Introduction
Breast carcinoma (BC) is the most prevalent cause of
cancer-related mortality in women worldwide (1,2),
both in low- and middle-income countries and approximately 1.67
million new cancer cases were diagnosed in 2012 (25% of all
cancers) (3). At present, the
treatment of breast cancer mainly involves surgery, radiotherapy
and chemotherapy; 75% of patients with breast cancer require
radiotherapy. Although the early treatment effects are good, the
treatment effects in general remain poor, the main reason being the
fact that the sensitivity of breast cancer to radiation is not
ideal. Therefore, the identification of methods with which to
improve the sensitivity to radiation of breast cancer are of utmost
importance.
Apoptosis (programmed cell death, type I) refers to
a constellation of characteristic changes leading directly to cell
death (4). Surface death
receptors, through the mitochondrial release of cytochrome
c, cellular stress and some treatments can trigger apoptosis
(5). Apoptosis was previously
considered the primary mechanism of radiation-induced cell death
(6).
Autophagy is a lysosomal-dependent self-digestion
process (7) which promotes cells
survival under certain types of stress, such as nutrient
starvation, reactive oxygen species (ROS), hypoxia, DNA damage and
the unfolded protein response (8).
However, excessive cell damage is beyond the limit of repair under
certain physiological conditions, and in this case, autophagy turns
into the programmed cell death mechanism (type-II) (9). Therefore, autophagy is considered to
be a 'double-edged sword' in the process of tumor development.
Deoxycytidine kinase (dCK) is an enzyme critical for
the phosphorylation of natural deoxyribonucleic acid (10–13).
This reaction is the first and rate-limiting step in
deoxyribo-nucleoside salvage, which produce and maintain a balanced
pool of deoxyribonucleoside triphosphates (dNTPs) for DNA synthesis
(14). dCK also promotes the
phosphorylation of ara-C, CNDAC and other nucleoside analogues;
these drugs can only be activated after phosphorylation, and then
inhibit tumor growth (15–18). The phosphorylation of dCK and
post-translational modification is crucial for its enzymatic
activity (19). dCK protein has
four phosphorylation sites, Thr-3, Ser-11, Ser-15 and Ser74
(20–23). dCK activity can be increased by
Ser-74 phosphorylation (24,25)
and this phosphorylation is required for the initiation of the G2/M
checkpoint (26). It has been
previously demonstrated that nucleoside analogs that exhibit
synergistic activity with radiotherapy are activated by dCK
(11). In our previous study, we
demonstrated that dCK regulated radiation-induced cell death
through apoptosis and autophagy in HeLa cells (27).
In this study, we aimed to analyze the roles of dCK
in ionizing radiation (IR)-induced apoptosis and autophagy in
breast cancer cells, in order to determine whether dCK participates
in the regulation of cell death induced by IR and to elucidate the
main underlying mechanisms.
Materials and methods
Cell line, antibodies and reagents
The human breast cancer cell lines, SKBR3
(HER2-like: estrogen receptor-negative/progesterone
receptor-negative, ErbB2-positive, ATCC® HTB-30™) and
MDA-MB-231 (basal-like: estrogen receptor/progesterone
receptor/ErbB2-negative, ATCC® HTB-26™), were purchased
from the American Type Culture Collection (ATCC, Manassas, VA, USA)
and cultured in Dulbecco's modified Eagle's medium (DMEM;
Gibco-BRL, Life Technologies, Gaithersburg, MD, USA) supplemented
with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin
(Invitrogen, Carlsbad, CA, USA) in glass Petri dishes at 37°C in a
5% CO2 incubator.
Anti-dCK (Cat. no. ab96599; diluted 1:500) antibody
was purchased from Abcam Inc. (Cambridge, MA, USA). Anti-MAPLC3
(Cat. no. 4108; diluted 1:500), anti-p-mammalian target of
rapamycin (p-mTOR; Cat. no. 2971; diluted 1:500), anti-p-Akt (Cat.
no. 9271; diluted 1:500), anti-p-P70S6K (Cat. no. 9205; diluted
1:500) and anti-poly(ADP-ribose) polymerase (PARP; Cat. no. 9542;
diluted 1:1,000) antibodies were purchased from Cell Signaling
Technology (Beverly, MA, USA). Anti-GAPDH (Cat. no. 5174; diluted
1:1,000) antibody was obtained from Santa Cruz Biotechnology (Santa
Cruz, CA, USA). Peroxidase-conjugated anti-mouse IgG (Cat. no.
7056; diluted 1:1,000) and peroxidase-conjugated anti-rabbit IgG
(Cat. no. 7054; diluted 1:1,000) were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). Fetal bovine serum (FBS),
3-methyladenine (3-MA, used for treatment at 2 mM for 48 h) and
rapamycin (used for treatment at 200 nM for 48 h) were purchased
from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA), ZVAD-FMK
(used for treatment at 20 µM for 48 h) was purchased from
Selleckchem (Houston, TX, USA) and the pSUPER retroviral vector was
obtained from OligoEngine (Seattle, WA, USA).
Radiation
An X-ray generator (X-RAD 320 ix, Precision X-ray
Inc., North Branford, CT, USA) was utilized to deliver radiation at
a dose rate of 1.0 Gy/min. All the cells were exposed to IR for 8
min.
Plasmids
Wild-type (WT; dCK-WT), dCK-S74A, dCK-S74E mutants
were kind gifts from Dr Bo Xu (Southern Research Institute,
Birmingham, AL, USA). shRNAs were designed according to the
instructions provided on the website 'www.idtdna.com'. The shRNAs were synthesized,
denatured, ligated to the pSUPER vector at the BglII and
HindIII sites. The primers used were as follows: dCK-WT
forward, 5′-tcactagtatggccaccccgcccaagagaagc-3′ and reverse,
5′-acgctcgatcacaaagatctcaaaaactctt-3′; dCK-S74A forward,
5′-cttacaatggcacagaaaaatggtgg-3′ and reverse,
5′-ccaccatttttctgtgccattgtaag-3′; dCK-S74E forward,
5′-cttacaatggaacagaaaaatggtgg-3′ and reverse,
5′-ccaccatttttctgttccattgtaag-3′;and dCK-shRNAs forward,
5′-gatctgtggttcctgaacctgttgttcaagagacaacaggttcaggaaccacttttta-3′
and reverse,
5′-agcttaaaaagtggttcctgaacctgttgtctcttgaacaacaggttcaggaaccaca-3′.
Establishment of cells in which dCK was
silenced
The pSUPER-dCK and the vector with a scrambled
sequence, i.e., pSUPER, were constructed in our laboratory. The
plasmids were transfected into 293T packaging cells (Cell Resource
Center of Shanghai Institutes for Biological Sciences, Chinese
Academy of Sciences, Shanghai, China) by calcium phosphate
co-precipitation, to produce pseudovirus particles [Ampho Pack
plasmid 10 µg, PsupershRNA plasmid 10 µg, 2 mol/l
CaCl2 31 µl, ddH2O to 250 µl
and 2X HEPES buffer salt solution (HBS) 250 µl]. Supernatant
containing pseudovirus particles was collected after 72 h and then
used to infect the SKBR3 cells and MDA-MB-231 cells together with
polybrene (8 µg/ml). Positive stable cell clones were
selected by growing the cells with puromycin (0.8 µg/ml) for
1 week.
Western blot analysis
The cells were harvested and lysed in RIPA lysis
buffer [HEPES (50 mM), sodium chloride (Nacl; 150 mM),
ethylenediaminetetraacetic acid (EDTA; 1 mM), egtazic acid (EGTA;
2.5 mM), sodium fluoride (NaF; 10 mM), dithiothreitol (DTT; 1 mM),
sodium orthovanadate (SV; 1 mM), phenylmethane sulfonyl fluoride
(PMSF; 1 mM), Nonidet P-40 (NP-40; 1%) and sodium dodecyl sulphate
(SDS; 0.1%)], and a 2 ml aliquot was mixed with 20 µl
protease inhibitor cocktail and the lysates was laid on ice for 5
min, followed by sonication.
The supernatant was then removed to another tube
following centrifugation at 15,000 x g for 10 min and lysate was
mixed with 5X SDS loading buffer (BioTeke, Beijing, China) and
heated to 95°C for 5 min. Of the total protein, 50 µg were
separated by SDS-PAGE, and transferred onto nitrocellulose
membranes which were then blocked in 5% non-fat dried milk in
Tris-buffered saline (TBS) and Tween-20 (10 mmol/l Tris, pH 7.5,
100 mmol/l NaCl and 0.1% Tween-20) at room temperature for 1.5 h,
and then incubated with appropriate primary antibody (anti-dCK
antibody, anti-MAPLC3 antibody, anti-p-mTOR antibody, anti-p-Akt
antibody, anti-p-P70S6K antibody, anti-PARP antibody and anti-GAPDH
antibody) overnight at 4°C, and horseradish peroxidase-conjugated
secondary antibodies (anti-mouse IgG or anti-rabbit IgG) at room
temperature for 1 h. Finally, the signals were visualized by using
the Pierce chemiluminescence detection system according to the
manufacturer's instructions (Santa Cruz Biotechnology); GAPDH
protein was used as a loading control. The intensity of the protein
bands was quantified using image software (Quantity One) and ratios
of specific bands to the loading control were analyzed.
Flow cytometric analysis
The cells were plated in 6-well plates and exposed
to IR. The cells were then collected at 48 h following IR and
washed 3 times in PBS. For apoptosis detection, the cells were
stained with PI (Sigma-Aldrich Chemical Co.) and FITC-labeled
Annexin V (Sigma-Aldrich Chemical Co.). For cell death analysis,
the cells were stained by trypan blue (Sigma-Aldrich Chemical Co.).
The stained cells were detected using a flow cytometer (BD
Biosciences, San Jose, CA, USA). The data were analyzed using
CellQuest software (BD Biosciences) and FlowJo software (Tree Star
Inc., Ashland, OR, USA).
Colony formation assay
The cells were seeded in 6-well plates in triplicate
using standard culture medium. After 24 h, the cells were
irradiated (0, 2, 4, 6 and 8 Gy). After 2 weeks, the cells were
fixed with 4% formaldehyde, and stained with 0.5% crystal violet
(Merck, Darmstadt, Germany). The number of colonies (>50 cells)
was counted using a TC20™ Automated Cell Counter (Bio-Rad
Laboratories, Shanghai, China) and normalized to the corresponding
non-irradiated control. Cell survival curves were made by the
multitarget click model of GraphPad Prism 5.0 (Systat Software, San
Jose, CA, USA).
Data calculation methods
For all figures, we took the comparison between the
pSUPER and dCK knockdown groups or we took the comparison between
the vector, dCK-WT, dCK-S74A and dCK-S74E groups into
consideration. For example, in Fig.
1B, we first want to detect the rate of cell death induced only
by IR. Therefore, the effects of IR on mortality were determined by
the mortality of the IR (8 Gy) group minus the mortality of the
control (0 Gy) group and then by dividing the control group. We
then compared the difference between the pSUPER and dCK knockdown
group to examine the role of dCK in IR-induced cell death.
Statistical analysis
All data were obtained from at least 3 independent
experiments. Statistical evaluations are presented as the means ±
SE. The significance of the differences between groups was
determined by one-way ANOVA, and a value of P<0.05 was
considered to indicate a statistically significant difference.
Results
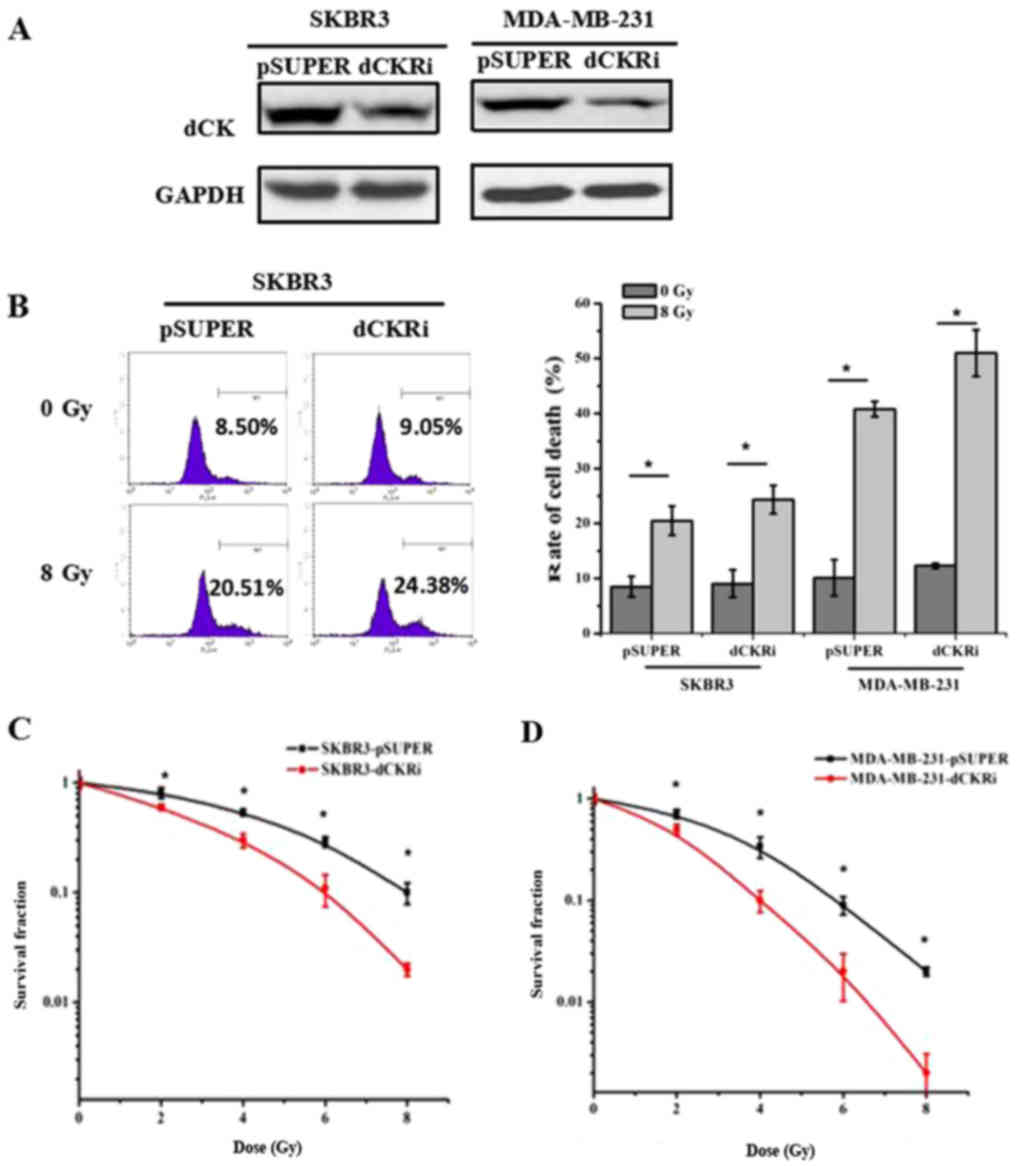
dCK decreases radiation-induced cell
death
In order to elucidate the roles of dCK in
radiation-induced cell death, dCK expression was knocked down in
the SKBR3 and MDA-MB-231 cells (Fig.
1A). We then determined that the knockdown of dCK enhanced
IR-induced cell death by 53% in the MDA-MB-231 cells and by 28% in
the SKBR3 cells as compared with the control cells (Fig. 1B). Colony formation assays
demonstrated that dCK knockdown increased radiosensitivity
(Fig. 1C and D). dCK S74A has a
serine 74 to alanine substitution, which abrogates phosphorylation,
and dCK S74E has a serine 74 to glutamic acid substitution, which
mimics phosphorylation. We re-introduced the empty vector,
wild-type dCK, dCK S74A or the S74E plasmid into the cells in which
dCK was knocked down (Fig. 1E). IR
increased the rate of cell death by 129, 90, 137 and 65% in the
SKBR3 cells transfected with vector, dCK WT, dCK S74A, dCK S74E
respectively, and by 367, 320, 356, 196% in MDA-MB-231 cells
transfected with vector, dCK WT, dCK S74A, dCK S74E respectively.
Thus, compared to the control cells exposed to IR, the
re-introduction of dCK WT, dCK S74A and dCK S74E decreased
IR-induced cell death by 39, -8 and 64% in the SKBR3 cells, and by
47, 11 and 171% in the MDA-MB-231 cells, respectively, suggesting
that dCK protects breast cancer cells from IR-induced cell death
(Fig. 1F and G).
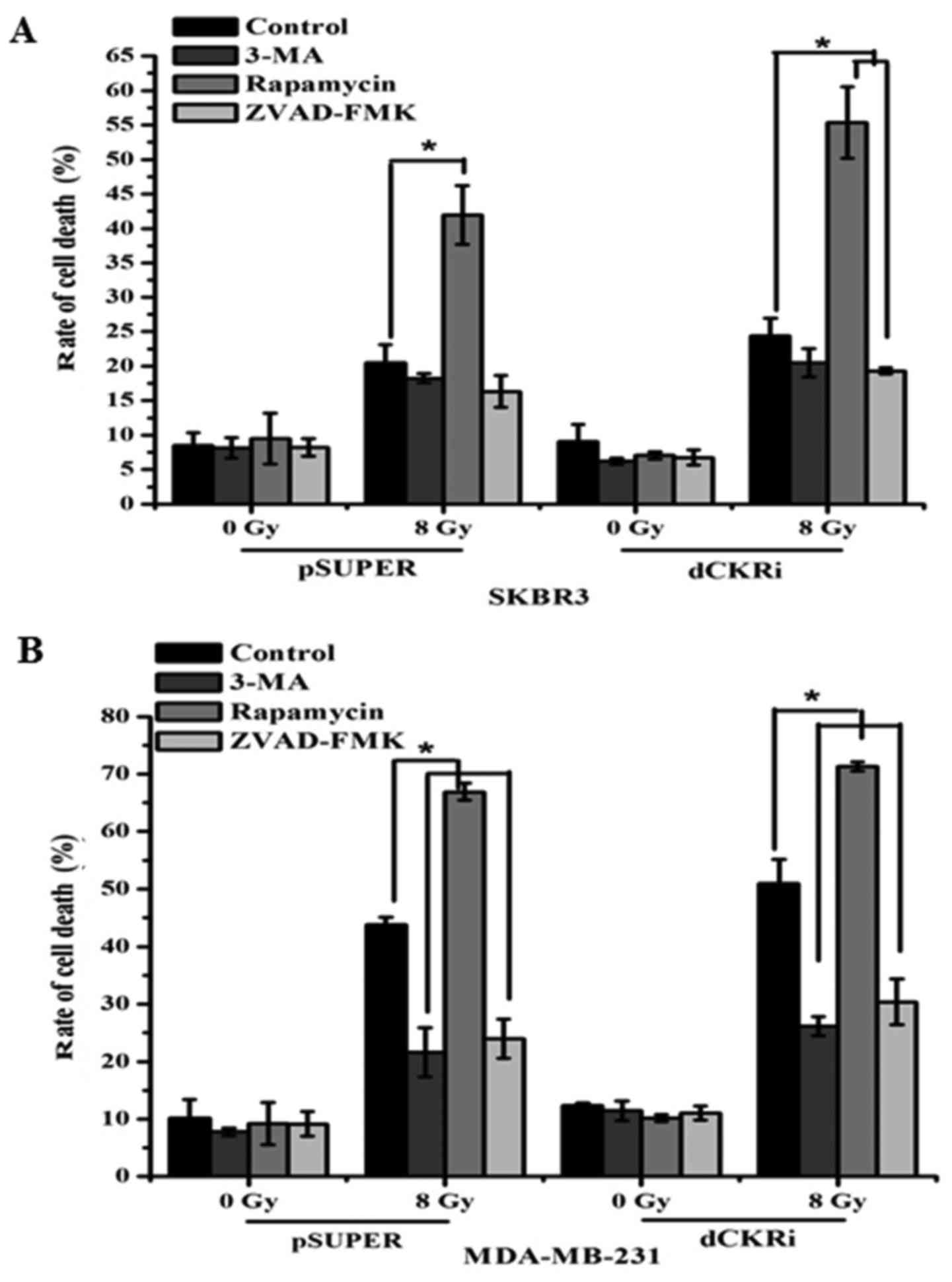
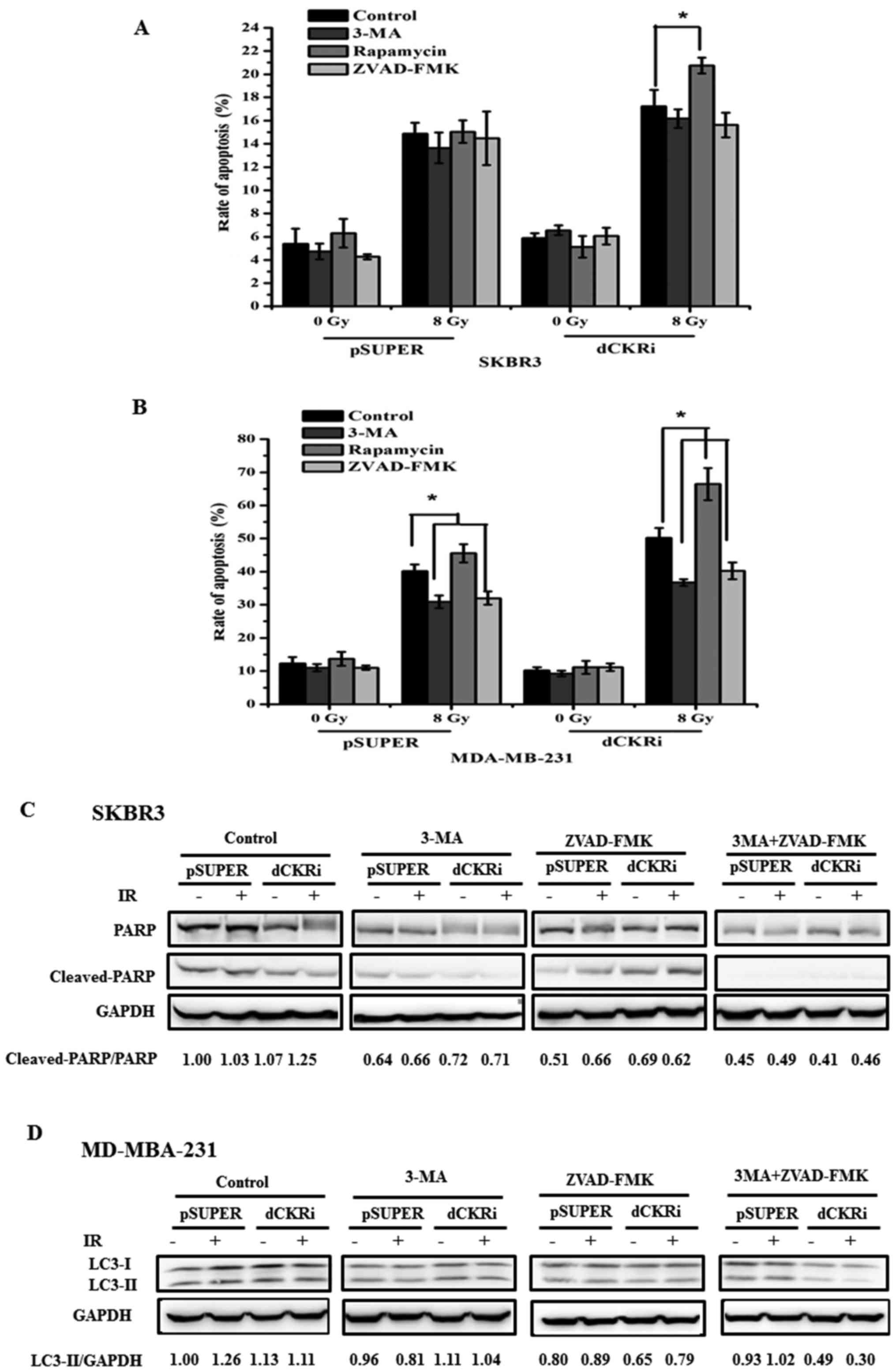
Autophagy and apoptosis can contribute to cell death
following exposure to IR. We found that IR increased cell death by
488% in the MDA-MB-231 cells. However, the knockdown ATG5 and
Beclin1 (BECN1; data not shown) only increased IR-induced cell
death by 247 and 211% (data not shown). Furthermore, we inhibited
autophagy and apoptosis with 3-MA and ZVAD-FMK, respectively, and
induced autophagy with rapamycin. We found that ZVAD-FMK decreased
IR-induced cell death by 21% and rapamycin increased IR-induced
cell death by 127% in the SKBR3 cells in which dCK was silenced. In
the SKBR3-pSUPER cells, ZVAD-FMK did not affect IR-induced cell
death and rapamycin increased IR-induced cell death by 104%
(Fig. 2A). Moreover, 3-MA and
ZVAD-FMK decreased IR-induced cell death by 51 and 43%, and
rapamycin increased IR-induced cell death by 53% in the
MDA-MB-231-pSUPER cells. 3-MA and ZVAD-FMK decreased IR-induced
cell death by 49 and 40%, and rapamycin increased IR-induced cell
death by 40% in the MDA-MB-231 cells in which dCK was silenced
(Fig. 2B). Thus, these data
indicated that both autophagy and apoptosis contributed to
IR-induced cell death (Fig.
2).
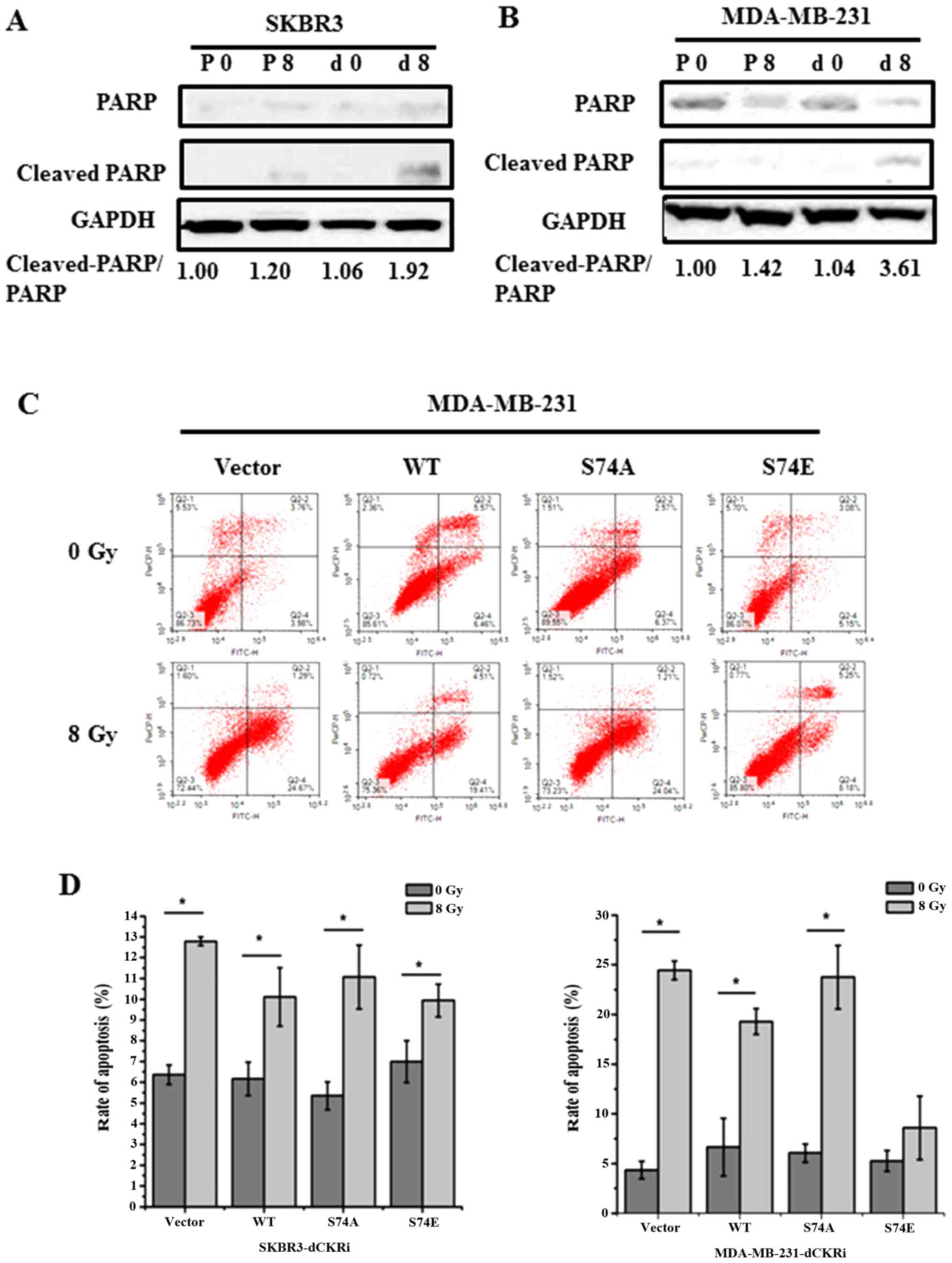
dCK suppresses IR-induced apoptosis
To confirm that dCK contributes to IR-induced
apoptosis, we knocked down dCK and then examined the expression of
PARP and cleaved-PARP in the MDA-MB-231 and SKBR3 cells (Fig. 3A and B). In Fig. 3A and B, P0, P8, d0 and d8 represent
pSUPER-transfected cells exposed to 0 Gy IR, and pSUPER-transfected
cells exposed to 8 Gy IR, cells in which dCK was knocked down and
exposed to 0 Gy IR, and cells in which dCK was knocked down cells
and exposed to 8 Gy IR, respectively. Our results revealed that the
knockdown of dCK increased the IR increased the ratio of cleaved
PARP/PARP by 247% in the MDA-MB-231 and by 81% in the SKBR3 cells,
suggesting that dCK was involved in IR-induced apoptosis.
Furthermore, empty vector, dCK-WT, dCK-S74A and dCK-S74E plasmids
were re-introduced into the cells in which dCK was knocked down and
the rate of apoptosis was detected. In the MDA-MB-231 cells,
compared to the empty vector group, the re-introduction of
dCK-S74A, dCK-WT and dCK-S74E decreased IR-induced apoptosis (early
apoptosis and late apoptosis) by 171, 273 and 383%, respectively.
In addition, in the SKBR3 cells, compared to the empty vector
group, the re-introduction of dCK-S74A, dCK-WT and dCK-S74E
decreased IR-induced apoptosis by −6, 37 and 59%. It was thus
suggested that phosphorylated dCK suppresses IR induced-apoptosis
(Fig. 3C and D).
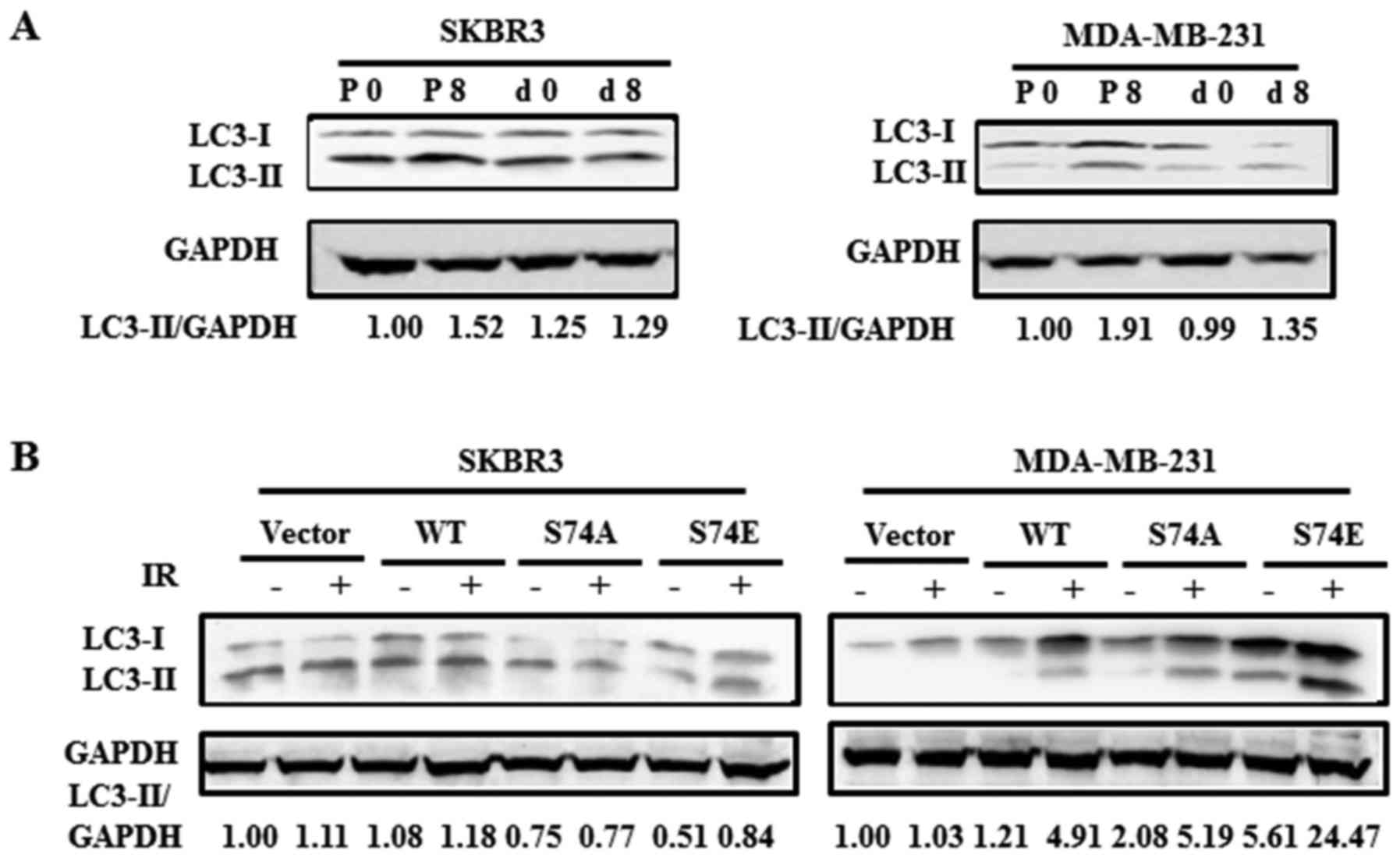
dCK promotes IR-induced autophagy
As is already known, the excessive induction of
autophagy, or when cell damage is beyond the limit of repair,
autophagy then turns into a programmed cell death mechanism
(type-II) (9,28–30).
In this study, we first used autophagy inhibitors to examine the
role of dCK in IR-induced autophagy. We found that 3-MA markedly
decreased LC3-II expression in the cells exposed to IR (data not
shown). Ammonium chloride (NH4Cl) is a lysosomal inhibitor which
can block organelle acidification and enable the assessment of the
autophagic flux (31). In this
study, western blot analysis revealed that LC3-II expression
increased in a time-dependent manner (data not shown), reaching
peak levels at 72 h following exposure to IR and the signal was
much higher in the MDA-MB-2321 cells treated with NH4Cl
and IR (data not shown).
To confirm that dCK contributes to IR-induced
autophagy, we knocked down dCK and then examined the expression of
LC3-I and LC3-II in the MDA-MB-231 and SKBR3 cells. The results
revealed that compared with the pSUPER-transfected cells, the
knockdown of dCK decreased the expression of LC3-II (Fig. 4A). In addition, in order to
investigate whether dCK S74 phosphorylation is associated with
IR-induced autophagy, we re-introduced dCK constructs into the
cells in which dCK was knocked down (Fig. 4B). Western blot analysis revealed
that in the SKBR3 cells, only dCK S74E increased the level of
LC3-II protein by 65% following exposure to IR compared to the
vector group. In the MDA-MB-231 cells, the re-introduction of
dCK-WT, dCK-S74E and dCK-S74A increased the levels of LC3-II by
306, 336 and 150% following exposure to IR, respectively and there
was no significant change with IR treatment in the empty vector
group cells. These results thus indicate that dCK S74
phosphorylation is involved in IR-induced autophagy.
Crosstalk between apoptosis and autophagy
following exposure to IR
Given that dCK plays an important role in both
IR-induced apoptosis and autophagy, we focused on the potential
association between apoptosis and autophagy. Cisplatin, a first
class anti-tumor drug widely used in the treatment of various types
of cancer, targets DNA to induce apoptosis through the
mitochondrial death pathway or Fas death pathway (32,33).
We thus used cisplatin as a positive control and found that
cisplatin-induced apoptosis was reversed by ZVAD-FMK (data not
shown).
In the SKBR3 cells in which dCK was knocked down,
rapamycin increased IR-induced apoptosis by 20% compared to the
control group; however, 3-MA and ZVAD-FMK failed to affect
apoptosis. There was also no significant difference in IR-induced
apoptosis in the cells transfected with the pSUPER control and
treated with rapamycin, 3-MA or ZVAD-FMK (Fig. 5A). In the MDA-MB-231 cells
transfected with the pSUPER control, 3-MA and ZVAD-FMK decreased
IR-induced apoptosis by 21 and 8%, while rapamycin increased
apoptosis by 13%. In the MDA-MB-231 cells in which dCK was knocked
down, 3-MA and ZVAD-FMK decreased the rate of apoptosis by 17 and
20%, while rapamycin increased it by 26% compared to the control
group (Fig. 5B). IR increased the
ratio of cleaved-PARP/PARP by 17% in the SKBR3 cells in which dCK
was silenced and this ratio did not change in the pSUPER
control-transfected cells. 3-MA decreased the cleaved-PARP/PARP
ratio in SKBR3-pSUPER and SKBR3-dCKRi cells following exposure to
IR. When the cells were treated with ZVAD-FMK, the
cleaved-PARP/PARP ratio decreased. Of note, the ratio of
cleaved-PARP/PARP was much lower in the cells treated with 3-MA +
ZVAD-FMK than in the cells treated with 3-MA or ZVAD-FMK alone
(Fig. 5C). It was suggested that
the suppression of autophagy decreased IR-induced apoptosis.
In the MDA-MB-231 cells, IR increased the expression
of LC3-II by 26% in the pSUPER control group; however, there was no
change in the cells in which dCK was knocked down. 3-MA decreased
LC3-II expression by 36% in the pSUPER-transfected cells and by 6%
in the cells in which dCK was knocked down following exposure to
IR. When the cells were treated with ZVAD-FMK, LC3-II expression
decreased in MDA-MB-231 cells in the pSUPER and dCK knockdown
group. Of note, the ratio of LC3-II was much lower in the cells (in
which dCK was knocked down cells) treated with 3-MA + ZVAD-FMK
compared with the cells treated with 3-MA or ZVAD-FMK alone in
(Fig. 5D). These findings
suggested that the suppression of apoptosis decreased IR-induced
autophagy. This suggests that a crosstalk exists between apoptosis
and autophagy following exposure to IR.
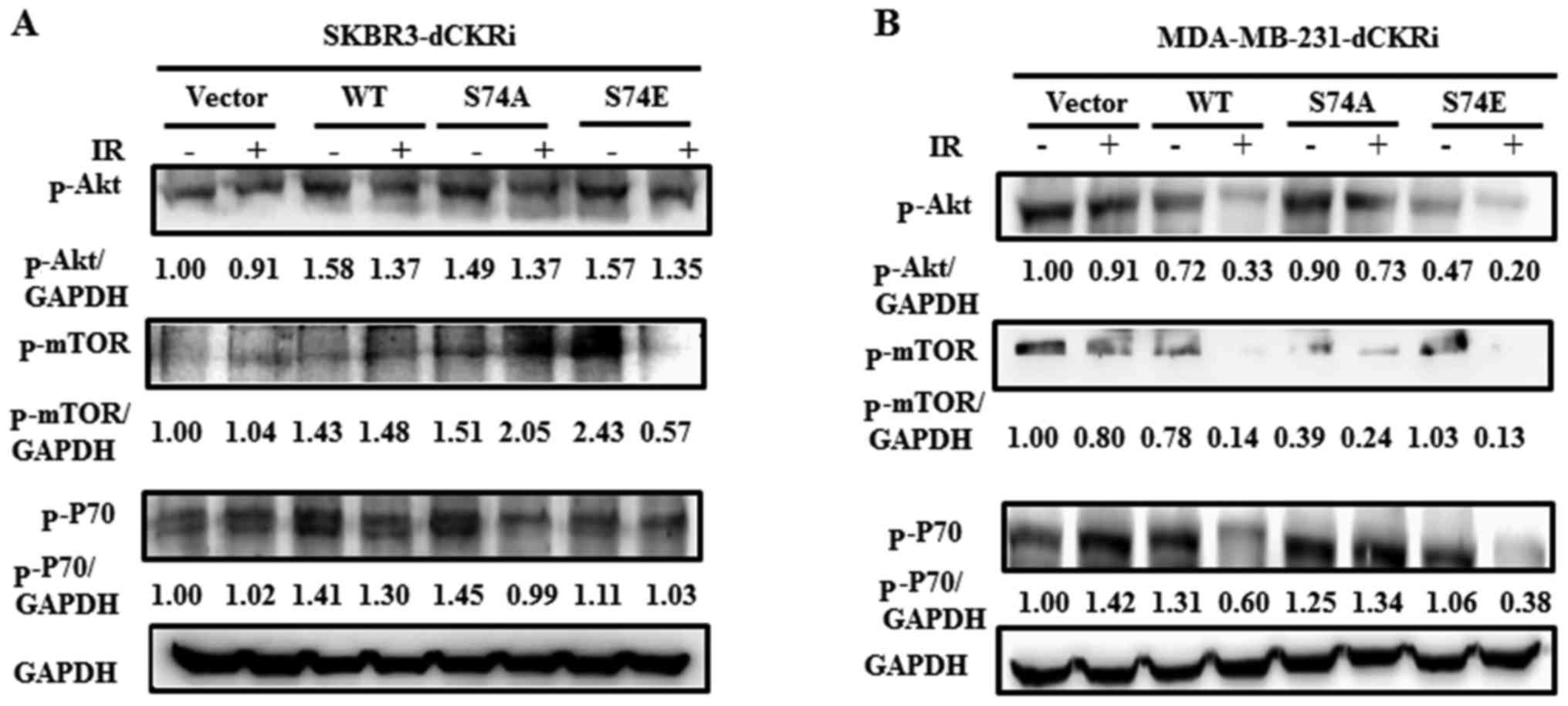
dCK regulates IR-induced autophagy
through the mTOR pathway
Since dCK plays an important role in IR-induced
autophagy, we wioshed to determine whether the Akt/mTOR/p70S6K
signaling pathway is involved in this process. As shown in Fig. 6A, in the SKBR3 cells, only dCK-S74E
decreased the expression of p-mTOR by 77% following exposure to IR.
In the MDA-MB-231 cells, dCK-WT significantly decreased the level
of p-Akt by 54%, that of p-mTOR by 82% and that of p-P70S6K by 54%.
dCK S74E significantly decreased the level of p-Akt by 57%, that of
p-mTOR by 87% and that of p-P70S6K by 64%. dCK-S74A slightly
decreased the level of p-Akt, p-mTOR and p-P70S6K by 19, 38 and 7%,
respectively following exposure to IR. However, the empty vector
only decreased the levels of p-Akt and p-mTOR by 9 and 20%,
respectively (Fig. 6B). These data
suggest that activated dCK inhibits the Akt/mTOR/P70S6K pathway and
promotes autophagy in breast cancer cells exposed to IR.
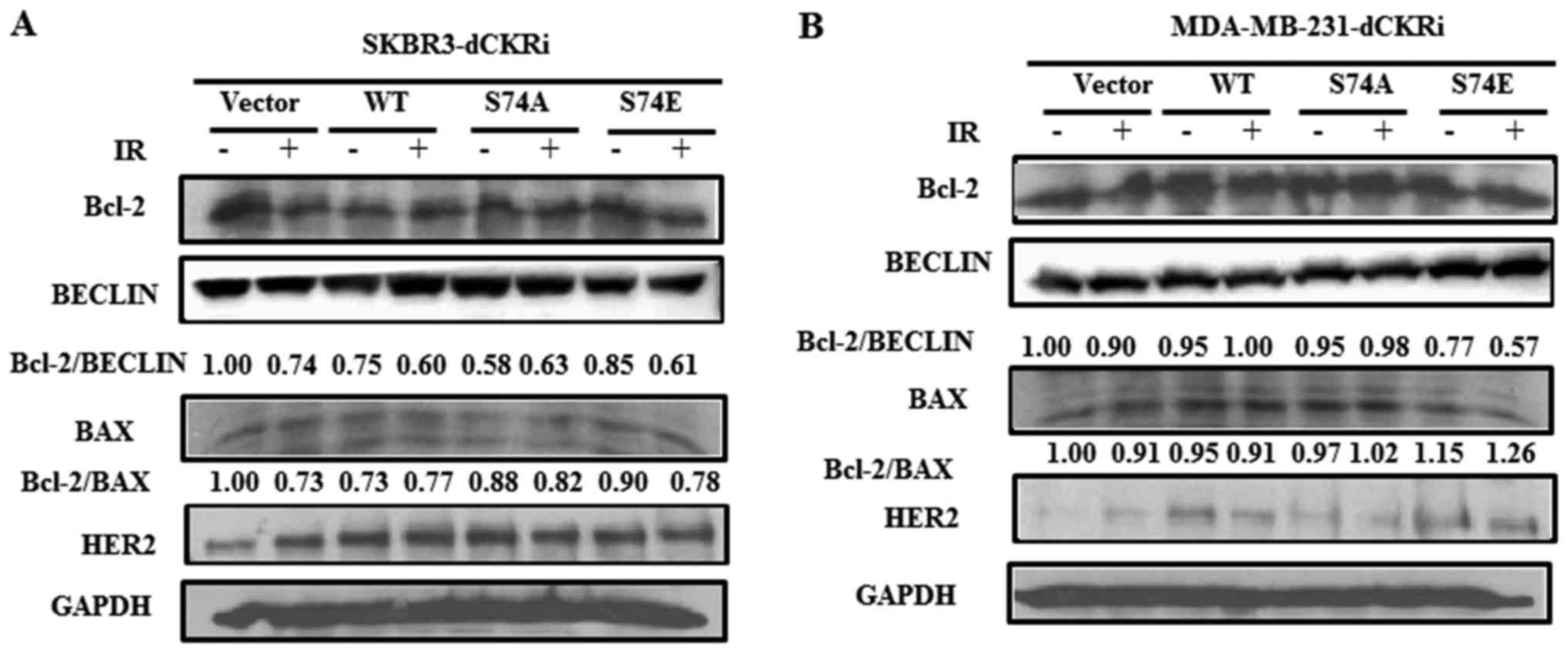
Activation of dCK regulates the binding
of Bcl-2 to BECN1 following exposure to IR
The complex formed by the autophagy-related protein,
BENC1, and the anti-apoptotic protein, Bcl-2, leads to the
inhibition of autophagy-associated cell death (34–37).
In this study, we thus determined the effect of dCK activation on
the interaction between Bcl-2 and BECN1 following exposure to IR.
As shown in Fig. 7, dCK-S74E
decreased the ratio of Bcl-2/BECN1 compared with the control group
in the SKBR3 and MDA-MB-231 cells; however, there was no change in
the ratio of Bcl-2/BAX, suggesting that activated dCK inhibits the
binding of Bcl-2/BECN1 and promotes autophagy following exposure to
IR.
Discussion
Breast cancer is one of the most common types of
cancer worldwide and is the leading cause of cancer-related
mortality among females (38,39).
Radiotherapy is a major strategy in the treatment of breast cancer.
It is known that many nucleoside analogs are used in combination
with radiotherapy, and dCK is required for the anti-tumor activity
of these nucleoside analogs (11,40).
IR can activate dCK and the phosphorylation of dCK is crucial for
its enzymatic activity (41).
In this study, we found that the knockdown of dCK
increased IR-induced cell death and apoptosis in both the
MDA-MB-231 and SKBR3 cells. We found that the MDA-MB-231 cells were
more sensitive to IR than the SKBR3 cells. This may be due to the
fact that the SKBR3 cells express HER2, but the MDA-MB-231 cells
are HER2-negative. HER2 overexpression promotes DNA damage repair
and results in resistance to radiation (42). In this study, we re-introduced
different dCK constructs into the SKBR3 and MDA-MB-231 cell lines
in which dCK was knocked down. dCK phosphor-mimetic S74E reversed
IR-induced cell death and apoptosis after the silencing of dCK,
while dCK S74A failed to do so, confirming the important role of
dCK S74 phosphorylation in cell death and apoptosis under IR
treatment.
Autophagy was traditionally considered to be a
protective mechanism, important for the removal of damaged proteins
and organelles, and conferring stress tolerance and enhancing cell
viability under adverse conditions (43–45).
However when cell damage is beyond the limits of repair under
certain physiological conditions, autophagy turns into the
programmed cell death mechanism (type-II) (9,45,46).
BECN1 is a key mediator of autophagy. It interacts with several
co-factors that regulate autophagy and its dysfunction has been
implicated in several disorders, including many types of human
cancer (36). Moreover, Atg5 is
covalently bound to Atg12 (47),
which is essential for the occurrence of autophagy (48). In this study, we found that the
knockdown of ATG5 and BECN1 decreased IR-induced cell death
compared with the control group in MDA-MB-231 cell models (data not
shown). Apoptosis and autophagic cell death are the most important
mechanisms of cell death. By using 3-MA and ZVAD-FMK to inhibit
autophagy or apoptosis and rapamycin to induce autophagy, our data
indicated that both the autophagy inhibitor and apoptosis inhibitor
led to a decrease in IR-induced cell death in the cells transfected
with the pSUPER control vector and in the cells in which dCK was
knocked down. Rapamycin increased IR-induced cell death
significantly in the cells in which dCK was knocked down,
indicating that both apoptosis and autophagy contribute to
IR-induced cell death.
We provide evidence that dCK increases LC3-II
expression in response to IR. Moreover, S74E noticeably increased
LC3-II expression compared with WT or S74A in response to IR,
indicating that dCK S74 phosphorylation promoted IR-induced
autophagy. It is very interesting that S74E inhibited IR-induced
total cell death and apoptosis and increased autophagic cell death
in the MDA-MB-231 cell line, suggesting that apoptosis plays a more
important role than autophagy in contributing to cell death. We
found that IR-induced apoptosis was inhibited by 3-MA and
IR-induced autophagy was inhibited by ZVAD-FMK. Treatment with 3-MA
+ ZVAD-FMK decreased apoptosis and autophagy more markedly than
3-MA or ZVAD-FMK alone, confirming the existence of a crosstalk
between apoptosis and autophagy following exposure to IR.
Rapamycin is a well-known inhibitor of mTOR, which
inhibits only some of the functions of mTORC1 (49–51).
The Akt/mTOR/p70S6K signaling pathway plays a key role in the
regulation of not only cell survival and proliferation, but also
autophagy (52,53). Given that
ataxia-telangiectasia-mutated (ATM) kinase can phosphorylate dCK on
serine 74 to activate it in response to IR (26), and that ATM can promote IR-induced
autophagy via the Akt/mTOR/P70S6K pathway (27), we hypothesized that dCK regulates
IR-induced autophagy via the Akt/mTOR/p70S6K pathway and that
dCK-S74 phosphorylation may participate in this mechanism. We found
that in the SKBR3 cells, only S74E suppressed p-mTOR expression
obviously following exposure to IR, suggesting that activated dCK
may participate in the mTOR pathway to regulate IR-induced
autophagy. In the MDA-MB-231 cells, both WT and S74E suppressed the
Akt/mTOR/p70S6K pathway significantly in response to IR. However,
dCK-S74A did not markedly affect the pathway following exposure to
IR, indicating that activated dCK may increase IR-induced autophagy
by inhibiting the Akt/mTOR/P70S6K pathway in MDA-MB-231 cells.
One of the key mechanisms for the control of
autophagy is the modulation of the the interaction between the
autophagic protein, BECN1 and the members of the anti-apoptotic
Bcl-2 family (e.g., Bcl-2, Bcl-xL and Mcl-1) (35,54,55).
This binding is regulated by a variety of proteins and compounds
that are able to enhance or inhibit the Bcl-2/BECN1 interaction in
order to repress or activate autophagy, respectively (37). In this study, we examined the
effects of dCK activation on the interaction between BCL2 and BECN1
following exposure to IR. The results of western blot analysis
revealed that dCK-S74E decreased the expression of Bcl-2 and
increased the expression of BECN1, and decreased the ratio of
Bcl-2/BECN1 in the MDA-MB-231 and SKBR3 cells, suggesting that
activated dCK inhibits the binding of Bcl-2/Beclin and promotes
autophagy following exposure to IR.
In conclusion, dCK was found to affect IR-induced
apoptosis and autophagy, and there was a switch from autophagy to
apoptosis in the SKBR3 and MDA-MB-231 cell lines. The
posphorylation of dCK at serine 74 increased autophagy through the
Akt/mTOR/p70S6K signaling pathway, and inhibited the binding of
Bcl-2/Beclin in response to IR in the MDA-MB-231 cell line.
Acknowledgments
We would like to thank Dr Bo Xu (Southern Research
Institute, Birmingham, AL, USA) for providing the dCK plasmids.
This study was supported by a NSFC grant (nos. 31370837, 81573082
and 81673092) and the Provincial Program of Science and Technology
of Jilin (20150101142JC).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
da Silveira WA, Palma PVB, Sicchieri RD,
Villacis RAR, Mandarano LRM, Oliveira TMG, Antonio HMR, Andrade JM,
Muglia VF, Rogatto SR, et al: Transcription factor networks derived
from breast cancer stem cells control the immune response in the
basal subtype. Sci Rep. 7:28512017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Volders JH, Haloua MH, Krekel NM, Meijer S
and van den Tol PM: Current status of ultrasound-guided surgery in
the treatment of breast cancer. World J Clin Oncol. 7:44–53. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rashedi I, Panigrahi S, Ezzati P, Ghavami
S and Los M: Autoimmunity and apoptosis - therapeutic implications.
Curr Med Chem. 14:3139–3151. 2007. View Article : Google Scholar
|
|
6
|
Singh A, Yashavarddhan MH, Kalita B,
Ranjan R, Bajaj S, Prakash H and Gupta ML: Podophyllotoxin and
rutin modulates ionizing radiation-induced oxidative stress and
apoptotic cell death in mice bone marrow and spleen. Front Immunol.
8:1832017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kroemer G, Mariño G and Levine B:
Autophagy and the integrated stress response. Mol Cell. 40:280–293.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen Y and Klionsky DJ: The regulation of
autophagy - unanswered questions. J Cell Sci. 124:161–170. 2011.
View Article : Google Scholar
|
|
10
|
Arnér ES and Eriksson S: Mammalian
deoxyribonucleoside kinases. Pharmacol Ther. 67:155–186. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee MW, Parker WB and Xu B: New insights
into the synergism of nucleoside analogs with radiotherapy. Radiat
Oncol. 8:2232013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Saiki Y, Yoshino Y, Fujimura H, Manabe T,
Kudo Y, Shimada M, Mano N, Nakano T, Lee Y, Shimizu S, et al: DCK
is frequently inactivated in acquired gemcitabine-resistant human
cancer cells. Biochem Biophys Res Commun. 421:98–104. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hodzic J, Giovannetti E, Diosdado B, Adema
AD and Peters GJ: Regulation of deoxycytidine kinase expression and
sensitivity to gemcitabine by micro-RNA 330 and promoter
methylation in cancer cells. Nucleosides Nucleotides Nucleic Acids.
30:1214–1222. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schwarzenberg J, Radu CG, Benz M, Fueger
B, Tran AQ, Phelps ME, Witte ON, Satyamurthy N, Czernin J and
Schiepers C: Human biodistribution and radiation dosimetry of novel
PET probes targeting the deoxyribonucleoside salvage pathway. Eur J
Nucl Med Mol Imaging. 38:711–721. 2011. View Article : Google Scholar :
|
|
15
|
Rivero A, Rapado I, Tomás JF, Montalbán C,
de Oña R, Paz-Carreira J, Canales M, Martínez R, Sánchez-Godoy P,
de Sevilla AF, et al: Relationship between deoxycytidine kinase
(DCK) genotypic variants and fludarabine toxicity in patients with
follicular lymphoma. Leuk Res. 35:431–437. 2011. View Article : Google Scholar
|
|
16
|
Réjiba S, Bigand C, Parmentier C and Hajri
A: Gemcitabine-based chemogene therapy for pancreatic cancer using
Ad-dCK:UMK GDEPT and TS/RR siRNA strategies. Neoplasia. 11:637–650.
2009. View Article : Google Scholar
|
|
17
|
Sigmond J, Bergman AM, Leon LG, Loves WJ,
Hoebe EK and Peters GJ: Staurosporine increases toxicity of
gemcitabine in non-small cell lung cancer cells: Role of protein
kinase C, deoxycytidine kinase and ribonucleotide reductase.
Anticancer Drugs. 21:591–599. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Si S, Liao Q, Zhao YP, Hu Y, Zhang Q and
You LL: Relationship between single nucleotide polymorphisms in the
deoxycytidine kinase gene and chemosensitivity of gemcitabine in
six pancreatic cancer cell lines. Chin Med J (Engl). 124:419–422.
2011.
|
|
19
|
Horie R, Nakamura O, Yamagami Y, Mori M,
Nishimura H, Fukuoka N and Yamamoto T: Apoptosis and antitumor
effects induced by the combination of an mTOR inhibitor and an
autophagy inhibitor in human osteosarcoma MG63 cells. Int J Oncol.
48:37–44. 2016. View Article : Google Scholar :
|
|
20
|
Smal C, Vertommen D, Bertrand L, Rider MH,
van den Neste E and Bontemps F: Identification of phosphorylation
sites on human deoxycytidine kinase after overexpression in
eucaryotic cells. Nucleosides Nucleotides Nucleic Acids.
25:1141–1146. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Amsailale R, Van Den Neste E, Arts A,
Starczewska E, Bontemps F and Smal C: Phosphorylation of
deoxycytidine kinase on Ser-74: Impact on kinetic properties and
nucleoside analog activation in cancer cells. Biochem Pharmacol.
84:43–51. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hazra S, Szewczak A, Ort S, Konrad M and
Lavie A: Post-translational phosphorylation of serine 74 of human
deoxycytidine kinase favors the enzyme adopting the open
conformation making it competent for nucleoside binding and
release. Biochemistry. 50:2870–2880. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Smal C, Ntamashimikiro S, Arts A, Van Den
Neste E and Bontemps F: Influence of phosphorylation of THR-3,
SER-11, and SER-15 on deoxycytidine kinase activity and stability.
Nucleosides Nucleotides Nucleic Acids. 29:404–407. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fedorko M: Effect of chloroquine on
morphology of cytoplasmic granules in maturing human leukocytes -
an ultrastructural study. J Clin Invest. 46:1932–1942. 1967.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Smal C, Vertommen D, Amsailale R, Arts A,
Degand H, Morsomme P, Rider MH, Neste EV and Bontemps F: Casein
kinase 1delta activates human recombinant deoxycytidine kinase by
Ser-74 phosphorylation, but is not involved in the in vivo
regulation of its activity. Arch Biochem Biophys. 502:44–52. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang C, Lee M, Hao J, Cui X, Guo X, Smal
C, Bontemps F, Ma S, Liu X, Engler D, et al: Deoxycytidine kinase
regulates the G2/M checkpoint through interaction with
cyclin-dependent kinase 1 in response to DNA damage. Nucleic Acids
Res. 40:9621–9632. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhong R, Xin R, Chen Z, Liang N, Liu Y, Ma
S and Liu X: The role of deoxycytidine kinase (dCK) in
radiation-induced cell death. Int J Mol Sci. 17:172016. View Article : Google Scholar
|
|
28
|
Kenific CM and Debnath J: Cellular and
metabolic functions for autophagy in cancer cells. Trends Cell
Biol. 25:37–45. 2015. View Article : Google Scholar
|
|
29
|
Rosenfeldt MT and Ryan KM: The multiple
roles of autophagy in cancer. Carcinogenesis. 32:955–963. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen HY and White E: Role of autophagy in
cancer prevention. Cancer Prev Res (Phila). 4:973–983. 2011.
View Article : Google Scholar
|
|
31
|
Martín-Acebes MA, Blázquez AB, de Oya NJ,
Escribano-Romero E, Shi PY and Saiz JC: A single amino acid
substitution in the core protein of West Nile virus increases
resistance to acidotropic compounds. PLoS One. 8:e694792013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pinato O, Musetti C and Sissi C: Pt-based
drugs: The spotlight will be on proteins. Metallomics. 6:380–395.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rebillard A, Lagadic-Gossmann D and
Dimanche-Boitrel MT: Cisplatin cytotoxicity: DNA and plasma
membrane targets. Curr Med Chem. 15:2656–2663. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Marquez RT and Xu L: Bcl-2:Beclin 1
complex: multiple, mechanisms regulating autophagy/apoptosis toggle
switch. Am J Cancer Res. 2:214–221. 2012.PubMed/NCBI
|
|
35
|
Erlich S, Mizrachy L, Segev O, Lindenboim
L, Zmira O, Adi-Harel S, Hirsch JA, Stein R and Pinkas-Kramarski R:
Differential interactions between Beclin 1 and Bcl-2 family
members. Autophagy. 3:561–568. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Decuypere JP, Parys JB and Bultynck G:
Regulation of the autophagic bcl-2/beclin 1 interaction. Cells.
1:284–312. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
39
|
Warburton AJ and Boone DN: Insights from
Global Analyses of Long Noncoding RNAs in Breast Cancer. Curr
Pathobiol Rep. 5:23–34. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Beyaert M, Starczewska E, Van Den Neste E
and Bontemps F: A crucial role for ATR in the regulation of
deoxycytidine kinase activity. Biochem Pharmacol. 100:40–50. 2016.
View Article : Google Scholar
|
|
41
|
Bunimovich YL, Nair-Gill E, Riedinger M,
McCracken MN, Cheng D, McLaughlin J, Radu CG and Witte ON:
Deoxycytidine kinase augments ATM-mediated DNA repair and
contributes to radiation resistance. PLoS One. 9:e1041252014.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
de Melo Gagliato D, Jardim DL, Marchesi MS
and Hortobagyi GN: Mechanisms of resistance and sensitivity to
anti-HER2 therapies in HER2+ breast cancer. Oncotarget.
7:64431–64446. 2016.PubMed/NCBI
|
|
43
|
Wu HM, Jiang ZF, Ding PS, Shao LJ and Liu
RY: Hypoxia-induced autophagy mediates cisplatin resistance in lung
cancer cells. Sci Rep. 5:122912015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Denton D, Nicolson S and Kumar S: Cell
death by autophagy: Facts and apparent artefacts. Cell Death
Differ. 19:87–95. 2012. View Article : Google Scholar :
|
|
45
|
Codogno P and Meijer AJ: Autophagy and
signaling: Their role in cell survival and cell death. Cell Death
Differ. 12(Suppl 2): 1509–1518. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Fimia GM and Piacentini M: Regulation of
autophagy in mammals and its interplay with apoptosis. Cell Mol
Life Sci. 67:1581–1588. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mizushima N, Sugita H, Yoshimori T and
Ohsumi Y: A new protein conjugation system in human. The
counterpart of the yeast Apg12p conjugation system essential for
autophagy. J Biol Chem. 273:33889–33892. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mizushima N, Noda T, Yoshimori T, Tanaka
Y, Ishii T, George MD, Klionsky DJ, Ohsumi M and Ohsumi Y: A
protein conjugation system essential for autophagy. Nature.
395:395–398. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
49
|
Klionsky DJ, Baehrecke EH, Brumell JH, Chu
CT, Codogno P, Cuervo AM, Debnath J, Deretic V, Elazar Z, Eskelinen
EL, et al: A comprehensive glossary of autophagy-related molecules
and processes (2nd edition). Autophagy. 7:1273–1294. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang MH, Jiang JZ, Cai YL, Piao LH and
Jin Z: Significance of dynamic changes in gastric smooth muscle
cell apoptosis, PI3K-AKT-mTOR and AMPK-mTOR signaling in a rat
model of diabetic gastroparesis. Mol Med Rep. 16:1530–1536. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Linke M, Fritsch SD, Sukhbaatar N,
Hengstschläger M and Weichhart T: mTORC1 and mTORC2 as regulators
of cell metabolism in immunity. FEBS Lett. 591:3089–3103. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Cao B, Li J, Zhou X, Juan J, Han K, Zhang
Z, Kong Y, Wang J and Mao X: Clioquinol induces pro-death autophagy
in leukemia and myeloma cells by disrupting the mTOR signaling
pathway. Sci Rep. 4:57492014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Saiki S, Sasazawa Y, Imamichi Y, Kawajiri
S, Fujimaki T, Tanida I, Kobayashi H, Sato F, Sato S, Ishikawa K,
et al: Caffeine induces apoptosis by enhancement of autophagy via
PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 7:176–187. 2011.
View Article : Google Scholar :
|
|
54
|
Lindqvist LM, Heinlein M, Huang DC and
Vaux DL: Prosurvival Bcl-2 family members affect autophagy only
indirectly, by inhibiting Bax and Bak. Proc Natl Acad Sci USA.
111:8512–8517. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Priault M, Hue E, Marhuenda F, Pilet P,
Oliver L and Vallette FM: Differential dependence on Beclin 1 for
the regulation of pro-survival autophagy by Bcl-2 and Bcl-xL in
HCT116 colorectal cancer cells. PLoS One. 5:e87552010. View Article : Google Scholar : PubMed/NCBI
|