Introduction
Melanoma is a type of cancer which occurs in
melanocytes. Melanocytes are responsible for the production of the
pigment melanin and are derived from the neural crest.
Approximately 90% of melanoma occurs in skin (cutaneous melanoma),
but it also rarely arises from the mucosal surfaces or areas which
neural cells migrate. Examples are the eye, intestine and mouth.
Malignant melanoma is very common among Caucasians (1). A total of 76,380 new cases of
melanoma and 10,130 related deaths were expected in 2016 in the
USA. Surgical resection, applications of immunotherapy, biologic
therapy, radiation therapy, or chemotherapy may improve survival
(2,3).
PKC belongs to the protein kinase enzyme family
which post-translationally modify other proteins by phosphorylation
of hydroxyl groups of serine and threonine amino acid residues, so
PKCs tend to be involved in many signal transduction cascades.
There are 15 PKC isozymes identified in humans; they are divided as
classical, novel and atypical (aPKCs). aPKCs contain two
structurally and functionally distinct isozymes: PKC-ι and PKC-ζ
which are involved in cell cycle progression, tumorigenesis,
survival and migration in many cancers (4–7).
Lung cancer cell proliferation is highly dependent on the PKC-ι
level through activation of the ERK1 pathway (6). Overexpression of PKC-ι plays an
important role in the leukemia chemoresistance (7). PKC-ι is also involved in glioma cell
proliferation; it regulates the phosphorylation of cyclin dependent
kinase activating kinase/cyclin dependent kinase 7 pathway
(8,9). Non-small lung cancer cell
proliferation is highly dependent on PKC-ι (10). aPKCs are involved in TGF-β induced
EMT by phosphorylating Par6 (11).
We believe that both aPKCs are involved in EMT process of melanoma
cells by regulating the formation of vimentin intermediate filament
(VIF) assembly. EMT is an important event of tumor progression
where the apicobasolaterally polarized cohesive epithelial cells of
epithelium detach from its basement and gain the ability of
mesenchymal cells, which move independently. EMT is characterized
by the loss of E-cadherin and gain of vimentin. During such
morphological changes of cells in EMT, cytoskeletal intermediate
filaments undergo a massive composition change by initiating the
expression of vimentin. Even though the role of vimentin in EMT is
not fully understood, some literature strongly supports the
relevance of VIF assembly in EMT as being part of tumorigenesis
(12,13).
Selzer et al (14) has reported a comprehensive
comparison of PKC isoform expression between normal melanocytes,
transformed melanocytes and melanoma cell lines. PKC-ι may play a
role in cellular malignancy as shown by its association with the
transformed phenotype of human melanomas in vivo and in
vitro. PKC-ι and PKC-ζ are overexpressed in both transformed
and malignant melanoma. In comparison, PKC-ζ was detected in low
levels in normal melanocytes while PKC-ι was not detected. We also
report that PKC-ι and PKC-ζ are overexpressed in SK-MEL-2 malignant
melanoma cell line compared to undetectable levels of PKC-ι and low
levels of PKC-ζ in PCS-200-013 normal melanocytes (15).
In the present study, we have investigated the
effects of the novel aPKC specific inhibitors ACPD and DNDA on the
proliferation, apoptosis, migration and invasion of two normal
melanocyte cell lines (PCS-200-013 and MEL-F-NEO) and two malignant
melanoma cell lines (SK-MEL-2 and MeWo). In this study, we showed
that both inhibitors can decrease the levels of total and
phosphorylated PKC-ζ and PKC-ι levels. We also found that both
inhibitors can decrease the levels of both phosphorylated and total
vimentin and increase E-cadherin levels which are associated with
EMT. Treatment with inhibitors altered the levels of CD44,
β-catenin, NF-κB p65, Par6, RhoA, AKT and PTEN whose roles are
established in either cell survival or metastasis (16–18).
Furthermore, we established that melanoma cells proliferate via
aPKC/AKT/NF-κB mediated pathway while inducing the EMT via
PKC-ι/Par6/RhoA pathway. We report that PKC-ι activates vimentin by
phosphorylation. We also report that treatment with ACPD/DNDA
induced apoptosis as shown by increasing caspase-3 levels,
decreasing Bcl-2 levels and changes in Poly(ADP-ribose) polymerase
(PARP) levels in addition to cell proliferation and downregulation
of EMT in melanoma cells.
Materials and methods
Materials
ACPD (product no. R426911) was purchased from
Sigma-Aldrich (St. Louis, MO, USA) and DNDA was obtained from the
National Institute of health (NIH; Bethesda, MD, USA). They were
dissolved in sterile distilled water (vehicle) before use.
Antibodies were purchased as follows. PKC-ι (cat. no. 610175) and
Bcl-2 (cat. no. 610538) were from BD Biosciences (San Jose, CA,
USA), PKC-ζ (cat. no. sc-17781), NF-κB p65 (cat. no. sc-372-G) and
caspase-3 (cat. no. sc-7272) from Santa Cruz Biotechnology (Santa
Cruz, CA, USA), phospho PKC-ζ (Thr 410) (cat. no. PA5-17837),
phospho PKC-ι (Thr 555) (cat. no. 44-968G), E-cadherin (cat. no.
701134), vimentin (cat. no. MA3-745) and myelin basic protein (MBP)
(cat. no. PA1-10008) from Thermo Fisher Scientific (Waltham, MA,
USA), phospho-vimentin (Ser39) (cat. no. 13614S), PTEN (cat. no.
9559S), phospho PTEN (Ser380) (cat. no. 9551), AKT (cat. no.
4691X), phospho AKT (Ser473) (cat. no. 4059S), PARP (cat. no. 9532)
and cleaved-PARP (cat. no. 9185) from Cell Signaling Technology
(Danvers, MA, USA). β-actinperoxidase (cat. no. A3854) from
Sigma-Aldrich. CD44 (cat. no. ab97478), RhoA (cat. no. ab54835) and
β-catenin (cat. no. ab16051) from Abcam (Cambridge, UK).
Phospho-MBP (Thr 125) (cat. no. 05-429) from EMD Millipore
(Billerica, MA, USA). Enhanced chemiluminescence solution (product
no. 34080) was purchased from Pierce (Rockford, IL, USA).
Dulbecco's phosphate-buffered saline without Mg2+ and
Ca2+ (DPBS) (product no. D8537) and Trypsin-EDTA
(ethylenediaminetetraacetic acid) solution (product no. T4049) were
purchased from Sigma-Aldrich. WST-1 reagent for cell proliferation
(cat. no. 11644807001) was purchased from Roche Diagnostics
(Mannhelm, Germany). Basement membrane extraction (BME) and
Calcein-AM solutions were purchased from Trevigen (Gaithersburg,
MD, USA) and Molecular Probes (Eugene, OR, USA), respectively.
Human small interfering RNA (siRNA) for PKC-ι (cat. no.
SR303741) and for PKC-ζ (cat. no. 303747) were purchased from
Origene Technologies Inc. (Rockville, MD, USA). Human recombinant
proteins PKC-ι (PV3183), PKC-ζ (P2273) and MBP (MBS717422) were
purchased from Thermo Fisher Scientific and MyBioSource (San Diego,
CA, USA), respectively.
Database preparation and molecular
docking
Database preparation was performed using the
National Cancer Institute/Developmental Therapeutics Program
(NCI/DTP) and molecular docking was performed using 'AutoDockTools'
and 'AutoDock Vina' programs by selecting structural pockets in
PKC-ι and PKC-ζ which were compatible with small drug like
molecules. PKC-ι and PKC-ζ structural pockets were identified based
on 'fpocket', a very fast open source protein pocket (cavity)
detection system based on Voronoi Tessellation. The detailed
procedure was performed as described in Pillai et al
(19).
Cell culture
PCS-200-013, SK-MEL-2 and MeWo cell lines were
purchased from the American Type Tissue Culture Collection (ATCC;
Rockville, MD, USA) and MEL-F-NEO cell line was purchased from
Zen-Bio, Inc. (Research Triangle Park, NC, USA). Furthermore, cells
were cultured at 37°C and 5% CO2.
Dermal cell basal medium (PCS-200-030) with
melanocyte growth kit (PCS-200-042) were used for PCS-200-013 and
melanocyte growth medium (MEL-2) were used for MEL-F-NEO cell
culturing according to the respective instruction manual. Eagle's
minimum essential media (EMEM) (90% v/v) with fetal bovine serum
(FBS) (10% v/v) and penicillin (5 µg/ml) were used for
SK-MEL-2 and MeWo cell culturing. All cell lines were seeded and
grown as monolayers in T25 or T75 flasks.
PKC activity assay
PKC activity assay was conducted by monitoring the
phosphorylation of myelin basic protein (MBP) (0.025 mg/ml), a
known substrate for PKCs. The detailed procedure was performed as
described in the study by Pillai et al (19) for both ACPD and DNDA on recombinant
PKC-ι and PKC-ζ (0.01 µg/µl) using a series of
inhibitor concentrations (0–10 µM). Samples then
fractionated by SDS-PAGE and immunoblotted. Kinase activity was
calculated based on the densitometry values of western blots
(WB).
Inhibition of expression of PKC-ι and
PKC-ζ with siRNA
SK-MEL-2 and MeWo cells (4×104) were
cultured in T25 flasks and treated with either siRNA (20 nM)
for PKC-ι or PKC-ζ or scrambled siRNA after 24 h
post-plating time and incubated for 48 h. Detailed procedure was
performed as described in the study by Win and Acevedo-Duncan
(20).
Inhibitor dose response curves for cell
viability
PCS-200-013, MEL-F-NEO, SK-MEL-2 and MeWo cells
(4×104) were cultured in T25 flasks and treated with
either an equal volume of sterile water (vehicle control) or ACPD
or DNDA (0.1–3.5 µM). Additional doses of sterile water or
ACPD or DNDA were supplied every 24 h during a 3-day incubation
period and cells were subsequently lifted using Trypsin-EDTA
solution (1.5 ml/flask) and neutralized with the equal volume of
media. Subsequently, live cells were counted using the Scepter, an
automated cell counter from Millipore (Billerica, MA, USA) at 24-h
intervals. Cell counts obtained from Scepter were compared with the
counts obtained from the Cellometer Vision from Nexcelom Bioscience
(Lawrence, MA, USA).
WST-1 assay for cell viability and
cytotoxicity
Approximately 4×103 cells/well
(PCS-200-013, MEL-F-NEO, SK-MEL-2 and MeWo) were cultured in a
96-well plate. After 24-h post plating time, fresh media were
supplied (200 µl/well) and treated with either an equal
volume of sterile water (vehicle control) or with the half maximal
inhibitory concentration (IC50) of ACPD (2.5 µM)
or DNDA (2.5 µM). This IC50 was obtained based on
the cell viability counts in the previous experiments. Additional
doses were supplied every 24 h during a 3-day incubation period. At
the end of the 3-day treatment, media were removed and fresh media
(100 µl) were added with
4-[3-(4-iodophenyl)-2-(4-nitropheny))-2H-5-tetrazolio]-1,3-benzene
disulfonate (WST-1) reagent (10 µl) to each well. The
absorbance was measured at 450 nm for every 1 h up to 6–10 h using
the Synergy HT microplate reader from BioTek Instruments Inc.
(Winooski, VT, USA).
Assays for cell migration and
invasion
Wound healing assay
The detailed procedure was performed for SK-MEL-2
and MeWo cells as described by O'Connell et al (21). Cells were treated with either
sterile water or ACPD or DNDA to achieve the final concentration of
2.5 µM and plates were incubated at 37°C and 5%
CO2. Photographs of wound closure were taken utilizing a
Motic AE31E microscope (×40 magnification) at 24-h intervals for 4
days.
Basement membrane extract (BME)
invasion assay (Boyden chamber assay)
This in vitro invasion assay was performed
for SK-MEL-2 and MeWo cells as described by O'Connell et al
(21). BME (0.5×) was used instead
of Matrigel. Crystal violet (0.5%) was used to stain the cells
adhered to the bottom of the lower chamber in order to visualize
the inhibition of invasion. Images of the stained cells were taken
from Motic AE31E microscope (×40 magnification).
Immunoprecipitation and western blot
analysis
Approximately 1×105 cells (SK-MEL-2 and
MeWo) were cultured in T75 flasks and 24 h post-plating, fresh
media were supplied and cells were treated with either an equal
volume of sterile water or ACPD or DNDA (2.5 µM). Additional
doses were supplied every 24 h during a 3-day incubation period.
Cells were then lifted using Trypsin and cell lysate were collected
either with cell lysis buffer (cat. no. C7027; Invitrogen) or IP
lysis buffer (cat. no. 87788; Thermo Fisher Scientific). The WB and
immunoprecipitation were performed as described in the study by Win
et al (22) and samples
were then fractionated by SDS-PAGE and immunoblotted.
Densitometry
The intensity of each WB band was measured using
'AlphaView' software for 'Fluorchem' systems developed by
ProteinSimple (San Jose, CA, USA) in which the background intensity
was subtracted from the intensity of each band to obtain the
corrected intensity of the proteins.
Statistical analysis
All data are presented as mean ± SD. Statistical
analysis was performed with one or two-way ANOVA followed by
Tukey's HSD test as multiple comparisons tests using the
'VassarStats' web tool for statistical analysis. P≤0.05 or P≤0.01
indicated statistical significance.
Results
Specific binding of ACPD and DNDA to
aPKCs
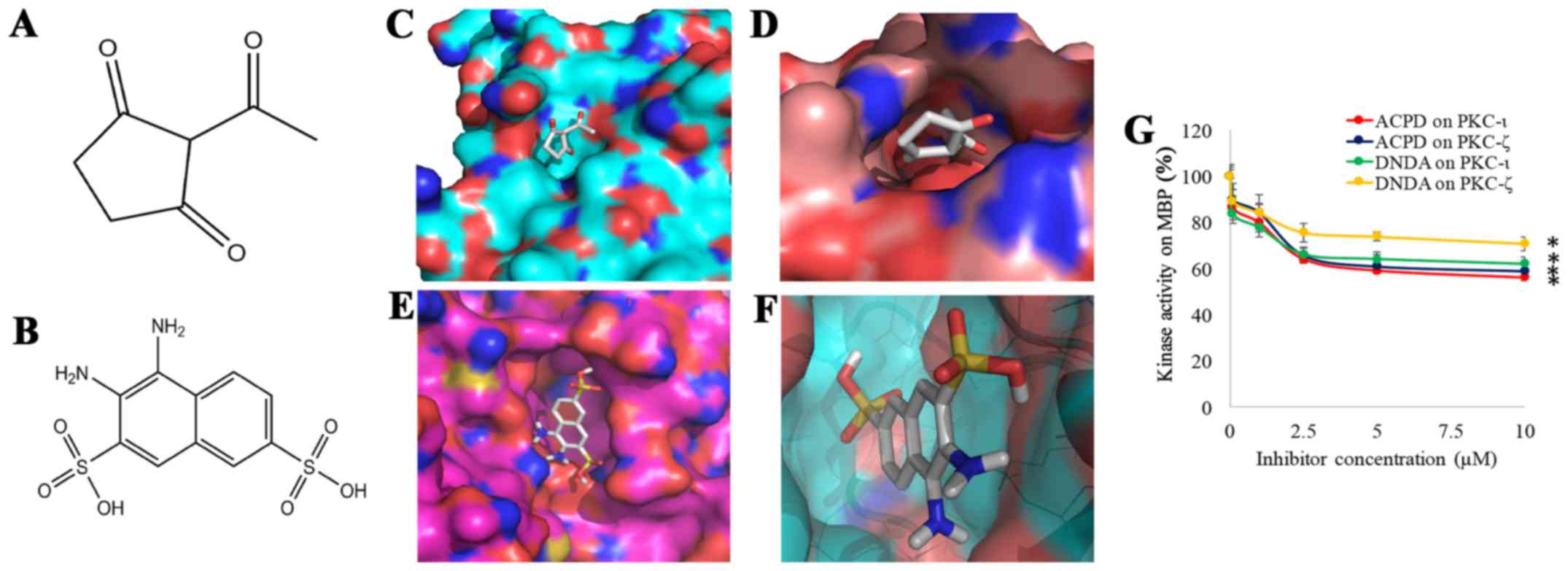
To establish the therapeutic potential of aPKCs,
ACPD (Fig. 1A) and DNDA (Fig. 1B) were identified based on
molecular docking (MD). Approximately 3×105 drug like
organic compounds (molecular weight <500 g/mol) in NCI/DTP, were
screened by positioning them in the structural pockets of PKC-ι and
PKC-ζ and then scored based on predicted polar and non-polar
interactions. ACPD was found to interact with amino acid residues
Gln 469, Ile 470, Lys 485 and Leu 488 of the catalytic domain of
PKC-ι (Fig. 1C) and Arg 265, Pro
267, Asp 269 and Lys 290 of the catalytic domain of PKC-ζ (Fig. 1D). DNDA interacts with amino acid
residues of Asp 339, Asp 382, Leu 385 and Thr 395 of the catalytic
domain of PKC-ι (Fig. 1E) and Asp
337, Asp 380, Leu 383 and Thr 393 of the catalytic domain of PKC-ζ
(Fig. 1F). Approximately -7
kcal/mol docking score was obtained for ACPD and DNDA separately
for PKC-ι and PKC-ζ for 4 different pockets. Sixteen pockets were
identified and tested for both PKC-ι and PKC-ζ separately and all
the pockets that scored above -6.5 kcal/mol were rejected to
identify these specific binding sites of the inhibitors. The
results here suggest that both ACPD and DNDA interact with PKC-ι
and PKC-ζ in a fairly equal manner.
Specific kinase activities of ACPD and
DNDA
Determination of specific activity of inhibitors was
essential since over 70% similarity is observed in the primary
structures of PKC-ι and PKC-ζ catalytic domains (5,23,24).
Specificity of ACPD was previously reported as it inhibits both
PKC-ι and PKC-ζ without affecting other PKC isoforms (25). Additionally, ACPD does not inhibit
other kinases such as AMPK, Akt2, FGFR1/2/3/4, mTOR, GSK3β,
IRAK1/4, JAK1/2, MEK1, ERK1/2, JNK1/2, PKA, Src, ROCK2 and PI3K
(26,27). This confirms our finding of ACPD in
molecular docking experiments that it did not show significant
binding to other kinases apart from PKC-ι and PKC-ζ. We conducted
kinase activity assay (in vitro) of ACPD and DNDA in order
to confirm our virtual screening data. Kinase activities of ACPD
and DNDA (Fig. 1G) were determined
for a series of concentrations (0.1–10 µM) using recombinant
active PKC-ι or PKC-ζ in the presence of MBP which is a well-known
substrate for PKCs (28). Both
compounds demonstrated significant inhibitions for both PKC-ι and
PKC-ζ under all tested concentrations. Both compounds showed
maximum inhibition for their 10 µM solutions as ACPD on
PKC-ι as 44% (P≤0.05), ACPD on PKC-ζ as 41% (P≤0.05), DNDA on PKC-ι
as 38% (P≤0.05) and DNDA on PKC-ζ as 29% (P≤0.05). This confirms
that DNDA also shows specific inhibition on PKC-ι and PKC-ζ in
addition to ACPD. These kinase activity data confirm our virtual
screening data.
Inhibitor dose-response curves
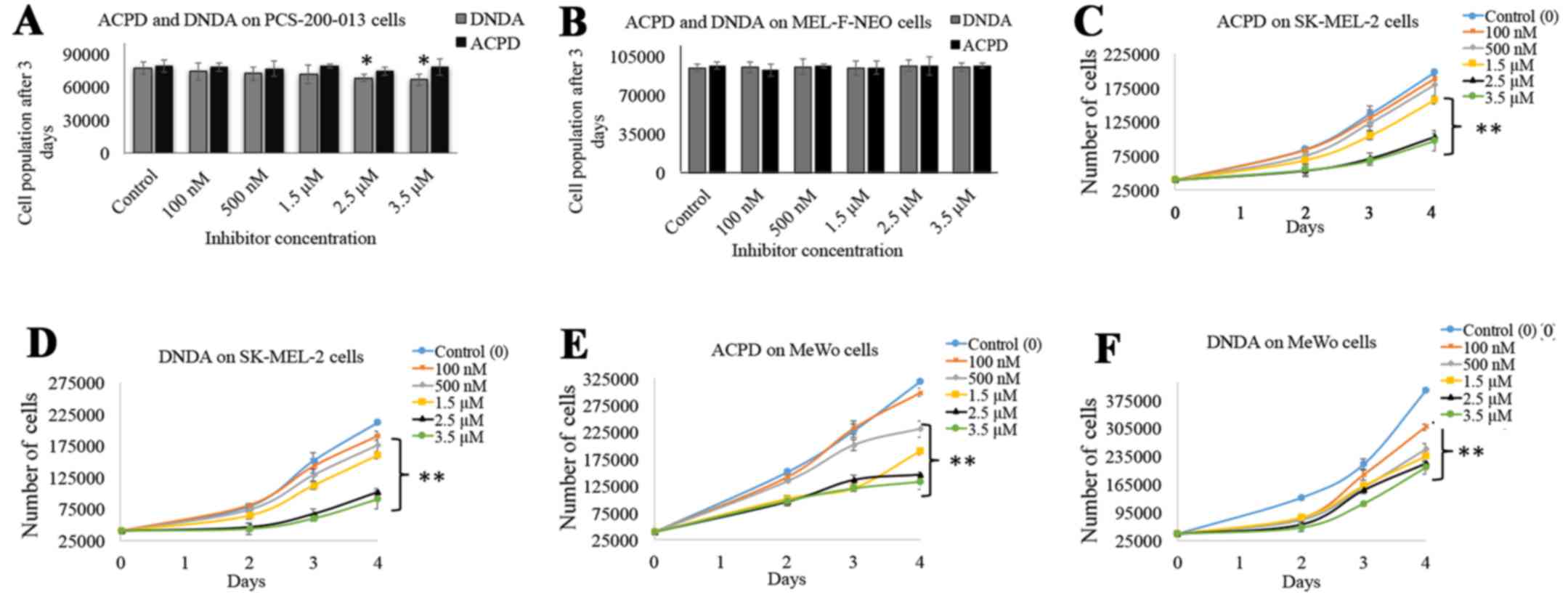
Dose curves for ACPD and DNDA were obtained to
investigate the effects on cell proliferation of normal and
malignant cell lines over a wide range of concentration. ACPD and
DNDA did not show a significant effect on PCS-200-013 (Fig. 2A) except 2.5 and 3.5 µM DNDA
treatments in which significant inhibitions were achieved (P≤0.05).
Similarly, neither inhibitor showed a significant inhibition for
MEL-F-NEO normal melanocyte cells (Fig. 2B). Both inhibitors significantly
decreased cell proliferation of SK-MEL-2 and MeWo upon increasing
the concentrations. ACPD decreased proliferation by 20% for 1.5
µM (P≤0.01), 48% for 2.5 µM (P≤0.01) and 51% for 3.5
µM (P≤0.01) (Fig. 2C) and
DNDA decreased 24% for 1.5 µM (P≤0.01), 52% for 2.5
µM (P≤0.01) and 57% for 3.5 µM (P≤0.01) (Fig. 2D) in the SK-MEL-2 cell line. ACPD
decreased proliferation by 41% for 1.5 µM (P≤0.01), 54% for
2.5 µM (P≤0.01) and 58% for 3.5 µM (P≤0.01) (Fig. 2E) and DNDA decreased proliferation
by 41% for 1.5 µM (P≤0.01), 46% for 2.5 µM (P≤0.01)
and 48% for 3.5 µM (P≤0.01) (Fig. 2F) in the MeWo cell line. These
results suggest that both inhibitors can effectively decrease the
cell population while not having a significant effect on normal
melanocytes. Based on these results the IC50 of ACPD and
DNDA for both drugs were found to be ~2.5 µM and this
concentration was used in later experiments.
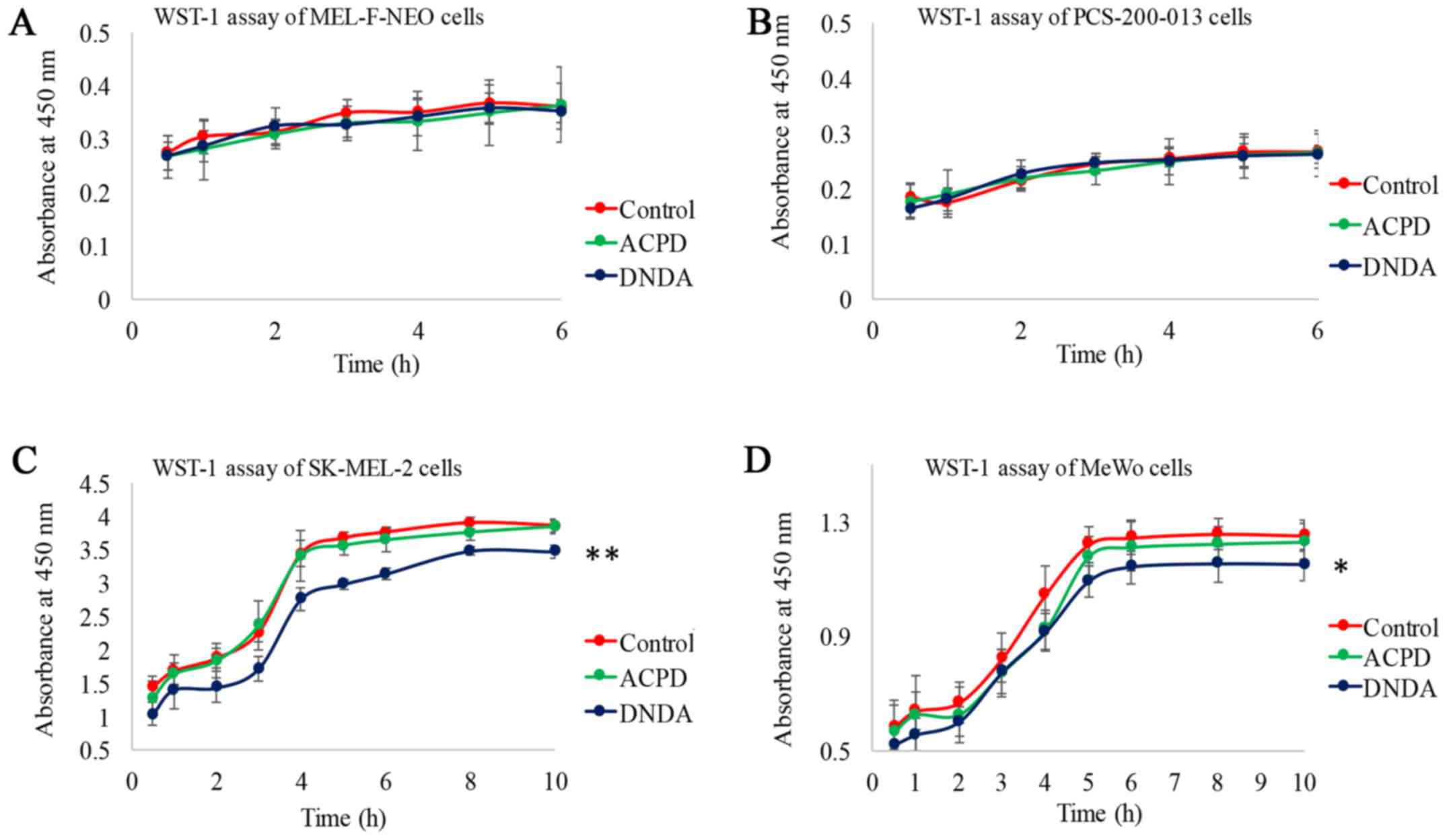
WST-1 assay for cell viability and
cytotoxicity
To determine the in vitro cytotoxicity of
ACPD and DNDA on normal and malignant cell lines, we performed a
WST-1 assay. The measured absorbance at 450 nm is directly
proportional to the number of viable cells present. Viable cells
produce a water-soluble formazan with WsT-1 as a result of their
mitochondrial dehydrogenase activity. WST-1 assay is preferred over
the 3-(4,S-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium
bromide (MTT) test. This is because MTT requires acidic isopropanol
to dissolve formazan, providing an additional toxicity to cells
(29). Neither inhibitor showed a
significant effect on normal melanocyte cells (Fig. 3A and B). ACPD did not show a
significant effect on either malignant cell line but DNDA showed a
significant decrease in absorption on SK-MEL-2 (P≤0.01) and on MeWo
(P≤0.05) (Fig. 3C and D). Results
indicate that both ACPD and DNDA appeared to be cytostatic thereby
inhibiting cell growth and proliferation. DNDA showed a mild
cytotoxicity to both melanoma cell lines in addition to its
cytostatic nature.
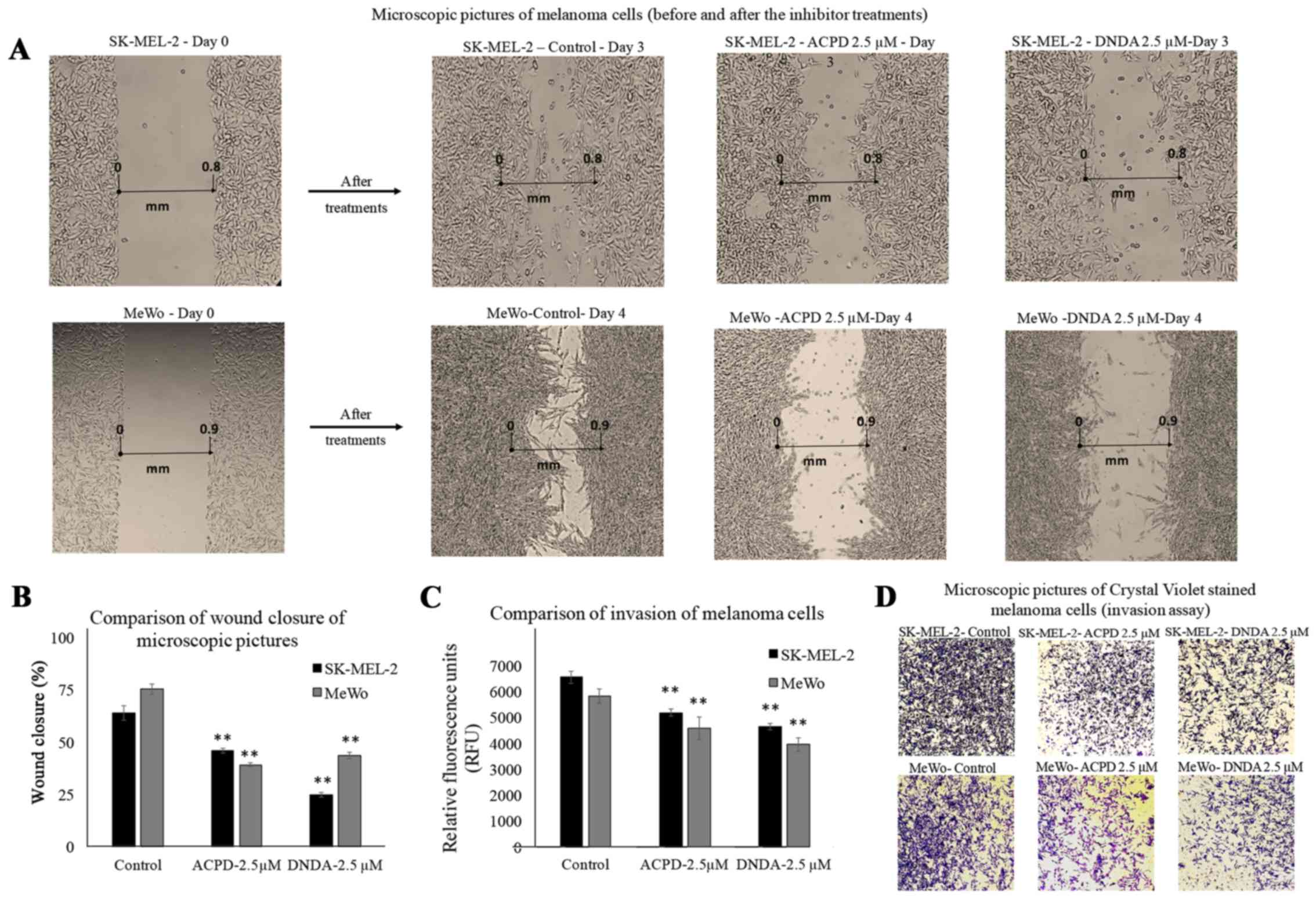
Wound healing assay
Wound healing assay was performed to investigate the
effect of ACPD and DNDA on malignant melanoma cell migration in
vitro. Wound healing assay is commonly used to investigate
in vitro migration of cancer cells (30). Photographs for each cell line are
compared as 'day 0' (starting point) and 'day 3' or 'day 4' for
both malignant cell lines (Fig.
4A). Cells treated with ACPD 2.5 µM and DNDA 2.5
µM were compared with their respective controls. The areas
of the scratch (wound) were calculated and compared to determine
the statistical significance (Fig.
4B). We found that both inhibitors significantly reduce the
wound closure (P≤0.01) of both cell lines compared to respective
controls. Results suggested that both drugs are equally effective
in reducing cell migration in vitro.
BME invasion assay
This invasion assay was performed to investigate the
effect of ACPD and DNDA on malignant melanoma cell invasion in
vitro. Although it is similar to Boyden chamber assay, it
avoids scraping off the Matrigel and staining to assess the number
of migrated cells through the filter. Hence, the method carries
less human error compared to a conventional Boyden chamber assay
(21). Migrated cells were stained
with a fluorescent marker; Calcein-AM. Live cells cleave the ester
(AM) of the molecule in order to produce fluorescence. Thus, the
amount of fluorescence accumulate in the bottom chamber is
proportional to the number of invaded cells. The relative
fluorescent units (excitation at 485 and emission at 528 nm) after
2 h of exposure were reported for inhibitor treatments for both
SK-MEL-2 and MeWo cell lines compared to controls (Fig. 4C). Invasion was significantly
reduced (P≤0.05) by 24 and 21% in ACPD (2.5 µM) treated
SK-MEL-2 and MeWo cells compared to its control. In DNDA (2.5
µM) treated samples, the invasion was significantly reduced
(P≤0.05) by 32% in both SK-MEL-2 and MeWo cell lines compared to
its control. Invaded cells on the bottom chamber were stained with
crystal violet and images were taken in randomly chosen fields as
the visual representation of the invasion assay (Fig. 4D).
Effect of ACPD and DNDA on aPKC levels in
malignant melanoma
Before analyzing the levels of aPKCs in inhibitor
treated melanoma cell lines using WB, we compared the protein
levels of aPKCs, E-cadherin, vimentin and Bcl-2 in MEL-F-NEO normal
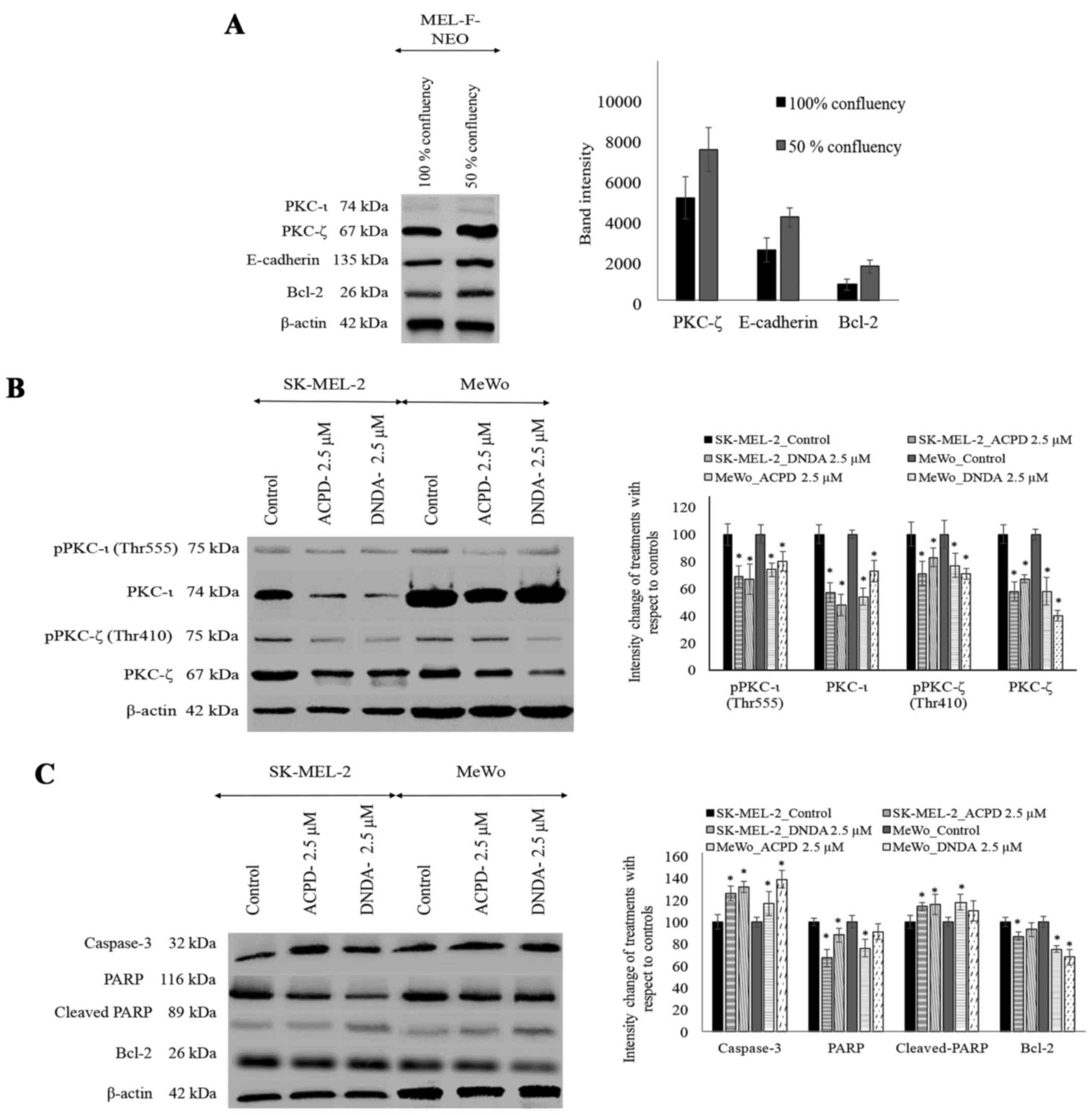
melanocytes at 100% confluency and 50% confluency. PKC-ι and
vimentin were not detected at either confluency level in MEL-F-NEO
cells. PKC-ζ, E-cadherin and Bcl-2 levels were found to be higher
in 50% confluency level compared to 100% confluency (Fig. 5A). β-actin was used as the internal
control to ensure that equal amounts of proteins were loaded in
each lane in the SDS-PAGE.
WB were performed to investigate the effect of ACPD
and DNDA on the expression of aPKCs on malignant melanoma (Fig. 5B). ACPD significantly reduced the
PKC-ι level by 43 and by 31% of pPKC-ι in SK-MEL-2 cell line and by
46% of PKC-ι and 26% of pPKC-ι in MeWo cells. DNDA significantly
decreased the levels of PKC-ι by 52%, pPKC-ι by 33% in SK-MEL-2 and
by 27% of PKC-ι and pPKC-ι by 20% in MeWo cells. Also, ACPD
significantly reduced the PKC-ζ level by 42 and by 29% of pPKC-ζ in
SK-MEL-2 cell line and by 42% of PKC-ζ and 23% of pPKC-ζ in MeWo
cells. DNDA significantly reduced the levels of PKC-ζ by 33%,
pPKC-ζ by only 17% in SK-MEL-2 and by 60% of PKC-ζ and pPKC-ζ by
29% in MeWo. All values (percent) were calculated and compared to
their respective control in WB (Fig.
5B; densitometry analysis) and significance was indicated as
P≤0.05. β-actin was used as the internal control to ensure that
equal amounts of proteins were loaded in each lane in the
SDS-PAGE.
Effect of ACPD and DNDA on apoptosis of
malignant melanoma
Since both inhibitors significantly inhibit melanoma
cell proliferation, we tested the potential of the inhibitors on
inducing apoptosis (Fig. 5C).
Caspase-3 levels significantly increased by 26 and 17% in ACPD
treated SK-MEL-2 and MeWo cells, respectively. Caspase-3 levels
significantly increased by 32 and 39% in DNDA treated SK-MEL-2 and
MeWo cells, respectively. PARP levels significantly decreased by 33
and by 24% in ACPD treated SK-MEL-2 and MeWo cells, respectively,
while cleaved-PARP levels significantly increased by 14 and 18%,
respectively. In DNDA treated samples, PARP levels significantly
decreased by 12 and by 9% in SK-MEL-2 and MeWo cells, respectively,
while cleaved-PARP levels significantly increased by 16 and 10%,
respectively. Similarly, Bcl-2 levels significantly decreased by 13
and by 25% in ACPD treated SK-MEL-2 and MeWo cells, respectively,
while in DNDA treated cells Bcl-2 levels decreased by 7 and by 32%
in SK-MEL-2 and MeWo cells, respectively. All values (percent) were
calculated compared to their respective control in WB (Fig. 5C; densitometry analysis) and
significance are indicated as P≤0.05. β-actin was used as the
internal control to ensure that equal amounts of proteins were
loaded in each lane in the SDS-PAGE.
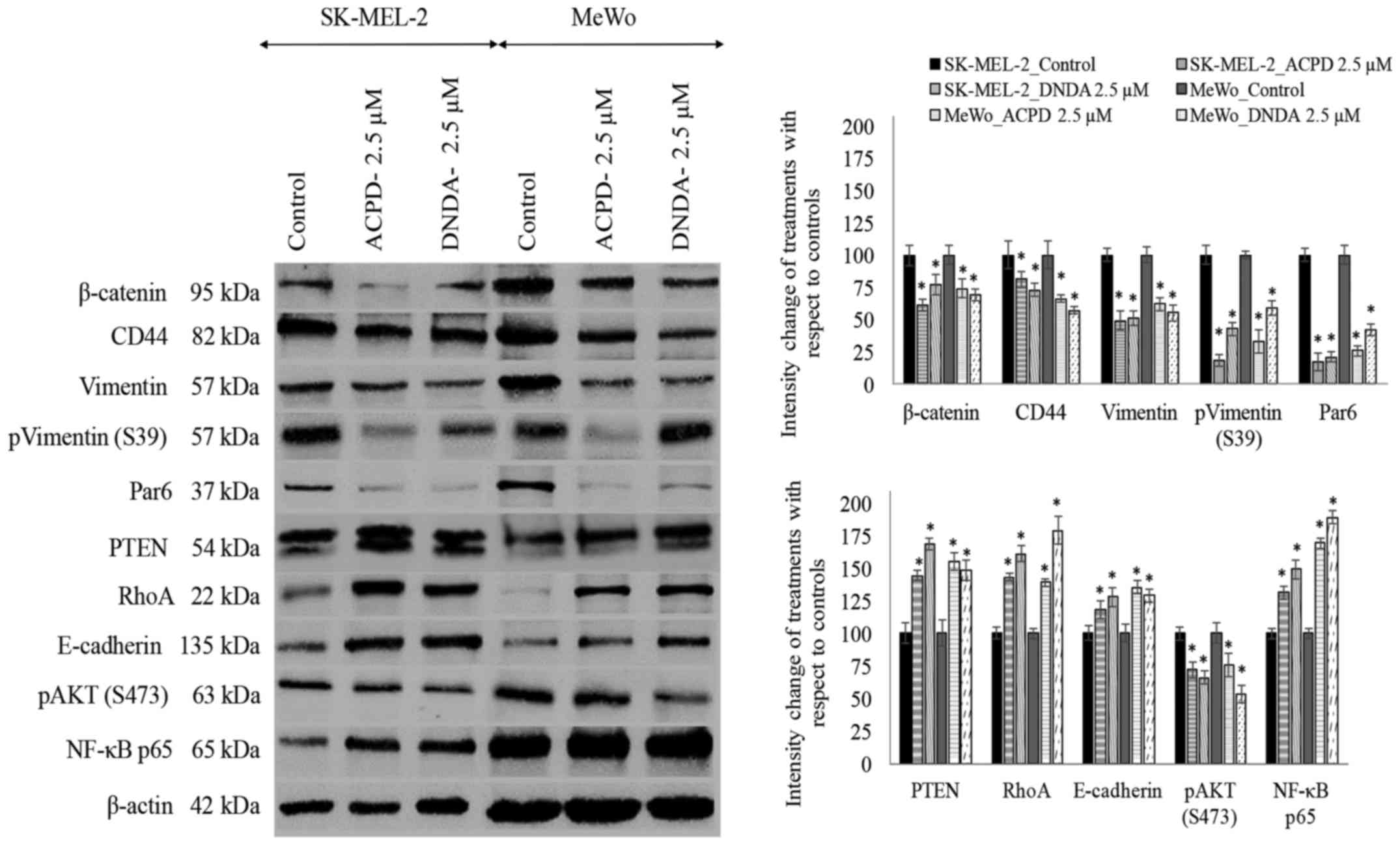
Effect of ACPD and DNDA on signaling
pathways and EMT
As shown in Fig. 6,
we also investigated the effects of ACPD and DNDA on EMT by
studying the signaling cascades crucial for cancer cell
proliferation, survival, migration and invasion. The purpose of the
analysis was to obtain a better understanding of how aPKCs are
involved in the progression of melanoma. We tested the effects of
ACPD and DNDA on proteins β-catenin, CD44, vimentin, Par6, PTEN,
RhoA, E-cadherin, AKT and NF-κB p65 to determine how Wnt signaling,
NF-κB signaling and AKT signaling are affected by aPKC inhibitors
during EMT stimulation.
β-catenin significantly decreased by 39 and 26% in
ACPD treated SK-MEL-2 and MeWo cells, respectively compared to 23
and 31% downregulation in DNDA treated samples. CD44 also decreased
significantly by 19 and 34% in ACPD treated SK-MEL-2 and MeWo
cells, in comparison to 27 and 43% downregulation in DNDA treated
samples. Vimentin levels significantly decreased by 51 and 38% in
ACPD treated SK-MEL-2 and MeWo cells, respectively compared to 49
and 45% decreases in DNDA treated samples. E-cadherin levels
increased significantly by 18 and 35% in ACPD treated SK-MEL-2 and
MeWo cells, while there were 28 and 29% increases in DNDA treated
samples. Notably, NF-κB p65 levels significantly increased by 31
and 69% in ACPD treated SK-MEL-2 and MeWo cells, and there were 49
and 89% increases in DNDA treated samples. Phospho vimentin (S473)
levels significantly decreased by 82 and 67% in ACPD treated
SK-MEL-2 and MeWo cells, respectively, compared to 57 and 41%
decreases in DNDA treated samples. Par6 levels significantly
decreased by 83 and 74% in ACPD treated SK-MEL-2 and MeWo cells,
respectively compared to 79 and 58% decreases in DNDA treated
samples. PTEN levels significantly increased by 44 and 55% in ACPD
treated SK-MEL-2 and MeWo cells, respectively, compared to 68 and
48% increases in DNDA treated samples. RhoA levels significantly
increased by 87 and 70% in ACPD treated SK-MEL-2 and MeWo cells,
respectively, compared to 80 and 66% increases in DNDA treated
samples. All values (percent) were calculated compared to their
respective controls in WB (Fig. 6;
densitometry analysis) and significance was indicated as P≤0.05.
β-actin was used as the internal control to ensure that equal
amounts of proteins were loaded in each lane in the SDS-PAGE.
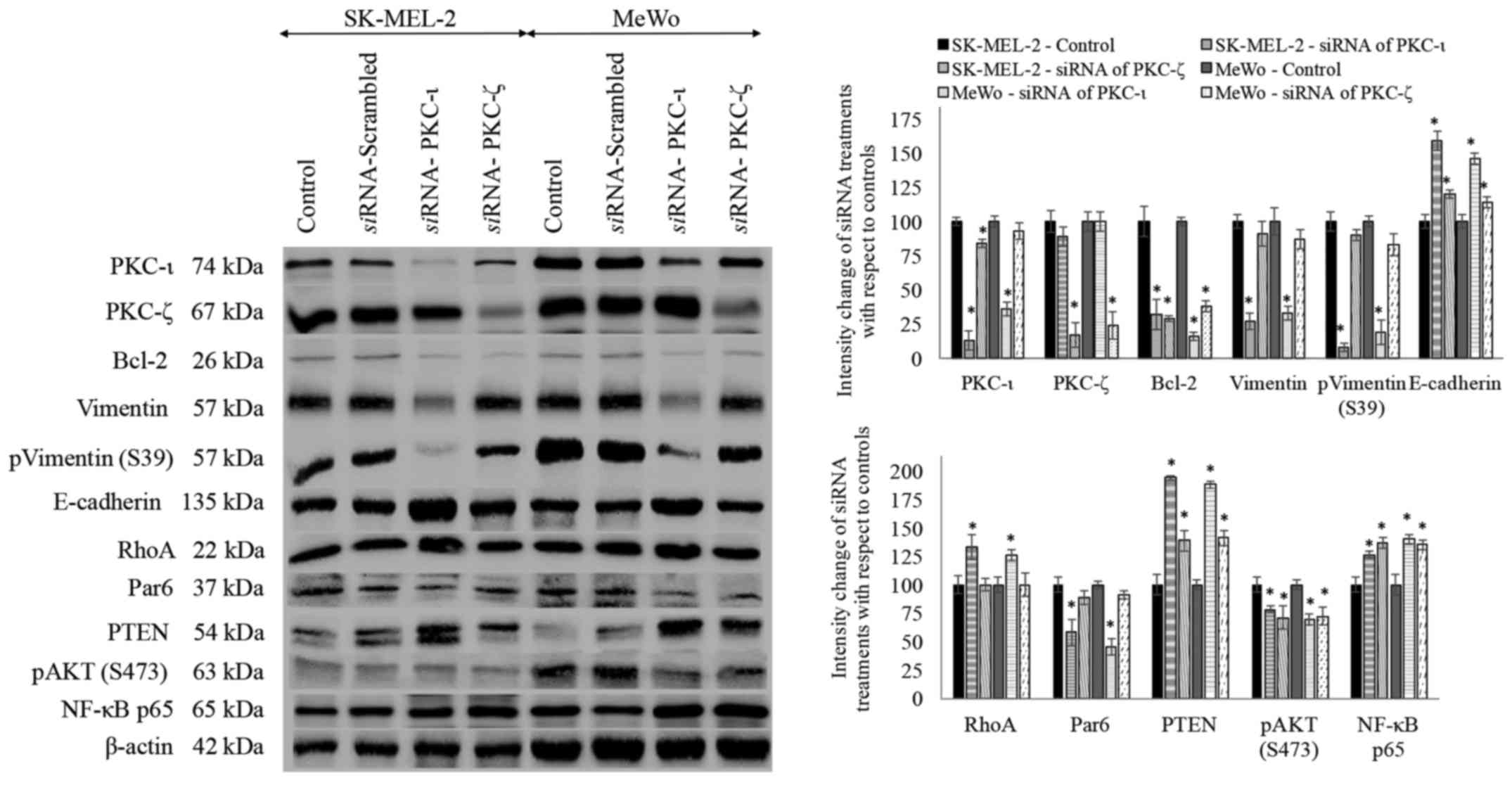
siRNA treatments for PKC-ι and PKC-ζ
Both melanoma cell lines were treated with
siRNA for PKC-ι and PKC-ζ to knock down the expression of
said proteins and subsequently investigated the levels of protein
expression for the proteins tested (Fig. 7). Scrambled siRNA was also
used in addition to the control and there was no significant
difference observed between the control and scrambled siRNA
treatments for the said proteins.
siRNA treatments of PKC-ι resulted in
significant decrease in PKC-ι level by 87 and 64% in SK-MEL-2 and
MeWo cell lines, respectively. PKC-ζ decreased by 11 and 0% which
is not significant, while Bcl-2 significantly decreased by 68 and
84%, vimentin decreased by 73 and 67%, phospho vimentin (S39)
significantly decreased by 92 and 81%, E-cadherin increased by 59
and 46%, RhoA increased by 33 and 26%, Par6 decreased by 42 and
55%, PTEN significantly increased by 94 and 88%, phospho AKT (S473)
decreased by 22 and 31% and NF-κB increased by 26 and 40% in PKC-ι
siRNA treated SK-MEL-2 and MeWo cells, respectively.
siRNA treatments of PKC-ζ resulted in
significant decrease in PKC-ζ level by 83 and 76% in SK-MEL-2 and
MeWo cell lines, respectively. PKC-ι decreased by 16 and 7% which
is not significant, Bcl-2 significantly decreased by 71 and 62%,
vimentin decreased only by 9 and 13%, which is not significant,
phospho vimentin (S39) only decreased by 10 and 17%, E-cadherin
significantly increased by 20 and 14%, Par6 decreased by 11 and 9%,
PTEN increased by 39 and 41%, phospho AKT (S473) decreased by 29
and 28% and NF-κB increased by 37 and 35% in PKC-ι siRNA
treated SK-MEL-2 and MeWo cells, respectively. RhoA levels of PKC-ζ
siRNA treated samples did not show a significant difference
to its control. All significance values are indicated as
P≤0.05.
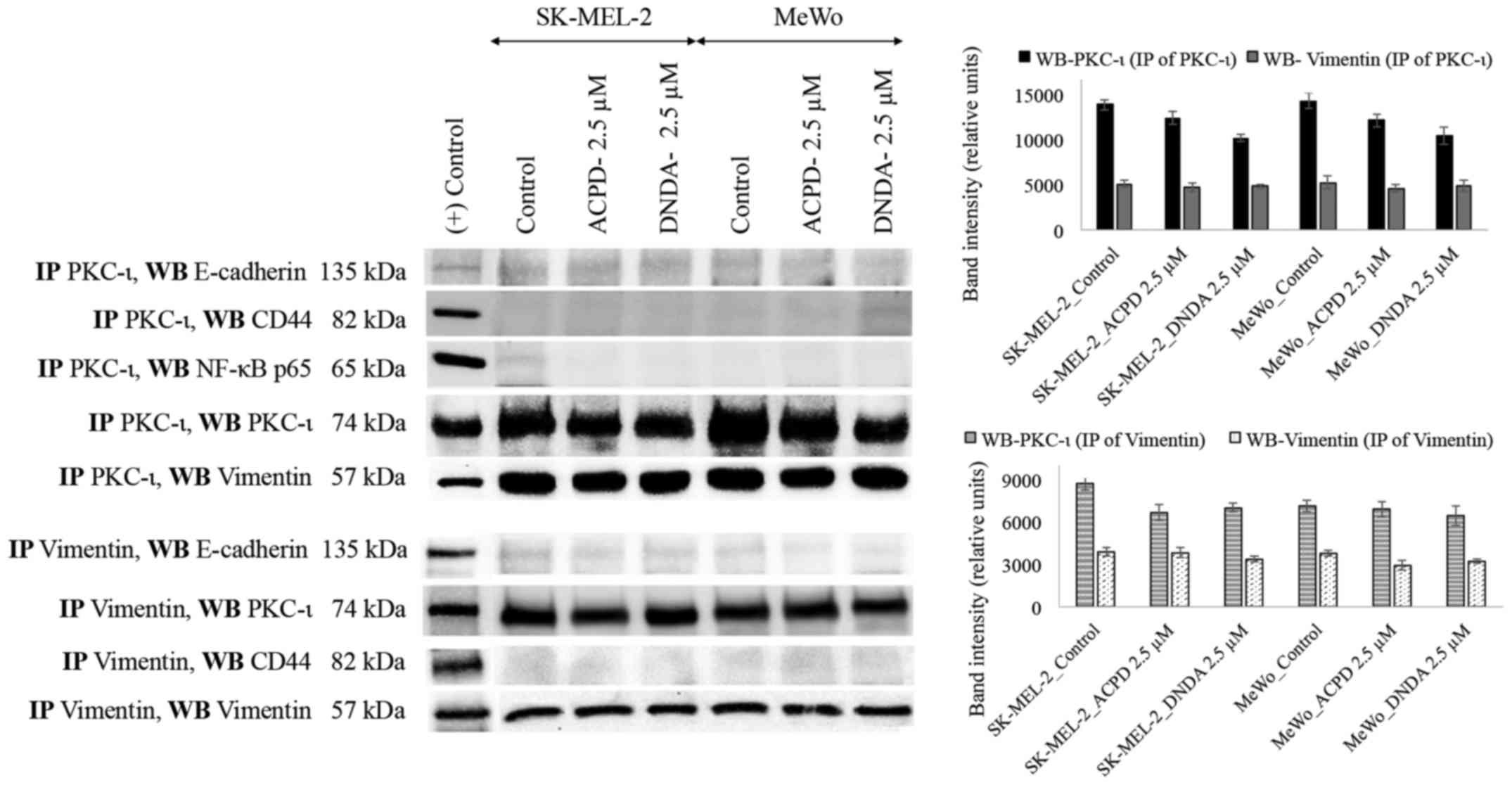
Association of PKC-ι and vimentin
We immunoprecipitated (IP) PKC-ι and PKC-ζ
separately and WB experiments were conducted independently for
E-cadherin, CD44, vimentin and NF-κB p65. PKC-ζ IP samples did not
show any association with any mentioned proteins. Only vimentin
immunoblot showed an association with PKC-ι IP samples (Fig. 8). This result suggests that PKC-ι
associate with vimentin. To confirm this association, vimentin was
immunoprecipitated, developed for said proteins and only PKC-ι was
associated with vimentin.
Discussion
PKC-ι and PKC-ζ both have a wide range of effects
and are overexpressed in many human cancers (4–7,31–33).
Common upstream elements can activate both, but they each perform
their own functions (34–37). The domains between PKC-ι and PKC-ζ
are largely conserved; given nearly 70% of the similarity, it was
important to identify inhibitors specific to aPKCs, and at the same
time determine how much each was inhibited. A previous study showed
the ACPD did not affect many upstream elements of the aPKC
activation pathways (25–27). The computational screening and
kinase activity assay data show that ACPD and DNDA are specific
inhibitors of aPKCs. ACPD showed the same effect as an inhibitor
for both PKC-ζ and PKC-ι in a relative sense. DNDA showed a better
action on PKC-ι than PKC-ζ (Fig.
1G).
Determination of cell viability and cytotoxicity of
malignant and normal cells after inhibitor treatments was
necessary, considering our ultimate intent was to determine if the
inhibitors had potential as therapeutic drugs. As observed in WST-1
experiments (Fig. 3A and D),
neither inhibitor had any significant effect on the normal
melanocyte cell viability. This in turn suggests that neither
inhibitor proved toxic to the normal cells. Both ACPD and DNDA are
cytostatic, rather than cytotoxic. Regardless, DNDA showed a mild
toxicity towards both malignant melanoma cell lines, suggesting
both inhibitors are effective against malignant cells without
harming normal cells. This confirms the previous data, which
suggested that malignant cells rely on aPKCs to remain viable
(8,7,38,39).
As observed in the WB on normal melanocytes (Fig. 5A), normal melanocytes produced no
detectable levels of PKC-ι, but they produced PKC-ζ. Malignant
melanoma cells (Fig. 5B)
overexpressed both PKC-ι and PKC-ζ. The total and phosphorylated
levels of PKC-ι and PKC-ζ significantly reduced upon treatments
with both ACPD and DNDA. Similarly, melanoma cell proliferation was
markedly reduced (Fig. 2C–F) upon
treatments while not observably affecting cell proliferation on
normal melanocyte cells (Fig. 2A and
B). This confirms that melanoma cellular functions are highly
dependent on aPKCs, but normal melanocytes do not depend on
aPKCs.
Previous studies have shown that overexpression of
aPKCs have an anti-apoptotic effect in many cancers (4–9,12).
We could determine whether treatment with the inhibitors could
induce apoptosis, we tested apoptotic markers through a WB
(Fig. 5C). Increase in caspase-3,
increase in PARP cleavage, and decrease in Bcl-2 all indicate
apoptosis stimulation (40–43).
Increase in caspase-3 levels is not always a direct indication of
induction of apoptosis due to the tight binding of cleaved
caspase-3 with X-linked inhibitor of apoptosis protein (XIAP). XIAP
undergoes auto-ubiquitilation, but this process delays apoptosis
until all XIAP is removed (44).
Owing to this, we also tested direct PARP and cleaved PARP levels
because PARP is a known downstream target for caspase-3. It cleaves
more upon inducing apoptosis (45). We also tested Bcl-2 levels since it
inhibits caspase activity by preventing xytochrome c release
from the mitochondria and/or by binding to the apoptosis-activating
factor (APAF-1). ACPD and DNDA treatments each depicted an increase
in apoptotic activity in both SK-MEL-2 and MeWo cell lines. The
data confirm some previous results showing aPKCs have an
anti-apoptotic effect. One major anti-apoptotic pathway is NF-κB
activation, which entails aPKC releasing NF-κB and NF-κB
translocates to the nucleus to promote cell survival (46). Notably, the inhibitors not only
reduced activity of aPKCs as evidenced by the kinase activity
assay, but they also reduced the aPKC expression as shown in
western blots. This suggests that aPKC activation plays a critical
role in aPKC expression, but also that regulation mechanisms
require further study.
In addition to apoptosis evasion, transformed
melanocytes and melanoma cells also proceed through EMT. In this
process, cells lose their epithelial characteristics to acquire the
behavior of mesenchymal cells, thereby promoting individual cell
migration and invasion from the primary site to distant organs. We
tested the levels of vimentin, which is a mesenchymal marker
produced in large quantities during EMT while cells inversely lose
their E-cadherin (12,13). We tested the expression of vimentin
in normal melanocytes, but we found it undetectable. Even though
the role of vimentin in EMT is not yet fully understood, vimentin
is very important in gaining rear-to-front polarity for mesenchymal
cells (47). We also tested
β-catenin, which is a part of adherin junctions and involved in
many functions, including coordination of cell-cell adhesion and
gene transcription (48).
β-catenin activation destabilizes the cell-cell junctions and
β-catenin translocates to the nucleus, continuously driving
transcription of targeted genes such as CD44. CD44 is a
transmembrane glycoprotein upregulated by Wnt5A, and plays the role
of an important mediator in tumor progression and cell invasion
(16,17,46).
Both ACPD and DNDA showed promising effects on EMT markers, and
E-cadherin levels increased upon treatments while CD44, β-catenin
and vimentin levels all decreased. We also tested the levels of
phosphorylated vimentin (S39), they decreased upon inhibitor
treatments. The data suggest that the aPKC inhibition slows or
possibly reverses EMT and supports the significant reduction
observed in both migration and invasion of malignant melanoma cells
upon treating ACPD and DNDA.
Immunoprecipitation and reverse immunoprecipitation
of PKC-ι and vimentin showed a strong direct association between
them (Fig. 8). To confirm
inhibitor effects on melanoma, we treated the cells with
siRNA for PKC-ι and PKC-ζ. Results revealed that upon
knocking down PKC-ι, total and phosphorylated vimentin levels
significantly decreased by 73 and 93% for SK-MEL-2 cells, as well
as 67 and 81% for MeWo cells. The effect of PKC-ζ knockdown on
vimentin is negligible compared to the large effect we observed in
PKC-ι knockdown. Our results suggest that both vimentin and PKC-ι
work together changing the polarity in cancer cell migration.
Vimentin activates upon the binding of PKC-ι and phosphorylates at
Ser39. It has been previously shown that Par6 can be phosphorylated
by aPKCs upon activation of TGF-β receptors, and activated Par6
stimulates EMT in A549 adinocarcinoma cells (11,49).
TGF-β activation stimulates degradation of RhoA and cells lose
E-cadherin while increasing vimentin. Both inhibitor treatments
increased the levels of E-cadherin and RhoA, indicating the
inhibition of PKC-ι or PKC-ζ or both can lead to complete stop or
reversal of melanoma EMT (Fig. 6).
To confirm the results, we tested the levels of Par6 and RhoA and
E-cadherin levels in siRNA treated cells (Fig. 7). Results revealed that PKC-ι
knockdown increased both E-cadherin and RhoA effectively compared
to the PKC-ζ knockdown. In PKC-ζ siRNA treatments, the RhoA
effect is only negligible, though its effect on E-cadherin is less
when one compares it to the PKC-ι knockdown. This suggests that
only PKC-ι is responsible for stimulating EMT. TGF-β stimulation
also activates the Wnt/β-catenin pathway; in that case, stabilized
β-catenin translocates to the nucleus and inhibits metastasis
suppressors in melanoma (16).
Previous research supports our observations here that negative
regulation of EMT is observed upon inhibition of aPKCs by ACPD and
DNDA. It has been previously shown that activated Vimentin inhibits
PTEN by increasing the phosphorylation of PTEN to enhance PI3K/AKT
activity which leads to cell differentiation and survival of
osteoblasts (50). This process
can also inhibit apoptosis via the NF-κB pathway (51). Increases in PTEN levels in both
ACPD and DNDA treated samples and siRNA treated samples for
PKC-ι and PKC-ζ strongly support this statement. Notably, increased
levels of PTEN in PKC-ζ treated siRNA samples suggested the
involvement of PKC-ζ. It has been published that PKC-ζ stimulates
IKK α/β, which ultimately stimulates translocation of NF-κB complex
into the nucleus resulting in increased cell survival (52). Notably, we found increases in NF-κB
p65 levels upon inhibitor treatments, but also upon both
siRNA treatments. This may seem illogical, but previous
studies have shown that expression of NF-κB p65 has a corresponding
increase in IκB, resulting in an auto-regulatory loop (52). It is possible that the upstream
signaling for NF-κB translocation is not being inhibited by
negative feedback since translocation has been inhibited
downstream, resulting in more NF-κB production. NF-κB is still
inhibited from translocating to the nucleus. This causes cell
mediated apoptosis and cell survival rate reductions. In summary
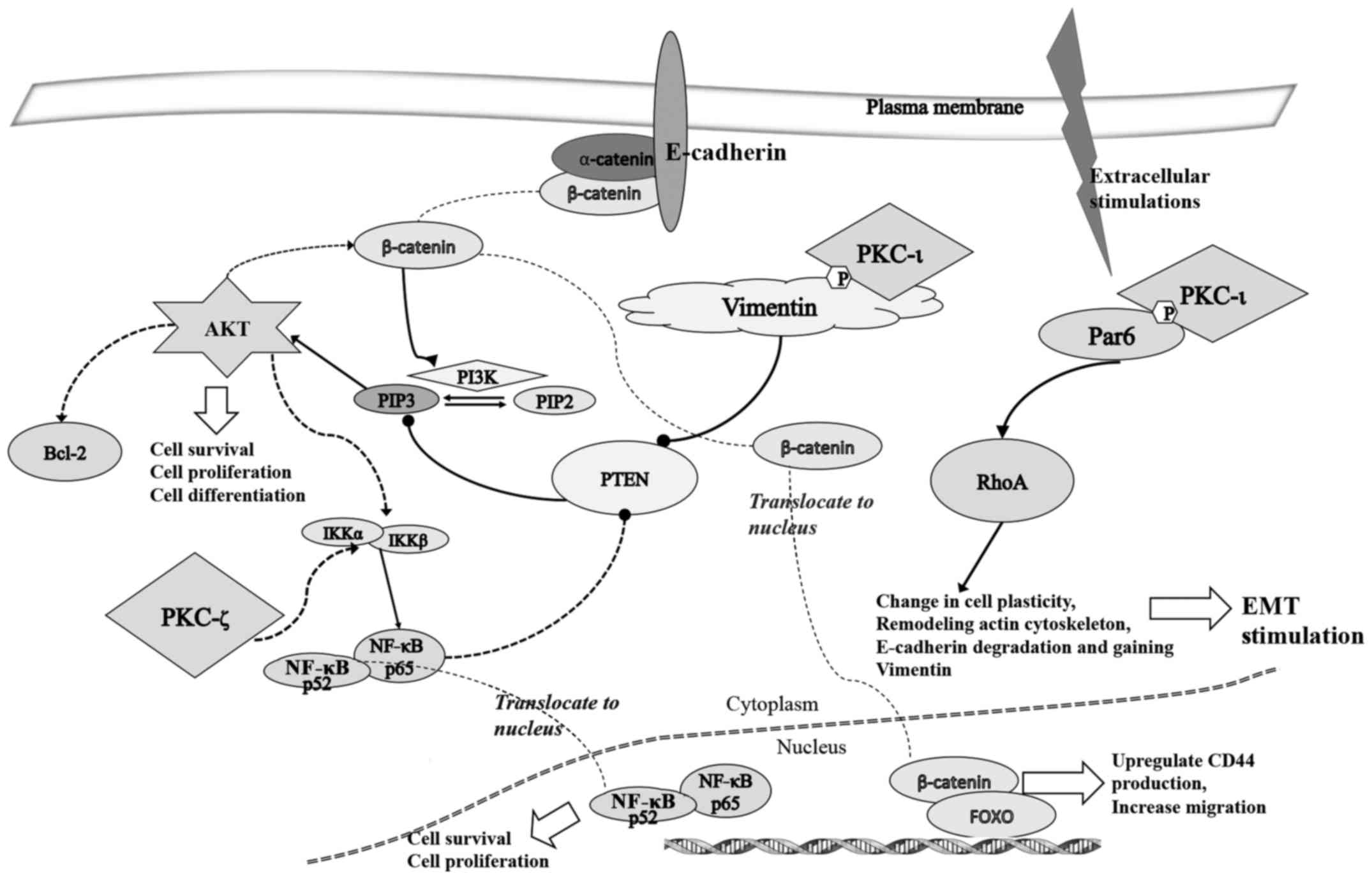
(Fig. 9), our results suggest that
PKC-ι is responsible for inducing EMT, while PKC-ζ is mainly
involved in NF-κB signaling to promote cell growth and survival. In
addition, PKC-ι is involved in stimulating AKT signaling by
inhibiting PTEN via vimentin activation. Activated vimentin induces
the phosphorylation of PTEN.
In conclusion, our results suggest that ACPD and
DNDA are effective aPKC inhibitors in melanoma cells, and do not
affect normal melanocytes. ACPD and DNDA can reduce the cell
proliferation, migration and invasion, while inducing apoptosis.
Results also show a direct relationship between vimentin, PKC-ι and
EMT. aPKC inhibitors (ACPD and DNDA) can reduce EMT progression, or
possibly reverse it by specifically inhibiting PKC-ι. Finally,
collected evidence suggest ACPD and DNDA could be potential
therapeutic drugs for malignant melanoma.
Acknowledgments
We acknowledge the generous financial contribution
from the Frederick H. Leonhardt, the Daniel Tanner Foundation, the
David R. Clare and Margaret C. Clare Foundation, the Elizabeth
Ireland Graves Foundation, the Harvey R. Chaplin, Carl C. Anderson,
Sr. and Marie Jo Anderson Charitable Foundation and the Charles and
Ann Johnson Foundation. Moreover, we thank Ms. Rekha Patel for her
excellent technical advice and Mr. Christopher Apostolatos and Ms.
Michelle Wilde for their help in critically editing the
manuscript.
References
|
1
|
Eggermont AM: Adjuvant ipilimumab in stage
III melanoma: New landscape, new questions. Eur J Cancer. 69:39–42.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hao M, Song F, Du X, Wang G, Yang Y, Chen
K and Yang J: Advances in targeted therapy for unresectable
melanoma: New drugs and combinations. Cancer Lett. 359:1–8. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Melanoma of the Skin - Cancer Stat Facts.
<http://seer.cancer.gov/statfacts/html/melan.html>,
03/30/2017.
|
|
4
|
Manning G, Whyte DB, Martinez R, Hunter T
and Sudarsanam S: The protein kinase complement of the human
genome. Science. 298:1912–1934. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Koivunen J, Aaltonen V and Peltonen J:
Protein kinase C (PKC) family in cancer progression. Cancer Lett.
235:1–10. 2006. View Article : Google Scholar
|
|
6
|
Regala RP, Weems C, Jamieson L, Khoor A,
Edell ES, Lohse CM and Fields AP: Atypical protein kinase C iota is
an oncogene in human non-small cell lung cancer. Cancer Res.
65:8905–8911. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Murray NR and Fields AP: Atypical protein
kinase C ι protects human leukemia cells against drug-induced
apoptosis. J Biol Chem. 272:27521–27524. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Acevedo-Duncan M, Patel R, Whelan S and
Bicaku E: Human glioma PKC-iota and PKC-betaII phosphorylate
cyclin-dependent kinase activating kinase during the cell cycle.
Cell Prolif. 35:23–36. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Patel R, Win H, Desai S, Patel K, Matthews
JA and Acevedo-Duncan M: Involvement of PKC-iota in glioma
proliferation. Cell Prolif. 41:122–135. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Regala RP, Thompson EA and Fields AP:
Atypical protein kinase C iota expression and aurothiomalate
sensitivity in human lung cancer cells. Cancer Res. 68:5888–5895.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gunaratne A, Thai BL and Di Guglielmo GM:
Atypical protein kinase C phosphorylates Par6 and facilitates
transforming growth factor β-induced epithelial-to-mesenchymal
transition. Mol Cell Biol. 33:874–886. 2013. View Article : Google Scholar :
|
|
12
|
Kim KK, Kugler MC, Wolters PJ, Robillard
L, Galvez MG, Brumwell AN, Sheppard D and Chapman HA: Alveolar
epithelial cell mesenchymal transition develops in vivo during
pulmonary fibrosis and is regulated by the extracellular matrix.
Proc Natl Acad Sci USA. 103:13180–13185. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wei J, Xu G, Wu M, Zhang Y, Li Q, Liu P,
Zhu T, Song A, Zhao L, Han Z, et al: Overexpression of vimentin
contributes to prostate cancer invasion and metastasis via src
regulation. Anticancer Res. 28(1A): 327–334. 2008.PubMed/NCBI
|
|
14
|
Selzer E, Okamoto I, Lucas T, Kodym R,
Pehamberger H and Jansen B: Protein kinase C isoforms in normal and
transformed cells of the melanocytic lineage. Melanoma Res.
12:201–209. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ratnayake WS and Acevedo-Duncan M: Use of
ACPD and ICA-1 as inhibitors of atypical protein kinase C-zeta (ζ)
and iota (ι) in metastasized melanoma cells. Cancer Res. 76(14
suppl): Abs 4569. 2016. View Article : Google Scholar
|
|
16
|
Dissanayake SK, Wade M, Johnson CE,
O'Connell MP, Leotlela PD, French AD, Shah KV, Hewitt KJ, Rosenthal
DT, Indig FE, et al: The Wnt5A/protein kinase C pathway mediates
motility in melanoma cells via the inhibition of metastasis
suppressors and initiation of an epithelial to mesenchymal
transition. J Biol Chem. 282:17259–17271. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weeraratna AT, Jiang Y, Hostetter G,
Rosenblatt K, Duray P, Bittner M and Trent JM: Wnt5a signaling
directly affects cell motility and invasion of metastatic melanoma.
Cancer Cell. 1:279–288. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qureshi R, Arora H and Rizvi MA: EMT in
cervical cancer: Its role in tumour progression and response to
therapy. Cancer Lett. 356:321–331. 2015. View Article : Google Scholar
|
|
19
|
Pillai P, Desai S, Patel R, Sajan M,
Farese R, Ostrov D and Acevedo-Duncan M: A novel PKC-ι inhibitor
abrogates cell proliferation and induces apoptosis in
neuroblastoma. Int J Biochem Cell Biol. 43:784–794. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Win HY and Acevedo-Duncan M: Role of
protein kinase C-iota in transformed non-malignant RWPE-1 cells and
androgen-independent prostate carcinoma DU-145 cells. Cell Prolif.
42:182–194. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
O'Connell MP, French AD, Leotlela PD and
Weeraratna AT: Assaying Wnt5A-mediated invasion in melanoma cells.
Methods Mol Biol. 468:243–253. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Win HY and Acevedo-Duncan M: Atypical
protein kinase C phosphorylates IKKalphabeta in transformed
non-malignant and malignant prostate cell survival. Cancer Lett.
270:302–311. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Selbie LA, Schmitz-Peiffer C, Sheng Y and
Biden TJ: Molecular cloning and characterization of PKC iota, an
atypical isoform of protein kinase C derived from insulin-secreting
cells. J Biol Chem. 268:24296–24302. 1993.PubMed/NCBI
|
|
24
|
Xiao H and Liu M: Atypical protein kinase
C in cell motility. Cell Mol Life Sci. 70:3057–3066. 2013.
View Article : Google Scholar
|
|
25
|
Sajan MP, Acevedo-Duncan ME, Standaert ML,
Ivey RA, Lee M and Farese RV: Akt-dependent phosphorylation of
hepatic FoxO1 is compartmentalized on a WD40/ProF scaffold and is
selectively inhibited by aPKC in early phases of diet-induced
obesity. Diabetes. 63:2690–2701. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sajan MP, Ivey RA III and Farese RV:
Metformin action in human hepatocytes: Coactivation of atypical
protein kinase C alters 5′-AMP-activated protein kinase effects on
lipogenic and gluconeogenic enzyme expression. Diabetologia.
56:2507–2516. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sajan MP, Ivey RA, Lee MC and Farese RV:
Hepatic insulin resistance in ob/ob mice involves increases in
ceramide, aPKC activity, and selective impairment of Akt-dependent
FoxO1 phosphorylation. J Lipid Res. 56:70–80. 2015. View Article : Google Scholar :
|
|
28
|
Wooten MW, Seibenhener ML, Zhou G,
Vandenplas ML and Tan TH: Overexpression of atypical PKC in PC12
cells enhances NGF-responsiveness and survival through an NF-kappaB
dependent pathway. Cell Death Differ. 6:753–764. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ukeda H, Maeda S, Ishii T and Sawamura M:
Spectro-photometric assay for superoxide dismutase based on
tetrazolium salt
3′-{1-[phenylamino)-carbonyl]3,4-tetrazolium]-bis(4-methoxy-6-nitro)benzenesulfonic
acid hydrate reduction by xanthine-xanthine oxidase. Anal Biochem.
251:206–209. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu S, Wang H, Pan H, Shi Y, Li T, Ge S,
Jia R, Zhang H and Fan X: ANRIL lncRNA triggers efficient
therapeutic efficacy by reprogramming the aberrant INK4-hub in
melanoma. Cancer Lett. 381:41–48. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Farese RV: Function and dysfunction of
aPKC isoforms for glucose transport in insulin-sensitive and
insulin-resistant states. Am J Physiol Endocrinol Metab.
283:E1–E11. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Suzuki A, Yamanaka T, Hirose T, Manabe N,
Mizuno K, Shimizu M, Akimoto K, Izumi Y, Ohnishi T and Ohno S:
Atypical protein kinase C is involved in the evolutionarily
conserved par protein complex and plays a critical role in
establishing epithelia-specific junctional structures. J Cell Biol.
152:1183–1196. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Du R, Lu KV, Petritsch C, Liu P, Ganss R,
Passegué E, Song H, Vandenberg S, Johnson RS, Werb Z, et al: HIF1α
induces the recruitment of bone marrow-derived vascular modulatory
cells to regulate tumor angiogenesis and invasion. Cancer Cell.
13:206–220. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Suzuki A, Akimoto K and Ohno S: Protein
kinase C lambda/iota (PKClambda/iota): A PKC isotype essential for
the development of multicellular organisms. J Biochem. 133:9–16.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mamidipudi V, Lin C, Seibenhener ML and
Wooten MW: Regulation of interleukin receptor-associated kinase
(IRAK) phosphorylation and signaling by iota protein kinase C. J
Biol Chem. 279:4161–4165. 2004. View Article : Google Scholar
|
|
36
|
Mah IK, Soloff R, Hedrick SM and Mariani
FV: Atypical PKC-iota controls Stem cell expansion via regulation
of the Notch pathway. Stem Cell Rep. 5:866–880. 2015. View Article : Google Scholar
|
|
37
|
Brady SC, Allan LA and Clarke PR:
Regulation of caspase 9 through phosphorylation by protein kinase C
zeta in response to hyperosmotic stress. Mol Cell Biol.
25:10543–10555. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shultz JC, Vu N, Shultz MD, Mba M-UU,
Shapiro BA and Chalfant CE: The Proto-oncogene PKCι regulates the
alternative splicing of Bcl-x pre-mRNA. Mol Cancer Res. 10:660–669.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
do Carmo A, Balça-Silva J, Matias D and
Lopes MC: PKC signaling in glioblastoma. Cancer Biol Ther.
14:287–294. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jänicke RU, Sprengart ML, Wati MR and
Porter AG: Caspase-3 is required for DNA fragmentation and
morphological changes associated with apoptosis. J Biol Chem.
273:9357–9360. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kroemer G: The proto-oncogene Bcl-2 and
its role in regulating apoptosis. Nat Med. 3:614–620. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Watson AJ, Askew JN and Benson RS:
Poly(adenosine diphosphate ribose) polymerase inhibition prevents
necrosis induced by H2O2 but not apoptosis. Gastroenterology.
109:472–482. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Soldani C and Scovassi AI:
Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: An update.
Apoptosis. 7:321–328. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
van Raam BJ, Drewniak A, Groenewold V, van
den Berg TK and and Kuijpers TW: Granulocyte colony-stimulating
factor delays neutrophil apoptosis by inhibition of calpains
upstream of caspase-3. Blood. 112:2046–2054. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cohen GM: Caspases: The executioners of
apoptosis. Biochem J. 326:1–16. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kasof GM, Lu JJ, Liu D, Speer B, Mongan
KN, Gomes BC and Lorenzi MV: Tumor necrosis factor-alpha induces
the expression of DR6, a member of the TNF receptor family, through
activation of NF-kappaB. Oncogene. 20:7965–7975. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mendez MG, Kojima S and Goldman RD:
Vimentin induces changes in cell shape, motility, and adhesion
during the epithelial to mesenchymal transition. FASEB J.
24:1838–1851. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chaw SY, Majeed AA, Dalley AJ, Chan A,
Stein S and Farah CS: Epithelial to mesenchymal transition (EMT)
biomarkers -E-cadherin, beta-catenin, APC and Vimentin - in oral
squamous cell carcinogenesis and transformation. Oral Oncol.
48:997–1006. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gunaratne A and Di Guglielmo GM: Par6 is
phosphorylated by aPKC to facilitate EMT. Cell Adhes Migr.
7:357–361. 2013. View Article : Google Scholar
|
|
50
|
Xi G, Shen X, Rosen CJ and Clemmons DR:
IRS-1 functions as a molecular scaffold to coordinate IGF-I/IGFBP-2
signaling during osteoblast differentiation. J Bone Miner Res.
31:1300–1314. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Brumatti G, Salmanidis M and Ekert PG:
Crossing paths: Interactions between the cell death machinery and
growth factor survival signals. Cell Mol Life Sci. 67:1619–1630.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Scott ML, Fujita T, Liou HC, Nolan GP and
Baltimore D: The p65 subunit of NF-kappa B regulates I kappa B by
two distinct mechanisms. Genes Dev. 7(7A): 1266–1276. 1993.
View Article : Google Scholar : PubMed/NCBI
|