Introduction
Drug repositioning is an innovative concept in the
drug discovery process whereby new indications that offer the
possibility of reduced time and risk for old drugs are sought
(1,2). Among them, several drugs to treat
metabolic syndrome have been repositioned to treat various cancers.
For example, simvastatin is a 3-hydroxy-3-mehtylglutaryl-coenzyme A
(HMG-CoA) reductase inhibitor and is often used to treat lipid
disorders. Recent studies have shown that simvastatin exhibits
anticancer effects by regulating proliferation, apoptosis, and
metastasis in various tumors (3–6).
Studies that have examined the underlying mechanisms of the
anticancer effects indicate that simvastatin inhibits
cyclin-dependent kinases (CDK) and cyclins (7) and promotes apoptosis by upregulating
the Notch1 gene (8) and inhibiting
Akt phosphorylation (9,10). Additionally, simvastatin also
enhances radiosensitization by suppressing BIRC5 (survivin) and
CTGF (connective tissue growth factor) in gastric cancer and
colorectal cancer (11).
Furthermore, several studies have shown that simvastatin triggers
caspase-dependent apoptosis via p53 activation (8,12),
but another study has shown that simvastatin induces apoptosis
through the JNK signaling pathway independent of p53 status
(13). However, the anticancer
mechanism of simvastatin relative to the p53 status remains
unclear.
Radiotherapy is a highly effective tool for cancer
treatment and is used to treat >50% of cancer patients (14). Although several factors such as
overexpression of DNA repair protein (15) and tumor microenvironment (16) affect the radiosensitivity, many
studies have focused on p53 status correlated with radiation
response (17–19). In unstressed cells, p53 is
maintained at low levels and controlled by mouse double minute 2
homolog (MDM2), which is a negative regulator of p53 (20). However, upon exposure to ionizing
radiation (IR), p53 is phosphorylated and subsequently inhibits the
p53-MDM2 interaction, which increases the expression of proteins
that induce apoptosis (e.g., bax and bcl-2), cell cycle arrest
(e.g., p21), and DNA damage repair (e.g., Rad51) (21). Therefore, p53 is a good prognostic
factor of radiosensitivity, and p53 mutations may promote
resistance to radiotherapy.
Radioresistant p53-deficient tumors usually express
the wild-type MDM2 protein because mutations in both MDM2 and p53
do not typically occur within one tumor (22). Wild-type MDM2 protein may retain
its role in cell cycle control, cell survival, and invasion
(23). Additionally, MDM2 is
frequently overexpressed in advanced cancer patients with invasive
and high-grade tumors (24). Thus,
inhibition of MDM2 may reinforce anticancer activities in
p53-deficient cancers. Recently, several studies have shown that
MDM2 inhibitors display broad anticancer activity in various
cancers. Feng et al reported that MI-219, a small-molecule
inhibitor of MDM2, sensitizes prostate cancer to radiotherapy in a
p53-dependent manner (25).
Another study has demonstrated that p53-independent induction of
p21 through MDM2 downregulation might contribute to anticancer
activity (26). Furthermore,
anti-MDM2 antisense oligonucleotides have chemosensitization and
radiosensitization effects in vitro and in vivo in
several human cancer models regardless of p53 status (27). However, the effects of combining
the MDM2 inhibitor with radiotherapy in p53-deficient and p53
wild-type cancers remain unclear.
In this study, we investigated whether the
combination of simvastatin and IR would radiosensitize
p53-deficient cancer cells. Furthermore, by focusing on the MDM2
and p53 interaction, we identified novel functions for simvastatin
as an MDM2 inhibitor and defined the impact of simvastatin and MDM2
on cellular radiosensitivity. We hope that our results contribute
to a scientific rationale for the clinical use of simvastatin in
combination with radiotherapy in patients with malignant cancers,
particularly p53-deficient cancers.
Materials and methods
Materials
HCT116 p53+/+ human colorectal cancer
cells were obtained from the Korean Cell Line Bank (Seoul, South
Korea), and HCT116 p53−/− human colorectal cancer cells
were kindly provided by Dr B. Vogelstein of Johns Hopkins
University. Simvastatin was obtained from Sigma-Aldrich Chemical
Corp. (St. Louis, MO, USA) and Cayman Chemical Corp. (Ann Arbor,
MI, USA). Roswell Park Memorial Institute (RPMI)-media, fetal
bovine serum (FBS), trypsin, and antibiotics were purchased from
Welgene (Seoul, Korea). Antibodies against MDM2, p53, cyclin D1,
survivin, p21, ku70, ku80, DNA-PKcs, p-ATM and GAPDH were purchased
from Santa Cruz (San Diego, CA, USA). Cyclin A2, pRb, p-cdc2
(CDK1), CDK2, cyclin B1, Rad50, FOXO3a, and E-cadherin antibodies
that were used in this study were obtained from Cell Signaling
Technology (Mississauga, ON, Canada). CDK4 and CDK6 antibodies were
purchased from Bethyl Laboratories (Montgomery, TX, USA). Rad51 and
ERCC1 antibodies were purchased from Abcam Bio (Cambridge, MA, USA)
and γH2AX was purchased from Millipore (Bedford, MA, USA).
Cell culture and treatments
HCT116 p53+/+ and p53−/− cells
were cultured in RPMI-1640 supplemented with 10% (v/v) FBS, 100
U/ml penicillin and 100 µg/ml streptomycin. All cells were
cultured in a humidified incubator under an atmosphere of 5%
CO2 at 37°C. Simvastatin was dissolved in 1 ml of DMSO
to a concentration of 12 mM and stored at 4°C. Cells were treated
with the indicated concentrations of simvastatin 24 h before
irradiation.
Irradiation
For in vitro experiments, the cells were
irradiated with a 137Cs γ-ray source (Atomic Energy of
Canada, Ltd., Chalk River, Ontario, Canada) at a dose rate of 2.67
Gy/min. For in vivo experiments, mice were irradiated using
a 60Co γ-ray source (Theratron 780; Atomic Energy of
Canada) with a 0.5 cm diameter bolus of tissue equivalent material
to allow for dose buildup.
Proliferation assays
The cells were seeded in a 96-well plate at a
density of 1×103 cells per well. Varying concentrations
of simvastatin (0–80 µM) were added to each well, and the
cells were incubated for 48 h, followed by addition of the
water-soluble tetrazolium (WST)-1 cytotoxicity assay reagent
(EZ-Cytox; Dogen, Seoul, Korea) according to the manufacturer's
instructions.
Clonogenic survival assays
Cells were trypsinized, seeded in 4 ml medium and
incubated overnight before the simvastatin treatment. Cells were
treated with 0–20 µM simvastatin for 48 h or 5 µM
simvastatin for 24 h before IR and then further incubated for 24 h.
After 12 days, cells were fixed for 30 min with 100% methanol and
stained with 0.4% crystal violet, followed by rinsing with tap
water. Colonies containing >50 cells were counted.
Xenograft tumors in athymic mice
Athymic Balb/c nude mice (4-week-old males) were
obtained from Nara Biothech Co. (Seoul, Korea) and maintained in a
laminar airflow cabinet under specific pathogen-free conditions.
HCT116 p53+/+ and p53−/− xenograft mouse
models were established by subcutaneous injection of
3×106 HCT116 p53+/+ or p53−/−
cells into the right thigh. When the tumor reached a volume of ~100
mm3, the mice were randomly divided into four groups
(n=5): control, simvastatin, IR, and simvastatin plus IR. The
simvastatin-treated groups (simvastatin alone and simvastatin plus
IR) were injected (intraperitoneally) once per day with 20 mg/kg.
When the tumor volume of the control group reached 150–180
mm3, the IR-treated groups (IR alone and simvastatin
plus IR) were treated with a single 5 Gy fraction of local-regional
irradiation using a 60Co irradiator. The tumor volume
(V) was calculated using the standard formula: V (mm3) =
π/6 × (smaller diameter)2 × (larger diameter). Mice were
euthanized by carbon dioxide (CO2) inhalation when the
average tumor volume of the control group reached 1,000
mm3.
Cell cycle analysis
After simvastatin (5 µM) exposure for 24 h,
cells were irradiated, incubated for 24 h, harvested, stained with
propidium iodide (1 mg/ml) according to the manufacturer's
protocol, and analyzed using a FACSCanto II flow cytometer
(Becton-Dickinson, Franklin Lakes, NJ, USA). A minimum of 10,000
cells per sample were counted, and data analysis was performed
using the CellQuest software.
Immunofluorescence
Immunofluorescent staining was performed to
determine the nuclear distributions of γH2AX and p-ATM foci in
HCT116 p53+/+ and p53−/− cells using image
analysis. Cells were grown on chamber slides 1 day prior to the
irradiation or simvastatin treatments. After simvastatin (5
µM) exposure for 24 h, cells were irradiated and incubated 1
or 24 h before harvest. Cells were fixed (4% paraformaldehyde in
phosphate-buffered saline, PBS, 10 min), permeabilized (0.6% Triton
X-100 in PBS, 10 min), and blocked (4% FBS in PBS, 1 h) and then
incubated overnight at 4°C or for >4 h at room temperature with
primary antibodies (1:500, anti-γH2AX, anti-p-ATM). The cells were
then washed with PBS and incubated for 1 h at room temperature in
the dark with appropriate fluorescein isothiocyanate (FITC)-labeled
secondary antibodies (1:500), including Alexa Fluor 488 goat
anti-mouse IgG (H+L) for γH2AX and p-ATM (green). The cells were
then washed with PBS, stained with DAPI (blue), and mounted onto
slides using fluorescence mounting medium. The slides were finally
examined using a fluorescence microscope with digital imaging
system (Olympus, Tokyo, Japan), and images were captured using a
charge-coupled device camera. For the quantitative analysis,
foci-positive cells were counted in ≥50 cells from randomly
captured images.
Western blot analysis
Cells were lysed with a radioimmunoprecipitation
assay (RIPA) buffer, and the proteins were separated by sodium
dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE) and
transferred to nitrocellulose membranes. The membranes were blocked
with 5% (v/v) skim milk in PBS with 0.1% Tween-20, incubated with
the indicated primary antibodies (1:1,000) and secondary antibodies
(1:1,000), and subsequently developed using an enhanced
chemiluminescence western blotting substrate (Cyanangen Srl,
Bologna, Italy) using the ImageQuant LAS-4000 mini (GE Healthcare,
Fairfield, CT, USA). The signal intensity of each band was measured
using the Multigauge V3.0 software (Fujifilm Life Science, Tokyo,
Japan).
Statistical analysis
All data are expressed as the mean ± standard error
of the mean (SEM). Statistical analysis was performed using
independent t-tests and one-way analysis of variance (ANOVA)
followed by Tukey's honest significant difference (HSD) test
through the Statistical Package for the Social Sciences (SPSS)
software (version 23.0, Chicago, IL, USA). The combination index
(CI) was calculated using the formula CI = (%A × %B) / (%AB × 100),
where %A and %B are the percent viability of simvastatin and IR
alone and %AB is the combination of them. Subadditivity was defined
as CI<1.0; additivity was defined as CI, 1.0; and
supra-additivity was defined as CI>1.0 (28).
Ethics statement
All animal study protocols and experiments were
approved by the Institutional Animal Care and Use Committee (IACUC)
of the Korean Institute of Radiological and Medical Sciences
(KIRAMS 2016-53).
Results
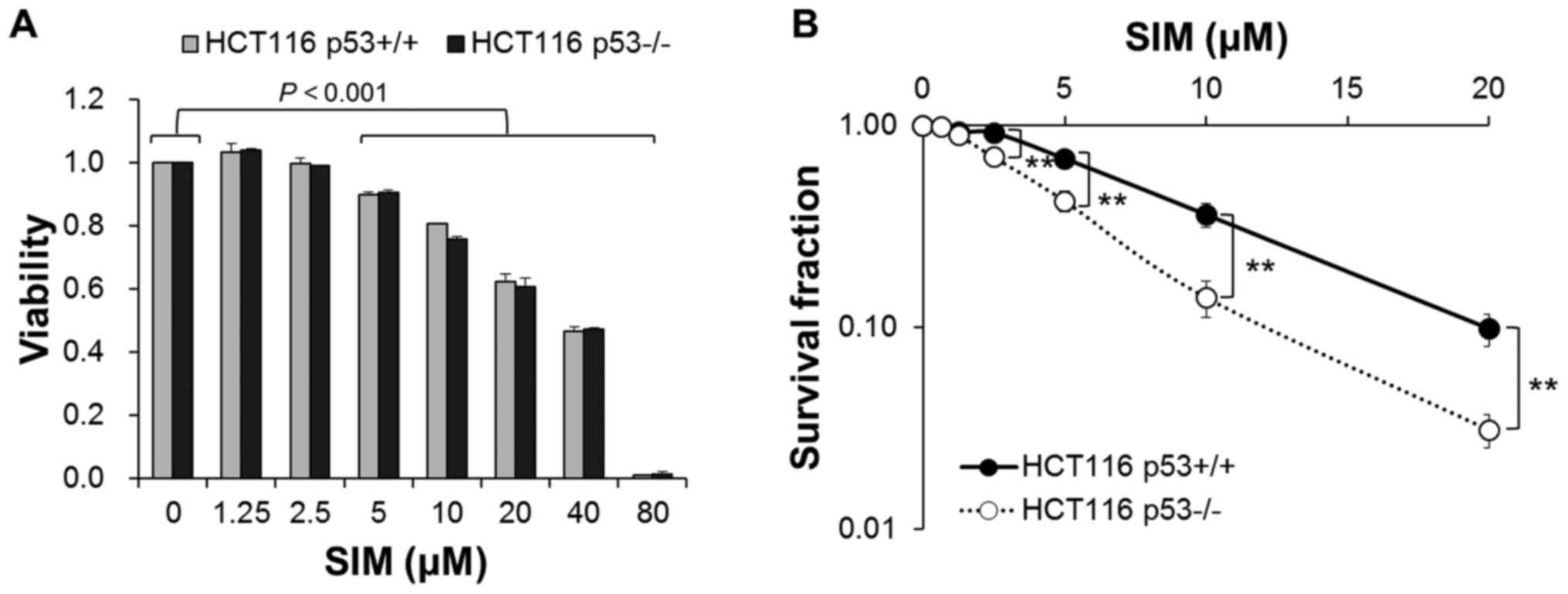
Simvastatin inhibits cell proliferation
in HCT116 p53 wild-type and p53-deficient cells
To investigate the effect of simvastatin on the cell
proliferation, HCT116 p53+/+ and p53−/− cells
were exposed to various concentrations of simvastatin (0–80
µM) for 48 h. Fig. 1A shows
that simvastatin inhibited the proliferation of both cells in a
concentration-dependent manner. Simvastatin decreased cell
proliferation by 89.9±0.8, 80.8±0.0, 62.2±2.6, 46.7±1.3 and
0.7±0.2% in HCT116 p53+/+ cells and 90.6±0.6, 75.6±0.9,
60.7±2.8, 47.2±0.6 and 1.3±0.6% in HCT116 p53−/− cells
at the concentrations of 5, 10, 20, 40 and 80 µM,
respectively (P<0.001). In addition, simvastatin weakened
cell-cell interaction and cell adherence in both cells (data not
shown). However, simvastatin had no differences in the cytotoxicity
between HCT116 p53+/+ and p53−/− cells.
Simvastatin induces lower clonogenic cell
survival in HCT116 p53-deficient than p53 wild-type cells
We also assessed p53-dependent clonogenicity by
simvastatin in HCT116 p53+/+ and p53−/−
cells. In HCT116 p53+/+ cells, simvastatin treatment
showed clonogenic survival fractions of 68.6±6.0, 36.2±4.9 and
9.9±1.8% at the concentrations of 5, 10 and 20 µM,
respectively (P<0.001) (Fig.
1B). Moreover, simvastatin induced clonogenic survival
fractions of 89.2±2.0, 69.6±4.0, 42.2±5.1, 14.1±2.9 and 3.1±0.6% in
HCT116 p53−/− cells at the concentrations of 1.25, 2.5,
5, 10 and 20 µM, respectively (P<0.001). Simvastatin
significantly decreased clonogenic cell survival of both groups of
cells in a concentration-dependent manner, and HCT116
p53−/− cells were more sensitive to clonogenic cell
death by simvastatin than HCT116 p53+/+ cells were
(P<0.01).
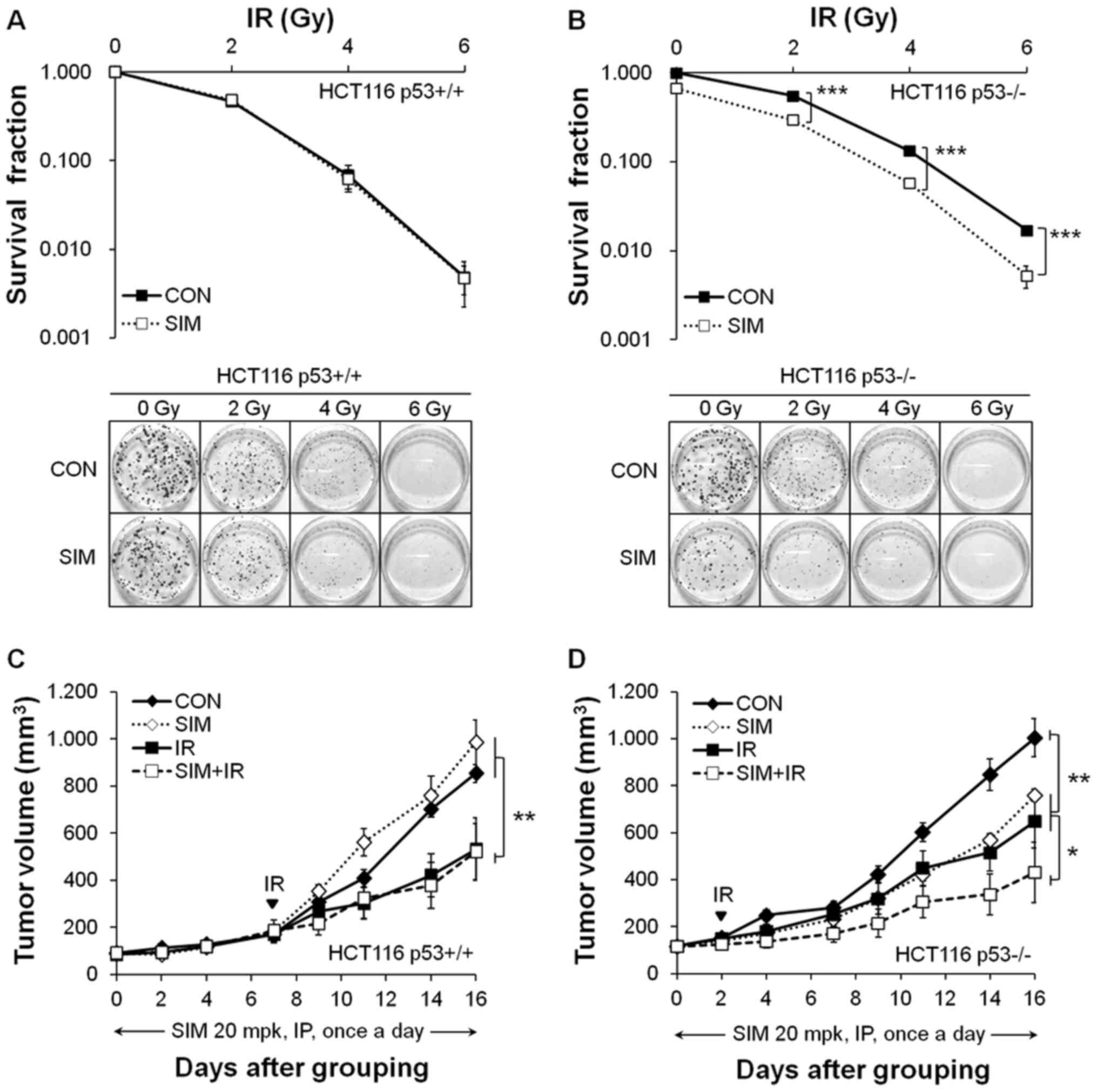
Simvastatin enhances the radiosensitivity
of HCT116 p53-deficient cells and xenograft tumors
To evaluate the radio-sensitization effects of
simvastatin in vitro, we treated HCT116 p53+/+
and p53−/− cells with 5 µM simvastatin for 24 h
followed by IR and then replaced the medium with fresh medium at 24
h after IR. In the clonogenic assays, the surviving fractions from
2 to 6 Gy IR were 46.9±3.4, 6.8±1.2 and 0.5±0.1% and the
combinations with simvastatin were 48.2±3.7, 6.2±1.0 and 0.5±0.1%
in HCT116 p53+/+ cells (Fig. 2A). In HCT116 p53−/−
cells, the surviving fractions from 2 to 6 Gy IR were 55.1±1.6,
13.3±0.5 and 1.7±0.2% and the combinations with simvastain were
44.2±1.4, 8.6±0.7 and 0.8±0.1% (Fig.
2B). Simvastatin plus IR significantly reduced the clonogenic
survival rate compared with IR alone in HCT116 p53−/−
cells (CI>1.0; P<0.01) but not in p53+/+ cells.
These results showed that the radiosensitizing effect of
simvastatin was only shown in HCT116 p53−/− cells.
We then investigated whether simvastatin enhanced
radiosensitivity in mice xenografted with HCT116 p53+/+
and p53−/− cells. When the mean tumor volume in the
control group reached ~1,000 mm3, simvastatin reduced
HCT116 p53−/− xenograft tumor growth (by 24.4±2.5%,
P<0.01; compared with control) but did not suppress HCT116
p53+/+ xenograft tumor growth (Fig. 2C and D). Moreover, simvastatin plus
IR enhanced HCT116 p53−/− xenograft tumor growth
inhibition more strongly than IR alone (IR, 35.4±11.2%; simvastatin
+ IR, 57.2±12.8%, P<0.05) but did not enhance p53+/+
xenograft tumor growth inhibition (IR, 37.5±15.4%; simvastatin +
IR, 38.8±14.0%). After the injection of simvastatin for 16 days,
there was no significant body weight loss in mice treated with
simvastatin (20 mg/kg/day) compared with the vehicle-treated
control group (data not shown). In summary, simvastatin enhanced
radiosensitivity of HCT116 p53−/− (CI>1.0), but not
p53+/+, cells and xenograft tumors.
Simvastatin reduces IR-induced G2/M
arrest of HCT116 p53-deficient and p53 wild-type cells
To examine the effect of simvastatin on IR-induced
cell cycle progression in the presence or absence of p53, we
analyzed cell cycle alterations by flow cytometry. As shown in
Fig. 3A, simvastatin markedly
attenuated IR-induced G2/M arrest and shifted HCT116
p53−/− and p53+/+ cells into the G1/S phases
(P<0.001). Specifically, simvastatin decreased the IR-induced
fraction of HCT116 p53−/− cells in the G2/M phase (IR,
76.8±1.1%; simvastatin + IR, 29.8±2.7%) compared with the fraction
of p53+/+ cells in G2/M (IR, 52.2±2.0%; simvastatin +
IR, 21.9±1.0%).
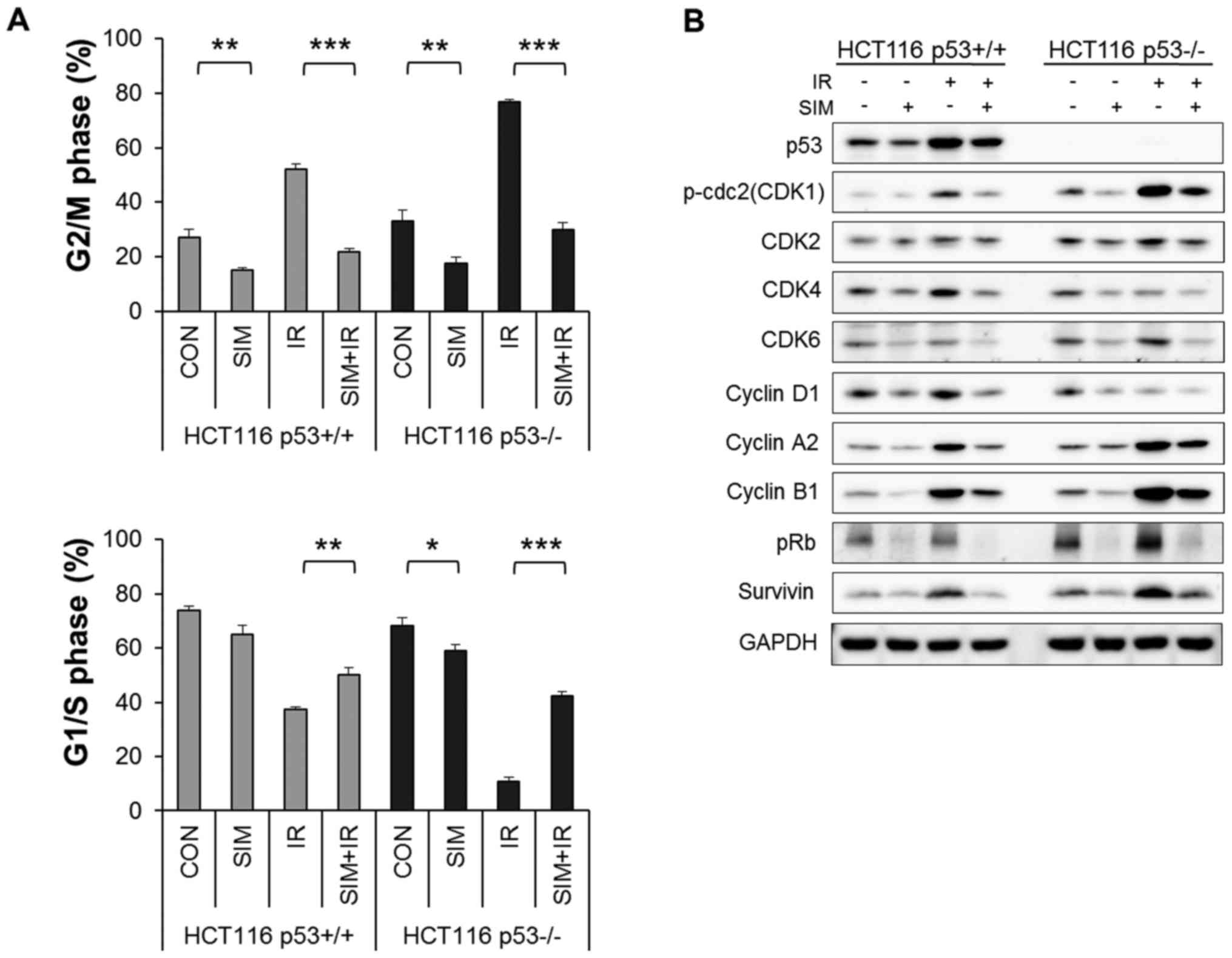 | Figure 3Simvastatin inhibits cell cycle
progression by significantly attenuating IR-induced G2/M arrest.
HCT116 p53+/+ and p53−/− cells were
pretreated with 5 µM simvastatin (SIM) for 24 h and then
irradiated with 6 Gy IR. (A) Flow cytometric cell cycle analysis of
PI incorporation was performed after treatment of cells with IR for
24 h. Values represent the mean ± SEM. Statistically significant
differences are shown; *P<0.05,
**P<0.01, ***P<0.001. (B) Western blot
analysis was performed to determine the protein expression levels
of the CDKs (CDK1, CDK2, CDK4 and CDK6), cyclins (cyclin D1, cyclin
A2, and cyclin B1), pRb, and survivin in HCT116 p53+/+
and p53−/− cells following treatment with simvastatin,
IR, or both. |
To confirm the decrease in IR-induced G2/M arrest by
simvastatin, we evaluated the expression levels of cell
cycle-regulated proteins, including cyclin-dependent kinases (CDK1,
CDK2, CDK4 and CDK6), cyclins (cyclin D1, cyclin A2 and cyclin B1),
pRb, and survivin, by western blotting. Fig. 3B shows that simvastatin decreased
all cell cycle-regulated proteins that we evaluated. Furthermore,
simvastatin plus IR significantly reduced the protein levels that
were upregulated after IR (CDK1, CDK2, cyclin A2, cyclin B1 and
survivin) in HCT116 p53+/+ and p53−/− cells
(P<0.05). However, simvastatin had no differences in the cell
cycle-regulated protein levels between the cell lines.
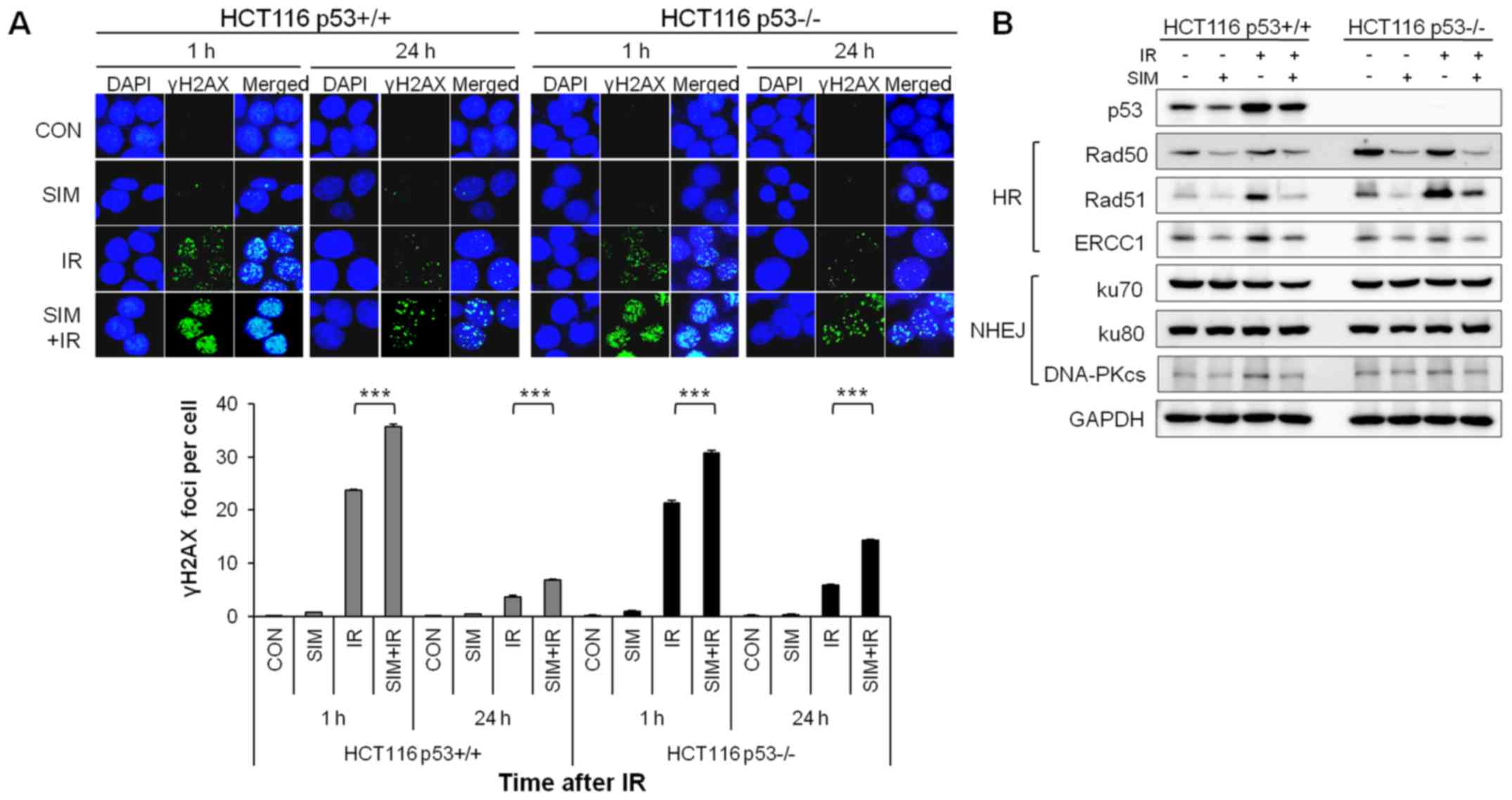
Simvastatin impaires DNA damage repair in
HCT116 p53-deficient cells more than in HCT116 p53 wild-type
cells
To analyze the effect of simvastatin on DNA damage,
we performed immunofluorescence staining of γH2AX, a marker of DNA
damage, and visualized the nuclear DNA using DAPI. At 1 h after IR,
simvastatin plus IR exhibited a significantly increased number of
γH2AX foci compared with IR alone in the HCT116 p53+/+
and p53−/− cells (Fig.
4A). Additionally, simvastatin plus IR consistently retained
the γH2AX foci for ≤24 h after IR in both cell lines, especially in
the HCT116 p53−/− cells (P<0.001). To investigate
whether simvastatin affected the DNA repair pathways, we examined
the expression levels of homologous recombination (HR) repair
proteins (Rad50, Rad51 and ERCC1) and non-homologous end joining
(NHEJ) repair proteins (ku70, ku80 and DNA-PKcs) in HCT116
p53+/+ and p53−/− cells by western blotting.
In the HR repair proteins, simvastatin decreased the Rad50, Rad51
and ERCC1 protein levels in HCT116 p53+/+ and
p53−/− cells (Fig. 4B).
Furthermore, IR alone significantly increased the expression of the
Rad51 protein compared with the control, whereas simvastatin plus
IR reduced the expression of the Rad51 protein compared with IR
alone in both cell lines. However, expression of NHEJ repair
proteins was not altered by simvastatin in either cell line. These
results indicated that simvastatin plus IR prolonged DNA damage and
reduced the HR repair protein levels relative to IR alone in HCT116
p53+/+ and p53−/− cells; these effects were
especially noticeable in HCT116 p53−/− cells.
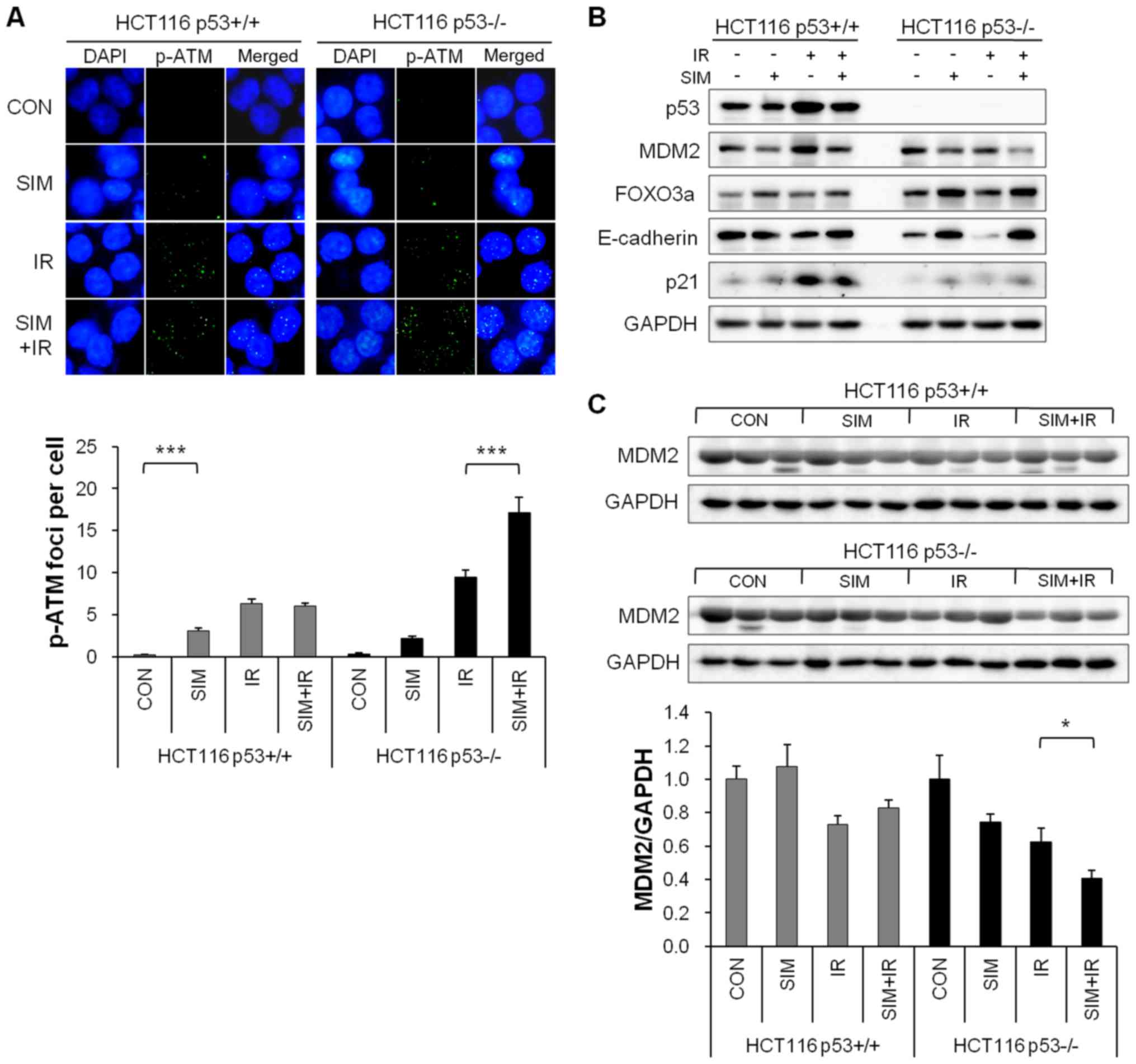
Simvastatin enhances the radiosensitizing
effect by reducing MDM2 expression in p53-deficient cells and
xenograft tumors
To investigate the role of simvastatin in the
response to radiation according to p53 status, we examined the
expression of MDM2-related proteins in HCT116 p53+/+ and
p53−/− cells and xenograft tumors.
First, we confirmed the p-ATM foci formation, which
is an upstream factor of MDM2 in HCT116 p53+/+ and
p53−/− cells. Simvastatin plus IR markedly elevated
IR-induced p-ATM foci formation compared with IR alone in HCT116
p53−/− cells (P<0.001) but not in p53+/+
cells (Fig. 5A).
We also evaluated the expression of MDM2 and
downstream factors of MDM2, including FOXO3a, E-cadherin, and p21,
which are tumor suppressor proteins. Regardless of p53 status,
simvastatin decreased the MDM2 protein level in both cell lines
(P<0.001). Interestingly, simvastatin strongly increased the
expression levels of FOXO3a, E-cadherin, and p21 in HCT116
p53−/− cells but not in p53+/+ cells
(Fig. 5B). Additionally,
simvastatin reduced the MDM2 protein level in HCT116
p53−/− xenograft tumors (P<0.05) but not in
p53+/+ xenograft tumors (Fig. 5C). In summary, we demonstrated that
simvastatin acted as an MDM2 inhibitor and enhanced the
radiosensitivity of HCT116 p53−/− cells by increasing
p-ATM foci formation and the protein levels of FOXO3a, E-cadherin,
and p21.
Discussion
Simvastatin, an HMG-CoA reductase inhibitor,
exhibits anti-proliferative, pro-apoptotic, anti-invasive, and
anti-metastatic properties in various cancers. Cancer cells are
more sensitive to the statins than normal cells because the levels
of HMG-CoA and low-density lipid receptors are elevated in cancer
cells (29). Furthermore,
simvastatin enhances the radiation responses of various cancer cell
types (30) and induces apoptosis
through p53-dependent (12) or
p53-independent pathways (13).
Although numerous studies have suggested that simvastatin may be
useful in cancer therapy, the radiosensitizing effect of
simvastatin in radioresistant cancer cells remains unelucidated. In
this study, we examined the anticancer and radiosensitizing effects
of simvastatin in p53-deficient cancer cells, focusing on the
underlying mechanisms of the p53-MDM2 interaction.
The anticancer effect of simvastatin on cell growth
and cell cycle progression has been studied extensively.
Simvastatin reduces cancer cell growth by inducing apoptosis
through upregulation of pro-apoptotic proteins (BAX, caspase-3,
caspase-8 and caspase-9) and downregulation of anti-apoptotic
protein (Bcl-2, Bcl-xl and XIAP) (3,4,6,8,12,30)
in the presence or absence of p53. Consistent with previous
studies, we found that simvastatin significantly inhibited the
proliferation and clonogenic survival of HCT116 p53 wild-type and
p53-deficient cells (Fig. 1). This
study specifically showed that p53-deficient cells were more
sensitive to simvastatin than HCT116 p53 wild-type cells. Moreover,
simvastatin stimulated cell cycle arrest through the reduction of
cyclin D1 and survivin in both cell lines, which is consistent with
previous reports (3,12). Furthermore, we demonstrated that
simvastatin also reduced the expression levels of CDKs (CDK4 and
CDK6), cyclin B1, and pRb in both cell lines (Fig. 3B). One interesting observation in
our study was that simvastatin had a new function of reducing HR
repair protein levels (Rad50, Rad51 and ERCC1) (Fig. 4B), and simvastatin consequently
promoted cancer cell death. These results highlighted the
anticancer effects of simvastatin in cancer cells, but the effects
did not differ between the HCT116 p53 wild-type and p53-deficient
cells.
Previous studies have shown that simvastatin induces
G1 arrest (7) and increases
IR-induced clonogenic cell death in gastric and colorectal cancer
cells, but these studies did not consider p53 status (11). In contrast to these previous
studies, we observed that simvastatin did not induce G1 arrest and
that simvastatin exhibited radiosensitizing effects only in HCT116
p53-deficient cells. Although simvastatin did not affect the
proportion of G1 phase in HCT116 p53 wild-type and p53-deficient
cells, simvastatin plus IR induced accumulation of G1 phase by
preventing IR-induced G2/M arrest, compared with IR alone; this was
especially apparent for HCT116 p53-deficient cells (Fig. 3A). Additionally, simvastatin
decreased IR-induced Rad51 and cell cycle regulatory proteins
including CDK1, CDK2, cyclin A2, cyclin B1, and survivin in both
cell lines (Fig. 3B).
Interestingly, although simvastatin did not solely induce DNA
damage, simvastatin combined with IR increased IR-induced DNA
damage and the duration of γH2AX foci expression in both cell
lines, especially the p53-deficient cell line (Fig. 4A). Collectively, these data suggest
that simvastatin-induced radiosensitization in p53-deficient cells
may be associated with cell cycle regulation and DNA damage repair
pathways.
Based on our data, we examined the mechanism by
which simvastatin enhances the anticancer effect and radiation
response more strongly in p53-deficient cancer cells than in
wild-type cancer cells. MDM2, which is negative regulator of p53,
has p53-dependent and p53-independent roles in the regulation of
cancer cells (31,32). MDM2 suppresses p53 activities in
p53 wild-type cancer cells while it promotes tumor formation and
progression in p53-deficient cancer cells (24). When cells are irradiated, IR
phosphorylates ATM, sequentially activates p53, and degrades the
MDM2 in p53 wild-type cancer cells (33–35),
whereas phosphorylated ATM directly reduced MDM2 proteins in
p53-deficient cancer cells; but the MDM proteins are consistently
present in p53-deficient cancer cells (36). By focusing on the dual actions of
the MDM2, we examined the differences of anticancer and
radiosensitivity between the p53 wild-type and p53-deficient cells.
In this study, we found that simvastatin exhibited a novel
function, namely, MDM2 inhibition, regardless of p53 status. After
the MDM2 protein reduction by simvastatin, the degraded factors by
MDM2 including tumor suppressor proteins FOXO3a, E-cadherin, and
p21 was increased, and simvastatin combined with IR increased the
formation of p-ATM foci compared with IR alone in p53-deficient but
not p53 wild-type cancer cells (Fig.
5). In p53 wild-type cancer, MDM2 inhibition by simvastatin may
be specific to activation of p53, which is a major function of
MDM2, and not specific to increasing downstream factors of MDM2.
Accordingly, simvastatin inhibited p53-independent functions of
MDM2 and thereby promoted anticancer effects and radiosensitization
of p53-deficient cancer cells.
In conclusion, simvastatin stimulated clonogenic
cell death by reducing cell cycle-related proteins and HR DNA
repair proteins in p53 wild-type cells and especially in
p53-deficient cancer cells. Additionally, simvastatin enhanced the
radiation response by reducing the MDM2 proteins levels and
inducing the FOXO3a, E-cadherin, and p21 protein levels in
p53-deficient cancer cells and xenografts. The exact mechanism of
MDM2 inhibition-induced apoptosis and metastasis by simvastatin
remains to be clarified. In the present study, we presented the
basis for broad therapeutic applicability of simvastatin as an
MDM2-based radiosensitizer for the treatment of p53-deficient
cancers.
Acknowledgments
This study was supported by a grant of the Korea
Institute of Radiological and Medical Sciences (KIRAMS), funded by
Ministry of Science, ICT and Future Planning, Republic of Korea
(no. 1711031807/50541-2016).
References
|
1
|
Novac N: Challenges and opportunities of
drug repositioning. Trends Pharmacol Sci. 34:267–272. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ashburn TT and Thor KB: Drug
repositioning: Identifying and developing new uses for existing
drugs. Nat Rev Drug Discov. 3:673–683. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shen YY, Yuan Y, Du YY and Pan YY:
Molecular mechanism underlying the anticancer effect of simvastatin
on MDA-MB-231 human breast cancer cells. Mol Med Rep. 12:623–630.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hoque A, Chen H and Xu XC: Statin induces
apoptosis and cell growth arrest in prostate cancer cells. Cancer
Epidemiol Biomarkers Prev. 17:88–94. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hindler K, Cleeland CS, Rivera E and
Collard CD: The role of statins in cancer therapy. Oncologist.
11:306–315. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Spampanato C, De Maria S, Sarnataro M,
Giordano E, Zanfardino M, Baiano S, Cartenì M and Morelli F:
Simvastatin inhibits cancer cell growth by inducing apoptosis
correlated to activation of Bax and down-regulation of BCL-2 gene
expression. Int J Oncol. 40:935–941. 2012. View Article : Google Scholar
|
|
7
|
Relja B, Meder F, Wilhelm K, Henrich D,
Marzi I and Lehnert M: Simvastatin inhibits cell growth and induces
apoptosis and G0/G1 cell cycle arrest in hepatic cancer cells. Int
J Mol Med. 26:735–741. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang X, Ma J, Xu J, Su Q and Zhao J:
Simvastatin induces growth inhibition and apoptosis in HepG2 and
Huh7 hepatocellular carcinoma cells via upregulation of Notch1
expression. Mol Med Rep. 11:2334–2340. 2015. View Article : Google Scholar
|
|
9
|
Fang Z, Tang Y, Fang J, Zhou Z, Xing Z,
Guo Z, Guo X, Wang W, Jiao W, Xu Z, et al: Simvastatin inhibits
renal cancer cell growth and metastasis via AKT/mTOR, ERK and
JAK2/STAT3 pathway. PLoS One. 8:e628232013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang T, Seah S, Loh X, Chan CW, Hartman M,
Goh BC and Lee SC: Simvastatin-induced breast cancer cell death and
deactivation of PI3K/Akt and MAPK/ERK signalling are reversed by
metabolic products of the mevalonate pathway. Oncotarget.
7:2532–2544. 2016. View Article : Google Scholar :
|
|
11
|
Lim T, Lee I, Kim J and Kang WK:
Synergistic effect of simvastatin plus radiation in gastric cancer
and colorectal cancer: Implications of BIRC5 and connective tissue
growth factor. Int J Radiat Oncol Biol Phys. 93:316–325. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Y, Xu SL, Wu YZ, Zhao MS, Xu WJ, Yang
HY and Li YX: Simvastatin induces caspase-dependent apoptosis and
activates P53 in OCM-1 cells. Exp Eye Res. 113:128–134. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Koyuturk M, Ersoz M and Altiok N:
Simvastatin induces apoptosis in human breast cancer cells: p53 and
estrogen receptor independent pathway requiring signalling through
JNK. Cancer Lett. 250:220–228. 2007. View Article : Google Scholar
|
|
14
|
Kumar S: Second malignant neoplasms
following radiotherapy. Int J Environ Res Public Health.
9:4744–4759. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Klein HL: The consequences of Rad51
overexpression for normal and tumor cells. DNA Repair (Amst).
7:686–693. 2008. View Article : Google Scholar
|
|
16
|
Rohwer N and Cramer T: Hypoxia-mediated
drug resistance: Novel insights on the functional interaction of
HIFs and cell death pathways. Drug Resist Updat. 14:191–201. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
El-Deiry WS: The role of p53 in
chemosensitivity and radiosensitivity. Oncogene. 22:7486–7495.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Böhnke A, Westphal F, Schmidt A, El-Awady
RA and Dahm-Daphi J: Role of p53 mutations, protein function and
DNA damage for the radiosensitivity of human tumour cells. Int J
Radiat Biol. 80:53–63. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gudkov AV and Komarova EA: The role of p53
in determining sensitivity to radiotherapy. Nat Rev Cancer.
3:117–129. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Alarcon-Vargas D and Ronai Z: p53-Mdm2 -
the affair that never ends. Carcinogenesis. 23:541–547. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Meek DW: Tumour suppression by p53: A role
for the DNA damage response? Nat Rev Cancer. 9:714–723.
2009.PubMed/NCBI
|
|
22
|
Ganguli G and Wasylyk B: p53-independent
functions of MDM2. Mol Cancer Res. 1:1027–1035. 2003.
|
|
23
|
Zhang Z and Zhang R: p53-independent
activities of MDM2 and their relevance to cancer therapy. Curr
Cancer Drug Targets. 5:9–20. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rayburn E, Zhang R, He J and Wang H: MDM2
and human malignancies: Expression, clinical pathology, prognostic
markers, and implications for chemotherapy. Curr Cancer Drug
Targets. 5:27–41. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Feng FY, Zhang Y, Kothari V, Evans JR,
Jackson WC, Chen W, Johnson SB, Luczak C, Wang S and Hamstra DA:
MDM2 inhibition sensitizes prostate cancer cells to androgen
ablation and radiotherapy in a p53-dependent manner. Neoplasia.
18:213–222. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Z, Wang H, Li M, Agrawal S, Chen X
and Zhang R: MDM2 is a negative regulator of p21WAF1/CIP1,
independent of p53. J Biol Chem. 279:16000–16006. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang Z, Wang H, Prasad G, Li M, Yu D,
Bonner JA, Agrawal S and Zhang R: Radiosensitization by antisense
anti-MDM2 mixed-backbone oligonucleotide in in vitro and in vivo
human cancer models. Clin Cancer Res. 10:1263–1273. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Duong HQ, You KS, Oh S, Kwak SJ and Seong
YS: Silencing of NRF2 reduces the expression of ALDH1A1 and ALDH3A1
and sensitizes to 5-FU in pancreatic cancer cells. Antioxidants.
6:62017. View Article : Google Scholar
|
|
29
|
Liao JK: Isoprenoids as mediators of the
biological effects of statins. J Clin Invest. 110:285–288. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hwang KE, Na KS, Park DS, Choi KH, Kim BR,
Shim H, Jeong ET and Kim HR: Apoptotic induction by simvastatin in
human lung cancer A549 cells via Akt signaling dependent
down-regulation of survivin. Invest New Drugs. 29:945–952. 2011.
View Article : Google Scholar
|
|
31
|
Shi D and Gu W: Dual roles of MDM2 in the
regulation of p53: Ubiquitination dependent and ubiquitination
independent mechanisms of MDM2 repression of p53 activity. Genes
Cancer. 3:240–248. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Q and Lozano G: Molecular pathways:
Targeting Mdm2 and Mdm4 in cancer therapy. Clin Cancer Res.
19:34–41. 2013. View Article : Google Scholar :
|
|
33
|
Chen L, Gilkes DM, Pan Y, Lane WS and Chen
J: ATM and Chk2-dependent phosphorylation of MDMX contribute to p53
activation after DNA damage. EMBO J. 24:3411–3422. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Perry ME: Mdm2 in the response to
radiation. Mol Cancer Res. 2:9–19. 2004.PubMed/NCBI
|
|
35
|
de Toledo SM, Azzam EI, Dahlberg WK,
Gooding TB and Little JB: ATM complexes with HDM2 and promotes its
rapid phosphorylation in a p53-independent manner in normal and
tumor human cells exposed to ionizing radiation. Oncogene.
19:6185–6193. 2000. View Article : Google Scholar
|
|
36
|
Perry ME: The regulation of the
p53-mediated stress response by MDM2 and MDM4. Cold Spring Harb
Perspect Biol. 2:a0009682010. View Article : Google Scholar : PubMed/NCBI
|