Introduction
Alpha-methylacyl-CoA racemase (AMACR) catalyzes the
reverse transition of (2R)-methylacyl-CoA and (2S)-methylacyl-CoA
(1,2) required for the β-oxidation of fatty
acids and the synthesis of bile acids (3,4). The
metabolism of fatty acids is activated in tumor tissue that is
associated with the increased production of AMACR, providing unique
benefits for the active proliferation of cancer cells (5,6). The
increased consumption of foods containing fatty acids, such as
animal fat, meat and milk, increases the risk of developing
prostate and intestinal cancer (7). By contrast, the inhibition of AMACR
production prevents the growth of malignant cells (8,9).
AMACR was identified and characterized in 1994 as the enzyme
involved in lipid metabolism (10), and its overexpression in prostate
adenocarcinoma, but not in benign prostate tissue was found in
2000–2002 (11–15). Thus, AMACR has been widely used as
a diagnostic marker for prostate cancer (16,17).
Recent publications have indicated the increased production of
AMACR, not only in prostate cancer, but also in tumors derived from
the epithelium of the intestines, kidneys, liver and embryonic stem
cells (6,18). Although the diagnostic value of
AMACR detection is evident, the mechanisms regulating an increase
in its levels in tumor cells, remain unexplored. The
transcriptional analysis of the AMACR proximal promoter has
resulted in the identification of the gene regulatory region
located between nucleotides −423 and −93, which is negatively
regulated by C/EBPα, p53 and NF-κB p50, but does not depend on the
signals mediated by the androgen receptor (19). The analysis of the AMACR promoter
region has not revealed the binding sites for proteins of the
LEF/TCF family; however, available data suggest that the increased
expression of AMACR in hepatocellular carcinoma is associated with
activating mutations of the β-catenin gene (CTNNB1), the
product of which transactivates the target genes via interaction
with proteins of the LEF/TCF family (20). It is known that β-catenin plays a
role in the regulation of the self-renewal of normal and tumor stem
cells of various tissues and is overproduced in cancers originating
from different organs (21).
Possibly, AMACR is a target of β-catenin, which causes an increase
in AMACR production in the case of CTNNB1 activating
mutations.
Carcinomas of the prostate, colon, kidney and liver,
in which the increased production of AMACR has been established,
are the leading causes of cancer-related mortality in different
parts of the world (22).
Therefore, AMACR is considered an important diagnostic marker of
cancer and a target of anticancer therapy. Currently, the search
for AMACR inhibitors is mainly focused on substances with substrate
specificity (9). Some authors have
presented the results of prostate cancer immunotherapy using
cytotoxic T lymphocytes that recognize AMACR (23). The possibility of the therapeutic
application of antibodies against AMACR has not yet been
investigated, at least to the best of our knowledge. This may be
associated with the intracellular localization of the protein and
its inaccessibility for antibodies. On the other hand, recent
studies have revealed the presence of AMACR in biological fluids of
patients suffering from cancer (24,25),
which indicates the obvious desirability for obtaining and for the
characterization of a wide variety of antibodies against AMACR.
The aim of this study was to obtain monoclonal
antibodies against human AMACR and to determine and map their
epitopes. The Biocompare database currently contains information
about 37 companies offering more than 400 different commercial
preparations of antibody against AMACR and their derivatives. These
preparations are supplied with the description of the different
abilities of the antibodies for their usage in various
applications, but do not contain characteristics of the epitopes
recognized by the antibody.
The first aim of the present study was to obtain the
hybridomas producing mouse monoclonal antibody against human AMACR.
As an antigen, we used the affinity purified recombinant human
protein encoded by the AMACR cDNA of 1,621 base pairs with an open
reading frame of 382 AA (GenBank Accession no. NM_014324). The
second aim was to describe the specific immune abilities of the
antibody and to detect the mimotopes recognized by the in-house
made and commercial antibody against AMACR using biopanning of the
phage peptide library Ph.D.-7C7 (BioLabs, Linden, NJ, USA) and the
sequencing of DNA from the selected phage particles. The third task
was to find the epitopes of human and mouse AMACR corresponding to
the phage mimotopes using a special Pepitope program (26), an algorithm PepSurf (27), and the crystal structure of AMACR
from Mycobacterium tuberculosis (MRC), PDB 1×74 (1). The final aim was to deliver the
antibodies prepared into live cells to estimate their biological
activities in cancer cells.
For this purpose, we prepared and characterized 20
murine hybridomas producing monoclonal antibodies recognizing human
and mouse AMACR. The antibodies produced by hybridomas 6H9 and 2A5
together with commercial 13H4 rabbit monoclonal anti AMACR antibody
were used for biopanning of phage peptide library followed by DNA
sequencing of the selected phage clones and mapping the
corresponding epitopes. We found that the epitopes recognized by
the antibodies 6H9 and 13H4 were formed, respectively, by sequences
G113, W114, R120, Q122, Q123, A124, G125, Y130, S132, L133, N134,
V189, W200 and W114, P119, R120, H126, I128, N129, Y130, S132,
L133, N134, G135, W200 in the MRC fold. These epitopes contain
approximately 50% identical AA and are localized in the region of
the enzyme catalytic center. When delivered into live HeLa cells
using cationic lipid-based PULSin reagent, specific antibodies
against AMACR were co-localized with peroxisomes. The in-house made
6H9 antibody exhibited a low level of co-localization compared to
the commercially available 63340 antibody, and did not inhibit the
growth rate of HeLa and T98G cells. On the whole, we generated
several clones of AMACR antibodies, and demonstrated that these
antibodies can be colonized into live cells. Currently, we are
testing the growth inhibitory properties of these antibodies.
Materials and methods
Animal care
A total of 5 BALB/с female mice (6–8 weeks old,
weighing 20–22 g; The Jackson Laboratory, Bar Harbor, ME, USA) were
used in all the experiments. The animals were maintained in the
animal facility at the Northwestern University (Chicago, IL, USA).
All experimental protocols were approved by the Institutional
Animal Care and Use Committee at the Northwestern University.
Cell lines and cell culture
The LNCaP human prostate cancer cell line, C3H10T1/2
(10T1/2; mouse embryonic fibroblasts) and the SP-2/0 mouse myeloma
cell line were obtained from the American Type Culture Collection
(ATCC, Manassas, VA, USA); 293T cells, MCF7 breast carcinoma cells,
HeLa cervical carcinoma cells, Caco2 colon carcinoma cells and T98G
human glioblastoma cells were obtained from the Culture Collection
of the Institute of Cytology RAS (St. Petersburg, Russia). The
Saos2 human osteosarcoma cells were kindly provided by Dr K. Helin
(European Institute of Oncology, Milan, Italy). The cells were
grown in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal
bovine serum (FCS) and gentamicin (50 μg/ml) in a
CO2 incubator under conditions of 5% CO2 and
100% humidity.
Preparation of hybridomas producing
monoclonal antibodies against AMACR: Induction and assessment of
the immune response
The BALB/c female mice were immunized by
intraperitoneal injections of 50 μg of affinity purified
recombinant AMACR in 150 μl of sterile phosphate-buffered
saline (PBS). As an adjuvant to the injection solution, 50
μg of the oligonucleotide 5′-TCC-ATG-ACG-TTC-CTG-ACG-TT-3′
were added (28). The injections
were repeated 5 times at intervals of 2 weeks. At 7 days after the
fourth injection, the animals were bled from the retro-orbital
sinus for the detection in their serum the antibodies against AMACR
by immunoblotting and enzyme-linked immunosorbent assay (ELISA) The
bleeding was performed under anesthesia [xylazine 10 mg/kg +
ketamine 80 mg/kg administered intraperitoneally (i.p.)].
Fusion of the immune splenocytes with
mouse myeloma cells, HAT selection, screening, cloning of
hybridomas and antibody isotyping
These procedures were carried out as previously
described (29). The SP-2/0
myeloma cell line was passaged 1 day prior to fusion to yield
1×107 cells the following day. The cells were
centrifuged for 5 min at, 200 × g at room temperature to remove
FCS, resuspended in 10 ml of DMEM/F12 without FCS and counted with
0.2% Trypan blue. Immune mouse spleen was removed under anesthesia
as mentioned above followed by 5–7 min with euthanasia and cervical
dislocation. The spleen was removed under sterile conditions and
perfused with DMEM/F12 medium without FCS using a 21G needle with a
10 ml syringe; the splenocytes were washed twice in 10 ml of
serum-free medium. For counting, 50 μl of the cell
suspension were mixed with 450 μl of 1% acetic acid and then
with 500 μl of 0.2% Trypan blue in PBS. Subsequently,
7×107 splenocytes and 1×107 myeloma cells
were mixed in a 50-ml tube, washed once by centrifugation as
described above, the supernatant was removed and the pellet was
resuspended by tapping the tube. The cells were placed in a water
bath at 37°C, mixed with 1 ml of warm 50% polyethylene glycol 1500
(Roche Diagnostics, Indianapolis, IN, USA) for 1 min with constant
stirring of the slurry. Immediately thereafter, the cells were
supplemented with 1 ml of serum-free medium and then for 3–4 min
with 3 ml of serum-free medium followed by the addition of 20 ml of
serum-free medium and 20 ml of medium with 15% FCS. The cells were
incubated for 30 min at 37°C, pelleted at 200 × g resuspended in 42
ml of DMEM/F12 with 15% FCS and 10% BM-Condimed H1 (Roche
Diagnostics), 1X HAT (Roche Diagnostics), and cultured at 5 ×
96-well plates of 100 μl per well. After 7 days, the growth
medium was exchanged with HAT-free medium. Within 3 days, the
hybridomas were screened for antibody production by ELISA. The
cells in the wells with positive reaction were cloned by
inoculating 100 cells in 96-well plates and the supernatants were
screened for antibody production. Cloning was continued until all
clones did not exhibit a positive reaction.
Monoclonal antibody isotyping
Antibody isotyping was performed using the mouse
hybridoma subisotyping kit purchased from Calbiochem (La Jolla, CA,
USA) according to the manufacturer's recommendations. Brifly,
separate wells of a 96-well ELISA plate were sequentially filled
with the goat anti-mouse IgGs, blocking solution, an the hybridoma
supernatants were diluted 10-fold, isotyping antisera were added,
and the wells were then visualized by TMB reagent and read at OD450
nm.
Detection of phage peptide mimotopes
recognized by antibodies against AMACR
Phage peptide sequences specifically recognized by
in-house made murine (clones 6H9 and 2A5) and the commercial rabbit
monoclonal (clone 13H4; Zeta Corp., Arcadia, CA, USA) antibody
against AMACR (mimotopes), were determined using a commercial phage
peptide library Ph.D.-7C7 (BioLabs) based on combinatorial
plurality of random 7-mer peptides fused to the pIII coat protein
of M13 phage (30). In this
library, the randomize peptide sequence is flanked by a pair of
cysteine residues. Under non-reducing conditions, the cysteines
form a disulfide crosslink, resulting in the phage display of
cyclized peptides. The library consists of 1.2×109
sequences amplified to produce ~200 copies of each sequence in 10
μl of the phage library.
Biopanning with antibodies in
solution
The selection of phage particles containing
mimotopes (biopanning), was performed by incubating the antibodies
coupled to protein A/G immobilized on agarose beads (Pierce,
Rockford, IL, USA). The E. coli ER2738 cells (included in
the Ph.D.-7C7 BioLabs kit) were grown in 5 ml of LB medium on a
shaker at 37°C until early logarithmic phase (OD600 to 0.01–0.05)
for titration and at the same time in 20 ml of LB medium for the
amplification of phage particles. Subsequently, 50 μl of 50%
protein A-agarose was resuspended in 1.5 ml microtube in 1 ml of
TBST [10 mM Tris-Cl (pH 8.0), 150 mM NaCl, 0.1% Tween-20] and
precipitated. The precipitate was washed in TBST by 3-fold
centrifugation for 5 min at 2,000 × g. Protein A-agarose was
resuspended in 1 ml of blocking buffer [0.1 M NaHCO3 (pH
8.6), 5 mg/ml bovine serum albumin (BSA), 0.02% NaN3],
incubated for 60 min at 4°C followed by stirring. Subsequently, 10
μl of the phage library in suspension (2×1011
Pfu) and 300 ng of anti-AMACR antibodies were mixed in 200
μl of TBST, incubated for 20 min at room temperature,
transferred to a 1.5 ml tube containing the affinity sorbent,
incubated for 15 min at room temperature with irregular stirring
and centrifuged for 5 min at 2,000 × g at room temperature. The
supernatant was removed and the sorbent was washed 10 times in 1 ml
of TBST and precipitated by centrifugation as described above. The
phage particles bound to protein A-agarose were eluted for 10 min
at room temperature in 1 ml of 0.2 M glycine-HCl (pH 2.2)
containing 1 mg/ml BSA. The eluate was centrifuged for 1 min at
13,000 × g, the supernatant was transferred to a new microtube,
neutralized by adding of 150 μl of 1 M Tris-HCl (pH 9.1) and
titrated on plates containing LB agar with
isopropyl-β-D-1-thiogalactopyranoside (IPTG) and
5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-Gal). The
eluted phage particles were amplified in 20 ml of ER2738 cell
culture in the logarithmic growth phase and incubated at 37°C for
4–5 h on a shaker. The culture of the bacterial cells was
centrifuged the following day for 10 min at 13,000 × g and 4°C, the
supernatant was transferred to new tube, and centrifugation was
repeated; 80% of the top layer of supernatant was transferred to
new tube, followed by the addition of a 1/6 volume of 20%
polyethylene glycol (PEG) 8000, containing 2.5 M NaCl; the solution
was left for 2 h at 4°C, the precipitate was spun down for 15 min
at 13,000 × g and 4°C, the pellet was resuspended in 1 ml of TBS
[50 mM Tris-Cl (pH 7.5), 150 mM NaCl, 0.2% NaN3],
centrifuged for 1 min at 13,000 × g, and the supernatant was
transferred to a new tube and titrated on plates with LB/IPTG/X-Gal
using the predicted titer levels of
10−8–10−11 Pfu. To do that, 200 μl of
bacterial cultures in microtubes were supplemented with 10
μl of phage particles in each dilution for 1–5 min, the
infected cells were transferred to tubes containing 3 ml of warmed
up to 45°C agarose, mixed and applied as the upper layer on the cup
agar plates containing IPTG/X-Gal; the plates were cooled for 5
min, inverted and incubated overnight at 37°C. The following day,
the numbers of blue colonies were counted and the phage titer was
determined using as a baseline titer of 2×1011 Pfu.
During the course of second round of phage concentration, we used
protein G-instead of protein A-agarose and the Tween-20
concentration in TBST was increased up to 0.5%. During third round
of phage selection, we used protein A-agarose and 0.5% Tween-20 for
washing. Finally, the phage particles from 10 different colonies
were transferred and amplified in separate tubes using overnight
culture of the ER2738 cells for 4–5 h at 37°С. The bacterial
cultures were then transferred to microtubes, centrifuged for 30
sec at 13,000 × g at +4°С, 500 μl of upper layer of the
supernatant was transferred into new tube, supplemented with 200
μl of PEG/NaCl, and the tube content was mixed by inverting
and left at room temperature for 10 min, centrifuged for 10 min at
13,000 × g; the supernatant was removed, the pellet was resuspended
in 100 μl of the buffer containing iodide chloride [10 mM
Tris-Cl (pH 8.0), 1 mM EDTA, 4M ICl], supplemented with 250
μl of ethanol for 10 min at room temperature and spun down
for 10 min; the supernatant was removed, the pellet was washed with
70% ethanol, dried and diluted in 30 μl of deionized water.
The phage DNA was sequenced using dye-labeled dideoxynucleotides in
the Sequencing Facility of the Northwestern University (Chicago,
IL, USA).
Antibody epitope mapping
This was performed using the program Pepitope
available online (http://pepitope.tau.ac.il) (26), the algorithm PepSurf (27) and the crystal structure of MRC, PDB
1×74 (1).
ELISA
In total, 100 μl of recombinant AMACR or BSA
as a negative control (10 μg/ml) in 0.05 M sodium carbonate
buffer (pH 9.6) were added into 1 well of a 96-well plate for ELISA
for 3 h at room temperature or overnight at +4°C. Unbound material
was removed by washing 3 times with PBST buffer (PBS containing
0.05% Tween-20). The unbound sites were saturated with the addition
of 3% BSA in PBST for 1 h. The plate was washed 3 times with PBST
and 100 μl of hybridoma supernatant, and diluted mouse
immune serum or BSA were added into 1 well for 1 h at room
temperature. The plate was then washed as described above and 100
μl of anti-mouse-HRP antibody was loaded into each well for
1 h at room temperature. To visualize peroxidase staining, 100
μl of tetramethylbenzidine (TMB) substrate (Sigma-Aldrich,
St. Louis, MO, USA) containing 0.01% hydrogen peroxide for 5–30 min
were added into each well. TMB was prepared from the 1 mg/ml stock
solution diluted 1:100 by 0.1 M sodium acetate buffer. The reaction
was terminated by the addition of 50 μl of 10% phosphoric
acid and the absorbance was read at 450 nm using a Dynex MRC TC
Revelation microplate reader (Dynex Technologies, Inc., Chantilly,
VA, USA).
Immunofluorescence staining
Intracellular proteins were stained as follows:
coverslips with spread cells grown in culture or attached to the
glass as a result of close contact with sections of various mouse
organs (kidneys, liver, heart, bladder and thymus, which were
obtained under the conditions described above in the section
entitled 'Animal care') were placed in 35-mm plates, washed with
PBS for 5 min, fixed with 4% paraformaldehyde for 15 min, and then
with 70% ethanol overnight at 4°C, treated with 0.2% Triton X-100
for 10 min and washed with PBS twice for 5 min. Unspecific binding
was blocked by 3% BSA with 0.1% Tween-20 for 1 h. The cells were
incubated with primary antibodies (rabbit monoclonal anti-AMACR,
cat. no. 13H4; Zeta Corp., Sierra Madre, CA, USA; mouse monoclonal
anti-AMACR, cat. no. ab63340; Abcam, Cambridge, UK; in-house made
mouse monoclonal antibodies against AMACR 6H9 and 2A5; antibody
dilutions were 1:50–1:200) in blocking solution for 1 h at room
temperature, washed 3 times with PBS for 5 min and incubated with
labeled species-specific antibodies [Cy3 goat anti-mouse IgG (H+L),
cat. no. A10521; and Cy5 goat anti-rabbit IgG (H+L), cat. no.
A10523, both from Molecular Probes, Eugene, OR, USA], washed 3
times with PBS for 5 min and mounted in Anti-Fade (Bio-Rad
Laboratories, Hercules, CA, USA) containing DAPI for nuclei
staining. Images were recorded on Leica TCS SP5 (Leica
Microsystems, Wetzlar, Germany) or Olympus FV3000 (Olympus, Tokyo,
Japan) confocal scanning microscopes using lasers at a 405, 488 and
561 nm wavelength.
Protein electrophoresis and
immunoblotting
Protein electrophoresis and immunoblotting were
performed as previously described (31). Electrophoresis was run on 8%
SDS-PAGE on mini or large vertical protein electrophoresis chambers
(Bio-Rad Laboratories). The probes for SDS-PAGE were prepared from
human prostate tissues and cells of established cell lines. The
prostate tissues were obtained from 5 male cancer patients, 60–70
years of age, subjected to radical prostatectomy in accordance with
the procedure approved by the Ethics Committee of the Northwestern
University Review Board (IRB) protocol. All patients provided
written informed consent. The prostate tissue probes included 5
samples of tumors and 4 samples of normal tissues adjacent to the
tumors obtained from the same patients.
Tissue samples of approximately 10 mg were placed in
tubes with 100 μl of lysis buffer containing 1% SDS, 50 mM
TrisCl (pH 6.8), 100 mM β-mercaptoethanole, protease and
phosphatase inhibitor cocktails (Sigma-Aldrich); treated with
Cole-Parmer 130 Watt Ultrasonic Processor (Cole-Parmer, Vernon
Hills, IL, USA) with 70% amplitude 6 times for 10 sec, incubated
for 0°C for 20 min, and centrifuged at 13, 000 × g for 15 min at
+4°C.
The cells grown on culture plates were washed twice
with PBS and removed with a plastic scrapper, sedimented by
centrifugation as described above and lysed with periodic
resuspending on ice for 30 min in 3 volumes (relative to the volume
of the dense cellular pellet) of buffer solution composed of 25 mM
Tris-HCl (pH 7.4), 250 mM NaCl, 0.25% NP-40, protease and
phosphatase inhibitor cocktails in 1:100 dilution. Cellular
extracts were centrifuged at 13,000 x g for 15 min at 4°C.
Supernatants were applied for further experiments. The probes were
equilibrated for protein content determined by Bradford reagent
(Bio-Rad Protein Assay kit II, cat. no. 5000002; Bio-Rad
Laboratories), transferred to micro-tubes with an equal volume of
loading buffer [5% SDS, 20% glycerol, 200 mM dithiotreitol, 120 mM
Tris-HCl (pH 6.8), 0.002% bromophenol blue] and boiled for 5 min in
a water bath. Probes in the volume of 25 μl containing 30–90
μg of total protein were loaded on one line of
polyacrylamide gel. Electrophoretically separated proteins were
transferred onto a PVDF membrane by semi-dry electrotransfer. The
proteins on the membrane were revealed using specific antibody
against AMACR described above and secondary Peroxidase AffiniPure
goat anti-mouse IgG (H+L) (cat. no. 115–035–003; Jackson
ImmunoResearch Laboratories, Inc., West Grove, PA, USA) and
anti-rabbit IgG, HRP-linked antibody, cat. no. 7074S; Cell
Signaling Technology, Danvers, MA, USA) and visualized with ECL™
Western Blotting Detection Reagent (cat. no. RPN2209;
Sigma-Aldrich).
AMACR immunoprecipitation assay
To evaluate the ability of the anti-AMACR antibody
to immunoprecipitate (IP)-specific antigen, we first prepared the
affinity matrix. Two 1.5 ml Eppendorf microtubes were loaded with
50 μl of 50% protein G-sepharose (Invitrogen, Carlsbad, CA,
USA), washed 5 times with 0.5 ml of PBS and supplemented with 1.25
ml of the serum-free hybridoma supernatant containing anti-AMACR
antibody 6H9 or with 2 μl of the immune serum from an
immunized with AMACR mouse in 1.25 ml of PBS. The supernatant was
prepared using ex-cell 610-HSF hybridoma serum-free medium (SAFC
Biosciences, Lenexa, KS, USA). The microtubes were rotated for 1.5
h at room temperature to couple the antibody to protein G-sepharose
and washed 5 times with the IP buffer containing 25 mM TrisCl (pH
7.5), 50 mM NaCl, 0.2% SDS, 1% NP-40, protease and phosphatase
inhibitor cocktails in 1:100 dilution. The prepared affinity matrix
was loaded with extracts from mouse liver containing 0.5–2 mg of
total protein. The tubes with IP were rotated overnight at 4°C to
couple the AMACR protein from the cell extracts to protein
G-sepharose, the affinity matrix was washed 5 times with the IP
buffer and twice with the same buffer without detergents. After a
final washing, the tubes containing the immune complexes were
loaded on 25 μl of SDS-PAGE loading buffer, boiled for 5 min
and centrifuged for 5 min at 13,000 x g. The supernatants were
loaded into 10% polyacrylamide gel to separate proteins under
reducing conditions. The separated proteins were transferred from
gel onto a PVDF membrane and visualized with ECL as described
above.
Affinity purification
The 6H9 and 2A5 hybridoma supernatants were prepared
by growing the cells for 15 days in Ex-Cell 610-HSF hybridoma
serum-free medium in CELLine 1000 flasks (Integra Biosciences,
Hudson, NH, USA). The supernatants were spun down for 30 min at 2,
000 × g, filtered through a 0.45-μm filter and evaluated for
protein concentration and antibody activity by ELISA and
immunoblotting. For affinity purification, the supernatants were
loaded into a 1 ml protein G-column (HiTrap Protein G HP; GE
Healthcare Life Sciences, Pittsburgh, PA, USA) equilibrated with 3
column volumes of binding buffer (0.02 M sodium phosphate, pH 7.0).
The column was washed with 10 column volumes of the binding buffer
to remove impurities and unbound material. The bound antibody was
eluted with 1 ml of elution buffer (0.1 M glycine-HCl, pH 2.7) into
a microtube containing 100 μl of neutralization buffer (1 M
Tris-HCl, pH 9.0). To exchange the elution buffer, 200 μl of
the sample was loaded on a Zeba Spin 0.5 ml desalting column
(Thermo Fisher Scientific, Waltham, MA, USA). Prior to desalting,
the column was washed 3 times by centrifugation with 300 μl
of PBS for 1 min at 1,500 × g. Subsequently, 200 μl of the
eluate was applied on the top of the resin and supplemented with 15
μl of the stacker. The sample recovery was performed by
spinning at 1,500 × g for 2 min, and the protein concentration was
estimated by Bradford assay.
Antibody delivery into cells and
estimation of their growth curves and proliferation rate
Antibody delivery into cells was performed using
PULSin reagent (Polyplus-transfection Inc., New York, NY, USA).
Subsequently, 6 wells of a 24-well plate were loaded with cover
glasses (one 8-mm glass into each well) and seeded with
2×105 exponentially growing HeLa or T98G cells in DMEM
containing 10% FCS. The following day, when the cell saturation
density was 70–80%, the cells were washed twice with PBS and the
growth medium in wells was exchanged to 900 μl of DME
serum-free medium. Three master mixes were prepared: i) 250
μl of 20 mM HEPES buffer (pH 7.4) and 2 μg of the
non-specific 2A5 antibody; ii) 250 μl of 20 mM HEPES buffer
(pH 7.4) and 2 μg of the specific 6H9 antibody against
AMACR; iii) 250 μl of 20 mM HEPES buffer (pH 7.4) and 2
μg of the specific 63340 antibody against AMACR purchased
from Abcam (Cambridge, UK). The mixes were filtered and
supplemented with 5 μl of PULSin reagent
(Polyplus-transfection Inc.) for 15 min at room temperature. Each
mixture was then divided in two and added to the wells with the
HeLa or T98G cells. Following 4 h of incubation at 37°C, the growth
medium was exchanged with fresh one containing 10% FCS and the
cells were kept in a CO2 incubator overnight. The
following day, the adherent cells on coverglasses were stained with
anti-mouse antibody conjugated with Cy3 dye (Invitrogen). In the
case of effective proteofection (≥80% exhibited positive
immunofluorescence with specific anti-AMACR antibody), the cells
were trypsinized, washed, counted and seeded into 15 wells of a
96-well plate at a density of 2×104 cells per well in
100 μl of culture medium. For growth curves, the number of
cells was evaluated in triplicate each day for the following 5 days
using counting chambers.
The proliferation rate was estimated using the Cell
Proliferation kit I MTT (Sigma-Aldrich) according to the
manufacturer's instructions. The cells treated with the antibodies
were seeded at a concentration of 5×103 cells/well in
100 μl culture medium in 9 wells of a 96-well plate and
incubated for 24 h. After the incubation period, 10 μl of
MTT labeling reagent (Cell Proliferation kit, REF 11465007001;
Roche Diagnostics, GmbH, Mannheim, Germany) were added to each well
and the cells were placed into a CO2 incubator for 4 h.
Subsequently, 100 μl of the solubilization solution were
added to each well and the plate was incubated overnight in a
CO2 incubator followed by measuring the optical density
(OD) at 570 nm using an ELISA reader (Fluorofot; Probanauchpribor
LLC, St. Petersburg, Russia).
Estimation of co-localization of antibody
to AMACR delivered into cells and peroxisome labeled with
fluorescent dye
The HeLa or T98G cells were labeled with CellLight
Peroxisome-GFP, BacMam 2.0 reagent (Molecular Probes, Eugene OR,
USA) as follows: The cells asynchronously growing on 60-mm cell
culture with 70% density were supplemented with 10 μl of
CellLight reagent (10 particles per cell) for 18 h. Co-localization
was evaluated the following day after staining the cells with mouse
monoclonal antibodies to AMACR (ab63340; Abcam, in-house made 6H9)
and anti-mouse antibody coupled to Cy3 as described above. For the
study of peroxisome co-localization with AMACR detected by the
antibody delivered into live cells, the cells with labeled
peroxisomes were transferred into the wells of a 24-well plate
containing coverglasses and proteofected the following day as
described above. Immunofluorescence was visualized using an Olympus
FV3000 fluorescence confocal microscope (Olympus) with 405, 488 and
561 nm lasers.
Statistical analysis
Statistical analysis was performed using Microsoft
Excel 2010 software. Each experiment was repeated at least 3
times.
Results
Preparation and characterization of
monoclonal antibodies against AMACR
The immunization of mice with recombinant human
AMACR and adjuvant oligonucleotide comprising the CpG residues
caused a rapid and strong immune response. In 1 week after the
fourth intraperitoneal injection of the antigen, the antibodies
which recognized AMACR were clearly detected in the mouse serum
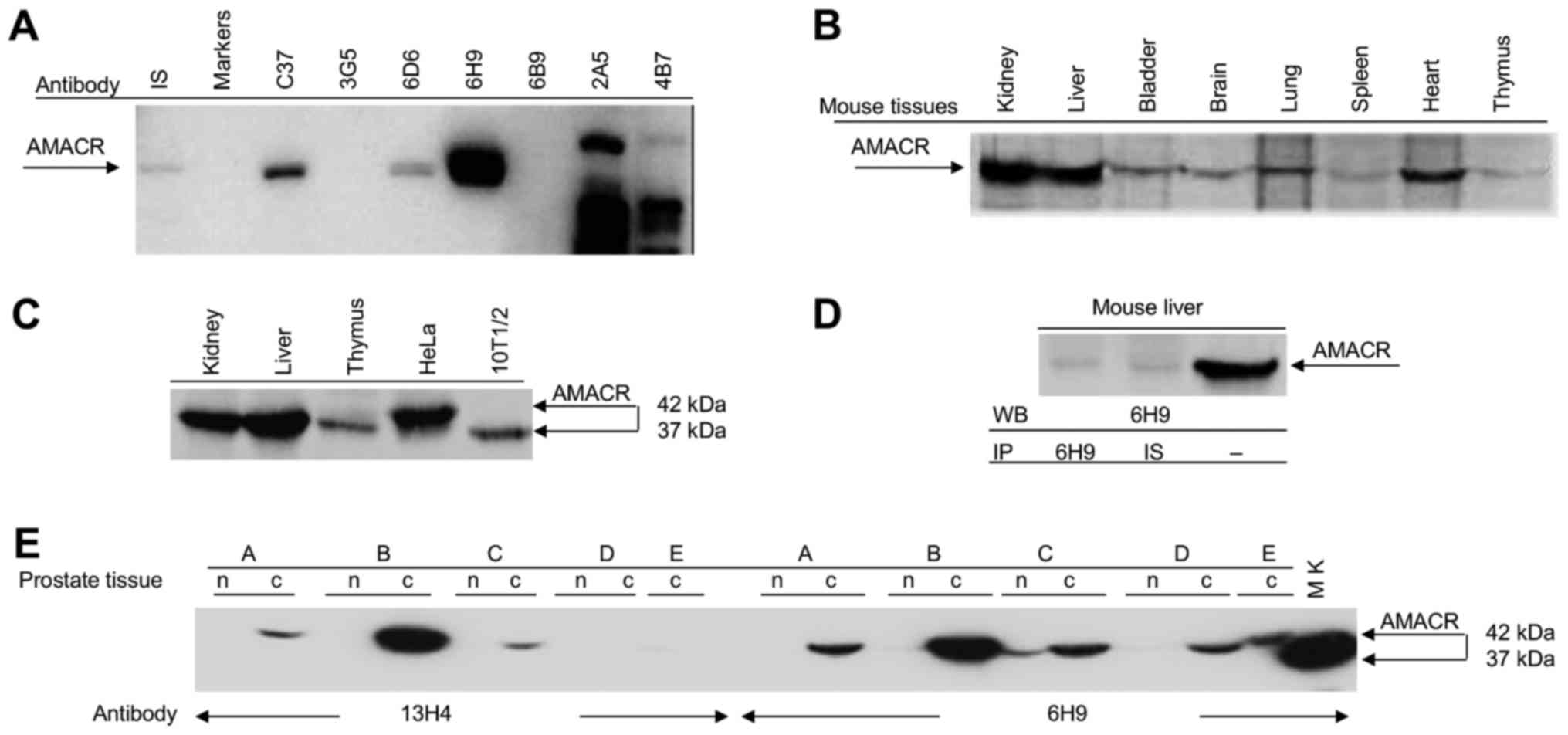
diluted 1:4,000 were by ELISA and immunoblotting (Fig. 1A). In the result of the fusion of
the immune splenocytes and myeloma cells followed by HAT selection
and screening by ELISA, we selected 20 hybridomas producing
antibodies which showed the signal exceeding 1.0 standard unit at
450 nm (>50% of the full scale value). In total, 5 of the 20
hybridomas produced antibodies of the IgG1 isotype, 4 of the IgG2b
isotype and 11 of the IgM isotype. The antibodies secreted by 4
different hybridomas bound the 1A AMACR isoform in both extracts
from the mouse kidney (37 kDa) and human prostate cell line LNCaP
(42 kDa), as shown by immunoblotting; the remaining 16 antibodies
did not bind any protein or interacted with several proteins The
blots in Fig. 1A shows that 3C7,
6D6, 6H9 antibodies bound a 42 kDa protein, 3G5, 6B9 do not
recognize any protein, while 2A5 and 4B7 recognize several
non-specific proteins in immunoblotting. For the following
experiments, we selected 6H9 antibody which a specifically
recognized 1A isoform of AMACR and 2A5 non-specific antibody as a
negative control for 6H9.
Evaluation of AMACR expression in various
tissues and cell lines of mouse and human origin by the in-house
made monoclonal antibodies
We found that in-house made antibody which bound
AMACR in extracts of LNCaP human prostate cells (Fig. 1A), also recognized this antigen by
immunoblotting in various mouse tissues. The highest levels of
AMACR expression were detected in the kidney, liver and heart;
however, in the bladder, brain, spleen and thymus, the antibody
bound small amounts of the protein (Fig. 1B). Human HeLa cells exhibited the
high levels of AMACR production similar to mouse liver and kidney
tissues; however, human AMACR migrated as a 42 kDa band, while all
mouse tissues contained the 37 kDa AMACR isoform (Figs. 1C and 2B). In immunoprecipitation assay, the
antibody to AMACR similar to the immune serum of mouse-donor
splenocytes used for hybridoma production bound minimal amounts of
AMACR from mouse liver in contrast to the high levels of this
protein detected by this antibody in immunoblotting (Fig. 1D). These results were confirmed by
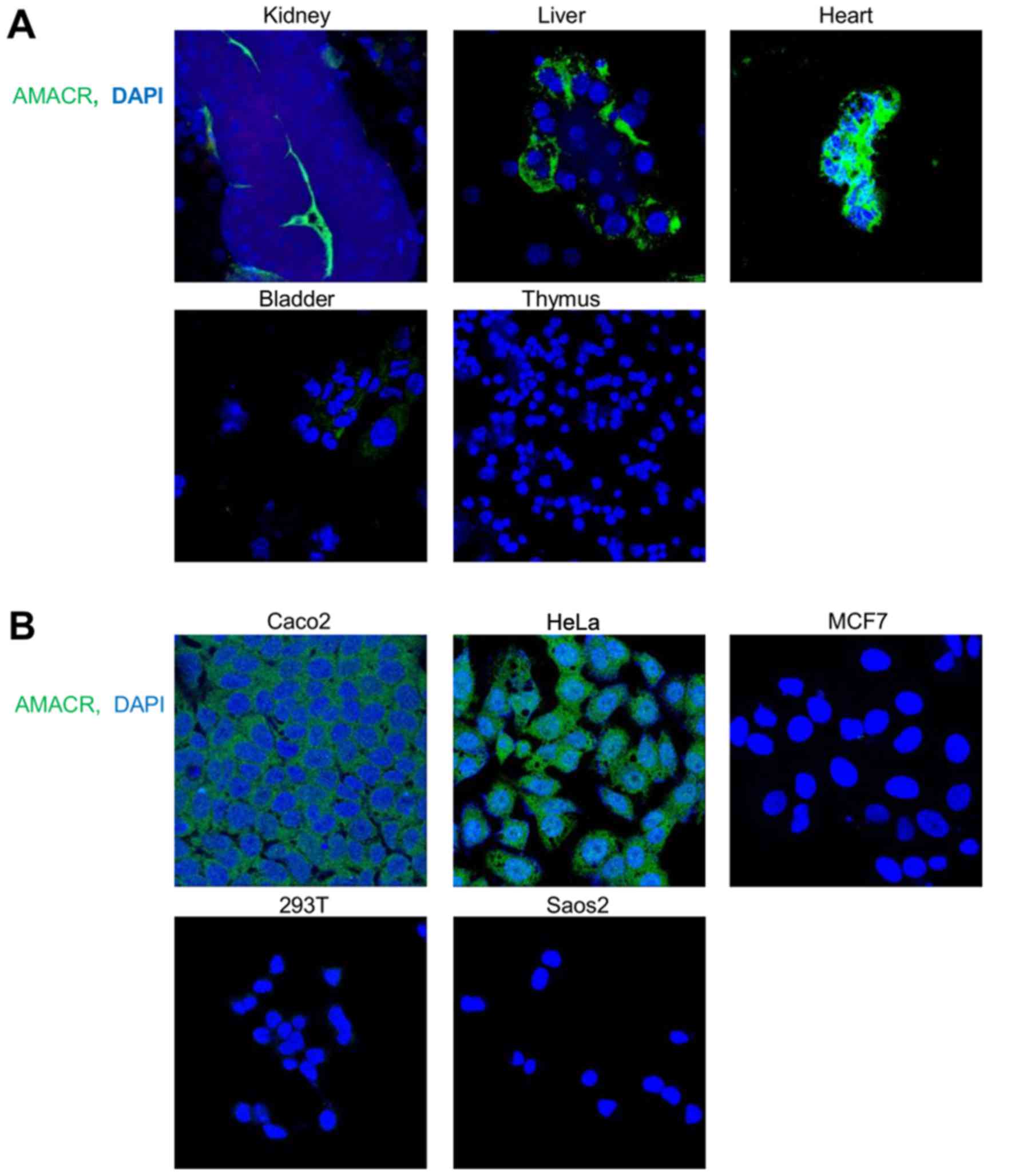
immunofluorescence staining of the mouse tissues performed with the
6H9 antibody (Fig. 2A). The cell
lines of human origin, similar to the mouse tissues, exhibited a
distinct AMACR expression dependent on their tissue origin. AMACR
was highly expressed in HeLa cervical carcinoma and Caco2 colon
carcinoma cells, in contrast to MCF7 breast carcinoma, 293T and
Saos2 osteosarcoma cells (Fig.
2B). To compare the specific activity of the in-house made 6H9
and commercial antibody, 13H4, we performed a site-by-site
immunoblot comparison of AMACR detection in probes from cancer or
normal prostate tissue from patients with prostate cancer. This
experiment revealed that the in-house made antibody, 6H9, possessed
a higher antigen sensitivity and detected AMACR in all 5 probes
from the cancer tissue of the patients with prostate cancer
compared with 3 from 5 probes detected by the commercial antibody,
13H4 (Fig. 1E).
Identification and characterization of
phage mimotopes recognized by the antibodies against AMACR
The antibodies, 6H9, 2A5 and 13H4, demonstrated a
high and reproducible signal in ELISA; however, they yielded
different images of antigen binding in immunoblotting. The 6H9 and
13H4 antibodies recognized the 42 kDa 1A human AMACR isoform, while
the 2A5 antibody bound several proteins with different molecular
mass (MM) (Fig. 1A). Given the
differences in the characteristics of the antibody/antigen
interaction in immunoblotting, we used the 2A5 antibody as a
negative control. Sequencing of the phage DNA from 10 different
clones obtained by biopanning and subsequent translation of the
phage DNA revealed that the 6H9 and 13H4 antibodies bound the same
AA sequence in 7 and 5 out of 10 cases, respectively. By contrast,
the 2A5 antibody recognized 10 different mimotopes in biopanning
(Table I).
 | Table ICharacterization of the phage
mimotopes and corresponding epitopes in MRC, human and mouse AMACR
recognized by the 6H9, 13Н4 and 2А5 antibodies. |
Table I
Characterization of the phage
mimotopes and corresponding epitopes in MRC, human and mouse AMACR
recognized by the 6H9, 13Н4 and 2А5 antibodies.
| Antibodies | Phage mimotopes and
their homologs in MRC, human and mouse AMACR | Number of fully
identical mimotopes in 10 selected phage | Number of identical
AА | Number of
conservative AA |
|---|
| 6H9 | | | | | | | | | | |
| Mimotope # 1 | L | K | W | G | V | H | W | 7/10 | | |
| MRC | L | R | W | G | V | N | W | | 5 | 5 |
| PGAA | 133 | 120 | 114 | 113 | 189 | 134 | 200 | | | |
| Human AMACR | L | F | F | G | V | S | W | | 4 | 5 |
| Mouse AMACR | L | F | F | G | V | S | W | | 4 | 5 |
| Mimotope # 2 | L | K | Y | G | Q | H | R | 1/10 | | |
| MRC | L | S | Y | G | Q | Q | R | | 5 | 5 |
| PGAA | 133 | 132 | 130 | 125 | 123 | 122 | 120 | | | |
| Human AMACR | L | A | Y | G | L | R | F | | 3 | 5 |
| Mouse AMACR | L | A | Y | G | V | K | F | | 3 | 5 |
| 13H4 | | | | | | | | | | |
| Mimotope # 1 | W | W | W | S | L | Q | P | 5/10 | | |
| MRC | | W | Y | S | L | R | P | | 4 | 4 |
| PGAA | | 114 | 130 | 132 | 133 | 120 | 119 | | | |
| Human AMACR | | F | Y | A | L | F | S | | 1 | 4 |
| Mouse AMACR | | F | Y | A | L | F | I | | 1 | 4 |
| Mimotope # 2 | H | K | S | L | G | T | W | 1/10 | | |
| MRC | H | N | S | L | G | N | W | | 5 | 6 |
| PGAA | 126 | 129 | 132 | 133 | 135 | 134 | 200 | | | |
| Human AMACR | H | N | A | L | G | S | W | | 4 | 6 |
| Mouse AMACR | Н | N | A | L | G | S | W | | | |
| 2А5 | | | | | | | | | | |
| Mimotope # 1 | F | P | T | F | P | N | Q | 1/10 | | |
| MRC | F | P | T | I | P | N | E | | 5 | 2 |
| PGAA | 319 | 328 | 318 | 313 | 311 | 309 | 310 | | | |
| Human AMACR | N | P | S | N | D | H | H | | 2 | 2 |
| Mouse AMACR | N | P | S | N | Q | H | H | | 2 | 2 |
| Mimotope # 2 | T | P | S | N | K | H | T | 1/10 | | |
| MRC | | P | T | N | R | H | | | 3 | 4 |
| PGAA | | 328 | 318 | 317 | 316 | 312 | | | | |
| Human AMACR | | P | S | G | R | H | | | 3 | 4 |
| Mouse AMACR | | P | S | A | R | H | | | 3 | 4 |
| Mimotope # 3 | N | N | I | L | P | М | T | 1/10 | | |
| MRC | N | E | I | F | P | М | Т | | 5 | 2 |
| PGAA | 309 | 310 | 313 | 319 | 328 | 329 | 318 | | | |
| Human AMACR | H | H | N | F | P | R | S | | 1 | 2 |
| Mouse AMACR | H | H | N | F | P | R | S | | 1 | 2 |
| Mimotope # 4 | W | G | F | P | Y | K | | 1/10 | | |
| MRC | W | G | Y | P | F | Q | | | 3 | 1 |
| PGAA | 326 | 325 | 319 | 328 | 318 | 327 | | | | |
| Human AMACR | V | D | I | P | F | S | | | 1 | 1 |
| Mouse AMACR | P | L | I | P | F | S | | | 1 | 1 |
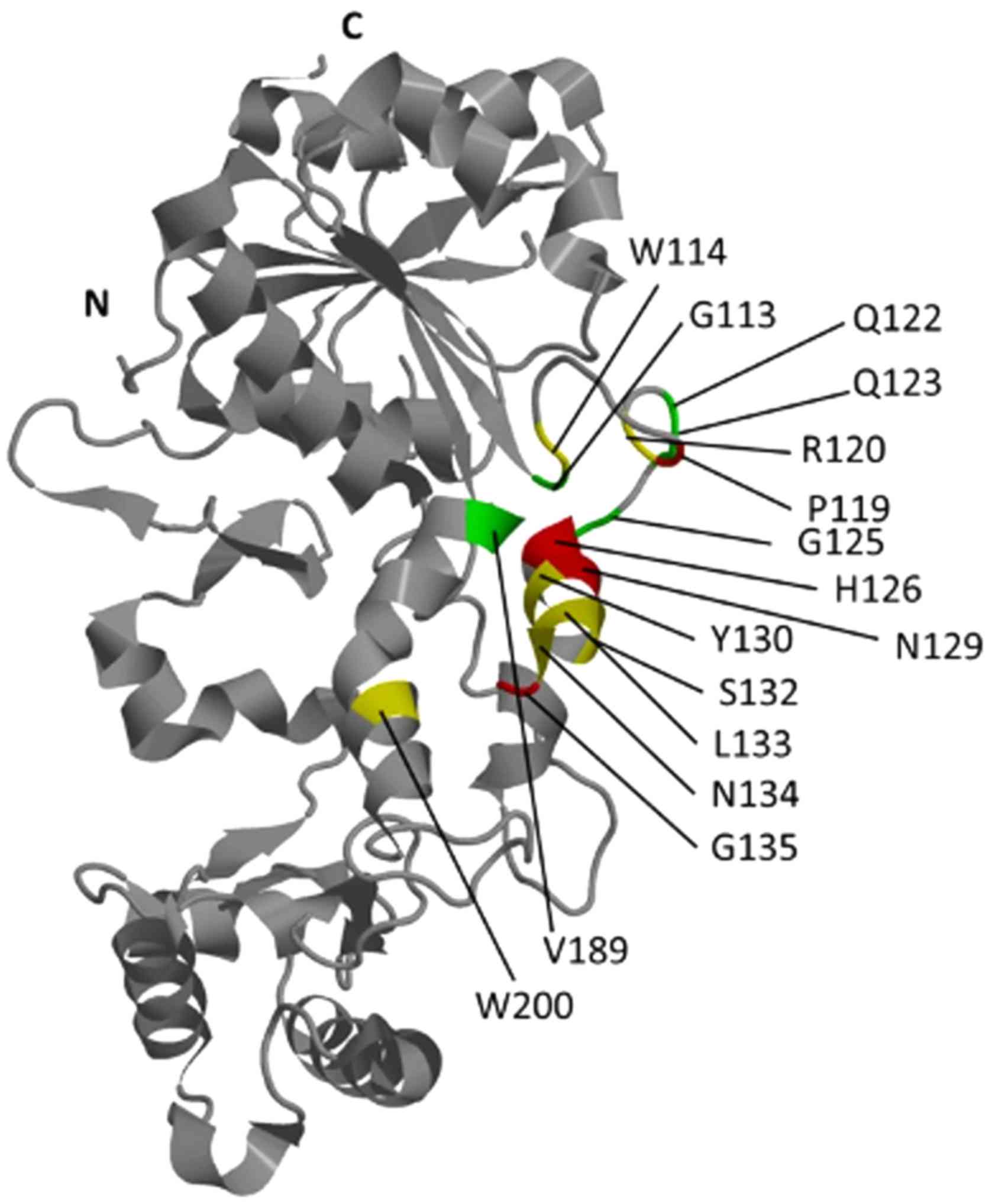
Mapping of 10 phage mimotopes selected with the 6H9
antibody revealed 3 clusters on the the surface of MRC, the AA
sequences, in which most closely corresponded to those in the phage
polypeptides. In accordance with the program, the most possible
conformational epitope (probability is 89.112 conventional units)
recognized by this antibody included AA G113, W114, R120, Q122,
Q123, A124, G125, Y130, S132, L133, N134, V189 and W200 of MRC.
This epitope was homologous to the phage mimotopes LKWGVHW and
LKYGQHR, identified, respectively, at 7 and 1 of the 10 phage
clones (Table I). To find the
epitope recognized by the 13H4 antibody, we used sequences of 9
phage mimotopes (mimotope KSLPXHS contains an unspecified residue
and was not included in the analysis). In total, 3 clusters of AA
were found for this antibody among which the greatest homology
(46.293 conventional units) showed the sequence which included
W114, P119, R120, H126, I128, N129, Y130, S132, L133, N134, G135
and W200 of MRC. This epitope was homologous to polypeptides
WWWSLQP and HKSLGTW, identified, respectively, in 5 and 1 from 9
phage clones (Table I). Antibody
2A5 interacted with 10 different mimotopes, from which the
sequences FPTFPNQ, TPSNKHT, NNILPMT, PWGFPYK revealed the greatest
degree of homology (probability is 37.089 conventional units) to
the epitope cluster of MRC comprising N309, E310, P311, H312, I313,
R316, N317, W318, F319, Y320, G325, W326, Q327, P328 and M329
(Fig. 3).
 | Figure 3Alignment of various AMACR
polypeptides: 1–2) LKWGVHW and LKYGQHR phage mimotopes detected by
6H9 antibody; 3–4) WWWSLQP and HKSLGTW phage mimotopes detected by
the 13H4 antibody; 5–8) FPTFPNQ, TPSNKHT, NNILPMT, PWGFPYK phage
mimotopes detected by the 2A5 antibody; 9) MRC, PDB 1×74, 10) human
AMACR, NP_055139.4; 11) mouse AMACR, NP_032563.2. (*), identical
AA; (:), highly conservative AA, more 0.5 point using matrix Gonnet
PAM 250, (−), low conservative AA, 0.5 and less point. WYSLRP,
SNKHP and WGFPYK epitope sequences are shorter than the
corresponding mimotopes on 1–2 AA which the program Pepitope did
not identified in MRC. |
All AA forming the epitopes recognized by the 6H9
and 13H4 antibodies were located in the region of MRC, composed of
helices α5 and α8, and unstructured sequence between helices α5 and
α6, helix α5 and sheet β5 (Fig.
4). The vast number of the AA forming these epitopes, 24 out of
27, were located between helices α5 and α6, helix α5 and sheet β5,
whereas in the region of helix α8, there were mapped only 3 AA
(Table I). The epitopes recognized
by the 6H9 and 13H4 antibodies indicated high levels of
conservatism, 71/69%, 50/38% and 71/77% of their AA were identical,
respectively, in MRC, human or mouse AMACR and conservative. The
epitopes detected by the 6H9 and 13H4 antibodies in human or mouse
AMACR were virtually identical (Table
I).
The epitopes recognized by the 2A5 antibody, which
was used as a negative control localized in the helix α13 and
sheets 10–11 of MRC, that represent a different region of the AMACR
fold compared with the region between helices α5 and α8, in which
the epitopes of the 6H9 and 13H4 antibodies were localized
(Fig. 3). Of note, despite the
high levels of identity of phage mimotopes and MRC epitopes
recognized by the 2A5 antibody (64% identity), these sequences
revealed low levels of conservatism (36%). When compared with
homologous sequences in mouse and human AMACR the levels of their
identity and conservatism were, respectively, 28 and 36% (Table I) (the percentage identity scores
were obtained by dividing the number of identical AA by the total
number of AA in all mimotopes).
Growth-associated activity of the 6H9
antibody against AMACR
To examine the growth-associated activity of the 6H9
antibody, we first tried to deliver the antibody into cells using a
liposome-based PULSin reagent (Polyplus-transfection). For these
experiments, we exploited HeLa human cervical adenocarcinoma cells,
which produce high levels of AMACR (Figs. 1C and 2B) and exhibit a high sensitivity in
antibody delivery with PULSin reagent (32). AMACR catalyzes the oxidation of
fatty acids and is localized in peroxisomes and mitochondria. The
6H9 antibody similar, to the commercially available 63340 antibody
(Abcam), detected abundant amounts of AMACR in HeLa cells (Fig. 5A). In HeLa cells treated with
paraformaldehyde, AMACR was mostly detected outside peroxisomes,
observed in both preparations stained with 6H9 or 63340 antibodies
(Fig. 5B). The distribution
pattern of specific 6H9 and 63340 antibodies proteofected into HeLa
cells was similar and differed from that of non-specific 2A5
antibody. While specific antibodies were distributed over the
entire cytoplasm compartment preferentially in the form of small
particles, non-specific antibody forms large cytoplasmic clusters,
often connected in chains near or around the nuclei (Fig. 5C). The levels of co-localization
with peroxisomes of different antibodies against AMACR proteofected
into HeLa cells depended on the type of antibody used. While
non-specific 2A5 antibody did not exhibit any co-localization
(Fig. 5D), 6H9 antibody, low level
and 63340 antibody, exhibited a greater level of co-localization
visible due to the change in the color of peroxisomes from green to
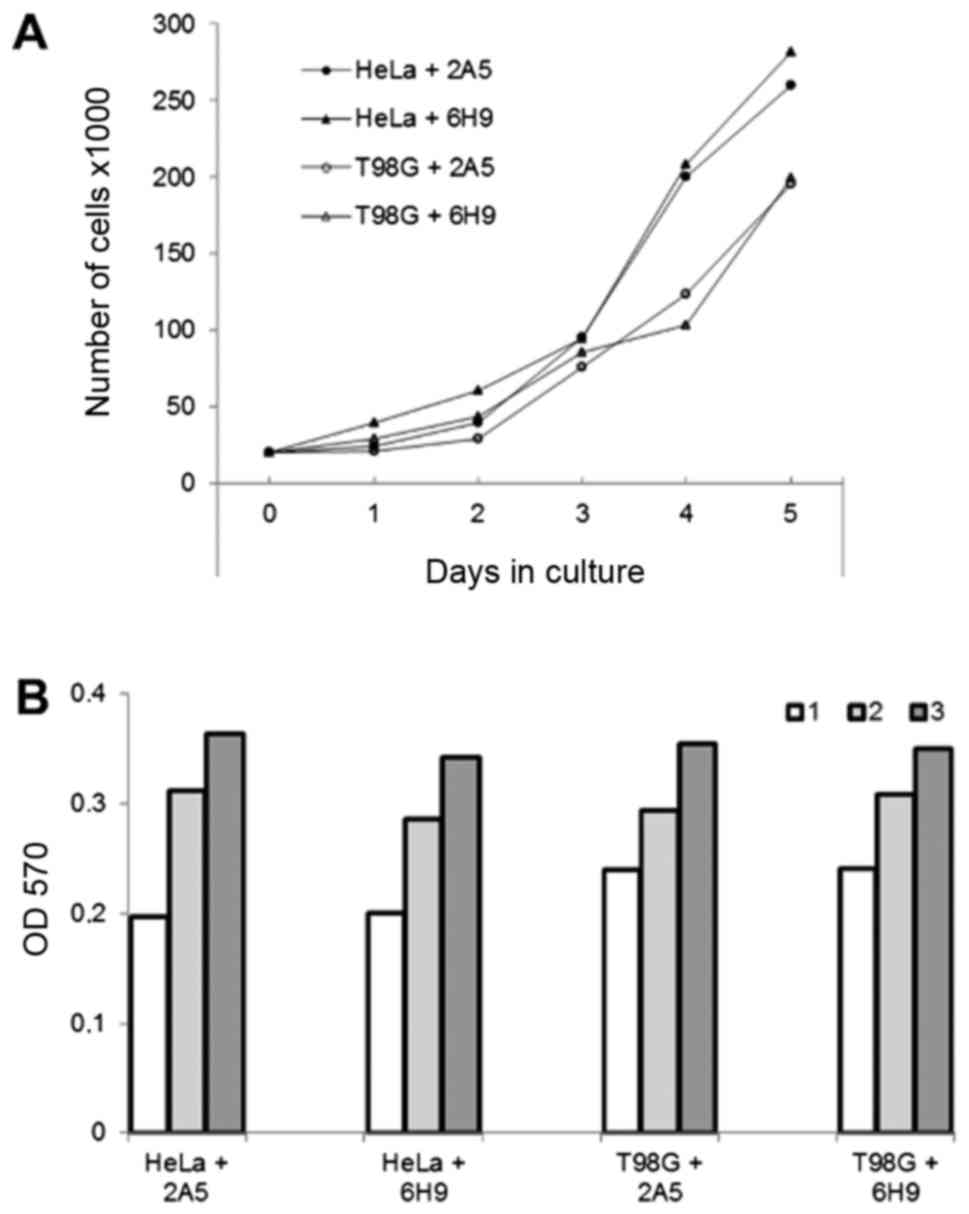
yellow (Fig. 5D). The growth rate
of the HeLa and T98G cells proteofected with antibody 6H9 or
non-specific antibody 2A5 was similar when determined using growth
curves or MTT assay (Fig. 6).
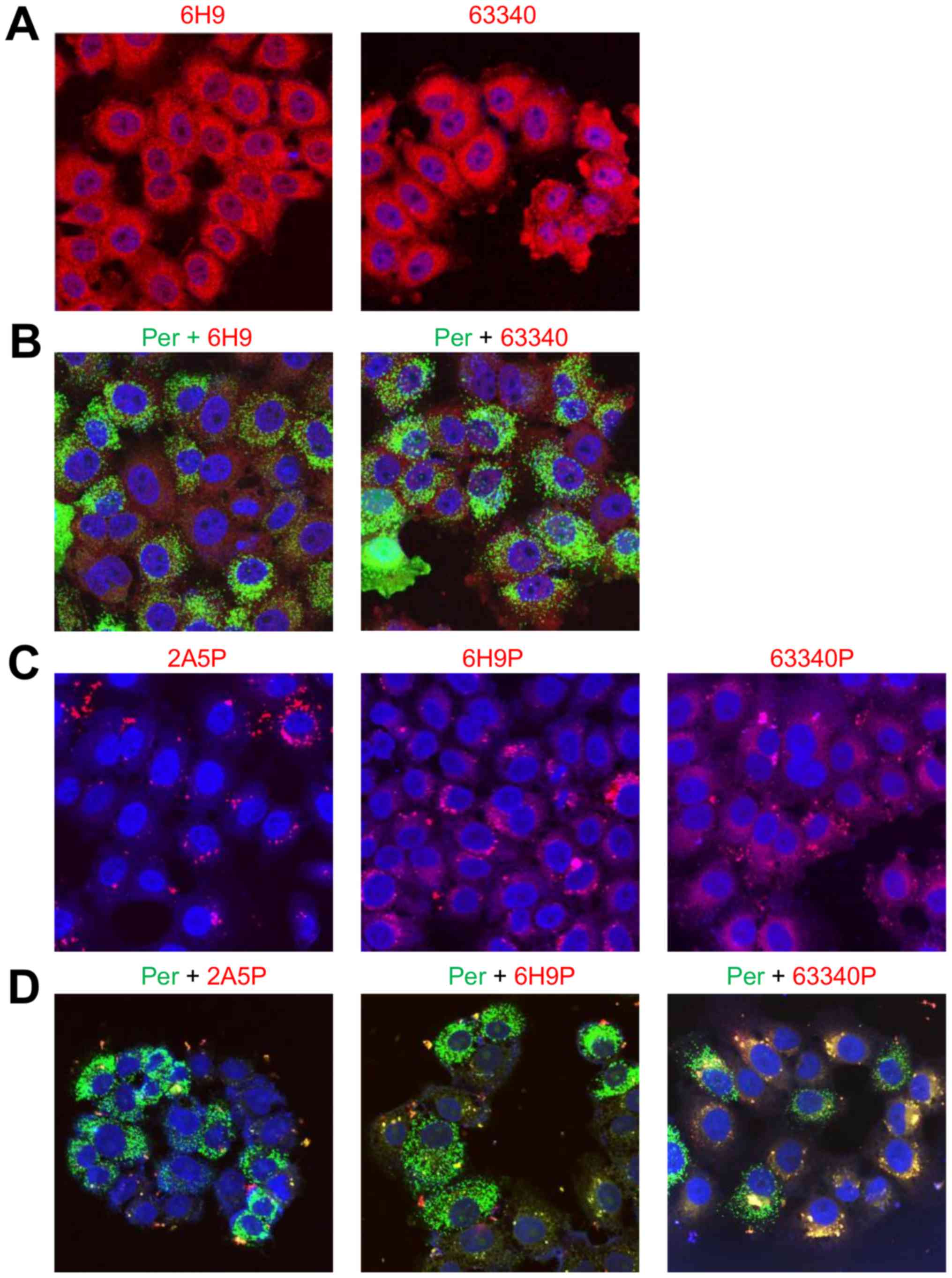 | Figure 5Immunofluorescent evaluation of AMACR
co-localization with peroxisomes in HeLa cells proteofected with
different antibodies against AMACR. (A) AMACR visualization,
accordingly, with in-house made 6H9 and commercial ab63340 (Abcam)
antibodies in fixed cells; (B) AMACR co-localization with
peroxisomes in fixed cells stained, accordingly, with 6H9 or
ab63340 antibodies; (C) cytoplasmic distribution of non-specific
2A5, specific 6H9 or ab63340 antibodies to AMACR proteofected into
live cells; (D) co-localization with peroxisomes of non-specific
2A5, specific 6H9 or ab63340 antibodies proteofected into live
cells. The positive staining is shown in red and yellow. The images
were recorded on Olympus FV3000 confocal scanning microscope using
lasers with 405, 488 and 561 nm wavelength, with a X40 objective;
the size of the images is 159 μm. P, proteofection; Per,
peroxisomes. Each experiment was repeated 3–5 times. |
Discussion
AMACR localizes in peroxisomes and mitochondria
(33), where it catalyzes the
racemization of (αR)-methylacyl-CoA esters required for the
synthesis of bile acids and ATP during subsequent β-oxidation
(34–36). Mouse AMACR is encoded by a single
gene and comprises at the N- and C-terminus the specific sequences
regulating input, respectively, in mitochondria and peroxisomes
(37). There are 10 different
isoforms of AMACR in humans, but only the 1A isoform encoding a
protein with the reading frame of 382 AA is functionally the most
important (38). Enzymatically,
AMACR, independently on cofactors, reversibly converts the R and S
asymmetric position of 2 carbon atom of the methyl group of
2-methyl-thioester. AMACR belongs to the family III of CoA
transferases (39), members of
which catalyze the transfer of a proton of the carbon atom directly
performed by two AA (1,2). In order to study the atomic structure
of the AMACR catalytic center, it was described as a model the
crystal structure of AMACR of MRC, which has 41 and 43% sequence
identity to the mouse and human protein, but in contrast to
mammals, MRC is expressed in an active form in a bacterial system
(1). In a native form, AMACR
exists as 89 kDa dimer, while the monomeric form MW is 39 kDa.
The monomeric form of AMACR is composed of two
domains: A large N-terminal and small C-terminal. Two linker
sequences, α8 and α12-β11, respectively connect these domains and
the C-terminal helix α14, in which a three-dimensional protein
structure associated with the N-domain. The small domain includes
residues 224–300 (β7-β9). The core of the large domain has an open
α/β structure consisting of 6 layers of the central parallel β
sheets (β1-β6), localized at the sides of the helices. The β5 and
β6 layers are connected by α5, α6 and α7 helices. The
characterization of the catalytic center of MRC by mutagenesis of
26 highly conserved AA showed that a substitution of alanine at 4
of these: Arg91, His126, Asp156 and Glu241, is accompanied by a
decrease in enzyme activity, but maintains the correct folding
(1). Three of the 4 named AA
localized in the N-terminal domain: Arg91, in α3; His126, in α5;
Asp156, in 7α helix; whereas Glu241 localized in the C-terminal
domain immediately after β8. The location of AA showing catalytic
activity gave reason to assume that the catalytic center of MRC was
located at the interface between the N- and C-domains, about the
N-terminal portion of helix α5 (1). This assumption was confirmed by
analyzing the structure of MRC co-crystallized with a variety of
substrates (2). It was found that
the conversion of isoforms R and S of AMACR substrates was carried
out by de- and reprotonation of the carbon atom by pair of amino
acids: His126/Asp156, which are located, in α5 and α7, respectively
through a mechanism called proton transfer 1.1 (2,39).
The active site of the enzyme involves additionally to
His126/Asp156 the conservative AA Asp 127, Tyr130, Asn152, Gly155,
Met188 and Glu241 (40). The
active enzyme site is located in the MRC dimer interface which is a
complex formed by the peptide chains of monomers (41). N-terminal portions of each monomer
folded in compact domains that are folded in extensive areas (AA
220–303) reaching the active site of another subunit of the dimer.
The 304–360 AA residues form an elongated C-terminal fragment
folding in the opposite direction, so that N- and C-terminal
portions are located next to each other. Asp156 played a role of
the catalytic base, whereas His126 and Glu241 of other monomer
(Glu241) form a pair which acted as the catalytic acid (2).
As a result of biopanning of the phage peptide
library with the in-house made monoclonal antibody 6H9, we selected
10 polypeptides, mimotopes. The homologous epitope in MRC
corresponding to the phage mimotopes formed a cluster comprising AA
G113, W114, R120, Q122, Q123, A124, G125, Y130, S132, L133, N134,
V189 and W200. Commercial rabbit monoclonal antibodies 13H4 against
AMACR recognized in the phage library 9 mimotopes, which were most
likely to lay in MRC cluster comprising AA W114, P119, R120, H126,
I128, N129, Y130, S132, L133, N134, G135 and W200. These
above-mentioned sequences represent conformational epitopes the
vast majority of AA residues of which are localized in the helix α5
and unstructured sequences between helices α5, α6, and layer β5
that is in the catalytic site of the enzyme (Fig. 2). In total, 6 AA residues (W114,
R120, Y130, L133, N134 and W200) are common in the epitopes
recognized by the 6H9 and 13H4 antibodies. These data suggest that
the 6H9 and 13H4 antibodies recognize similar conformational
sequence in the region of AMACR catalytic center (Figs. 3 and 4). From 13 and 12 AA, which form,
correspondingly, the 6H9 and 13H4 antibodies epitopes in MRC, 10
and 9 are identical or conserved, while 10 and 8 of those are
identical in human and mice (Fig.
3). These levels of identity correspond to the antibodies
specificity in immunoblotting and immunofluorescence, which bound
AMACR in various human and mouse tissues and cell lines (Figs. 1 and 2A and B).
AMACR is overexpressed in carcinoma of the prostate,
colon, kidney and other tissues (6,18),
while the inhibition of its enzyme activity prevents the growth of
cancer cells (8,9). AMACR is regarded as a potential
target of anticancer therapy. Modern anti-AMACR drugs mostly
represent substances with substrate specificity (9). In this study, to the best of our
knowledge, we present evidence for the first time to indicate that
mouse in-house made and rabbit commercial monoclonal antibody
against AMACR recognizes the epitopes which are mapped in the
catalytic center of the molecule. Our results suggest that antibody
against AMACR may modify its enzymatic activity and may potentially
inhibit the proliferation of cancer cells overexpressing AMACR. To
evaluate this possibility, we tried to deliver antibody to AMACR in
live HeLa cells followed by an estimation of their co-localization
with peroxisomes in which AMACR performs its function. AMACR was
abundantly produced by HeLa cells and evenly detected in
immunofluorescence by the in-house made 6H9 and commercially
available 63340 antibody purchased from Abcam (Fig. 5A). In fixed cells, AMACR did not
exhibit co-localization with peroxisomes (Fig. 5B), possibly as the antigen
conformation in peroxisomes induced by paraformaldehyde. This
suggestion is supported by results of following experiments, in
which the same antibodies proteofected into live cells were at
least partly co-localized with peroxisomes (Fig. 5C and D). For antibody delivery into
live cells, we used cationic lipid-based PULSin reagent which forms
a noncovalent complex with antibody and releases the antibody in
cytoplasm in its native functional form (32). The delivery of non-specific 2A5
antibody to HeLa cells resulted in the formation of big cytoplasmic
clusters which, presumably, represent endosomes with accumulated
antibody (Fig. 5C). Specific
antibody to AMACR delivered into live cells distributed evenly
(Fig. 5C) and at least partly
co-localized with peroxisomes (Fig.
5D). The in-house made 6H9 antibody exhibited a low level of
co-localization compared to commercially available 63340 antibody
(Fig. 5D). This may be associated
with the low capability of 6H9 antibody to precipitate AMACR from
solution (Fig. 1D) due to a low
affinity to the native antigen conformation. In accordance with
this suggestion, 6H9 antibody when delivered into HeLa or T98G
cells, did not inhibit their proliferation rate estimated by the
detection of growth curves or MTT assay (Fig. 6). On the whole, our data suggest
that antibodies (namely ab63340) against AMACR represent potential
therapeutic drugs with which to inactivate the protein and inhibit
cancer cell proliferation. Further studies are warranted in order
to explore possibilities for preparation of antibody recognizing
AMACR with high affinity in native conformation and for efficient
antibody delivery to its target.
Acknowledgments
Immunofluorescence data acquisition and analysis
were performed through the Institute of Cytology Cell Imaging
Shared Resource. We thank Dr I. Popova, Manager of the Recombinant
Protein Production Core at the Northwestern University, Chicago,
IL, USA for excellent technical assistance. The Saos2 human
osteosarcoma cells were kindly provided by Dr K. Helin (European
Institute of Oncology, Milan, Italy). This study was supported by
the Russian Research Foundation grant no. 14-50-00068 and the
Russian Foundation for Basic Research grant no. 16-04-00251 (to
B.V. Popov).
References
|
1
|
Savolainen K, Bhaumik P, Schmitz W, Kotti
TJ, Conzelmann E, Wierenga RK and Hiltunen JK: Alpha-methylacyl-CoA
racemase from Mycobacterium tuberculosis. Mutational and structural
characterization of the active site and the fold. J Biol Chem.
280:12611–12620. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bhaumik P, Schmitz W, Hassinen A, Hiltunen
JK, Conzelmann E and Wierenga RK: The catalysis of the 1,1-proton
transfer by alpha-methyl-acyl-CoA racemase is coupled to a movement
of the fatty acyl moiety over a hydrophobic, methionine-rich
surface. J Mol Biol. 367:1145–1161. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lloyd MD, Darley DJ, Wierzbicki AS and
Threadgill MD: Alpha-methylacyl-CoA racemase--an 'obscure'
metabolic enzyme takes centre stage. FEBS J. 275:1089–1102. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lloyd MD, Yevglevskis M, Lee GL, Wood PJ,
Threadgill MD and Woodman TJ: α-Methylacyl-CoA racemase (AMACR):
Metabolic enzyme, drug metabolizer and cancer marker P504S. Prog
Lipid Res. 52:220–230. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baron A, Migita T, Tang D and Loda M:
Fatty acid synthase: A metabolic oncogene in prostate cancer? J
Cell Biochem. 91:47–53. 2004. View Article : Google Scholar
|
|
6
|
Jiang Z, Fanger GR, Woda BA, Banner BF,
Algate P, Dresser K, Xu J and Chu PG: Expression of
alpha-methylacyl-CoA racemase (P504s) in various malignant
neoplasms and normal tissues: Astudy of 761 cases. Hum Pathol.
34:792–796. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rohrmann S, Platz EA, Kavanaugh CJ, Thuita
L, Hoffman SC and Helzlsouer KJ: Meat and dairy consumption and
subsequent risk of prostate cancer in a US cohort study. Cancer
Causes Control. 18:41–50. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Carnell AJ, Hale I, Denis S, Wanders RJ,
Isaacs WB, Wilson BA and Ferdinandusse S: Design, synthesis, and in
vitro testing of alpha-methylacyl-CoA racemase inhibitors. J Med
Chem. 50:2700–2707. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Carnell AJ, Kirk R, Smith M, McKenna S,
Lian LY and Gibson R: Inhibition of human α-methylacyl CoA racemase
(AMACR): A target for prostate cancer. ChemMedChem. 8:1643–1647.
2013.PubMed/NCBI
|
|
10
|
Schmitz W, Fingerhut R and Conzelmann E:
Purification and properties of an alpha-methylacyl-CoA racemase
from rat liver. Eur J Biochem. 222:313–323. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu J, Stolk JA, Zhang X, Silva SJ,
Houghton RL, Matsumura M, Vedvick TS, Leslie KB, Badaro R and Reed
SG: Identification of differentially expressed genes in human
prostate cancer using subtraction and microarray. Cancer Res.
60:1677–1682. 2000.PubMed/NCBI
|
|
12
|
Jiang Z, Woda BA, Rock KL, Xu Y, Savas L,
Khan A, Pihan G, Cai F, Babcook JS, Rathanaswami P, et al: P504S: A
new molecular marker for the detection of prostate carcinoma. Am J
Surg Pathol. 25:1397–1404. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Luo J, Zha S, Gage WR, Dunn TA, Hicks JL,
Bennett CJ, Ewing CM, Platz EA, Ferdinandusse S, Wanders RJ, et al:
Alpha-methylacyl-CoA racemase: A new molecular marker for prostate
cancer. Cancer Res. 62:2220–2226. 2002.PubMed/NCBI
|
|
14
|
Zhou M, Chinnaiyan AM, Kleer CG, Lucas PC
and Rubin MA: Alpha-Methylacyl-CoA racemase: A novel tumor marker
over-expressed in several human cancers and their precursor
lesions. Am J Surg Pathol. 26:926–931. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rubin MA, Zhou M, Dhanasekaran SM,
Varambally S, Barrette TR, Sanda MG, Pienta KJ, Ghosh D and
Chinnaiyan AM: alpha-Methylacyl coenzyme A racemase as a tissue
biomarker for prostate cancer. JAMA. 287:1662–1670. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiang Z, Woda BA, Wu CL and Yang XJ:
Discovery and clinical application of a novel prostate cancer
marker: Alpha-methylacyl CoA racemase (P504S). Am J Clin Pathol.
122:275–289. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Adley BP and Yang XJ: Application of
alpha-methylacyl coenzyme A racemase immunohistochemistry in the
diagnosis of prostate cancer: A review. Anal Quant Cytol Histol.
28:1–13. 2006.PubMed/NCBI
|
|
18
|
Kapoor S: AMACR: An emerging diagnostic
and prognostic tool in systemic malignancies. Int Urol Nephrol.
45:439–440. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen W, Wu W, Zhao J, Yu C, Liu W, Jiang A
and Zhang J: Molecular cloning and preliminary analysis of the
human alpha-methylacyl-CoA racemase promoter. Mol Biol Rep.
36:423–430. 2009. View Article : Google Scholar
|
|
20
|
Sekine S, Ogawa R, Ojima H and Kanai Y:
Overexpression of α-methylacyl-CoA racemase is associated with
CTNNB1 mutations in hepatocellular carcinomas. Histopathology.
58:712–719. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nusse R and Clevers H: Wnt/β-catenin
signaling, disease, and emerging therapeutic modalities. Cell.
169:985–999. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bosetti C, Bertuccio P, Malvezzi M, Levi
F, Chatenoud L, Negri E and La Vecchia C: Cancer mortality in
Europe, 2005–2009, and an overview of trends since 1980. Ann Oncol.
24:2657–2671. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Honma I, Torigoe T, Hirohashi Y, Kitamura
H, Sato E, Masumori N, Tamura Y, Tsukamoto T and Sato N and Sato N:
Aberrant expression and potency as a cancer immunotherapy target of
alpha-methylacyl-coenzyme A racemase in prostate cancer. J Transl
Med. 7:1032009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lin PY, Cheng KL, McGuffin-Cawley JD,
Shieu FS, Samia AC, Gupta S, Cooney M, Thompson CL and Liu CC:
Detection of alpha-methylacyl-CoA racemase (AMACR), a biomarker of
prostate cancer, in patient blood samples using a nanoparticle
electrochemical biosensor. Biosensors (Basel). 2:377–387. 2012.
View Article : Google Scholar
|
|
25
|
Nickens KP, Ali A, Scoggin T, Tan SH,
Ravindranath L, McLeod DG, Dobi A, Tacha D, Sesterhenn IA,
Srivastava S, et al: Prostate cancer marker panel with single cell
sensitivity in urine. Prostate. 75:969–975. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mayrose I, Penn O, Erez E, Rubinstein ND,
Shlomi T, Freund NT, Bublil EM, Ruppin E, Sharan R, Gershoni JM, et
al: Pepitope: Epitope mapping from affinity-selected peptides.
Bioinformatics. 23:3244–3246. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mayrose I, Shlomi T, Rubinstein ND,
Gershoni JM, Ruppin E, Sharan R and Pupko T: Epitope mapping using
combinatorial phage-display libraries: A graph-based algorithm.
Nucleic Acids Res. 35:69–78. 2007. View Article : Google Scholar :
|
|
28
|
Liu HM, Newbrough SE, Bhatia SK, Dahle CE,
Krieg AM and Weiner GJ: Immunostimulatory CpG oligodeoxynucleotides
enhance the immune response to vaccine strategies involving
granulocyte-macrophage colony-stimulating factor. Blood.
92:3730–3736. 1998.PubMed/NCBI
|
|
29
|
Popov B and Kaczmarek L: Antibody raised
to the short sequence from the zinc-finger domain of the EGR-1
recognizes 102 KD protein in mouse fibroblasts. Biochem Mol Biol
Int. 32:39–47. 1994.PubMed/NCBI
|
|
30
|
Negi SS and Braun W: Automated detection
of conformational epitopes using phage display Peptide sequences.
Bioinform Biol Insights. 3:71–81. 2009. View Article : Google Scholar
|
|
31
|
Petrov NS and Popov BV: Study of Wnt2
secreted by A-549 cells in paracrine activation of β-catenin in
co-cultured mesenchymal stem cells. Biochemistry (Mosc).
79:524–530. 2014. View Article : Google Scholar
|
|
32
|
Weill CO, Biri S and Erbacher P: Cationic
lipid-mediated intracellular delivery of antibodies into live
cells. Biotechniques. 44:Pvii–Pxi. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Amery L, Fransen M, De Nys K, Mannaerts GP
and Van Veldhoven PP: Mitochondrial and peroxisomal targeting of
2-methylacyl-CoA racemase in humans. J Lipid Res. 41:1752–1759.
2000.PubMed/NCBI
|
|
34
|
Pedersen JI, Veggan T and Björkhem I:
Substrate stereospecificity in oxidation of (25S)-3 alpha, 7 alpha,
12 alpha-trihydroxy-5 beta-cholestanoyl-CoA by peroxisomal
trihydroxy-5 beta-cholestanoyl-CoA oxidase. Biochem Biophys Res
Commun. 224:37–42. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hiltunen JK and Qin Y: beta-oxidation -
strategies for the metabolism of a wide variety of acyl-CoA esters.
Biochim Biophys Acta. 1484:117–128. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Russell DW: The enzymes, regulation, and
genetics of bile acid synthesis. Annu Rev Biochem. 72:137–174.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kotti TJ, Savolainen K, Helander HM, Yagi
A, Novikov DK, Kalkkinen N, Conzelmann E, Hiltunen JK and Schmitz
W: In mouse alpha -methylacyl-CoA racemase, the same gene product
is simultaneously located in mitochondria and peroxisomes. J Biol
Chem. 275:20887–20895. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ouyang B, Leung YK, Wang V, Chung E, Levin
L, Bracken B, Cheng L and Ho SM: α-Methylacyl-CoA racemase spliced
variants and their expression in normal and malignant prostate
tissues. Urology. 77:249.e1–249.e7. 2011. View Article : Google Scholar
|
|
39
|
White WB, Coleman JP and Hylemon PB:
Molecular cloning of a gene encoding a 45,000-dalton polypeptide
associated with bile acid 7-dehydroxylation in Eubacterium sp
strain VPI 12708. J Bacteriol. 170:611–616. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sharma S, Bhaumik P, Schmitz W, Venkatesan
R, Hiltunen JK, Conzelmann E, Juffer AH and Wierenga RK: The
enolization chemistry of a thioester-dependent racemase: The 1.4 Å
crystal structure of a reaction intermediate complex characterized
by detailed QM/MM calculations. J Phys Chem B. 116:3619–3629. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li X, Zheng QC and Zhang HX: Quantum
chemical modeling of 1,1-proton transfer reaction catalyzed by a
cofactor-independent α-methylacyl-CoA racemase. Int J Quantum Chem.
112:619–624. 2012. View Article : Google Scholar
|