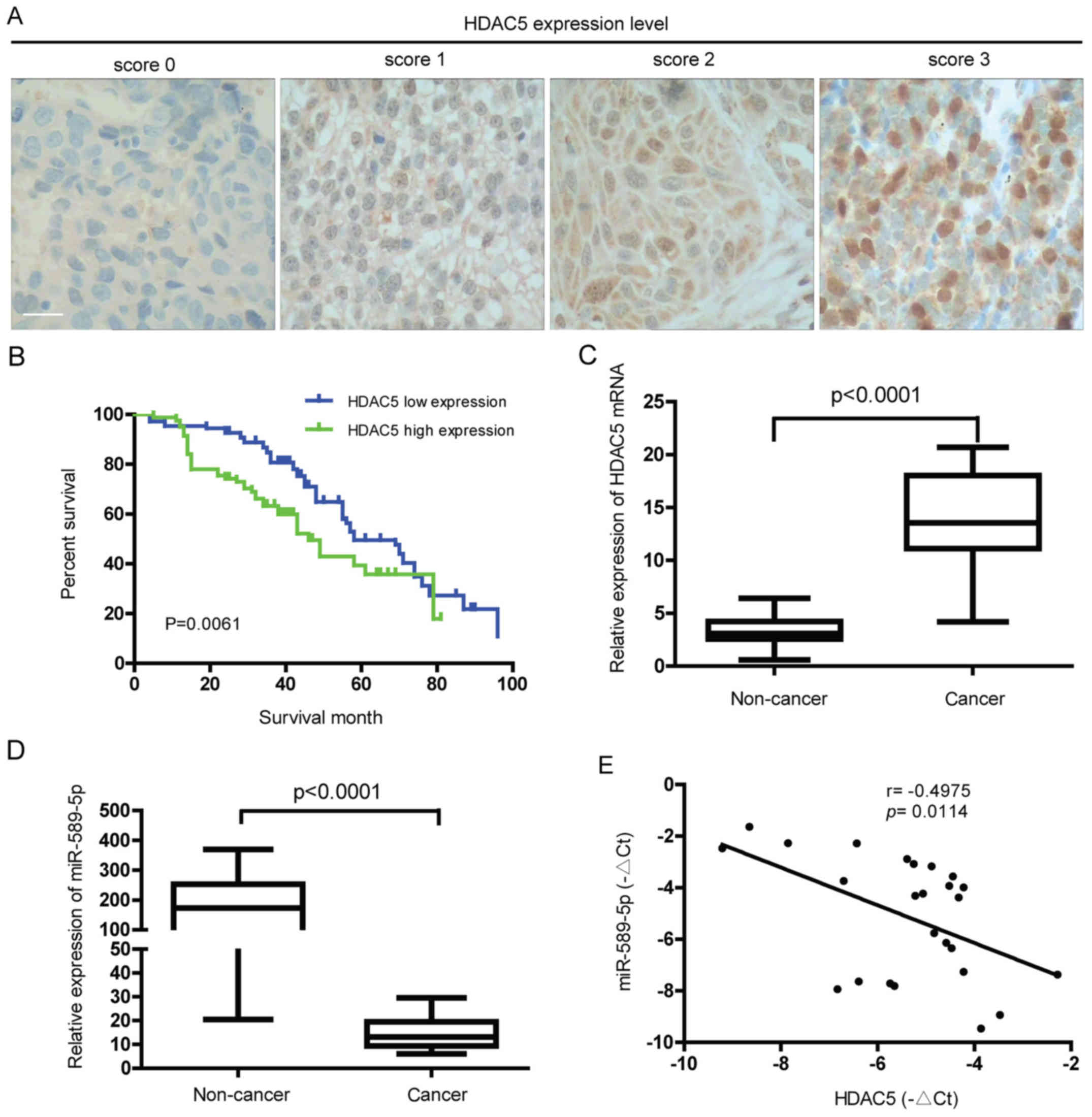
To characterize the expression level of HDAC5 in
NSCLC, immunohistochemical analysis of tumor tissues from NSCLC
patients was performed. As demonstrated in Fig. 1A, different protein levels of HDAC5
were observed in the NSCLC tissues from different patients. Further
study showed that high expression of HDAC5 was positively
correlated with lymph node metastasis, differentiation status and
TNM stage (P<0.05) but not with gender, age, tumor size and
histological type (P>0.05) (Table
I). Also, the 5-year overall survival (OS) rate of HDAC5 high
expression group was significantly lower than that of HDAC5 low
expression group (13.41 vs 26.85%, P=0.0061) (Fig. 1B). Ectopic expression of HDAC5 mRNA
was found in NSCLC but not in normal lung tissues (Fig. 1C). Consistent with previous study,
miR-589-5p, known as a tumor suppressor gene, was downregulated in
NSCLC compared to normal tissues (Fig.
1D). Notably, a statistically negative correlation between
HDAC5 mRNA and miR-589-5p was confirmed in NSCLC specimens
(Fig. 1E). The above findings
reveal that overexpression of HDAC5 found in NSCLC indicates a poor
prognosis, and may be associated with loss of miR-589-5p.
 | Table ICorrelation of the expression of
HDAC5 with clinicopathological features in non-small cell lung
cancer tissues. |
Table I
Correlation of the expression of
HDAC5 with clinicopathological features in non-small cell lung
cancer tissues.
| Total | − | HDAC5 expression
| P-value |
|---|
| + | ++ | +++ |
|---|
| 190 | 24 | 69 | 72 | 25 | |
| Gender | | | | | | 0.7352 |
| Male | 132 | 18 | 51 | 45 | 18 | |
| Female | 58 | 6 | 18 | 27 | 7 | |
| Age (years) | | | | | | 0.6278 |
| <57 | 88 | 15 | 29 | 34 | 10 | |
| ≥57 | 102 | 9 | 40 | 38 | 15 | |
| Tumor size
(cm) | | | | | | 0.1027 |
| ≤3 | 76 | 10 | 39 | 19 | 8 | |
| >3 | 114 | 14 | 30 | 53 | 17 | |
| Histological
type | | | | | | 0.2856 |
| Squamous cell
carcinoma | 63 | 8 | 25 | 21 | 9 | |
|
Adenocarcinoma | 106 | 14 | 36 | 44 | 12 | |
| Mixed/other | 21 | 2 | 8 | 7 | 4 | |
| Lymph node
metastasis | | | | | | 0.0262a |
| No | 84 | 14 | 42 | 21 | 7 | |
| Yes | 106 | 10 | 27 | 51 | 18 | |
| Differentiation
status | | | | | | 0.0085a |
| Well | 48 | 13 | 25 | 7 | 3 | |
| Moderate | 85 | 7 | 36 | 34 | 8 | |
| Poor | 57 | 4 | 8 | 31 | 14 | |
| TNM stage | | | | | | 0.0011a |
| I/II | 147 | 21 | 58 | 49 | 8 | |
| III/IV | 43 | 3 | 11 | 23 | 17 | |
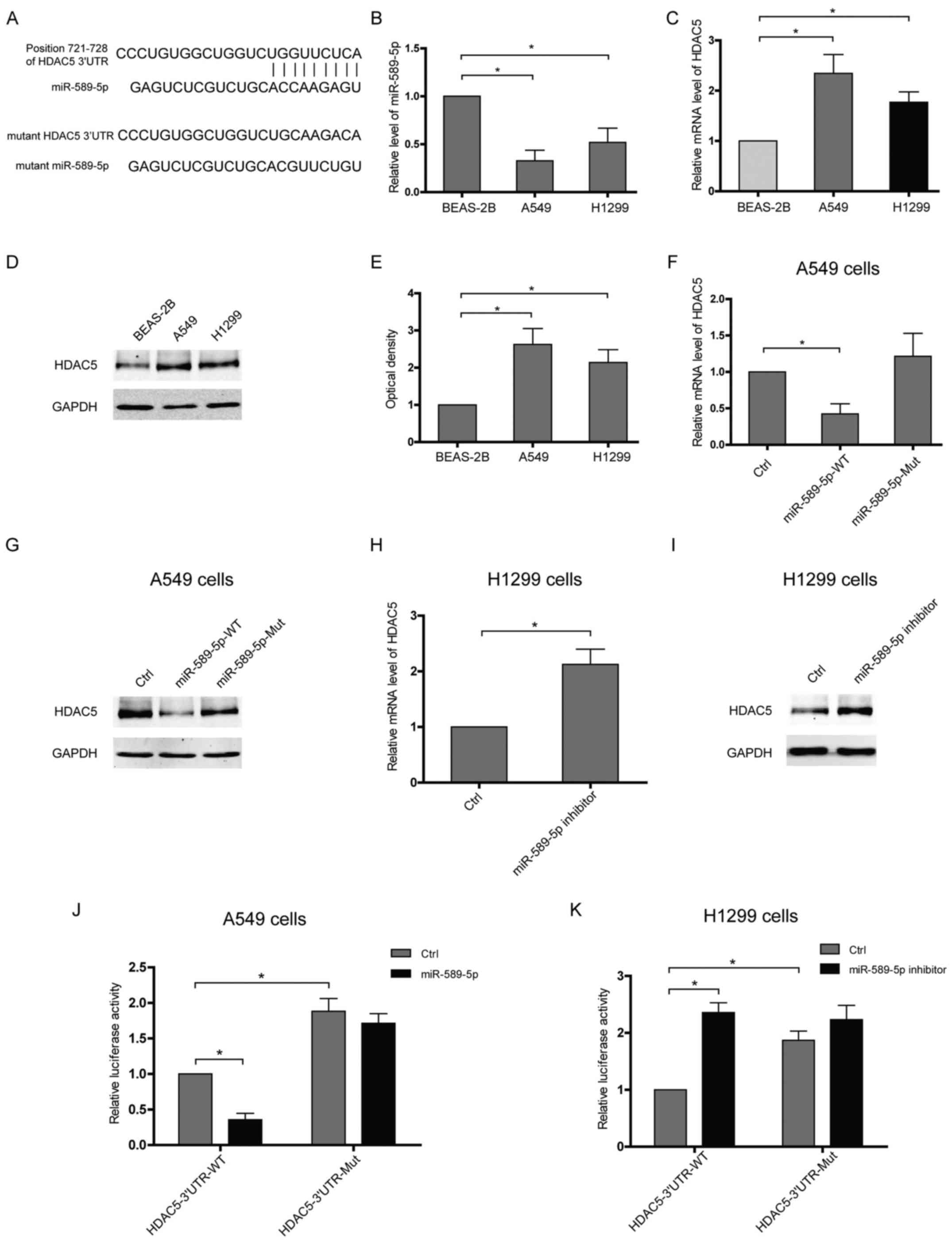
miR-589-5p targets the 3′UTR of HDAC5 and
then reduces HDAC5 expression in NSCLC cells
To verify HDAC5 as a target of miR-589-5p, software
TargetScan was applied to predict putative interaction and a
possible binding site within the 3′UTR of HDAC5 was found to
hybridize with miR-589-5p (Fig.
2A). Furthermore, the expressions of miR-589-5p and HDAC5 in
lung epithelial or NSCLC cells were examined. Upregulation of HDAC5
accompanied with decreased miR-589-5p was observed in NSCLC cells
compared with lung epithelial cells (Fig. 2B–E). The expressions of HDAC5 at
both mRNA and protein level were attenuated in NSCLC cells
transfected with wild-type miR-589-5p, but not with the mutant
(Fig. 2F and G). Knockdown of
miR-589-5p in lung epithelial cells using an anti-miRNA inhibitor
specific for miR-589-5p resulted in a significant upregulation of
HDAC5 (Fig. 2H and I). To
determine whether HDAC5 is directly targeted by miR-589-5p in
NSCLC, HDAC5 3′UTR fragment containing a wild-type or mutant
binding site was cloned into luciferase reporter vector and the
inhibitory effect of miR-589-5p was measured by luciferase
activity. It was found that the lucif-erase signal of wild-3′UTR
was reactively decreased by miR-589-5p, but the mutant was not
(Fig. 2J). Consistently, only the
luciferase reporter with intact 3′UTR was responsive to miR-589-5p
inhibitor (Fig. 2K). Hence, these
data indicate that miR-589-5p negatively regulates HDAC5 expression
via targeting the 3′UTR of HDAC5 mRNA.
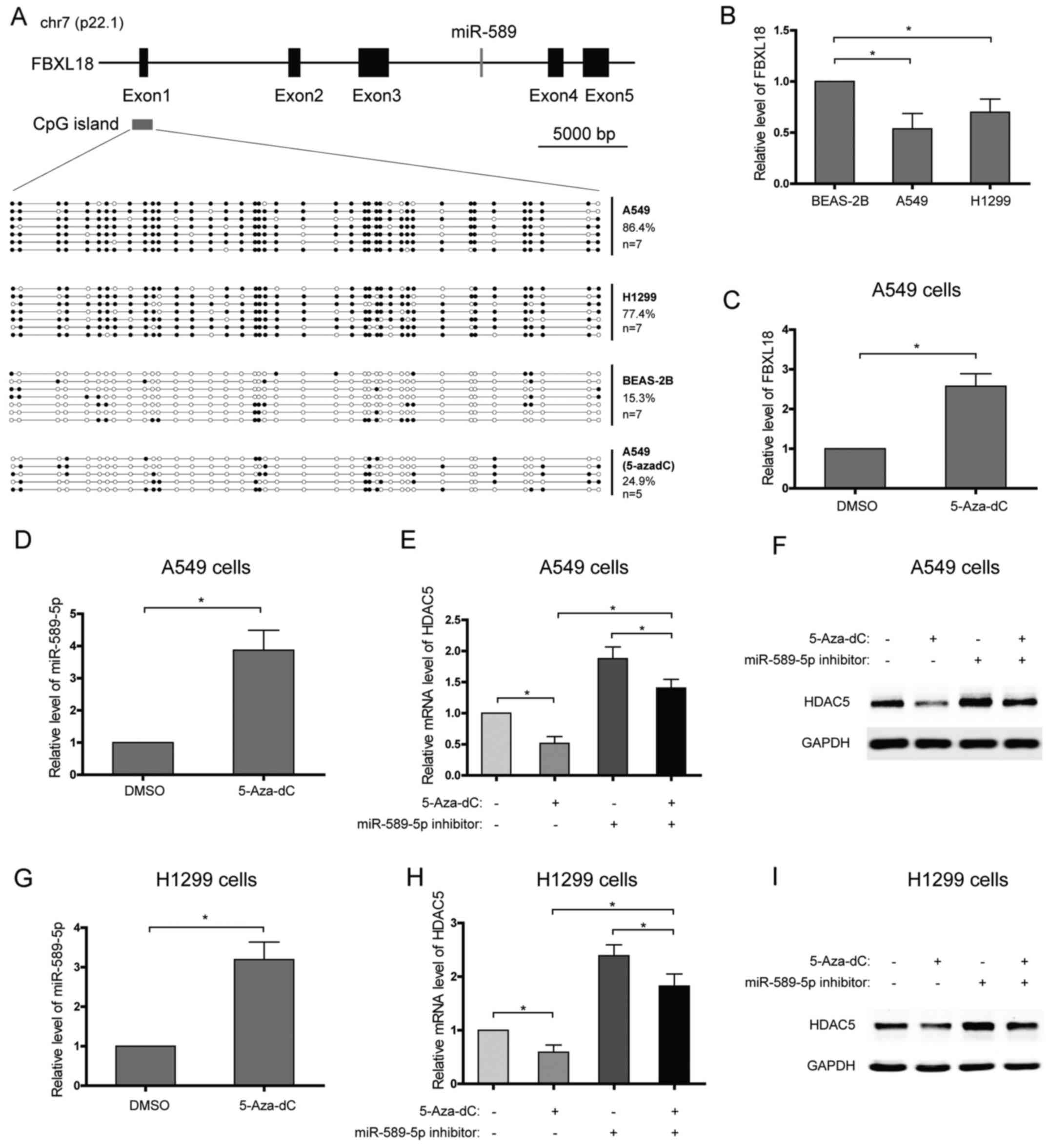
Promoter hypermethylation of the miR-589
gene decreases the level of miR-589-5p and promotes HDAC5
expression
miR-589 is an intronic microRNA which is located at
7p22.1 and within intron 3 of the F-Box and leucine rich repeat
protein 18 (FBXL18) gene, and they are in the same transcriptional
direction (Fig. 3A). Further
analysis showed that the expression patterns of miR-589 and its
host gene, FBXL18, in NSCLC cell lines or after 5-Aza-dC treatment
seemed to be similar, which suggested that they may share the same
promoter (Fig. 3B–D). In order to
characterize the DNA meth-ylation profile of the FBXL18/miR-589
gene promoter, DNA bisulfate conversion coupled to sequencing was
performed in NSCLC and lung epithelial cells. A CpG island was
predicted using UCSC genome browser and focused on (Fig. 3A). High DNA methylation levels of
the CpG island, with 86.4% of methylated CpGs in A549 cells and
77.4% in H1299 cells, were detected with DNA bisulfate conversion
(Fig. 3A). In contrast, low DNA
methylation level, with 15.3% of methylated CpGs, was found in the
lung epithelial BEAS-2B cells (Fig.
3A). To confirm that the promoter hypermethylation of the
miR-589 gene enhances the transcriptional repression of miR-589-5p
and then increases HDAC5 expression, NSCLC cells were treated with
DNA methylation inhibitor, 5-Aza-2′-deoxycytidine (5-Aza-dC), and
the expression of HDAC5 was analyzed. The results showed that
>60% of the CpGs were demethylated in A549 cells treated with
5-Aza-dC (Fig. 3A). In line with
this, miR-589-5p level was found to increase after 5-Aza-dC
treatment (Fig. 3D and G). As
expected, 5-Aza-dC observably suppressed the expression of HDAC5 in
NSCLC cells, while miR-589-5p specific inhibitor weakened the
suppression (Fig. 3E, F, H and I).
Therefore, these findings suggest that silence of miR-589-5p in
NSCLC is due to DNA hypermethylation.
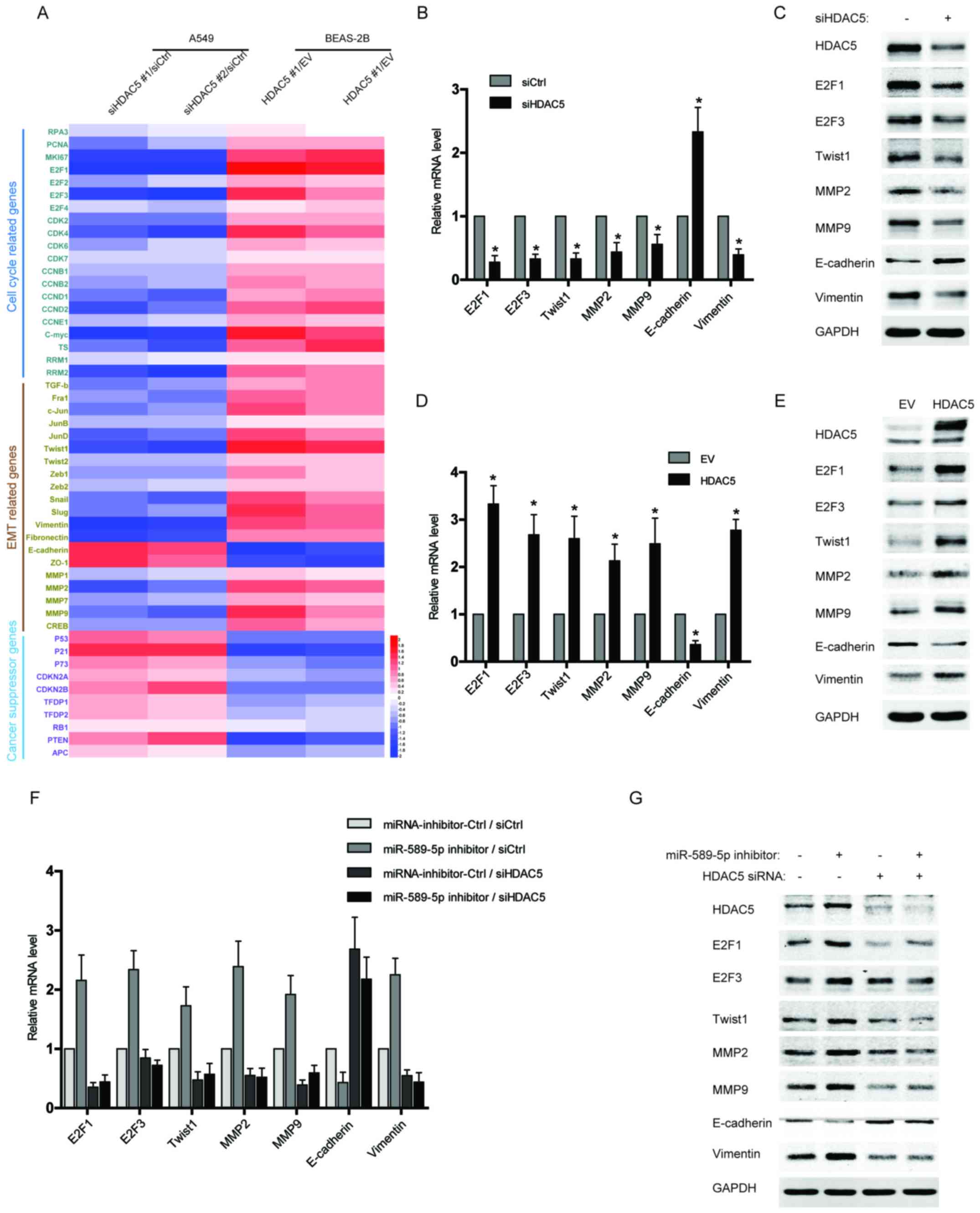
Upregulation of HDAC5 induces the
expression of proliferation and EMT-related genes in NSCLC
cells
Since HDAC5 has been reported to play a crucial role
in tumor progression and metastases, we examined the expression of
several gene clusters associated with cell cycle and EMT by qPCR
array. As shown in Fig. 4A, the
data from qPCR array revealed that multiple cell cycle or EMT
drivers, especially E2F1, E2F3, Twist1, MMP2, MMP9, and Vimentin,
were robustly increased in the BEAS-2B cells with HDAC5
overexpression, but reduced in HDAC5-silenced NSCLC cells compared
with control. Moreover, several tumor suppressor genes were also
analysed in the qPCR array, and their expression was repressed by
HDAC5 overexpression but activated after HDAC5 knockdown (Fig. 4A). The results of qPCR array were
further confirmed by independent qPCR and western blotting
(Fig. 4B–E). To determine the
significance of miR-589-5p/HDAC5 pathway in regulating cell
proliferation and EMT, the expressions of the above factors were
examined in the NSCLC cells transfected with miR-589-5p inhibitor
and HDAC5 siRNA. It was found that knockdown of HDAC5 completely
reversed the promotive effect of miR-589-5p inhibitor on the
expression of cell cycle or EMT drivers (Fig. 4F and G). Therefore, the evidence
demonstrates that miR-589-5p/HDAC5 pathway plays a crucial role in
regulating the cell cycle and EMT of NSCLC cells.
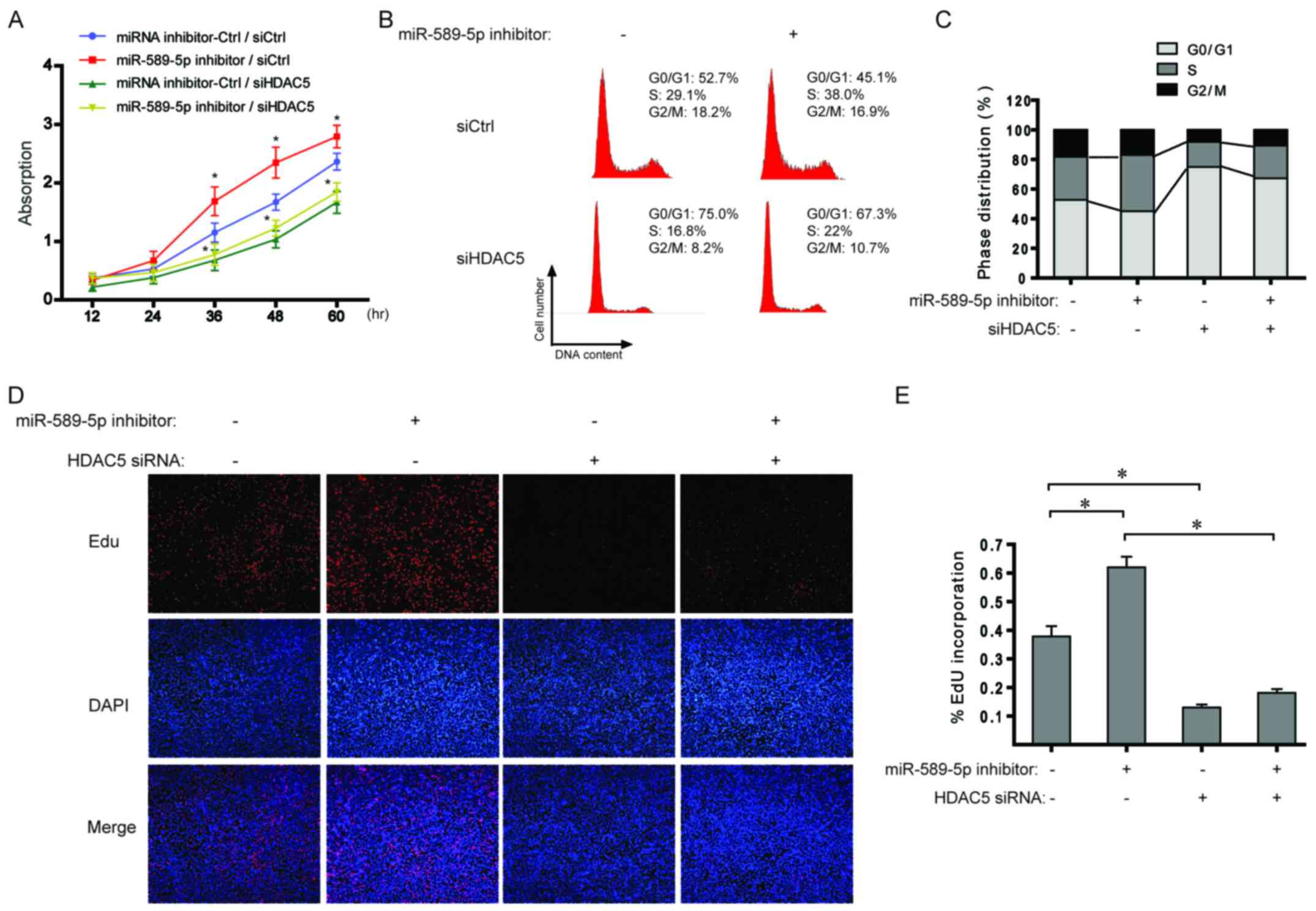
miR-589-5p/HDAC5 pathway regulates the
proliferation of NSCLC cells
With regard to the data from Fig. 4, the effect of miR-589-5p/HDAC5
pathway on cell proliferation was explored in NSCLC cells. As shown
in Fig. 5A, miR-589-5p inhibitor
boosted cell proliferation and additional knockdown of HDAC5
completely suppressed the induced proliferation. Cell cycle
analysis indicated that inhibiting miR-589-5p led to a significant
increase in the ratio of S phase cells, but a decrease after
transfection with HDAC5 siRNA (Fig. 5B
and C). Similarly, EdU incorporation assay revealed that the
percentage of cells with incorporated EdU was significantly
increased when treated with miR-589-5p inhibitor; however,
knockdown of HDAC5 impaired the effect (Fig. 5D and E). The above results indicate
a regulatory role of miR-589-5p/HDAC5 pathway in the proliferation
of NSCLC cells.
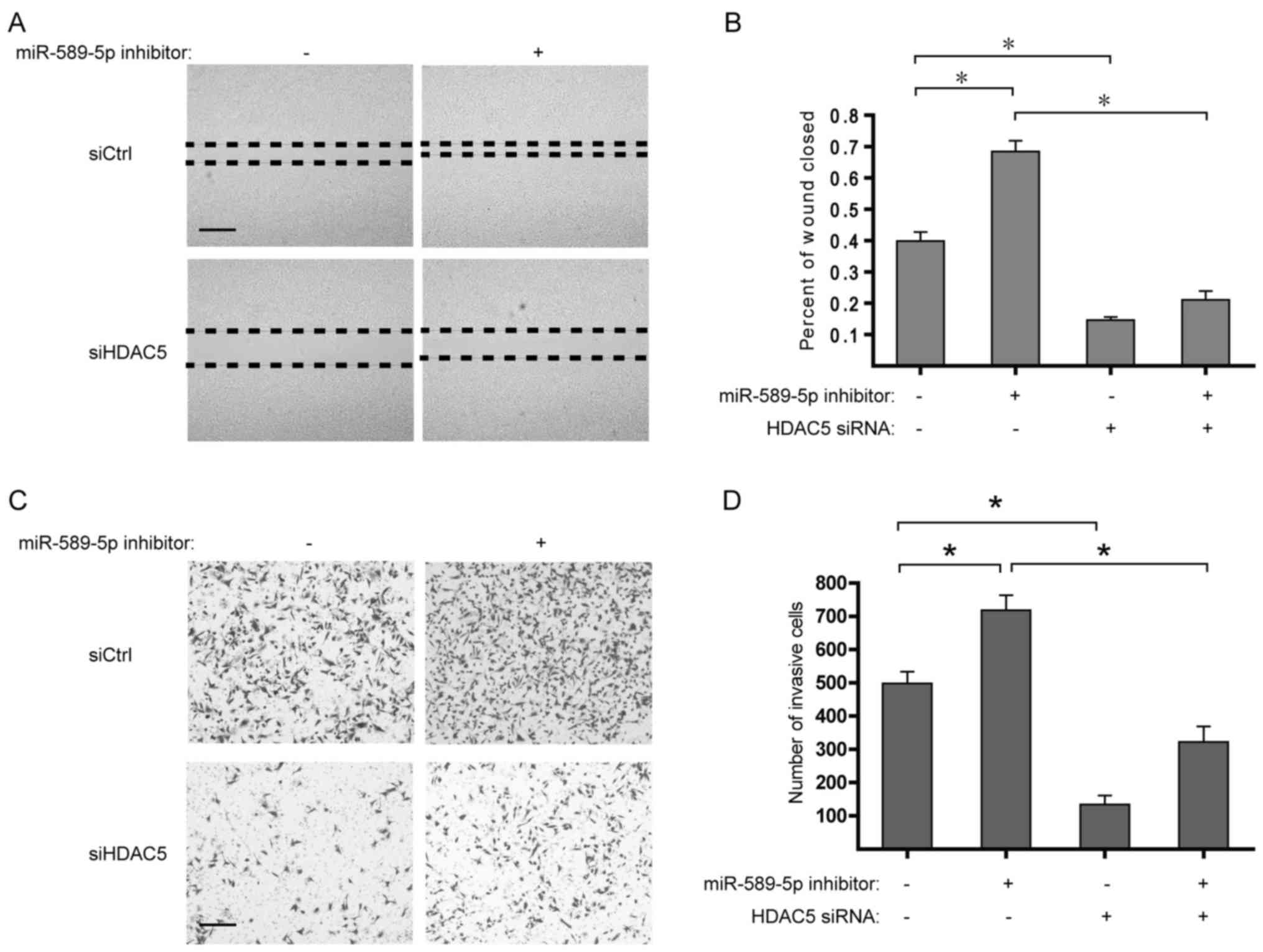
miR-589-5p/HDAC5 pathway modulates the
migration and invasion of NSCLC cells
Considering the upregulation of EMT-related genes
induced by HDAC5 overexpression, it is worth studying the role of
miR-589-5p/HDAC5 pathway in the migration and invasion of NSCLC. It
was found that suppression of miR-589-5p markedly strengthened the
migratory and invasive capabilities of NSCLC cells, whereas
deprivation of HDAC5 determined the reverse in the malignant
phenotypes (Fig. 6). Taken
together, these findings suggest that deregulation of
miR-589-5p/HDAC5 pathway displays a critical role in the migration
and invasion of NSCLC.
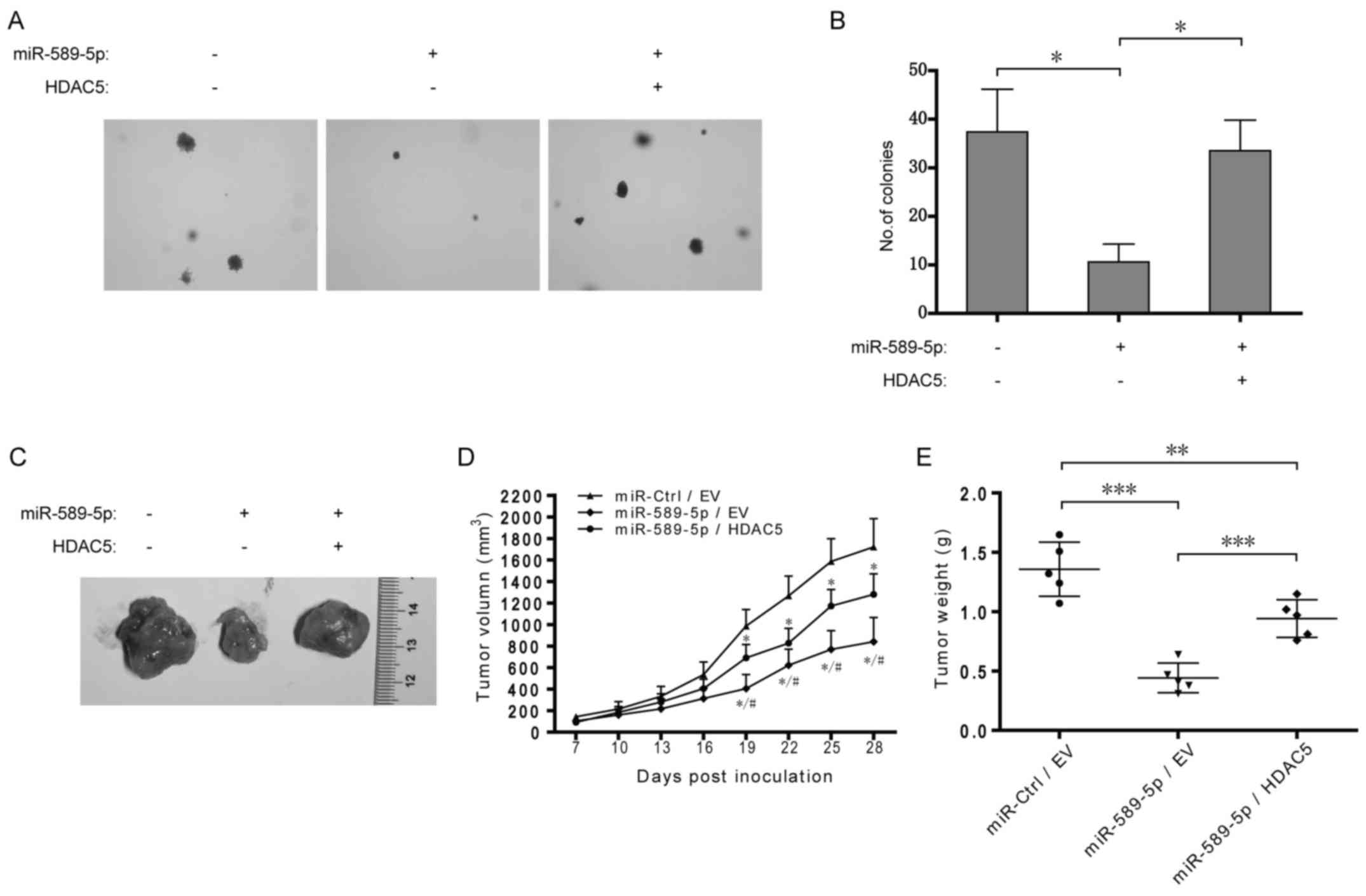
Dysregulation of miR-589-5p/HDAC5 pathway
promotes NSCLC tumorigenesis
To further determine the role of miR-589-5p/HDAC5
pathway in tumorigenesis, A549 cells with stable overexpression of
miR-589-5p and HDAC5 were constructed and then examined by colony
formation and xenograft assays. As shown in Fig. 7A and B, overexpression of
miR-589-5p resulted in the reduction of anchorage-independent
colony formation in soft agar in A549 cells, but additional
exogenous HDAC5 rescued the reduction. Xenograft assay demonstrated
that overexpression of miR-589-5p significantly decreased A549
xenograft growth in nude mice while simultaneous HDAC5
overexpression partially retrieved the growth (Fig. 7C–E). Taken together, these data
indicate a crucial role of miR-589-5p/HDAC5 pathway in NSCLC
tumorigenicity in vitro and in vivo.
Discussion
Over the past decade, epigenetic dysregulation
including DNA methylation patterns, histone modifications and
microRNA regulation has been reported to result in the initiation
and progression of lung cancer (13,14).
As the most studied epigenetic regulatory mechanism, DNA
methylation plays a crucial role in suppressing gene expression and
maintaining normal epigenetic regulatory processes (15). However, in cancer cells, CpG
islands located at the promoters of tumor suppression genes are
highly methylated leading to transcriptional repression (16). Similarly, as an important tumor
suppressor miRNA, miR-589-5p was found drastically decreased in
NSCLC compared with normal lung tissues. Further analysis indicated
that miR-589-5p silence was relevant for a strong gain of DNA
methylation in the miR-589 gene promoter. Consistently, treatment
with methylation inhibitor (5-Aza-dC) achieved restoration of
miR-589-5p expression in NSCLC cells. Although the mechanisms
underlying the promoter hypermethylation of the miR-589 gene were
not the focus of the present study, previous studies described some
putative factors involved in deregulated DNA methylation in lung
cancer. DNA methyltransferases (DNMTs) are responsible for
producing methylated CpG and aberrant DNMT expression is implicated
in the pathogenesis of lung cancer (17,18).
Upregulation of DNMTs silences different tumor suppressors by
promoter hypermethylation in lung cancer (18). Therefore, DNA methylation has
become a therapeutic target which can be disrupted by DNMT
inhibitors, such as decitabine and 5-Aza-dC (19).
In addition to DNA methylation, histone
modifications including acetylation, phosphorylation, and
methylation also contributes to epigenetic alterations in lung
cancer. Although the roles of HDAC family in tumorigenesis have not
been fully understood, HDAC inhibitors are emerging as novel
anticancer agents due to their ability to kill cancer cells by
inducing cell cycle arrest, autophagy, and apoptosis (20–22).
Here, accompanied with decreased miR-589-5p level, a higher
expression of HDAC5 was observed in NSCLC compared with normal lung
tissues. Further evidence showed that miR-589-5p significantly
repressed the expression of HDAC5 by targeting the 3′UTR of HDAC5
mRNA in NSCLC cells. As an important epigenetic modulator, HDAC5
promotes cell growth, migration, and invasion of breast cancer and
high levels of HDAC5 are closely related with poor survival in
human brain cancer patients (23,24).
With regard to miR-589-5p/HDAC5 pathway found in the study, it is
reasonable to hypothesize that HDAC5 may function as a critical
target of deregulated miR-589-5p, which promotes NSCLC
aggressiveness. As expected, the results demonstrated that HDAC5
mediated the malignant phenotypes including proliferation,
migration, invasion, and tumorigenicity induced by silence of
miR-589-5p in NSCLC cells.
HDAC5 has a broad range of downstream genes,
including some important transcription factors or modulators, such
as p53, CDK, and DLL4 (25,26).
The diverse functions of HDAC5 depend on its downstream genes which
regulate different cellular biological processes. For this reason,
we extended our studies to examine the expression profiling of gene
clusters associated with cell cycle and EMT by qPCR array. The
screening results showed that the cell cycle and EMT drivers were
widely downregulated after depletion of HDAC5 but upregulated by
HDAC5 overexpression. Specially, cell cycle drivers, such as E2F1
and E2F3, as well as EMT-inducing transcription factors (EMT-TFs)
like Twist1 were markedly induced after HDAC5 overexpression. In
lung cancer, abnormality in E2F1 expression has been described and
associated with poor patient survival (27). As a well-known transcription
factor, E2F1 transactivates various downstream effectors, such as
ribonucleotide reductase m2 (RRM2) and thymidylate synthase (TS),
which lead to tumor proliferation in NSCLC (28,29).
Of note, it was found that E2F1 could activate the transcription of
DNMT3A which specially resulted in an increased methylation level
and suppression of tumor suppressor genes (TSGs) (30). Therefore, inhibition of miR-589-5p
by DNA methylation may be maintained via a positive feedback,
miR-589-5p/HDAC5/E2F1/DNMT3A loop, in NSCLC. As a well-documented
member of EMT-TFs, Twist1 has been reported to induce
epithelial-mesenchymal transition and metastasis in NSCLC (31). In addition, aberrant expression of
Twist1 determines lung cancer chemoresistance and poor survival
(32). Consistent with upregulated
Twist1, Increased mesenchymal marker (Vimentin) and decreased
epithelial marker (E-cadherin) indicated HDAC5-induced EMT.
However, in contrast with above gene clusters, the
expressions of TSGs were negatively regulated by HDAC5 in NSCLC or
lung epithelial cells. Among these TSGs, p21 and PTEN were
significantly suppressed by HDAC5. Decreased PTEN expression level,
which occurs in up to 70% NSCLC patients, is associated to lower
survival (33). Loss of PTEN
expression was found to accelerate the EMT and development of lung
cancer by activating PI3K/AKT pathway (34,35).
Thus, the expression profiles of HDAC5 downstream effectors need to
be deeply investigated to understand the roles of HDAC5 in lung
cancer.
Therefore, we analyzed the effects of
miR-589-5p/HDAC5 pathway on malignant phenotypes, viz
proliferation, migration, invasion, and tumorigenicity. Consistent
with the above results, HDAC5 inhibition conferred the function of
miR-589-5p as a tumor suppressor in NSCLC cells. In hepato-cellular
carcinoma, knockdown of HDAC5 inhibits HCC cell proliferation and
tumorigenicity in nude mice (25).
Besides, suppression of HDAC5 sensitizes glioma cells to
chemotherapeutics by preventing EMT (36). However, overexpression of HDAC5
inhibits tumor cell growth and induces apoptosis in the
osteosarcoma U2OS cells (37).
Thus, distinct behavior of HDAC5 towards cell fate decisions in
different cancer types are worth studying in the future.
In conclusion, the present study demonstrates a poor
prognosis in NSCLC patients with high HDAC5 expression and a
negative correlation between miR-589-5p and HDAC5 in NSCLC
specimens. Further analysis showed that HDAC5 is directly targeted
by miR-589-5p, and hyper-methylation-mediated silence of miR-589-5p
results in the aberrant high expression of HDAC5 in NSCLC cells.
Moreover, miR-589-5p/HDAC5 pathway regulates the expression of cell
cycle and EMT-related gene clusters and consequently determines the
capabilities of proliferation, migration, invasion, and
tumorigenicity in NSCLC cells. Therefore, abnormal miR-589-5p/HDAC5
pathway caused by hypermethylation contributes to the
aggressiveness of NSCLC.
References
|
1
|
Álvarez-Fernández C and Esteban-González
E: Current status of EGFR/ErbB inhibitors in non-small cell lung
carcinoma. Med Clin (Barc). 146(Suppl 1): 2–6. 2016.In Spanish.
View Article : Google Scholar
|
|
2
|
Juergens RA, Wrangle J, Vendetti FP,
Murphy SC, Zhao M, Coleman B, Sebree R, Rodgers K, Hooker CM,
Franco N, et al: Combination epigenetic therapy has efficacy in
patients with refractory advanced non-small cell lung cancer.
Cancer Discov. 1:598–607. 2011. View Article : Google Scholar
|
|
3
|
Martin M, Kettmann R and Dequiedt F: Class
IIa histone deacetylases: Conducting development and
differentiation. Int J Dev Biol. 53:291–301. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mottet D, Pirotte S, Lamour V, Hagedorn M,
Javerzat S, Bikfalvi A, Bellahcène A, Verdin E and Castronovo V:
HDAC4 represses p21 (WAF1/Cip1) expression in human cancer cells
through a Sp1-dependent, p53-independent mechanism. Oncogene.
28:243–256. 2009. View Article : Google Scholar
|
|
5
|
Feng GW, Dong LD, Shang WJ, Pang XL, Li
JF, Liu L and Wang Y: HDAC5 promotes cell proliferation in human
hepatocellular carcinoma by up-regulating Six1 expression. Eur Rev
Med Pharmacol Sci. 18:811–816. 2014.PubMed/NCBI
|
|
6
|
Cacan E: Histone deacetylase-1-mediated
suppression of FAS in chemoresistant ovarian cancer cells.
Anticancer Res. 36:2819–2826. 2016.PubMed/NCBI
|
|
7
|
Liu J, Gu J, Feng Z, Yang Y, Zhu N, Lu W
and Qi F: Both HDAC5 and HDAC6 are required for the proliferation
and metastasis of melanoma cells. J Transl Med. 14:72016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang L, Li H, Ren Y, Zou S, Fang W, Jiang
X, Jia L, Li M, Liu X, Yuan X, et al: Targeting HDAC with a novel
inhibitor effectively reverses paclitaxel resistance in non-small
cell lung cancer via multiple mechanisms. Cell Death Dis.
7:e20632016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang K, Zhang H, Zhou X, Tang WB, Xiao L,
Liu YH, Liu H, Peng YM, Sun L and Liu FY: miRNA589 regulates
epithelial-mesenchymal transition in human peritoneal mesothelial
cells. J Biomed Biotechnol. 2012:6730962012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li W, Wang W, Ding M, Zheng X, Ma S and
Wang X: MiR-1244 sensitizes the resistance of non-small cell lung
cancer A549 cell to cisplatin. Cancer Cell Int. 16:302016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang X, Jiang P, Shuai L, Chen K, Li Z,
Zhang Y, Jiang Y and Li X: miR-589-5p inhibits MAP3K8 and
suppresses CD90+ cancer stem cells in hepatocellular
carcinoma. J Exp Clin Cancer Res. 35:1762016. View Article : Google Scholar
|
|
12
|
Dávalos-Salas M, Furlan-Magaril M,
González-Buendía E, Valdes-Quezada C, Ayala-Ortega E and
Recillas-Targa F: Gain of DNA methylation is enhanced in the
absence of CTCF at the human retinoblastoma gene promoter. BMC
Cancer. 11:2322011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brzeziańska E, Dutkowska A and Antczak A:
The significance of epigenetic alterations in lung carcinogenesis.
Mol Biol Rep. 40:309–325. 2013. View Article : Google Scholar
|
|
14
|
Balgkouranidou I, Liloglou T and Lianidou
ES: Lung cancer epigenetics: Emerging biomarkers. Biomarkers Med.
7:49–58. 2013. View Article : Google Scholar
|
|
15
|
Miranda TB and Jones PA: DNA methylation:
The nuts and bolts of repression. J Cell Physiol. 213:384–390.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mehta A, Dobersch S, Romero-Olmedo AJ and
Barreto G: Epigenetics in lung cancer diagnosis and therapy. Cancer
Metastasis Rev. 34:229–241. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Patel K, Dickson J, Din S, Macleod K,
Jodrell D and Ramsahoye B: Targeting of 5-aza-2′-deoxycytidine
residues by chromatin-associated DNMT1 induces proteasomal
degradation of the free enzyme. Nucleic Acids Res. 38:4313–4324.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Damiani LA, Yingling CM, Leng S, Romo PE,
Nakamura J and Belinsky SA: Carcinogen-induced gene promoter
hypermethylation is mediated by DNMT1 and causal for transformation
of immortalized bronchial epithelial cells. Cancer Res.
68:9005–9014. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Forde PM, Brahmer JR and Kelly RJ: New
strategies in lung cancer: Epigenetic therapy for non-small cell
lung cancer. Clin Cancer Res. 20:2244–2248. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu ZH, Li J, Xia J, Jiang R, Zuo GW, Li
XP, Chen Y, Xiong W and Chen DL: Ginsenoside 20 (s)-Rh2 as potent
natural histone deacetylase inhibitors suppressing the growth of
human leukemia cells. Chem Biol Interact. 242:227–234. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang J, Ng S, Wang J, Zhou J, Tan SH,
Yang N, Lin Q, Xia D and Shen HM: Histone deacetylase inhibitors
induce autophagy through FOXO1-dependent pathways. Autophagy.
11:629–642. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jang SM, Kang EJ, Kim JW, Kim CH, An JH
and Choi KH: Transcription factor Sox4 is required for
PUMA-mediated apoptosis induced by histone deacetylase inhibitor,
TSA. Biochem Biophys Res Commun. 438:445–451. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li A, Liu Z, Li M, Zhou S, Xu Y, Xiao Y
and Yang W: HDAC5, a potential therapeutic target and prognostic
biomarker, promotes proliferation, invasion and migration in human
breast cancer. Oncotarget. 7:37966–37978. 2016.PubMed/NCBI
|
|
24
|
Milde T, Oehme I, Korshunov A,
Kopp-Schneider A, Remke M, Northcott P, Deubzer HE, Lodrini M,
Taylor MD, von Deimling A, et al: HDAC5 and HDAC9 in
medulloblastoma: Novel markers for risk stratification and role in
tumor cell growth. Clin Cancer Res. 16:3240–3252. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fan J, Lou B, Chen W, Zhang J, Lin S, Lv
FF and Chen Y: Downregulation of HDAC5 inhibits growth of human
hepatocellular carcinoma by induction of apoptosis and cell cycle
arrest. Tumour Biol. 35:11523–11532. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
He P, Liang J, Shao T, Guo Y, Hou Y and Li
Y: HDAC5 promotes colorectal cancer cell proliferation by
up-regulating DLL4 expression. Int J Clin Exp Med. 8:6510–6516.
2015.PubMed/NCBI
|
|
27
|
Gorgoulis VG, Zacharatos P, Mariatos G,
Kotsinas A, Bouda M, Kletsas D, Asimacopoulos PJ, Agnantis N,
Kittas C and Papavassiliou AG: Transcription factor E2F-1 acts as a
growth-promoting factor and is associated with adverse prognosis in
non-small cell lung carcinomas. J Pathol. 198:142–156. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Grossi F, Dal Bello MG, Salvi S, Puzone R,
Pfeffer U, Fontana V, Alama A, Rijavec E, Barletta G, Genova C, et
al: Expression of ribonucleotide reductase subunit-2 and
thymidylate synthase correlates with poor prognosis in patients
with resected stages I-III non-small cell lung cancer. Dis Markers.
2015:3026492015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Noro R, Miyanaga A, Minegishi Y, Okano T,
Seike M, Soeno C, Kataoka K, Matsuda K, Yoshimura A and Gemma A:
Histone deacetylase inhibitor enhances sensitivity of
non-small-cell lung cancer cells to 5-FU/S-1 via down-regulation of
thymidylate synthase expression and up-regulation of p21
(waf1/cip1) expression. Cancer Sci. 101:1424–1430. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tang YA, Lin RK, Tsai YT, Hsu HS, Yang YC,
Chen CY and Wang YC: MDM2 overexpression deregulates the
transcriptional control of RB/E2F leading to DNA methyltransferase
3A overexpression in lung cancer. Clin Cancer Res. 18:4325–4333.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li L and Wu D: miR-32 inhibits
proliferation, epithelial-mesenchymal transition, and metastasis by
targeting TWIST1 in non-small-cell lung cancer cells. Onco Targets
Ther. 9:1489–1498. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ávila-Moreno F, Armas-López L,
Álvarez-Moran AM, López-Bujanda Z, Ortiz-Quintero B,
Hidalgo-Miranda A, Urrea-Ramírez F, Rivera-Rosales RM,
Vázquez-Manríquez E, Peña-Mirabal E, et al: Overexpression of MEOX2
and TWIST1 is associated with H3K27me3 levels and determines lung
cancer chemoresistance and prognosis. PLoS One. 9:e1141042014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pérez-Ramírez C, Cañadas-Garre M, Molina
MA, Faus-Dáder MJ and Calleja-Hernández MA: PTEN and PI3K/AKT in
non-small-cell lung cancer. Pharmacogenomics. 16:1843–1862. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li J, Yang S, Yan W, Yang J, Qin YJ, Lin
XL, Xie RY, Wang SC, Jin W, Gao F, et al: MicroRNA-19 triggers
epithelial-mesenchymal transition of lung cancer cells accompanied
by growth inhibition. Lab Invest. 95:1056–1070. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yun F, Jia Y, Li X, Yuan L, Sun Q, Yu H,
Shi L and Yuan H: Clinicopathological significance of PTEN and
PI3K/AKT signal transduction pathway in non-small cell lung cancer.
Int J Clin Exp Pathol. 6:2112–2120. 2013.PubMed/NCBI
|
|
36
|
Liu Q, Sun Y, Zheng JM, Yan XL, Chen HM,
Chen JK and Huang HQ: Formononetin sensitizes glioma cells to
doxorubicin through preventing EMT via inhibition of histone
deacetylase 5. Int J Clin Exp Pathol. 8:6434–6441. 2015.PubMed/NCBI
|
|
37
|
Huang Y, Tan M, Gosink M, Wang KK and Sun
Y: Histone deacetylase 5 is not a p53 target gene, but its
overexpression inhibits tumor cell growth and induces apoptosis.
Cancer Res. 62:2913–2922. 2002.PubMed/NCBI
|