Introduction
The retrovirus, human T-cell leukemia virus type 1
(HTLV-1), is associated with adult T-cell leukemia/lymphoma (ATLL)
and chronic inflammatory disorders such as HTLV-1-associated
myelopathy/tropical spastic paraparesis (HAM/TSP), uveitis and
arthritis (1). Globally,
approximately 20 million people are estimated to be infected by
HTLV-1, and 90% of them remain asymptomatic carriers during their
lives (2). ATLL is a highly
aggressive malignancy of mature CD4+ T cells and
develops only after a long period of latency, usually several
decades (1). Despite advances in
the development of novel therapeutic agents, the prognosis of ATLL
remains poor, and there is no definite cure for HAM/TSP (1). Many patients acquire resistance to
chemotherapeutic agents, leading to treatment failures. A higher
viral load in individuals with HTLV-1 infection increases their
risk of developing ATLL and HAM/TSP (3,4).
Therefore, reduction of the number of HTLV-1-infected T cells is
crucial in the prevention and treatment of HTLV-1-associated
diseases.
HTLV-1 induces oncogenesis through deregulation of
selected cellular signaling pathways. In the virally infected T
cells, activation of nuclear factor-κB (NF-κB), activator protein-1
(AP-1) and Akt results in upregulation of expression of a large
number of cellular genes involved in cell proliferation, survival
and metastasis (5,6). Thus, any new therapeutic agent must
be designed to target multiple signal transduction pathways for
maximum therapeutic benefits.
Natural phytochemicals, present in the human diet
with cancer preventive and anticancer properties, are suitable
alternatives (7). The plant
polyphenol, butein (3,4,2′,4′-tetrahydroxychalone), has been
traditionally used as an oriental medicine in Eastern Asia, and has
various biological functions (8,9).
Butein affects apoptosis, cell proliferation and the cell cycle in
diverse cancers (8,9). The preliminary results of a clinical
trial on the effects of butein-containing flavonoids in gastric
cancer patients showed it was safe with good tolerability, and
produced marked decrease in tumor size (10). Thus, butein seems to be a potent
anticancer agent. Several previous reports have suggested the
involvement of NF-κB, Akt, extracellular signal-regulated kinase,
p38 kinase and p53 in the induction of cell cycle arrest at
G1 or G2/M and apoptosis, and inhibition of
metastasis by butein in various cell lines (8,9,11–13).
Therefore, we hypothesized that butein may act as a chemopreventive
and/or chemotherapeutic agent against HTLV-1-associated
diseases.
We report herein that butein inhibits cell growth
and stimulates apoptosis of HTLV-1-infected T cells by inhibiting
NF-κB, AP-1 and Akt activities and signaling pathways. Importantly,
in a xenograft model, butein inhibited HTLV-1-infected T
cell-induced tumor growth. Our study revealed for the first time,
butein effect on HTLV-1-infected T cells both in vitro and
in vivo, and the molecular mechanism by which butein
exhibited anti-ATLL effects.
Materials and methods
Reagents used
Butein (cat. no. B3803) was purchased from Tokyo
Chemical Industry Co. (Tokyo, Japan), dissolved in dimethyl
sulfoxide (DMSO) and used for the treatment of cells. The broad
spectrum caspase inhibitor, Z-VAD-FMK (cat. no. G7232), was
purchased from Promega Corp. (Madison, WI, USA). Antibodies against
survivin (cat. no. 2808), Akt (cat. no. 9272), IκB kinase (IKK)α
(cat. no. 2682), IKKβ (cat. no. 2684), phospho-Akt (Thr308) (cat.
no. 13038), phospho-Akt (Ser473) (cat. no. 4060), phospho-IκBα
(Ser32 and 36) (cat. no. 9246), phospho-IKKα/β (Ser176/180 and
Ser177/181) (cat. no. 2694), RelA (cat. no. 8242), phospho-RelA
(Ser536) (cat. no. 3033), cleaved caspase-3 (cat. no. 9664), -8
(cat. no. 9496), -9 (cat. no. 9501) and poly(ADP-ribose) polymerase
(PARP) (cat. no. 9541) were purchased from Cell Signaling
Technology, Inc. (Beverly, MA, USA). Antibodies against Bcl-2 (cat.
no. MS-597), CDK2 (cat. no. MS-617), CDK4 (cat. no. MS-299), CDK6
(cat. no. MS-398), cyclin E (cat. no. MS-870), retinoblastoma
protein (pRb) (cat. no. MS-107) and actin (cat. no. MS-1295) were
obtained from Neomarkers, Inc. (Fremont, CA, USA). Antibodies
against XIAP (cat. no. M044-3), cyclin D1 (cat. no. K0062-3), heat
shock protein (HSP)70 (cat. no. SR-812C), phospho-pRb (Ser780)
(cat. no. M054-3S) were purchased from Medical & Biological
Laboratories, Co. (Aichi, Japan). Antibodies against cyclin D2
(cat. no. sc-593), c-IAP2 (cat. no. sc-7944), IκBα (cat. no.
sc-371), JunB (cat. no. sc-46) and JunD (cat. no. sc-74), and NF-κB
subunits p50 (cat. no. sc-114X), p52 (cat. no. sc-298X), RelA (cat.
no. sc-109X), c-Rel (cat. no. sc-70X) and RelB (cat. no. sc-226X),
and AP-1 subunits c-Fos (cat. no. sc-52X), FosB (cat. no. sc-48X),
Fra-1 (cat. no. sc-605X), Fra-2 (cat. no. sc-604X), c-Jun (cat. no.
sc-45X), JunB (cat. no. sc-46X) and JunD (cat. no. sc-74X) for
supershift assay were from Santa Cruz Biotechnology, Inc. (Santa
Cruz, CA, USA). Antibody against HSP90 (cat. no. 610418) was from
BD BioSciences (San Jose, CA, USA).
Cell culture
The HTLV-1-infected MT-4 and HUT-102, and
ATLL-derived TL-OmI T-cell lines, were cultured in RPMI-1640 medium
(cat. no. 30264-56, Nacalai Tesque, Inc., Kyoto, Japan)
supplemented with 10% heat-inactivated fetal bovine serum
(Biological Industries, Kibbutz Beit Haemek, Israel) and 1%
penicillin/streptomycin (cat. no. 09367-34, Nacalai Tesque, Inc.).
Cells were maintained in a 37°C, 5% CO2 humidified
incubator. The study also included peripheral blood mononuclear
cells (PBMCs) obtained from a healthy donor. PBMC proliferation was
induced with 20 µg/ml of phytohemagglutinin (PHA) (cat. no.
L8754, Sigma-Aldrich Co., St. Louis, MO, USA).
Cell proliferation and cytotoxic
assay
The cell proliferative and toxic effects of butein
were determined by the water-soluble tetrazolium (WST)-8 uptake
method. Cells were plated (1×104 cells per well) in
96-well microtiter plates in triplicate and treated with various
concentrations of butein. After incubation for 24–72 h, 10
µl of the WST-8 reagent (cat. no. 07553-44, Nacalai Tesque,
Inc.) was added to each well. After 4 h, WST-8 reduction was
measured at 450 nm using a 680 XR microplate reader (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Values were normalized to
untreated control samples. The 50% growth inhibitory concentration
(IC50) value was calculated by dotting the data points
to a logistic curve using the CalcuSym software (version 2.0;
Biosoft, Cambridge, UK).
Analysis of cell apoptosis
Cells were treated with vehicle or butein for 24–72
h and then permeabilized by incubation on ice for 20 min with 100
µg/ml of digitonin, and treated with
phycoerythrin-conjugated APO2.7 antibody (cat. no. IM2088, Beckman
Coulter, Inc., Marseille, France) for 15 min at room temperature.
After staining with APO2.7 antibody, apoptosis was determined by
Epics XL flow cytometry (Beckman Coulter, Inc., Brea, CA, USA). In
addition, for analysis of morphological changes in nuclei, cells
were stained by 10 µg/ml of Hoechst 33342 (cat. no.
346-07951, Dojindo Molecular Technologies, Inc., Kumamoto, Japan)
and observed under a Leica DMI6000 microscope (Leica Microsystems,
Wetzlar, Germany).
In vitro measurement of caspase
Activity
Caspase activity was measured using Colorimetric
Caspase Assay kits (cat. nos. 4800, 4805 and 4810, Medical &
Biological Laboratories, Co.). Briefly, cell extracts were
recovered using the cell lysis buffer supplied with the kit and
assessed for caspase-3, -8 and -9 activities using colorimetric
probes. The assay kits are based on detection of chromophore
ρ-nitroanilide after cleavage from caspase-specific labeled
substrates. Colorimetric readings were performed in an automated
microplate reader.
Cell cycle analysis
Cells were stained with the CycleTEST Plus DNA
Reagent kit (cat. no. 340242, Becton-Dickinson Immunocytometry
Systems, San Jose, CA, USA). The cell cycle distribution was
analyzed for 10,000 collected cells by an Epics XL flow cytometry
that uses the MultiCycle software (version 3.0; Phoenix Flow
Systems, San Diego, CA, USA). The population of nuclei at each
phase of the cell cycle was determined and apoptotic cells with
hypodiploid DNA content were detected in the
sub-G0/G1 region.
Western blot analysis
Whole cell extracts were prepared by subjecting
butein-treated cells to lysis in lysis buffer [62.5 mM Tris-HCl (pH
6.8) (cat. no. 35434-21, Nacalai Tesque, Inc.), 2% sodium dodecyl
sulfate (cat. no. 31607-65, Nacalai Tesque, Inc.), 10% glycerol
(cat. no. 17045-65, Nacalai Tesque, Inc.), 6% 2-mercaptoethanol
(cat. no. 21438-82, Nacalai Tesque, Inc.) and 0.01% bromophenol
blue (cat. no. 021-02911, Wako Pure Chemical Industries, Osaka,
Japan)]. Lysates were spun to remove insoluble material. The
supernatants were collected and kept at -80°C. Protein
concentrations were determined using a commercial kit (DC Protein
Assay, cat. no. 5000116JA, Bio-Rad Laboratories, Inc.). Equal
amounts of protein extracts (20 µg) were resolved on sodium
dodecyl sulfate-polyacrylamide gels (SDS-PAGE). After
electrophoresis, the proteins were electrotransferred onto
polyvinylidene difluoride membranes (cat. no. IPVH00010EMD,
Millipore, Darmstadt, Germany), blotted with the relevant
antibodies. Finally, blots were hybridized with horseradish
peroxidase-conjugated secondary anti-mouse (cat. no. 7076, Cell
Signaling Technology, Inc.) or anti-rabbit IgG antibody (cat. no.
7074, Cell Signaling Technology, Inc.) and developed using an
enhanced chemiluminescence reagent (cat. no. RPN2232, Amersham
Biosciences Corp., Piscataway, NJ, USA).
RT-PCR
TRIzol reagent (cat. no. 15596026, Invitrogen Life
Technologies, Carlsbad, CA, USA) was used to extract total RNA from
cells. RNA (1 µg) was reverse transcribed into cDNA using a
PrimeScript RT-PCR kit (cat. no. RR014A, Takara Bio Inc., Otsu,
Japan). The sequence-specific primers used for RT-PCR were as
follows: for IκBα, forward 5′-GCCTGGACTCCATGAAAGAC-3′, reverse
5′-CAAGTG GAGTGGAGTCTGCTGCAGGTTGTT-3′; and for GAPDH, forward
5′-GCCAAGGTCATCCATGACAACTTTGG-3′, reverse
5′-GCCTGCTTCACCACCTTCTTGATGTC-3′. PCR amplifications were performed
in a programmable thermal cycler. Results were analyzed at 30
cycles to obtain results at exponential levels of
amplification.
Electrophoretic mobility shift assay
(EMSA)
To determine NF-κB and AP-1 activation, we prepared
nuclear extracts from butein-treated cells and performed EMSA, as
previously described (14).
Nuclear extracts were incubated with 32P-labeled probes.
The top strand sequences of the oligonucleotide probes or
competitors were as follows: for a typical NF-κB element of the
interleukin-2 receptor α chain (IL-2Rα) gene,
5′-GATCCGGCAGGGGAATCTCCCTCTC-3′ and for the
consensus AP-1 element of the IL-8 gene,
5′-GATCGTGATGACTCAGGTT-3′. The above
underlined sequences are the NF-κB and AP-1 binding sites,
respectively. In competition experiments, nuclear extracts were
preincubated for 15 min with 100-fold excess of unlabeled
oligonucleotides. For supershift assays, nuclear extracts were
incubated with antibodies against NF-κB or AP-1 subunits for 45 min
at room temperature before the complex was analyzed by EMSA. The
dried gels were visualized.
Xenograft tumor model
Five-week-old female C.B-17/Icr-severe combined
immune deficient (SCID) mice were obtained from Kyudo, Co. (Tosu,
Japan). To induce malignancy, 1×107 HUT-102 cells
suspended in sterile RPMI-1640 medium (200 µl) were
inoculated subcutaneously into the postauricular region of SCID
mice. The mice were divided at random into two treatment groups
(n=6, each). Butein was solubilized in soybean oil (cat. no.
190-03776, Wako Pure Chemical Industries) and administered at a
dose of 0.7 mg/kg intraperitoneally three times a week, and the
treatment was continued for 27 days, beginning on the day after
cell inoculation. The control group received the vehicle only. The
tumor diameter was measured weekly with a shifting caliper and
tumor volume was calculated. The mice were also weighed weekly,
beginning on day 0. All mice were sacrificed on day 28. The tumors
were excised and their weight measured. At the end of the study,
blood samples were collected and the sera were separated by
centrifuging blood and then stored at −80°C until assayed for
secreted soluble IL-2Rα [soluble cluster of differentiation 25
(sCD25)] and sCD30. This experiment was performed according to the
Guidelines for Animal Experimentation of the University of the
Ryukyus (Nishihara, Japan), and was approved by the Animal Care and
Use Committee of the University of the Ryukyus (reference no.
5837).
Morphological analysis of tumor tissues
and terminal deoxynucleotidyl transferase deoxyuridine triphosphate
nick end labeling (TUNEL) assay
The tumor specimens were collected from the control
mice group and 0.7 mg/kg butein group, fixed in formalin (Wako Pure
Chemical Industries) solution, dehydrated through graded ethanol
series (Japan Alcohol Selling Co., Tokyo, Japan) and embedded in
paraffin (cat. no. 09620, Sakura Finetek Japan Co., Tokyo, Japan).
The paraffin-embedded specimens of ATLL tumors were stained with
hematoxylin and eosin (H&E, cat. nos. 234-12 and 1159350025,
Merck, Darmstadt, Germany) and examined histologically. Analysis of
DNA fragmentation by TUNEL testing was performed using a commercial
kit (cat. no. 11684817910, Roche Applied Science, Penzberg,
Germany). Cells were examined under a light microscope (Axioskop 2
Plus) with an Achroplan 40×/0.65 lens (both from Zeiss,
Hallbergmoos, Germany). Images were acquired with an AxioCam 503
color and AxioVision LE64 software (Zeiss).
Biomarker analysis
Serum concentrations of human sCD25 (cat. no.
DR2A00, R&D Systems, Inc., Minneapolis, MN, USA) and human
sCD30 (cat. no. SK00582-01, Aviscera Bioscience, Inc., Santa Clara,
CA, USA) were measured by enzyme-linked immunosorbent assay
(ELISA), according to the protocol supplied by the
manufacturer.
Statistical analysis
Values are shown as mean ± standard deviation (SD).
Data of two groups were compared by the Student's t-test.
Differences were considered significant at P<0.05.
Results
Effects of butein on cell proliferation
and cytotoxicity of HTLV-1-infected T-cell lines
To determine the in vitro effects of butein
in ATLL cells, two HTLV-1-infected T-cell lines (MT-4 and HUT-102)
and an ATLL-derived T-cell line (TL-OmI) were cultured in the
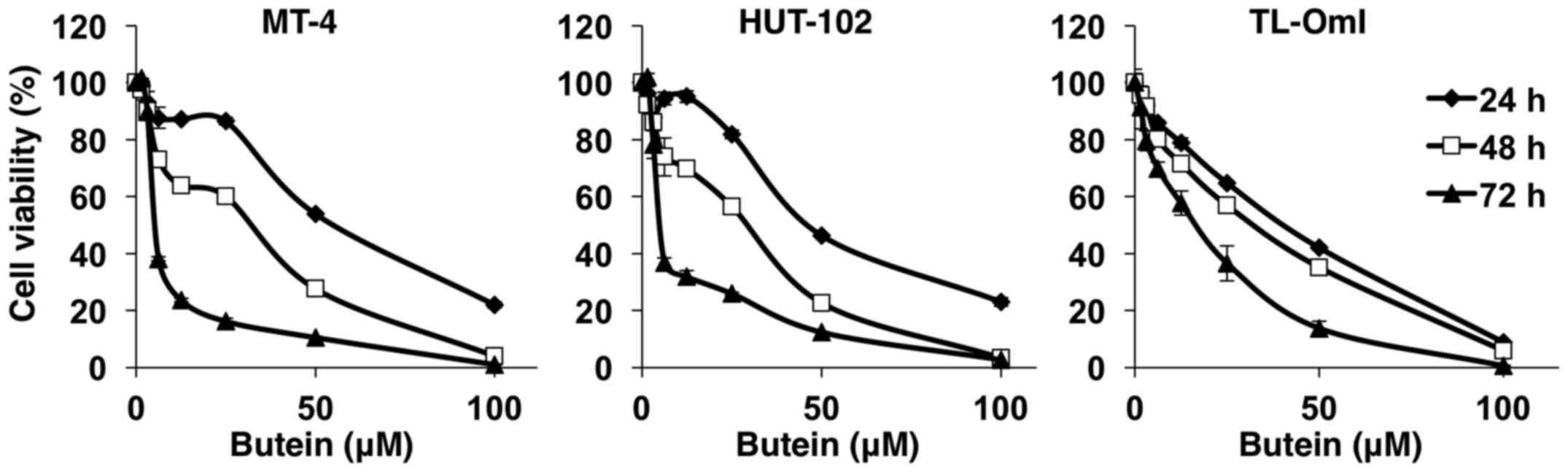
presence of butein (1.56–100 µM). As shown in Fig. 1, 24–72 h culture with butein
significantly reduced cell proliferation and viability in a
concentration- and time-dependent manner, with IC50
values at 72 h of 7.0 µM for HUT-102, 7.9 µM for MT-4
and 9.8 µM for TL-OmI. In contrast, the IC50 was
27.8 µM for normal PBMCs. Butein suppressed the
proliferative response of PBMCs to the T cell mitogen PHA, with
IC50 of 18.5 µM. Although the IC50
values for HTLV-1-infected T-cell lines were lower than those for
normal PBMCs, obvious selective cytotoxic activities of butein on
HTLV-1-infected T-cell lines have not been demonstrated.
Butein simultaneously induces cell
apoptosis and cell cycle arrest
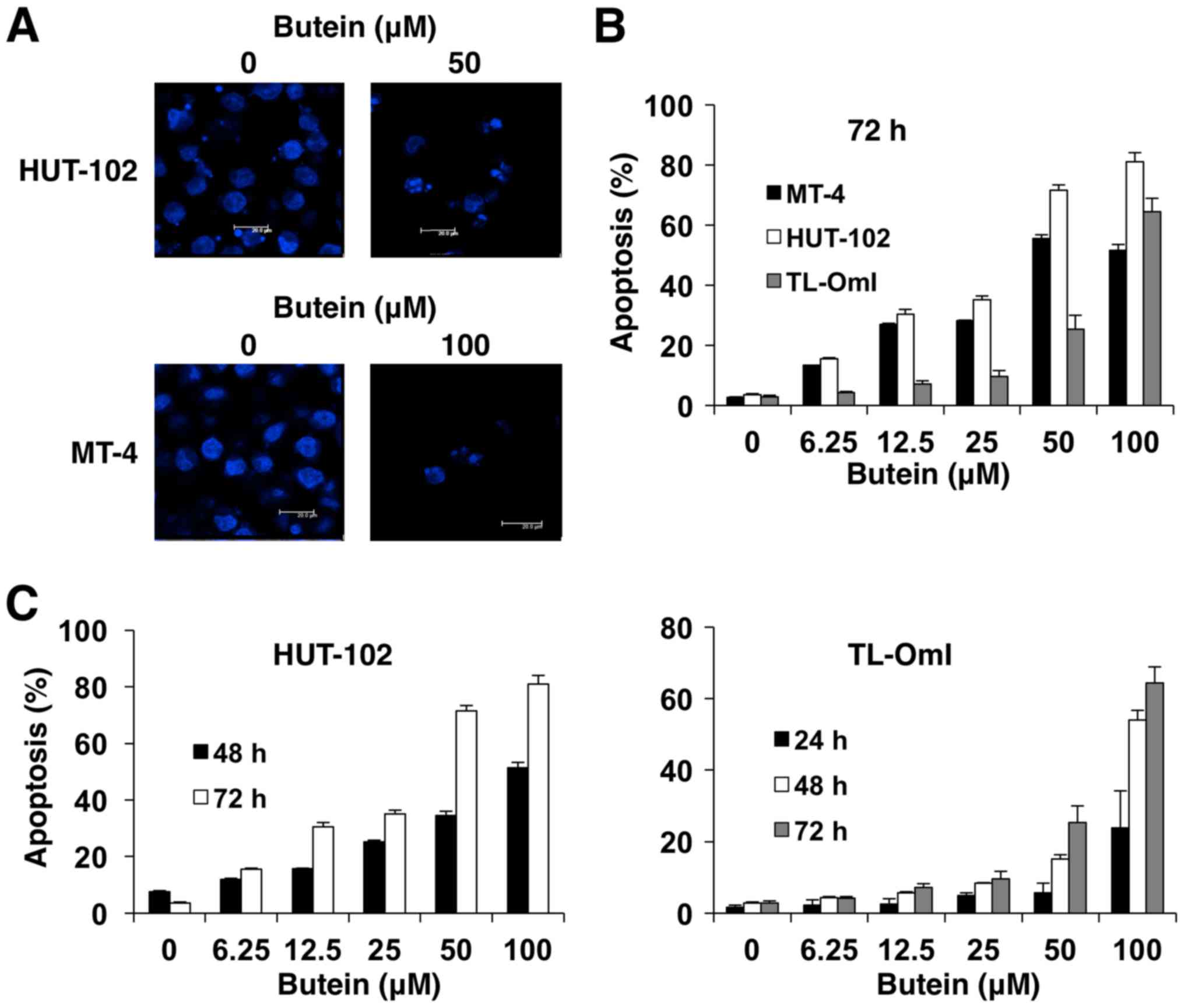
First, the effect of butein on cell morphology was
examined using microscopy. Cells cultured with butein for 48 h
demonstrated morphologies characteristic of apoptosis, such as
chromatin condensation and nuclear fragmentation (Fig. 2A). To further investigate the mode
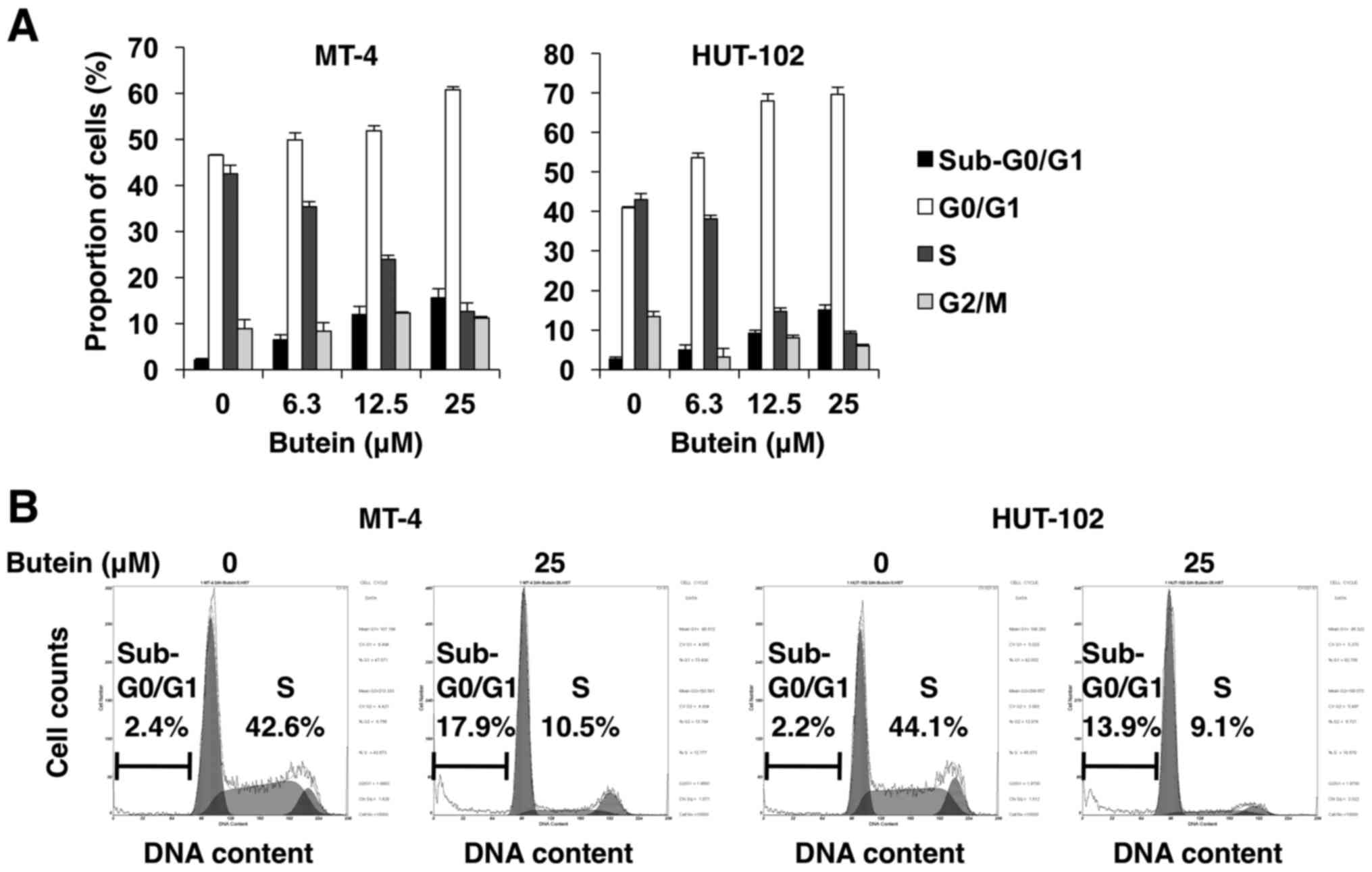
of butein-induced cell death, HTLV-1-infected T-cell lines were
treated with 6.3–25 µM butein for 24 h. Butein induced
concentration-dependent increases in the
sub-G0/G1 population (Fig. 3A and B). We also assessed the
expression of 38-kDa mitochondrial membrane antigen APO2.7 during
apoptosis (15). Apoptotic cells
(APO2.7-positive) increased after treatment with butein
concentration- and time-dependently (Fig. 2B and C). Collectively, these
results indicate that butein induces apoptosis of HTLV-1-infected
T-cell lines. In addition to apoptosis, the same experiment also
showed that butein dose-dependently increased the percentage of
cells in the G0/G1 phase and also reduced the
percentage of cells in the S phase (Fig. 3A and B). Considered together, the
results showed the arrest of more cells at the
G0/G1 phase of the cell cycle.
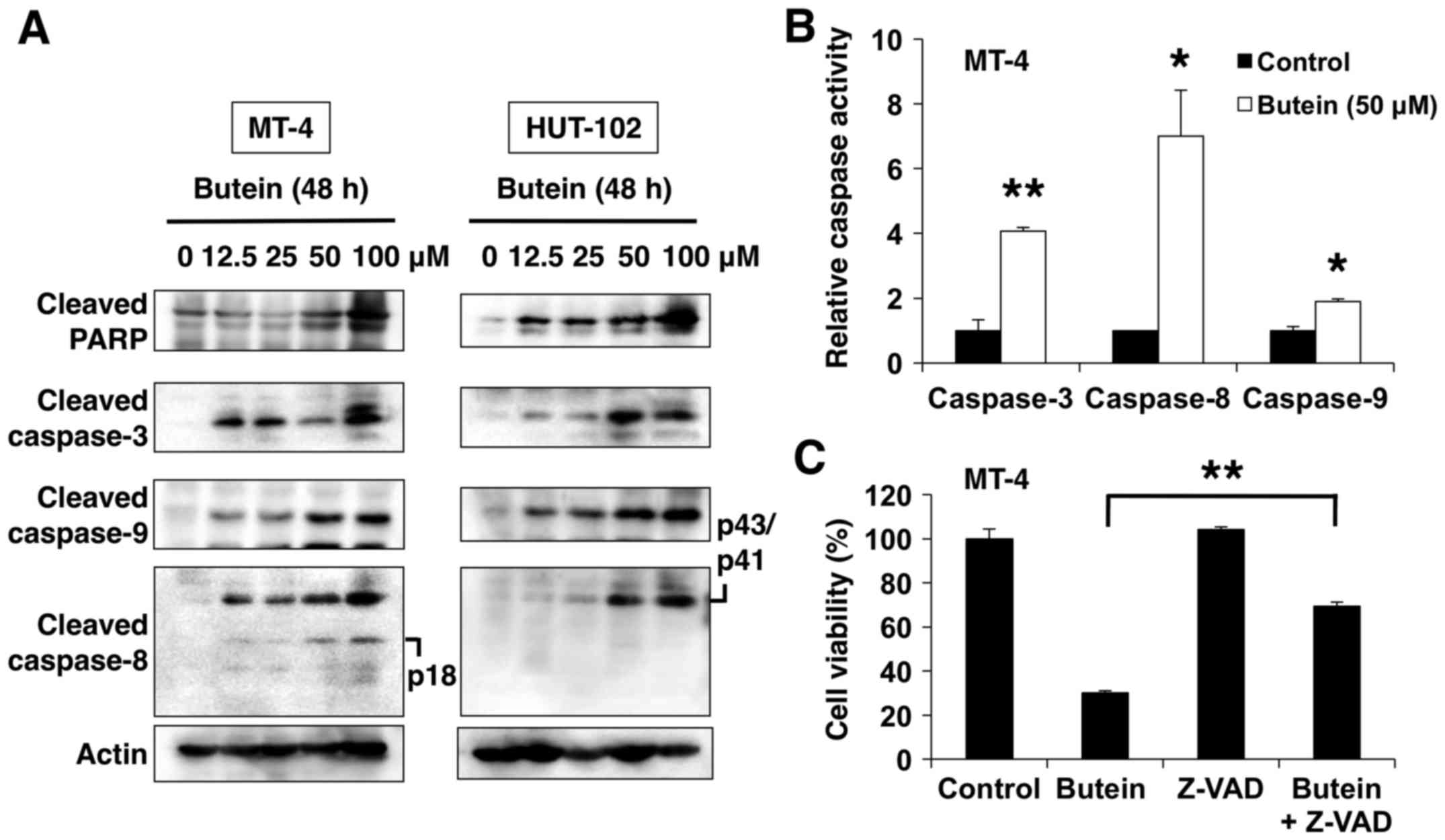
Butein induces caspase activation
The apoptotic process is executed by a member of the
highly conserved cysteine proteases, caspases. Initiator caspase-8
and -9 or effector caspase-3 are synthesized as inactive proenzymes
and become finally activated by proteolytic cleavage (16). To identify the mechanism underlying
butein-induced apoptosis of HTLV-1-infected T cells, we
investigated cleavage of caspase-3, -8 and -9, and also PARP, a
substrate for caspase-3. Exposure of MT-4 and HUT-102 cells to
butein (12.5–100 µM for 48 h) caused concentration-dependent
increase in the cleavage of caspase-3, -8 and -9, and PARP
(Fig. 4A), as well as increase in
activities of caspase-3, -8 and -9 (Fig. 4B). To further explore the
significance of the latter activation, a broad spectrum caspase
inhibitor, Z-VAD-FMK, was used to suppress the effect of butein.
Pretreatment of cells with Z-VAD-FMK partially attenuated
butein-induced inhibition of cell viability (Fig. 4C), suggesting that butein-mediated
cytotoxic effect is mediated, at least in part, through a
caspase-dependent apoptosis pathway.
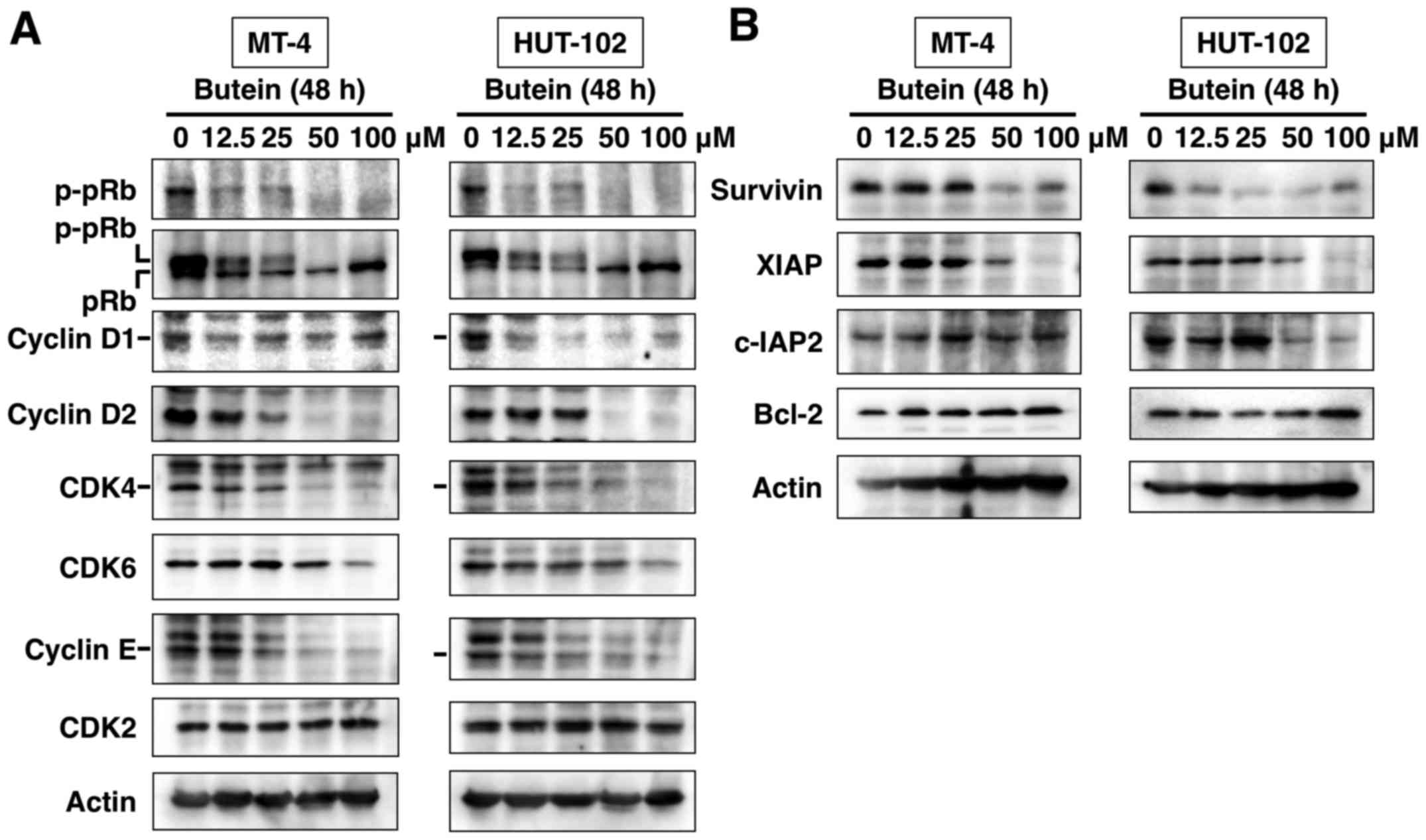
Effects of butein on regulatory proteins
involved in cell cycle regulation and apoptosis
Cell cycle progression is tightly regulated by
interaction between CDKs and cyclins (17). To identify the molecular mechanism
underlying butein-induced G0/G1 arrest, cells
were cultured with butein (12.5–100 µM) for 48 h. They were
harvested for protein extraction and western blot analysis. As
shown in Fig. 5A, levels of CDK4,
CDK6, cyclin D2 and cyclin E proteins were significantly decreased
in butein-treated MT-4 and HUT-102 cells, compared to DMSO-treated
cells. Butein also inhibited the expression of cyclin D1 in HUT-102
but not in MT-4 cells. However, butein had no significant effect on
CDK2 protein level. Furthermore, butein induced dephosphorylation
of pRb protein (Fig. 5A, top
panels) and changed the hyperphosphorylated form to a
hypophosphorylated form of pRB (Fig.
5A, second panels). The deregulation of apoptosis is often
studied by the expression of anti-apoptotic proteins. There was a
dose-dependent decrease in the protein expression of survivin and
XIAP, but not Bcl-2, in butein-treated MT-4 and HUT-102 cells.
Butein also inhibited the expression of c-IAP2 in HUT-102 but not
in MT-4 cells (Fig. 5B). The
effects of butein seem to slightly differ dependent on cell
lines.
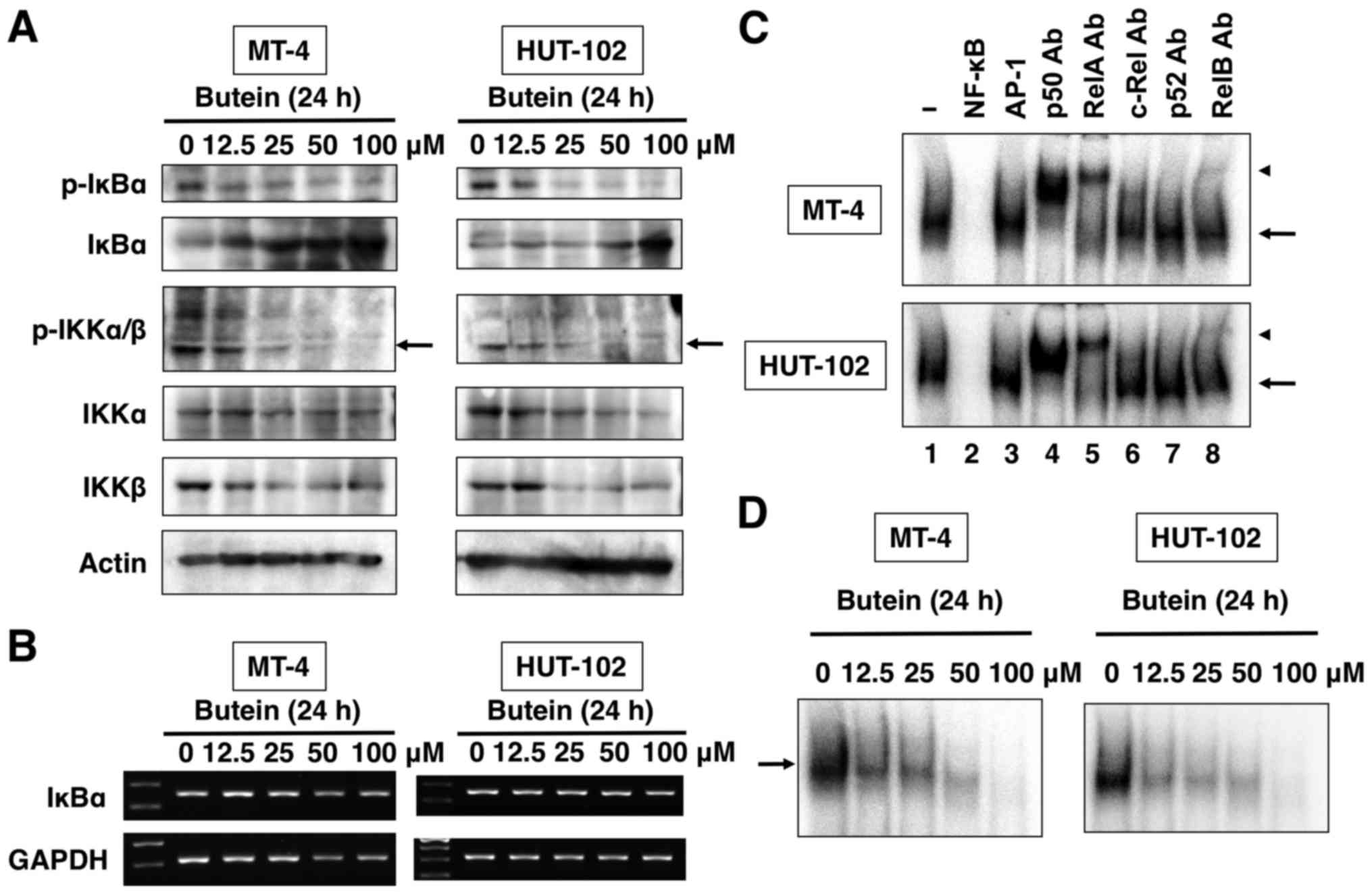
Butein inhibits NF-κB in HTLV-1-infected
T-cell lines
NF-κB forms a family of transcription factors that
regulate numerous biological processes, including immune response,
inflammation, cell growth, survival and development. The NF-κB
proteins are usually sequestered in the cytoplasm by a family of
inhibitors, including IκBα. IκBα degradation is mediated through
its phosphorylation by IKK, a trimeric complex composed of two
catalytic subunits, IKKα and IKKβ, and a regulatory subunit, IKKγ
(18). Incubation of MT-4 and
HUT-102 cells with butein resulted in significant dose-dependent
inhibition of phosphorylation and upregulation of IκBα protein
(Fig. 6A). To determine whether
butein-mediated upregulation of IκBα is due to transcriptional
activation of IκBα, we examined the expression of IκBα mRNA in
cells treated with butein by RT-PCR. As shown in Fig. 6B, butein treatment had no influence
on IκBα mRNA, suggesting that butein induces the inhibition of
degradation of IκBα protein. Next, we measured total and
phospho-protein levels of IKKα and IKKβ to evaluate the possible
inhibitory mechanism of butein on IκBα protein phosphorylation.
Immunoblot analysis demonstrated butein dose-dependent suppression
of phospho-protein levels of IKKα and IKKβ in MT-4 and HUT-102
cells. Total protein levels of both IKKα and IKKβ were reduced in
HUT-102 cells, while total protein level of IKKβ alone was reduced
in MT-4 cells (Fig. 6A). Thus, the
effects of butein seem to slightly differ dependent on cell
lines.
NF-κB is a complex of proteins, in which various
combinations of NF-κB proteins constitute active NF-κB heterodimers
that bind to specific DNA sequences. Thus, to show that the band
visualized by EMSA in HTLV-1-infected T-cell lines was indeed
NF-κB, nuclear extracts from MT-4 and HUT-102 cells were incubated
with antibodies to the NF-κB subunits and analyzed by EMSA. The
results showed shifts of the bands to higher molecular masses
(Fig. 6C), suggesting that the
activated complex consisted of p50, RelA and RelB. The addition of
excess unlabeled NF-κB caused complete disappearance of the band,
whereas the addition of AP-1 oligonucleotide had no effect on the
DNA binding. We also investigated whether butein could inhibit
constitutive NF-κB in HTLV-1-infected T-cell lines. MT-4 and
HUT-102 cells were cultured in the presence of different
concentrations of butein for 24 h and then analyzed for NF-κB
activation. Butein dose-dependently inhibited constitutive NF-κB
activation in HTLV-1-infected T-cell lines (Fig. 6D). These results indicate that
butein can suppress constitutively active NF-κB in HTLV-1-infected
T cells.
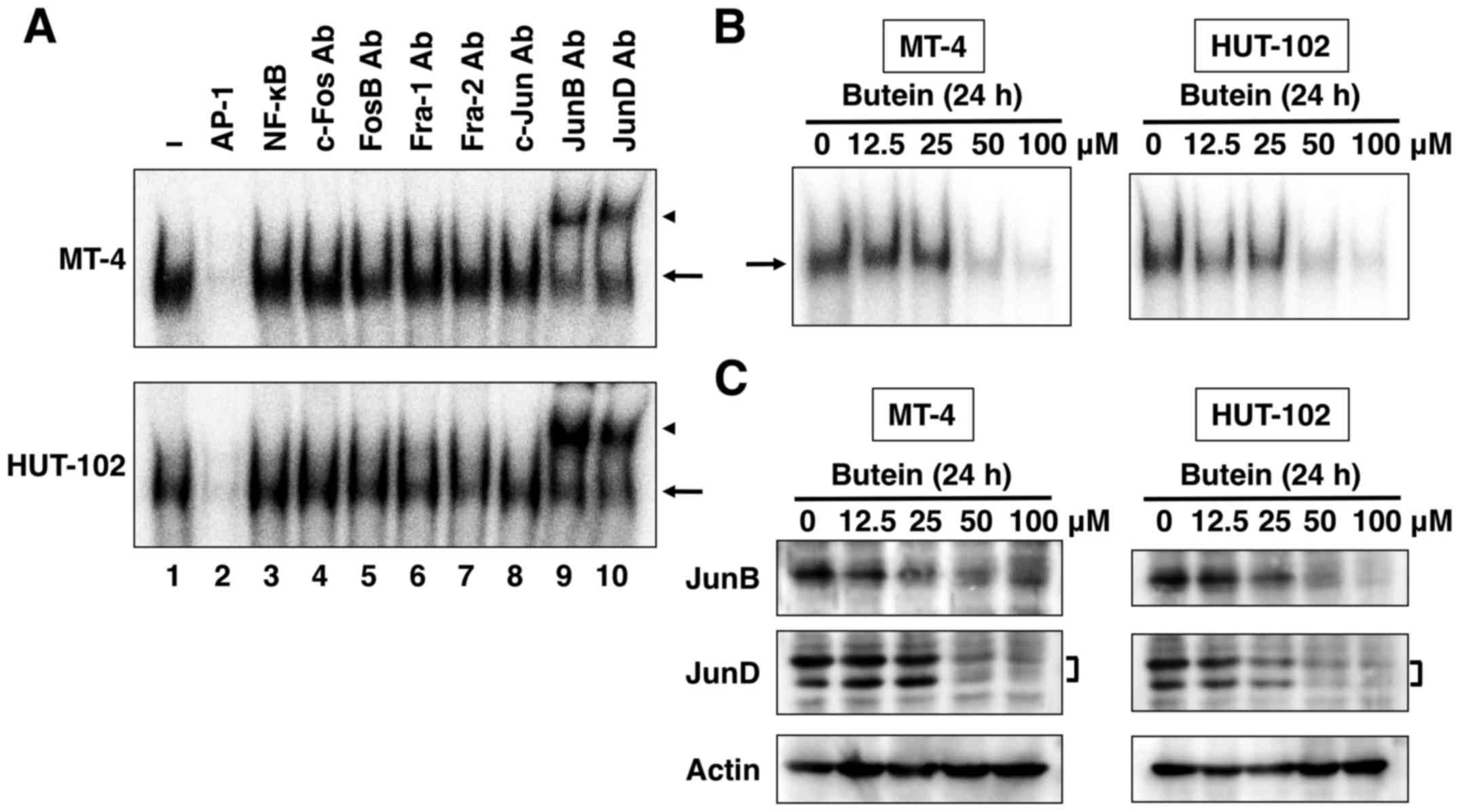
Butein suppresses AP-1 activity
AP-1 is a group of dimeric transcription factor
complexes composed of members of the Fos (c-Fos, FosB, Fra-1 and
Fra-2) and Jun (c-Jun, JunB and JunD) families, which play central
roles in the proliferation and transformation of T cells (6). EMSA showed supershifted band for
AP-1, as detected with specific antibodies to JunB and JunD in MT-4
and HUT-102 cells (Fig. 7A). As
shown in Fig. 7B, butein produced
a significant and dose-dependent suppression of AP-1 activation in
HTLV-1-infected T-cell lines. As shown in Fig. 7A, JunB and JunD are functional
components of AP-1 transcription factor in HTLV-1-infected T-cell
lines. The experiment demonstrated that butein inhibited AP-1
signaling pathway through dose-dependent suppression of JunB and
JunD protein levels (Fig. 7C).
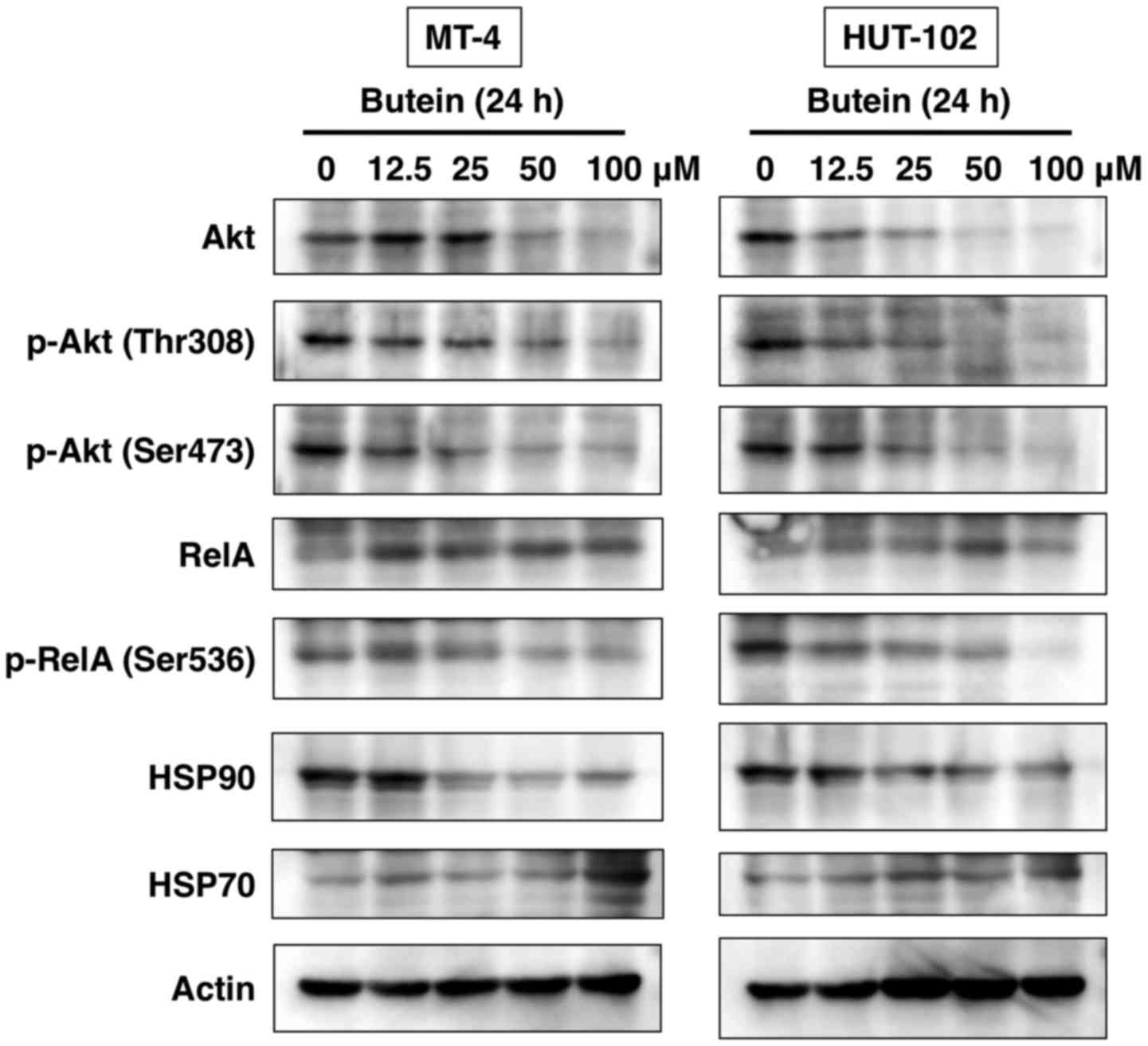
Butein inhibits Akt and HSP90 protein
expression in HTLV-1-infected T-cell lines
Akt is a component of an essential pathway for cell
survival and growth during development and carcinogenesis. It
regulates cell survival by directly targeting XIAP and by
indirectly modulating survivin levels (19,20).
Akt also regulates cell cycle and proliferation by indirectly
modulating the levels of cyclin D1, cyclin D2 and cyclin E
(21). Since butein decreased the
levels of XIAP, survivin, cyclin D1, cyclin D2 and cyclin E in the
treated cells, we determined its effect on Akt protein. Butein at
50–100 µM for MT-4 and 12.5–100 µM for HUT-102 cells,
caused a dose-dependent inhibition of Akt protein expression.
Treatment of MT-4 cells with butein at 50–100 µM and HUT-102
cells with butein at 12.5–100 µM caused a dose-dependent
inhibition in Akt phosphorylation at Thr308. Butein at 12.5–100
µM for MT-4 and 25–100 µM for HUT-102 cells, also
inhibited the phosphorylation of Akt at Ser473 (Fig. 8). RelA is phosphorylated at Ser536
by Akt and such phosphorylation enhances RelA trans-activation
potential (22). Butein also
caused inhibition of RelA phosphorylation (Fig. 8). The molecular chaperone HSP90 is
involved in the stabilization and conformational maturation of many
signaling proteins that are deregulated in cancers. HSP90 client
proteins include signaling proteins (IKKs and Akt) and cell cycle
regulators (CDKs) (23).
Therefore, we investigated the effect of butein on the expression
of HSP90. As shown in Fig. 8,
butein reduced HSP90 protein levels, but increased HSP70
levels.
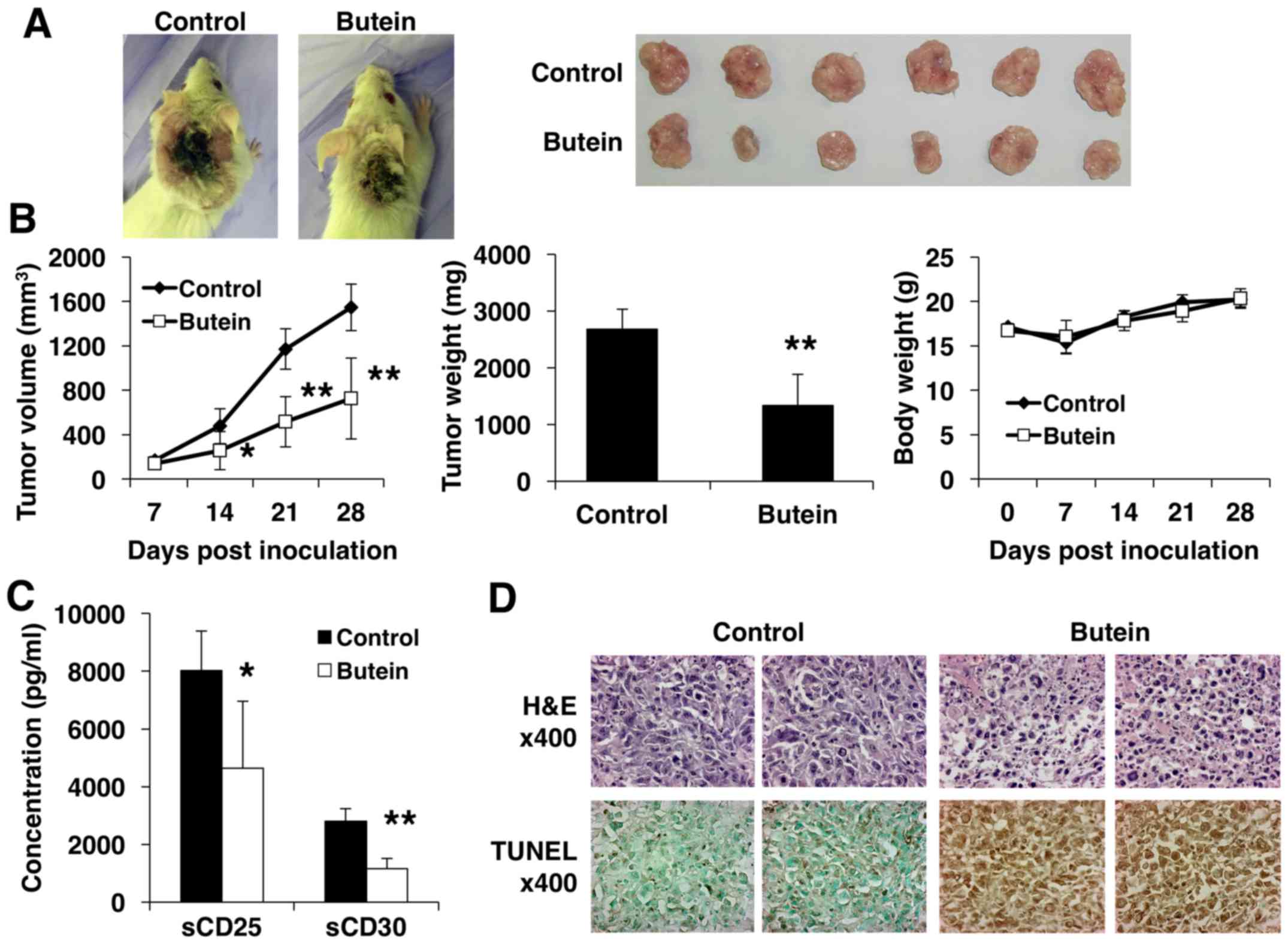
Butein suppresses growth of
HTLV-1-infected T-cell line in SCID mice
Butein significantly inhibited tumor growth in SCID
mice inoculated with HTLV-1-infected HUT-102 cells (Fig. 9A, left panels). In the control
group, the average tumor volume of 1,545 mm3 was reached
in 28 days after HUT-102 cell inoculation (Fig. 9B, left panel). At the same time
point, the average tumor volume in the butein-treated group was 727
mm3. Tumor tissues were removed, photographed (Fig. 9A, right panel), and weighed. The
mean tumor weight of butein-treated mice was significantly lower
than that of vehicle-treated mice (Fig. 9B, middle panel). In addition, the
body weight of mice remained stable with no significant differences
between the two groups (Fig. 9B,
right panel). At post-inoculation day 28, quantitative ELISA used
to determine the circulating levels of surrogate tumor markers
sCD25 (24) and sCD30 (25) secreted by HUT-102 tumor xenografts
showed 42% and 59% decrease in their levels, respectively, in
butein-treated mice, compared with the control group (Fig. 9C). Considered together, our results
showed that treatment with butein resulted in significant decreases
in serum sCD25 and sCD30 of SCID mice.
Finally, H&E and TUNEL staining assays were
performed to assess whether the inhibitory effect on tumor growth
induced by butein was due to an increase in apoptotic cell death.
H&E staining showed a significant increase in apoptotic tumor
cells in butein-treated mice, compared with the control group,
which were characterized by cytoplasmic condensation, chromatin
hyperchromatism and condensation, and nuclear fragmentation. TUNEL
assays confirmed these findings and demonstrated increased number
of TUNEL-positive tumor cells in butein-treated mice (Fig. 9D).
Discussion
ATLL is classified into four clinical subtypes based
on the clinical manifestations: acute, lymphoma, chronic and
smoldering (26). Acute, lymphoma
and unfavorable chronic types are considered the aggressive forms
of ATLL, while favorable chronic and smoldering types are indolent
ATLL (26). Various dietary and
lifestyle factors may be involved in the development and
progression of ATLL. Aggressive ATLL carries poor prognosis, with
only 24% for 3-year overall survival rate in patients on standard
combination chemotherapy (27).
Therefore, there is a great need for novel therapeutic approaches
for ATLL. Chemotherapeutic agents are usually highly toxic to
normal tissues. To reduce the number of HTLV-1-infected T cells,
natural products are suitable alternatives since they have minimal
side-effects compared to synthetic drugs.
In this study, we showed that butein decreased cell
viability of HTLV-1-infected T-cell lines. The ability of the cells
to divide is regulated by two classes of molecules, CDKs, a family
of serine/threonine kinases and their binding partners, cyclins.
The progress of the cells division process occurs through the
interaction of various cyclins with their respective CDK subunits
(28). Quiescent cells enter the
cell cycle after mitogenic stimulation and upregulation of cyclins
D and E during the G1 phase of the cell cycle (29). Cyclin D is associated with CDK4 and
CDK6 (30), while cyclin E is
associated with CDK2 (31). Thus,
cyclins and CDKs play important roles in DNA synthesis and cell
division. In contrast, pRb controls cell cycle progression at the
G1 to S transition in response to certain signals for
growth inhibition (32). pRb is
phosphorylated by catalytic subunits of CDK4/CDK6 and CDK2
complexed with specific regulatory subunits of cyclins D and E,
respectively. Treatment of HTLV-1-infected T-cell lines with butein
reduced the expression levels of cyclin D1, D2, E, CDK4, CDK6 and
phospho-pRb. Based on these findings, it is highly possible that
butein causes G1 arrest by blocking cyclin D-CDK4/6- and
cyclin E-CDK2-mediated pRb phosphorylation, which is crucial to the
progression of the cell cycle at the G1 to S
transition.
The process of apoptosis is controlled by various
interrelated pathways, which ensure that caspases, the proteolytic
initiators, and executioners of apoptosis, are triggered only in
cells requiring termination. We have shown in the present study the
activation of caspase-3, -8 and -9 during the application of butein
and that the addition of a general caspase inhibitor partially
decreased butein-mediated cell death. These data provide evidence
that butein-induced apoptosis was at least in part
caspase-dependent in HTLV-1-infected T-cell lines. Butein also
downregulated the protein expression of survivin, XIAP and c-IAP2,
which are possible upstream factors of caspase proteins.
NF-κB can modulate the transcriptional activation of
genes associated with cell proliferation, metastasis, tumor
promotion and suppression of apoptosis (33,34).
Hence, agents capable of suppressing NF-κB activation are
therapeutically promising in inhibiting carcinogenesis. Our results
suggest that the effects of butein on NF-κB were mediated through
inhibition of phosphorylation and the associated subsequent
proteolysis of IκBα since butein blocked phosphorylation and
degradation of IκBα. Furthermore, butein inhibited total and
phospho-protein levels of IKKα and IKKβ, and phosphorylation of
RelA. Treatment of HTLV-1-infected T-cell lines with butein further
inhibited NF-κB-regulated gene products, survivin, XIAP, c-IAP2,
cyclin D1, cyclin D2, CDK4 and CDK6.
Akt occupies a key regulatory node in the
phosphoinositide 3-kinase pathway, below which the pathway branches
significantly to influence a wide range of cellular processes that
promote cell cycle progression, cell growth and resistance to
apoptosis by modulating the levels of survivin, XIAP, cyclin D1,
cyclin D2 and cyclin E (19–21).
Our results demonstrated that butein decreased total and
phospho-protein levels of Akt in HTLV-1-infected T-cells. Akt is
known to phosphorylate RelA at Ser536 through an IKKα- or
IKKβ-dependent mechanism (35).
Butein may inhibit phosphorylation of RelA through inactivation of
Akt and IKKs. In addition, AP-1 regulates the expression and
function of various cell cycle regulators, such as cyclin D1 and
cyclin E (36). JunB is also
controlled by NF-κB (37). Thus,
NF-κB and AP-1 can also collaborate in ATLL, depending on a variety
of factors. Butein inhibited AP-1 signaling pathway via reduction
of JunB and JunD. Considered together, these findings suggest that
the effects of butein are mediated through a diverse range of
molecular targets, which collectively result in inhibition of cell
growth and survival.
Of note, butein profoundly inhibited HSP90 protein.
Previous studies showed that HSP90 inhibitors simultaneously
inactivated and destabilized several oncoproteins (e.g., IKKs, Akt
and CDKs) in cancer cells (23,38).
On the other hand, inhibition of HSP90 alone induces upregulation
of another chaperone, HSP70 (38).
Our results also showed that butein increased the expression of
HSP70. HSP70 is known to be anti-apoptotic, and therefore the
induction of HSP70 may ultimately limit the efficacy of HSP90
inhibitors (38). Collectively,
these results suggest that butein also inhibits HSP90, thereby
affecting various signaling pathways involved in the regulation of
cell growth and survival.
To establish the relevance of these in vitro
findings to the intact animal, we implanted SCID mice with
HTLV-1-infected T cell line HUT-102. Treatment with butein
significantly decreased HUT-102 growth in these mice and
significantly reduced serum levels of sCD25 and sCD30 without
affecting body weight. The results also showed upregulation of
TUNEL-positive cells undergoing early apoptosis in tissues of mice
treated with butein, relative to the control mice.
In conclusion, we demonstrated in the present study
the effects of butein, a plant polyphenol, on the growth of
HTLV-1-infected T-cell lines in both in vitro and in an
in vivo animal model. Although this study warrants further
investigation on ATLL primary samples, the results suggest that
butein is a potentially valuable chemopreventive and/or
chemotherapeutic agent against ATLL.
Acknowledgments
We thank Fujisaki Cell Center, Hayashibara
Biochemical Laboratories, Inc. (Okayama, Japan) for providing
HUT-102. This work was supported in part by JSPS KAKENHI grant
numbers, 25830085 and 25461428, and a grant from the Mishima Kaiun
Memorial Foundation.
References
|
1
|
Gonçalves DU, Proietti FA, Ribas JG,
Araújo MG, Pinheiro SR, Guedes AC and Carneiro-Proietti AB:
Epidemiology, treatment, and prevention of human T-cell leukemia
virus type 1-associated diseases. Clin Microbiol Rev. 23:577–589.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gessain A and Cassar O: Epidemiological
aspects and world distribution of HTLV-1 infection. Front
Microbiol. 3:3882012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Okayama A, Stuver S, Matsuoka M, Ishizaki
J, Tanaka G, Kubuki Y, Mueller N, Hsieh CC, Tachibana N and
Tsubouchi H: Role of HTLV-1 proviral DNA load and clonality in the
development of adult T-cell leukemia/lymphoma in asymptomatic
carriers. Int J Cancer. 110:621–625. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nagai M, Usuku K, Matsumoto W, Kodama D,
Takenouchi N, Moritoyo T, Hashiguchi S, Ichinose M, Bangham CR,
Izumo S, et al: Analysis of HTLV-I proviral load in 202 HAM/TSP
patients and 243 asymptomatic HTLV-I carriers: High proviral load
strongly predisposes to HAM/TSP. J Neurovirol. 4:586–593. 1998.
View Article : Google Scholar
|
|
5
|
Mori N: Cell signaling modifiers for
molecular targeted therapy in ATLL. Front Biosci (Landmark Ed).
14:1479–1489. 2009. View
Article : Google Scholar
|
|
6
|
Hall WW and Fujii M: Deregulation of
cell-signaling pathways in HTLV-1 infection. Oncogene.
24:5965–5975. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Surh Y-J: Cancer chemoprevention with
dietary phytochemicals. Nat Rev Cancer. 3:768–780. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yadav VR, Prasad S, Sung B and Aggarwal
BB: The role of chalcones in suppression of NF-κB-mediated
inflammation and cancer. Int Immunopharmacol. 11:295–309. 2011.
View Article : Google Scholar
|
|
9
|
Gupta SC, Kim JH, Prasad S and Aggarwal
BB: Regulation of survival, proliferation, invasion, angiogenesis,
and metastasis of tumor cells through modulation of inflammatory
pathways by nutraceuticals. Cancer Metastasis Rev. 29:405–434.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee S-H, Choi W-C, Kim K-S, Park J-W, Lee
S-H and Yoon S-W: Shrinkage of gastric cancer in an elderly patient
who received Rhus verniciflua Stokes extract. J Altern Complement
Med. 16:497–500. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Woo S-M, Choi YK, Kim AJ, Cho S-G and Ko
S-G: p53 causes butein-mediated apoptosis of chronic myeloid
leukemia cells. Mol Med Rep. 13:1091–1096. 2016.
|
|
12
|
Zhang L, Yang X, Li X, Li C, Zhao L, Zhou
Y and Hou H: Butein sensitizes HeLa cells to cisplatin through the
AKT and ERK/p38 MAPK pathways by targeting FoxO3a. Int J Mol Med.
36:957–966. 2015.PubMed/NCBI
|
|
13
|
Bai X, Ma Y and Zhang G: Butein suppresses
cervical cancer growth through the PI3K/AKT/mTOR pathway. Oncol
Rep. 33:3085–3092. 2015.PubMed/NCBI
|
|
14
|
Mori N and Prager D: Transactivation of
the interleukin-1alpha promoter by human T-cell leukemia virus type
I and type II Tax proteins. Blood. 87:3410–3417. 1996.PubMed/NCBI
|
|
15
|
Zhang C, Ao Z, Seth A and Schlossman SF: A
mitochondrial membrane protein defined by a novel monoclonal
antibody is preferentially detected in apoptotic cells. J Immunol.
157:3980–3987. 1996.PubMed/NCBI
|
|
16
|
Khan N, Afaq F and Mukhtar H: Apoptosis by
dietary factors: The suicide solution for delaying cancer growth.
Carcinogenesis. 28:233–239. 2007. View Article : Google Scholar
|
|
17
|
Lim S and Kaldis P: Cdks, cyclins and
CKIs: Roles beyond cell cycle regulation. Development.
140:3079–3093. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hayden MS and Ghosh S: Shared principles
in NF-kappaB signaling. Cell. 132:344–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dan HC, Sun M, Kaneko S, Feldman RI,
Nicosia SV, Wang H-G, Tsang BK and Cheng JQ: Akt phosphorylation
and stabilization of X-linked inhibitor of apoptosis protein
(XIAP). J Biol Chem. 279:5405–5412. 2004. View Article : Google Scholar
|
|
20
|
Vaira V, Lee CW, Goel HL, Bosari S,
Languino LR and Altieri DC: Regulation of survivin expression by
IGF-1/mTOR signaling. Oncogene. 26:2678–2684. 2007. View Article : Google Scholar
|
|
21
|
Zhang X, Tang N, Hadden TJ and Rishi AK:
Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta.
1813:1978–1986. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sizemore N, Leung S and Stark GR:
Activation of phosphatidylinositol 3-kinase in response to
interleukin-1 leads to phosphorylation and activation of the
NF-kappaB p65/RelA subunit. Mol Cell Biol. 19:4798–4805. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kamal A, Boehm MF and Burrows FJ:
Therapeutic and diagnostic implications of Hsp90 activation. Trends
Mol Med. 10:283–290. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kamihira S, Atogami S, Sohda H, Momita S,
Yamada Y and Tomonaga M: Significance of soluble interleukin-2
receptor levels for evaluation of the progression of adult T-cell
leukemia. Cancer. 73:2753–2758. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nishioka C, Takemoto S, Kataoka S,
Yamanaka S, Moriki T, Shoda M, Watanabe T and Taguchi H: Serum
level of soluble CD30 correlates with the aggressiveness of adult
T-cell leukemia/lymphoma. Cancer Sci. 96:810–815. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shimoyama M: Diagnostic criteria and
classification of clinical subtypes of adult T-cell
leukaemia-lymphoma. A report from the Lymphoma Study Group
(1984–87). Br J Haematol. 79:428–437. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tsukasaki K, Utsunomiya A, Fukuda H,
Shibata T, Fukushima T, Takatsuka Y, Ikeda S, Masuda M, Nagoshi H,
Ueda R, et al Japan Clinical Oncology Group Study JCOG9801:
VCAP-AMP-VECP compared with biweekly CHOP for adult T-cell
leukemia-lymphoma: Japan Clinical Oncology Group Study JCOG9801. J
Clin Oncol. 25:5458–5464. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Morgan DO: Cyclin-dependent kinases:
Engines, clocks, and microprocessors. Annu Rev Cell Dev Biol.
13:261–291. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lew DJ, Dulić V and Reed SI: Isolation of
three novel human cyclins by rescue of G1 cyclin (Cln) function in
yeast. Cell. 66:1197–1206. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bates S, Bonetta L, MacAllan D, Parry D,
Holder A, Dickson C and Peters G: CDK6 (PLSTIRE) and CDK4 (PSK-J3)
are a distinct subset of the cyclin-dependent kinases that
associate with cyclin D1. Oncogene. 9:71–79. 1994.PubMed/NCBI
|
|
31
|
Koff A, Giordano A, Desai D, Yamashita K,
Harper JW, Elledge S, Nishimoto T, Morgan DO, Franza BR and Roberts
JM: Formation and activation of a cyclin E-cdk2 complex during the
G1 phase of the human cell cycle. Science. 257:1689–1694. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Weinberg RA: The retinoblastoma protein
and cell cycle control. Cell. 81:323–330. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ben-Neriah Y and Karin M: Inflammation
meets cancer, with NF-κB as the matchmaker. Nat Immunol.
12:715–723. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jost PJ and Ruland J: Aberrant NF-kappaB
signaling in lymphoma: Mechanisms, consequences, and therapeutic
implications. Blood. 109:2700–2707. 2007.
|
|
35
|
Viatour P, Merville MP, Bours V and
Chariot A: Phosphorylation of NF-kappaB and IkappaB proteins:
Implications in cancer and inflammation. Trends Biochem Sci.
30:43–52. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hess J, Angel P and Schorpp-Kistner M:
AP-1 subunits: Quarrel and harmony among siblings. J Cell Sci.
117:5965–5973. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pahl HL: Activators and target genes of
Rel/NF-kappaB transcription factors. Oncogene. 18:6853–6866. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Drysdale MJ, Brough PA, Massey A, Jensen
MR and Schoepfer J: Targeting Hsp90 for the treatment of cancer.
Curr Opin Drug Discov Devel. 9:483–495. 2006.PubMed/NCBI
|