The worldwide incidence of cutaneous melanoma has
been increasing annually at a more rapid rate compared to any other
type of cancer (1). In 2012,
232,000 new cases of melanoma and 55,000 deaths were registered
worldwide, ranking 15th among most common cancers worldwide
(2). The incidence of cutaneous
melanoma varies greatly between countries and these different
incidence patterns are ascribed to variations in racial skin
phenotype, as well as differences in sun exposure. Moreover, unlike
other solid tumors, melanoma mostly affects young and middle-aged
individuals (median age at diagnosis, 57 years). The incidence
increases linearly after the age of 25 years until the age of 50
years, and then decreases, particularly in the female sex. When
analyzing incidence data in relation to sex, women are more
frequent in younger aged groups, while the male sex prevails from
the age of 55 onwards (3).
Ultraviolet (UV) light radiation from sunlight is
the main environmental risk factor for melanoma skin cancer
development (4–6). The increased risk of melanoma due to
sun exposure is directly associated with the UV level and in
particular to the UV-B spectrum (5). In addition, sun exposure patterns and
timing have been associated in a number of studies with an
increased risk of melanoma. In particular, intense and intermittent
sun exposure (typical of sunburn history) is associated with a
higher risk compared to a chronic continuous pattern of sun
exposure that is more frequently associated with actinic keratosis
and non-melanoma skin cancers (7–10).
Furthermore, a history of sunburn in childhood or adolescence is
associated with the highest risk of developing melanoma and
individuals experiencing >5 episodes of severe sunburn have a
2-fold increased risk (8,11). UV-A exposure from artificial
sources has been also linked to an increased risk of developing
melanoma. The follow-up of patients with psoriasis receiving UV-A
radiation phototherapy, as well as in individuals using sunbeds has
revealed an increased risk of melanoma in this population (12,13).
Specifically, several studies and a meta-analysis have demonstrated
a positive association between the risk of developing melanoma and
the amount of sunbed usage, particularly from a young age, thus
raising a major public health issue (12,14,15).
UV light from sunbeds has been formally classified as a human
carcinogen (14). No other
environmental factors, including tobacco/smoke addiction, have been
associated with melanoma (1).
In addition, host risks factors, such as the number
of congenital and acquired melanocytic nevi, genetic susceptibility
and a family history play a central role in the development of
melanoma (16–18). Approximately 25% of melanoma cases
arise on a pre-existing nevus (19). In this context, not only the total
number of nevi, but also the size and type of nevi, are
individually associated with an increased risk of melanoma
(20–23).
As regards genetic susceptibility, the polymorphisms
of the melanocortin 1 receptor (MC1R) gene, are responsible
for the different human skin-color phenotypes. Individuals with
characteristics, such as red hair, a light complexion and light
eyes exhibit a low pigmentation, with a consequent heightened
sensitivity to UV exposure (24).
Approximately 7–15% of melanoma cases occur in patients with a
family history of melanoma (25).
However, true hereditary melanoma (i.e., multi-generational,
unilateral lineage, multiple primary lesions and early onset of the
disease) are infrequent; the familial clustering of the disease is
considered to be responsible for the presence of a transmitted
genetic mutation (25,26). Over the past years, melanomas have
also been found to arise in families that are generally prone to
specific patterns of malignancies, such as the familial atypical
multiple mole-melanoma syndrome (FAMMM syndrome) and its variant,
the melanoma-astrocytoma syndrome (MAS) (26). Germline mutations in
cyclin-dependent kinase inhibitor 2A (CDKN2A or p16) and,
less common, mutations in cyclin-dependent kinase 4 (CDK4)
are the most frequent genetic abnormalities identified in these
families (26–28). Other inherited conditions
associated with an increased risk of developing melanoma are
xeroderma pigmentosum, familial retinoblastoma, Lynch syndrome type
II and Li-Fraumeni cancer syndrome (25).
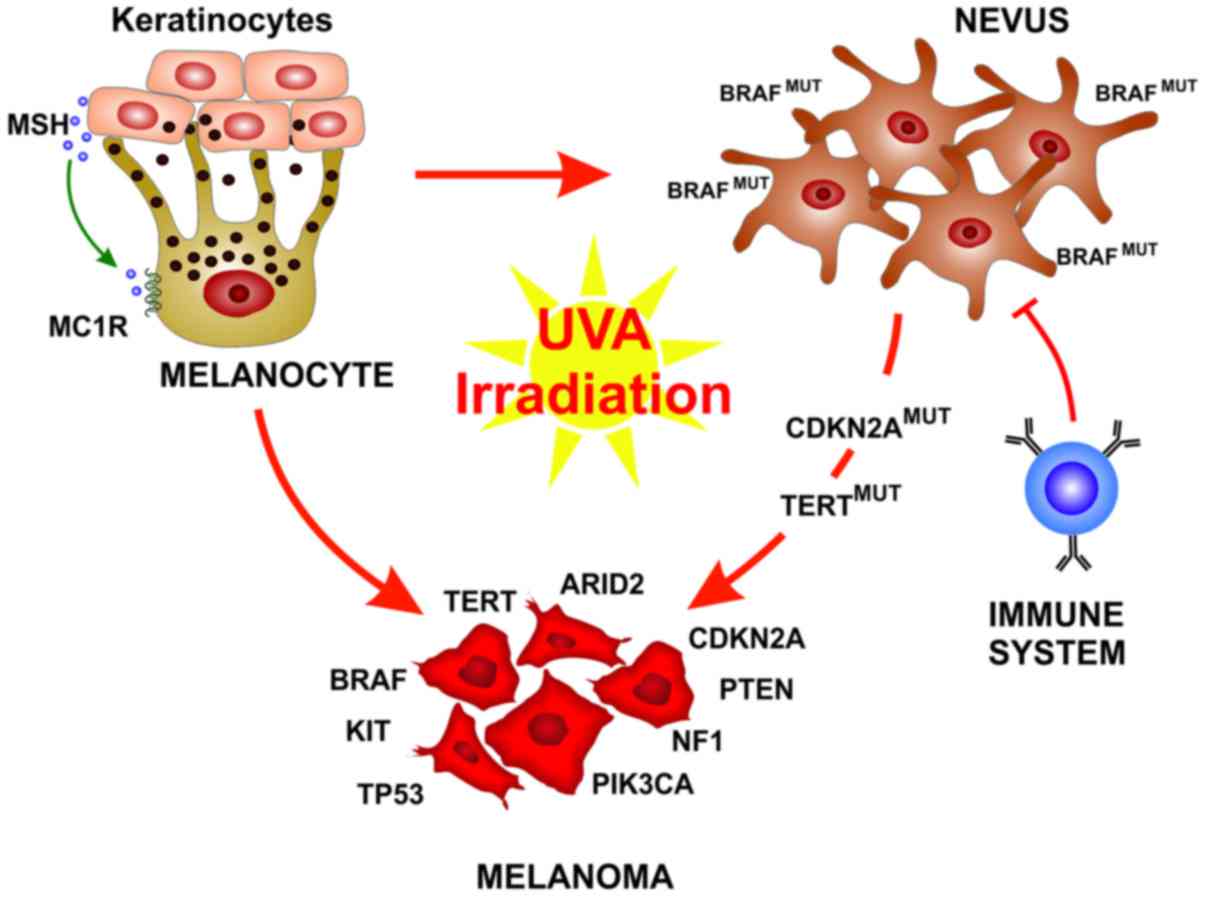
Melanocytes are neural crest-derived cells that can
be found principally in the basal epidermis and in hair follicles,
along mucosal surfaces, meninges and in the choroidal layer of the
eye (29). In response to
UV-induced DNA damage, skin keratinocytes produce the melanocyte
stimulating hormone (MSH) that binds the melanocortin receptor 1
(MC1R) on the melanocytes that than produce and release melanin.
The melanin pigment ultimately operates as a shield for UV
radiation, thus preventing further DNA alteration (30).
Cutaneous melanoma can be generally classified in
the Caucasian population by its origin from chronically or
intermittent sun-exposed skin that translate into different sites
of origin, a degree of cumulative UV exposure, age at diagnosis,
types of oncogenic drivers and mutational load (9). Indeed, melanomas in chronically
sun-exposed skin usually appear in older-aged individuals (>55
years), on chronically sun-exposed areas, such as the head and
neck, as well as the dorsal region of the upper extremities. The
main genetic drivers are B-Raf proto-oncogene (BRAF),
neurofibromin 1 (NF1) and NRAS mutations, and usually
melanomas associated with chronically sun-exposed skin have a high
mutational load related to UV exposure (9,31,32).
On the other hand, melanoma associated with intermittent
sun-exposed skin cases arise in younger-aged individuals (<55
years), on less sun-exposed areas, such as the trunk and proximal
extremities, and are usually associated with BRAFV600E
and a lower mutational load (31,32).
Over the past years, a deeper understanding of
melanoma development and biology has been reached. It has become
clear that the development of fully-evolved melanoma from
pre-neoplastic lesions is not represented by a single evolutionary
pattern. Each melanoma subtype can evolve from different precursor
lesions, and can involve different gene mutations and stage of
transformation (33). An
interesting finding is that BRAF is mutated in up to 80% of benign
nevi, resulting in limited melanocyte proliferation through the
oncogene-mediated activation of cell senescence (34,35).
These nevi remain indolent for decades also due to immune
surveillance (36). Therefore,
oncogenic BRAF alone is not sufficient for melanoma
development and rarely benign nevi further progress to melanoma
(33,37). When this usually occurs, it is
associated with the acquisition of subsequent mutations in key
genes, such as TERT or CDKN2A. On the other hand,
melanomas associated with chronic sun-exposed skin usually do not
arise form pre-existing nevi, but from melanomas in situ or
dysplastic lesions and carry a different set of mutations (33). Histological characterization is the
current mainstay of melanocytic neoplasia diagnosis and the
definition of their malignant potential. However, histopathology is
sometimes associated with the equivocal characterization of these
lesions, leading to their improper risk stratification (38). The increasing understanding of the
biological determinants of melanoma evolution and their potential
integration in the management of melanoma patients may lead to an
improve diagnosis and the earlier recognition of lesions at an
increased risk of progression, thus improving patient risk
stratification (Fig. 1).
Cutaneous melanoma is one of the most aggressive
forms of skin cancer and one of the leading causes of
cancer-related mortality due its metastatic power. Several studies
have demonstrated that melanoma spreading is the result of genetic
mutations and tumor microenvironmental alterations, characterized
by the overexpression of proteins able to favor tumor invasion and
surrounding infiltration (39–44).
In particular, a key role is played by the overexpression of matrix
metalloproteinases (MMPs), particularly MMP-9 and MMP-2, that
induces the degradation of the components of the extracellular
matrix, thus favoring tumor cell infiltration and spreading through
the bloodstream (40–42). The overexpression of these proteins
and tumor microenvironmental alterations are mediated by genetic
alterations and the dysregulation of the nuclear factor (NF)-κB
pathways. It has been demonstrated that MMP-9 overexpression
observed in melanoma is caused by intragenic methylation phenomena
that lead to protein overexpression (42). Furthermore, it has also been
demonstrated that NF-κB induces the overexpression of MMP-9 by the
activation of osteopontin (OPN), another protein of the tumor
microenvi-ronment, thus playing a fundamental role in the
development and progression of melanoma (43,44).
Apart from tumor microenvironmental alterations,
melanomas are associated with one of the greatest burdens of
somatic genetic alterations of all human tumors (45,46).
The most frequent somatic mutations in chronically or intermittent
sun-exposed skin melanomas affect genes that control central
cellular process, such us proliferation (BRAF, NRAS
and NF1), growth and metabolism [phosphatase and tensin
homolog (PTEN) and KIT proto-oncogene receptor tyrosine
kinase (KIT)], resistance to apoptosis [tumor protein p53
(TP53)], cell cycle control [cyclin-dependent kinase
inhibitor 2A (CDKN2A)] and replicative lifespan [telomerase
reverse transcriptase (TERT)] (47,48).
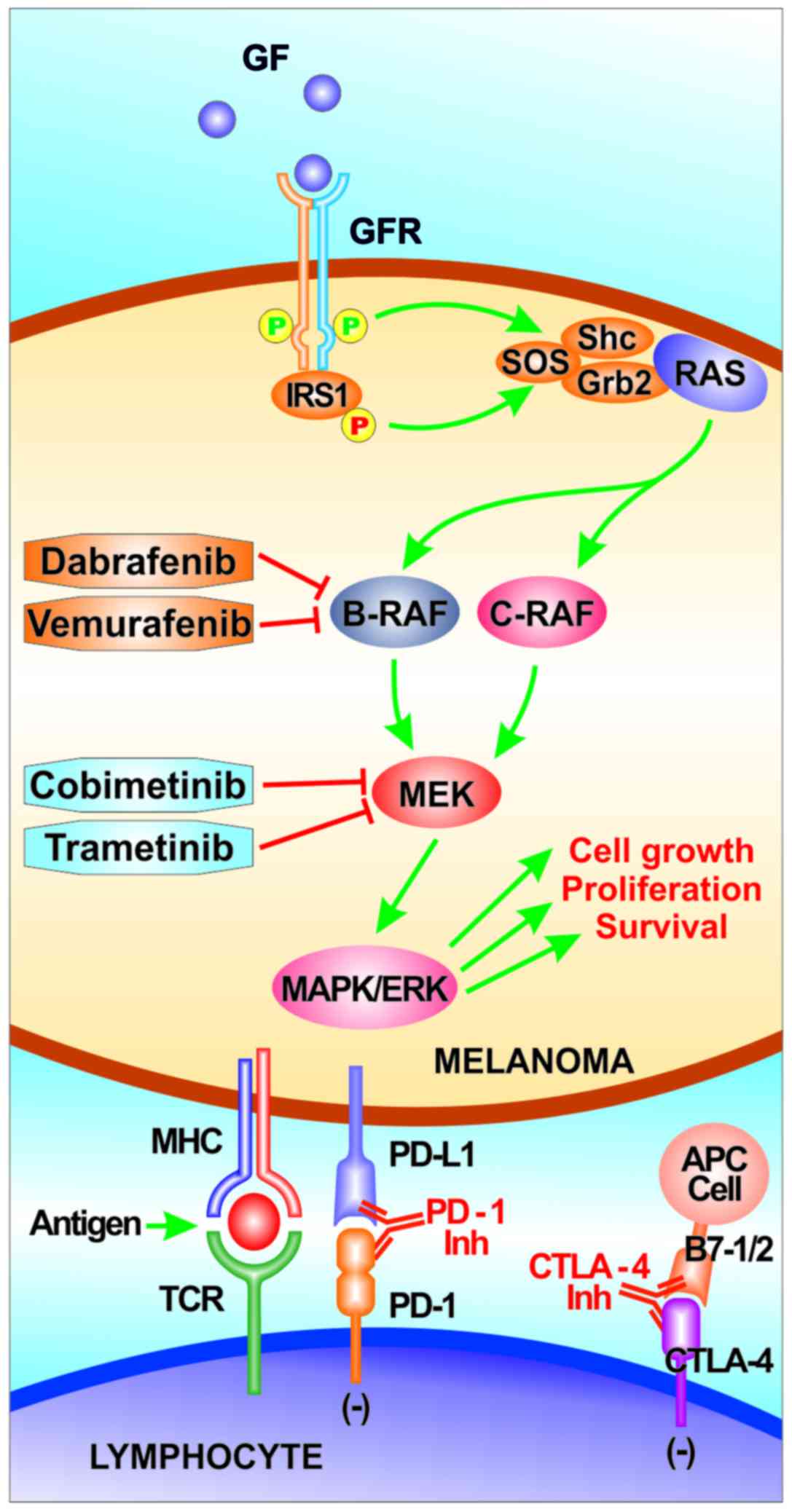
These genomic alterations typically lead to the aberrant activation
of two main signaling pathways in melanoma: The RAS/RAF/MEK/ERK
signaling cascade [also known as the mitogen-activated protein
kinase (MAPK) pathway] and the phosphoinositol-3-kinase (PI3K)/AKT
pathway (49).
The MAPK pathway is physiologically involved in the
transduction of extracellular signals, such as growth factors and
hormones, to the nucleus, leading to the expression of genes that
are central drivers of cell proliferation, differentiation and
survival (50,51). In addition, it has been shown that
MAPK activation is a critical player in the biology of different
types of cancer and is the most frequent pathway aberrantly
activated in melanoma (52). The
PI3K pathway is normally involved in cellular homeostasis and its
activation has been demonstrated to be central in different cancer
types, including melanoma where it is the second most frequently
activated pathway (53,54).
Up to 90% of melanomas exhibit an aberrant MAPK
pathway activation and this is a central step in melanoma
development, being responsible for cell cycle deregulation and
apoptosis inhibition (50,55,56).
Among the different mechanisms responsible for abnormal MAPK
pathway signaling in melanoma, the most frequent genetic
abnormalities are, by far, BRAF mutations (37,47).
Indeed, 37 to 50% of melanomas carry a somatic mutation in the
BRAF gene with the highest frequency in cutaneous melanomas
derived from intermittent sun exposure damage (approximately 60%
carry a BRAF mutation) (31).
Usually, BRAF mutations detected in cutaneous melanoma are missense
mutations that determine amino acid substitution at valine 600.
Approximately 80–90% of BRAF mutations are V600E (valine to
glutamic acid), while 5–12% are valine to lysine substitution
(V600K) and ≤5% are V600D (valine to aspartic acid) or V600R
(valine to arginine) (57,58).
BRAF protein is a serine/threonine protein kinase of
766 amino acids organized in three domains: Two with regulatory
function and one catalytic domain responsible for MEK
phosphorylation (59). The
catalytic domain is also responsible for maintaining the protein in
its inactive conformation, through a hydrophobic interaction
between the 'so-called' glycine-rich loop and the activation
segment, making it inaccessible for ATP binding (59). In the BRAFV600E
mutation, hydrophobic valine is replaced by polar, hydrophilic
glutamic acid, resulting in an abnormal flip of the catalytic
domain that generates a constitutive active conformation with a
kinase activity 500-fold higher than wild-type BRAF kinase
(60,61). Most of the non-V600E BRAF mutations
act similarly through the alteration of glycine-rich loop and
activation segment interaction, thus increasing BRAF kinase
activity (61).
The second most common cause of aberrant signaling
through the MAPK pathway in cutaneous melanoma is represented by
NRAS activating mutations. NRAS is mutated in 15–30% of
melanomas and in the majority of cases, these mutations are
missense mutations of codon 12, 13 or 61 (the latter account for
80% of all NRAS mutations in melanoma) (31,62).
Mutations of these codons lead to the prolongation of the
NRAS-active GTP-bound state, thus abnormally maintaining NRAS
signaling through both the MAPK and the PI3K pathways (47,63,64).
Importantly, NRAS and BRAF mutations are considered
mutually exclusive; however, co-mutations can rarely occur
(approximately 0.5% in treatment-naïve patients) (64).
NF1 is a tumor suppressor gene mutated in 10–15% of
melanoma cases and is the third most frequently mutated gene in
melanoma (65,66). The NF1 protein regulates the RAS
family by converting the active RAS-guanosine triphosphate
(RAS-GTP) to the inactive RAS-guanosine diphosphate (RAS-GDP),
thereby inhibiting downstream RAS signaling (67). Therefore, NF1 loss-of-function
determines the hyperactivation of NRAS protein and thus, increased
MAPK and PI3K pathways signaling (65,67,68).
NF1 genomic alterations are more frequent in melanomas associated
with chronically sun-exposed skin and are usually associated with a
high number of various genomic mutations, including co-occurence
with BRAF or NRAS mutations (68,69).
The receptor tyrosine kinase KIT is physiologically
involved in melanoma proliferation and survival through the
PI3K/AKT and the RAS/RAF/MEK/ERK pathways. Somatic activating
mutations in this gene have been found in 2–8% of all malignant
melanomas and are more frequent in acral melanomas and with
melanoma arising on intermitted sun-exposed skin (70,71).
Even though a conclusive model of recurrent
alterations leading to metastatic progression has yet to be
elucidated, β-catenin-mediated WNT signaling activation has been
shown to be associated with metastatic dissemination, as well as
melanoma formation (37,81). CTNNB1 (β-catenin) gene
mutations are detected in 2–4% of malignant melanomas and act
through the stabilization of β-catenin and increased transcription
of TCF/LEF-responsive target genes (82).
The majority of patients with newly-diagnosed
melanoma have early-stage disease. For these patients, surgical
excision represents the treatment of choice and is curative in the
majority of cases (83). However,
some patients will later relapse with disseminated disease, while
approximately 10% of melanoma cases are diagnosed at an advanced
stage, and are unresectable or already metastatic. Among stage IV
tumors, approximately one-third have visceral and brain involvement
at diagnosis, with a severe prognosis and lower probability to have
a sustained response to treatment (84). For patients facing advanced-stage
disease, melanoma treatment has been revolutionized since 2011,
with the approval of several therapeutic agents. These agents
include RAF and MEK kinase inhibitors, as well as immune checkpoint
inhibitors [anti-cytotoxic T-lymphocyte-associated antigen 4
antibodies (anti-CTLA4) and anti-programmed cell death protein 1
antibody (anti-PD1)]. Indeed, in the advanced-stage setting,
anti-PD1 and anti-CTLA4 antibodies (such as nivolumab,
pembrolizumab and ipilimumab), as well as selective BRAF inhibitors
(vemurafenib and dabrafenib) alone and/or in combination with MEK
inhibitors (cobimetinib and trametinib) have shown promising
results in clinical trials (Table
I) (85–93). Currently, only the presence of
BRAFV600E mutation is evaluated in the clinical setting,
as it is essential to drive the appropriate treatment strategy.
Other driver mutations, such as NRAS, NF1,
CKIT, CDKN2A and PTEN, have not yet been
included in standard clinical practice. However, the identification
of these genomic alterations can identify patients who may benefit
of experimental approach in clinical trials.
Immunotherapy and kinase inhibitors are nowadays the
backbone of systemic therapy, while chemotherapy is considered a
second-line, or even further, treatment option (94–96)
(Fig. 2). Anti-PD1 antibodies and,
with lower magnitude anti-CTLA4 therapeutic agents, offer lower
response rates, but potentially long durable responses (85,91,92).
In BRAFV600E melanoma, there has been a reasonable
approach to the use of BRAF inhibitors with MEK inhibitors. The
combination has led to high response rates (70%) and a rapid
response induction and symptom control, with a progression-free
survival of approximately 12 months (89,90,93).
To date, however, there are no available data from prospective
trials on the optimal choice for frontline treatment and treatment
sequence, at least to the best of our knowledge. Nivolumab and
pembrolizumab have shown to be effective on BRAF mutant melanoma
after BRAF inhibitor resistance has risen, but there are no similar
data for ipilimumab or for BRAF-inhibitor therapy in those with
primary or secondary resistance to anti-PD-1 therapy (97–99).
Currently, the combination of two different immune checkpoint
inhibitors or the combination anti-PD1/anti-CTLA4 with targeted
therapy must be considered an experimental approach in clinical
trials. Each strategy has a clear benefit and basic research has
demonstrated significant synergistic effects that need to be
weighted with the potential increase in toxicity (100,101).
The inclusion of patient characteristics
[biochemical parameters of melanoma kinetics, such us lactate
dehydrogenase (LDH)] and expected toxicity profile, as well as
comorbidities and patient personal preferences are central elements
to be taken into account for frontline treatment strategy
definition. In this rapidly evolving landscape, it is of great
importance the participation of patients in randomized clinical
trials.
The identification of biomarkers that can predict
patient benefits towards specific treatment strategies is a central
goal of cancer research. BRAF mutations, particular
BRAFV600E, is a typical predictive marker of response to
RAF inhibitors. However, these patients almost invariably develop
disease progression after a variable period of time and some
patients can display primary resistance to BRAF (+/− MEK)
inhibitors. Several studies have described the central role of
acquired genetic mutations affecting the Ras/Raf/MEK/ERK and
PI3K/PTEN/Akt/mTOR signaling pathways in inducing resistance to
both chemotherapy and targeted therapy in melanoma and other tumor
types (102–104). In particular, the mechanisms
responsible for BRAF (+/− MEK) inhibitor resistance can be divided
into genomic (NRAS/KRAS mutation 20%, BRAF
splice variants 16%, BRAF amplification 13%, MEK1/2
mutation 7%, bypass track mutations 11%), immunologic (epigenetic
and transcriptomic changes of molecules involved in antigen
presenting mechanisms) and a combination of both (105,106).
Currently, the detection of the mechanisms
responsible for BRAF and MEK inhibitor resistance is not part of
standard clinical practice; however, the development of
non-invasive techniques for tumor mutational status assessment may
lead to more rapid changes in this setting (107). The technique termed 'liquid
biopsy' enables the detection of tumor-derived circulating
cell-free DNA (ctDNA) in the plasma and is emerging as a promising
blood-based biomarker for monitoring the melanoma disease status.
Several studies have indicated that BRAFV600E detection
through ctDNA prior to the commencement of treatment is predictive
of the response to BRAF kinase inhibitors, and that high basal
ctDNA levels are associated with a lower response rate and
progression-free survival (107–109). Moreover, ctDNA is an indicator of
tumor burden and tumor dynamics, and it has been demonstrated that
an increase in ctDNA levels during treatment is indicative of
disease progression and acquired resistance (107,109). Notably, ctDNA can be used also
for the detection of mutations responsible for resistance to BRAF
targeted therapies and in the future, this can be used to guide
subsequent treatment strategies (107,109).
Immune checkpoint inhibitors are associated with a
low overall response rate (ORR). This has driven considerable
research efforts in the identification of biomarkers able to
predict which patients will more likely benefit from these
treatments. At this point, PD-L1 immunohistochemistry on tumor
specimens is not a candidate marker for PD-1 inhibitor treatment
response, due to the extremely heterogeneous results obtained from
clinical trials (88,99). Several other predictive biomarkers
are currently under investigation. The specific components of
melanoma microenvironment and in particular the CD8+ T
cell activation, through IFN-γ gene expression signature, has been
associated with immunotherapy response (110,111). Moreover, several studies have
demonstrated the mechanisms through which specific genomic
alterations can drive immune checkpoint resistance through the
alteration of antigen-presenting mechanisms and IFN-γ production
(112–114). Recently, in humans, it has been
demonstrated that specific gut microbiota compositions can drive
differential responses to immune checkpoint inhibitors (115–117). This is not only a new intriguing
field of research for immunotherapy biomarkers, but it also paves
the way for the potential modulation of human gut microbiota
composition to improve the immunotherapy response. As regards the
identification of complex biological interactions among different
pathways and their interplay with the immune system, bioinformatics
has yielded promising results. In this context, computational
models can simulate biochemical, metabolic and immune mediated
interactions and characterize how they are potentially involved in
melanoma development (118,119). Overall, computational approaches
may potentially lead to the identification of novel therapeutic
targets and may accelerate the drug discovery process (120).
Marked improvements in melanoma treatment have been
achieved over the past decade. The tireless efforts of researchers
have shed light on essential mechanisms involved in melanoma
biology, paving the way for targeted treatment and immunotherapy.
However, melanoma remains a lethal type of cancer, particularly
when diagnosed at an advanced stage. Further elucidation of
melanoma biology and evolution also in presence of
treatment-selective pressure represent a central goal of cancer
research in this field and may ultimately improve patient care and
prognosis.
Not applicable.
This study was supported by the Lega Italiana per la
Lotta contro i Tumori (LILT) and the grant entitled: Identification
of cancer driver genes for novel diagnostics and therapeutic
strategies - Piano per la ricerca 2016–2018 - Linea di intervento 2
- University of Catania, Department of Biomedical and
Biotechnological Sciences.
Not applicable.
GCL and LF wrote the manuscript and were involved in
data collection. JAM, RS and AZ contributed to enriching the
knowledge concerning the genesis of melanoma, the current medical
treatment and the predictive biomarkers tested for the diagnosis of
melanoma. SC was involved in the creation of the figures. ML, SC
and DAS conceived the review and revised the manuscript. All
authors edited the manuscript and have approved its final
version.
Not applicable.
Not applicable.
DAS is the Editor-in-Chief for the journal, but had
no personal involvement in the reviewing process, or any influence
in terms of adjudicating on the final decision, for this article.
The remaining authors have no competing interests.
|
1
|
Ali Z, Yousaf N and Larkin J: Melanoma
epidemiology, biology and prognosis. EJC Suppl. 11:81–91. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
3
|
Rastrelli M, Tropea S, Rossi CR and
Alaibac M: Melanoma: Epidemiology, risk factors, pathogenesis,
diagnosis and classification. In Vivo. 28:1005–1011.
2014.PubMed/NCBI
|
|
4
|
Gilchrest BA, Eller MS, Geller AC and Yaar
M: The pathogenesis of melanoma induced by ultraviolet radiation. N
Engl J Med. 340:1341–1348. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pennello G, Devesa S and Gail M:
Association of surface ultraviolet B radiation levels with melanoma
and nonmelanoma skin cancer in United States blacks. Cancer
Epidemiol Biomarkers Prev. 9:291–297. 2000.PubMed/NCBI
|
|
6
|
Falzone L, Marconi A, Loreto C, Franco S,
Spandidos DA and Libra M: Occupational exposure to carcinogens:
Benzene, pesticides and fibers (Review). Mol Med Rep. 14:4467–4474.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nelemans PJ, Groenendal H, Kiemeney LA,
Rampen FH, Ruiter DJ and Verbeek AL: Effect of intermittent
exposure to sunlight on melanoma risk among indoor workers and
sun-sensitive individuals. Environ Health Perspect. 101:252–255.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Elwood JM and Jopson J: Melanoma and sun
exposure: An overview of published studies. Int J Cancer.
73:198–203. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Candido S, Rapisarda V, Marconi A,
Malaponte G, Bevelacqua V, Gangemi P, Scalisi A, McCubrey JA,
Maestro R, Spandidos DA, et al: Analysis of the
B-RafV600E mutation in cutaneous melanoma patients with
occupational sun exposure. Oncol Rep. 31:1079–1082. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gandini S, Sera F, Cattaruzza MS, Pasquini
P, Picconi O, Boyle P and Melchi CF: Meta-analysis of risk factors
for cutaneous melanoma: II. Sun exposure. Eur J Cancer. 41:45–60.
2005. View Article : Google Scholar
|
|
11
|
White E, Kirkpatrick CS and Lee JA:
Case-control study of malignant melanoma in Washington state. I.
Constitutional factors and sun exposure. Am J Epidemiol.
139:857–868. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lazovich D, Vogel RI, Berwick M, Weinstock
MA, Anderson KE and Warshaw EM: Indoor tanning and risk of
melanoma: A case-control study in a highly exposed population.
Cancer Epidemiol Biomarkers Prev. 19:1557–1568. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Archier E, Devaux S, Castela E, Gallini A,
Aubin F, Le Maître M, Aractingi S, Bachelez H, Cribier B, Joly P,
et al: Carcinogenic risks of psoralen UV-A therapy and narrowband
UV-B therapy in chronic plaque psoriasis: A systematic literature
review. J Eur Acad Dermatol Venereol. 26(Suppl 3): 22–31. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
International Agency for Research on
Cancer Working Group on artificial ultraviolet (UV) light and skin
cancer: The association of use of sunbeds with cutaneous malignant
melanoma and other skin cancers: A systematic review. Int J Cancer.
120:1116–1122. 2007.
|
|
15
|
Wehner MR, Chren MM, Nameth D, Choudhry A,
Gaskins M, Nead KT, Boscardin WJ and Linos E: International
prevalence of indoor tanning: A systematic review and
meta-analysis. JAMA Dermatol. 150:390–400. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bauer J and Garbe C: Acquired melanocytic
nevi as risk factor for melanoma development. A comprehensive
review of epidemiological data. Pigment Cell Res. 16:297–306. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Russo AE, Torrisi E, Bevelacqua Y,
Perrotta R, Libra M, McCubrey JA, Spandidos DA, Stivala F and
Malaponte G: Melanoma: Molecular pathogenesis and emerging target
therapies (Review). Int J Oncol. 34:1481–1489. 2009.PubMed/NCBI
|
|
18
|
Hawkes JE, Truong A and Meyer LJ: Genetic
predisposition to melanoma. Semin Oncol. 43:591–597. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bevona C, Goggins W, Quinn T, Fullerton J
and Tsao H: Cutaneous melanomas associated with nevi. Arch
Dermatol. 139:1620–1624; discussion 1624. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Seykora J and Elder D: Dysplastic nevi and
other risk markers for melanoma. Semin Oncol. 23:682–687.
1996.PubMed/NCBI
|
|
21
|
Watt AJ, Kotsis SV and Chung KC: Risk of
melanoma arising in large congenital melanocytic nevi: A systematic
review. Plast Reconstr Surg. 113:1968–1974. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gandini S, Sera F, Cattaruzza MS, Pasquini
P, Abeni D, Boyle P and Melchi CF: Meta-analysis of risk factors
for cutaneous melanoma: I. Common and atypical naevi. Eur J Cancer.
41:28–44. 2005. View Article : Google Scholar
|
|
23
|
Olsen CM, Zens MS, Stukel TA, Sacerdote C,
Chang YM, Armstrong BK, Bataille V, Berwick M, Elwood JM, Holly EA,
et al: Nevus density and melanoma risk in women: A pooled analysis
to test the divergent pathway hypothesis. Int J Cancer.
124:937–944. 2009. View Article : Google Scholar :
|
|
24
|
Dessinioti C, Antoniou C, Katsambas A and
Stratigos AJ: Melanocortin 1 receptor variants: Functional role and
pigmentary associations. Photochem Photobiol. 87:978–987. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Goldstein AM and Tucker MA: Genetic
epidemiology of cutaneous melanoma: A global perspective. Arch
Dermatol. 137:1493–1496. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Soura E, Eliades PJ, Shannon K, Stratigos
AJ and Tsao H: Hereditary melanoma: Update on syndromes and
management: Genetics of familial atypical multiple mole melanoma
syndrome. J Am Acad Dermatol. 74:395–407; quiz 408–410. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gruis NA, van der Velden PA, Sandkuijl LA,
Prins DE, Weaver-Feldhaus J, Kamb A, Bergman W and Frants RR:
Homozygotes for CDKN2 (p16) germline mutation in Dutch familial
melanoma kindreds. Nat Genet. 10:351–353. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zuo L, Weger J, Yang Q, Goldstein AM,
Tucker MA, Walker GJ, Hayward N and Dracopoli NC: Germline
mutations in the p16INK4a binding domain of CDK4 in familial
melanoma. Nat Genet. 12:97–99. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kanitakis J: Anatomy, histology and
immunohistochemistry of normal human skin. Eur J Dermatol.
12:390–399; quiz 400–401. 2002.PubMed/NCBI
|
|
30
|
Lin JY and Fisher DE: Melanocyte biology
and skin pigmentation. Nature. 445:843–850. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Curtin JA, Fridlyand J, Kageshita T, Patel
HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Bröcker EB, LeBoit PE, et
al: Distinct sets of genetic alterations in melanoma. N Engl J Med.
353:2135–2147. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bastian BC: The molecular pathology of
melanoma: An integrated taxonomy of melanocytic neoplasia. Annu Rev
Pathol. 9:239–271. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shain AH and Bastian BC: From melanocytes
to melanomas. Nat Rev Cancer. 16:345–358. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pollock PM, Harper UL, Hansen KS, Yudt LM,
Stark M, Robbins CM, Moses TY, Hostetter G, Wagner U, Kakareka J,
et al: High frequency of BRAF mutations in nevi. Nat Genet.
33:19–20. 2003. View
Article : Google Scholar
|
|
35
|
Leonardi GC, Accardi G, Monastero R,
Nicoletti F and Libra M: Ageing: From inflammation to cancer. Immun
Ageing. 15:12018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Speeckaert R, van Geel N, Vermaelen KV,
Lambert J, Van Gele M, Speeckaert MM and Brochez L: Immune
reactions in benign and malignant melanocytic lesions: Lessons for
immunotherapy. Pigment Cell Melanoma Res. 24:334–344. 2011.
View Article : Google Scholar
|
|
37
|
Gray-Schopfer V, Wellbrock C and Marais R:
Melanoma biology and new targeted therapy. Nature. 445:851–857.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Farmer ER, Gonin R and Hanna MP:
Discordance in the histopathologic diagnosis of melanoma and
melanocytic nevi between expert pathologists. Hum Pathol.
27:528–531. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chiriboga L, Meehan S, Osman I, Glick M,
de la Cruz G, Howell BS, Friedman-Jiménez G, Schneider RJ and Jamal
S: Endothelin-1 in the tumor microenvironment correlates with
melanoma invasion. Melanoma Res. 26:236–244. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Moro N, Mauch C and Zigrino P:
Metalloproteinases in melanoma. Eur J Cell Biol. 93:23–29. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sandri S, Faião-Flores F, Tiago M,
Pennacchi PC, Massaro RR, Alves-Fernandes DK, Berardinelli GN,
Evangelista AF, de Lima Vazquez V, Reis RM and Maria-Engler SS:
Vemurafenib resistance increases melanoma invasiveness and
modulates the tumor microenvironment by MMP-2 upregulation.
Pharmacol Res. 111:523–533. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Falzone L, Salemi R, Travali S, Scalisi A,
McCubrey JA, Candido S and Libra M: MMP-9 overexpression is
associated with intragenic hypermethylation of MMP9 gene in
melanoma. Aging (Albany NY). 8:933–944. 2016. View Article : Google Scholar
|
|
43
|
Lee KR, Lee JS, Kim YR, Song IG and Hong
EK: Polysaccharide from Inonotus obliquus inhibits migration and
invasion in B16-F10 cells by suppressing MMP-2 and MMP-9 via
downregulation of NF-κB signaling pathway. Oncol Rep. 31:2447–2453.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Guarneri C, Bevelacqua V, Polesel J,
Falzone L, Cannavò PS, Spandidos DA, Malaponte G and Libra M: NF-κB
inhibition is associated with OPN/MMP 9 downregulation in cutaneous
melanoma. Oncol Rep. 37:737–746. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA Jr and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Akbani R, Akdemir KC, Aksoy BA, Albert M,
Ally A, Amin SB, Arachchi H, Arora A, Auman JT, Ayala B, et al:
Cancer Genome Atlas Network: Genomic classification of cutaneous
melanoma. Cell. 161:1681–1696. 2015. View Article : Google Scholar
|
|
47
|
Hodis E, Watson IR, Kryukov GV, Arold ST,
Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C,
et al: A landscape of driver mutations in melanoma. Cell.
150:251–263. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Krauthammer M, Kong Y, Ha BH, Evans P,
Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, et
al: Exome sequencing identifies recurrent somatic RAC1 mutations in
melanoma. Nat Genet. 44:1006–1014. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chappell WH, Steelman LS, Long JM, Kempf
RC, Abrams SL, Franklin RA, Bäsecke J, Stivala F, Donia M, Fagone
P, et al: Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors:
Rationale and importance to inhibiting these pathways in human
health. Oncotarget. 2:135–164. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wellbrock C, Karasarides M and Marais R:
The RAF proteins take centre stage. Nat Rev Mol Cell Biol.
5:875–885. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Raman M, Chen W and Cobb MH: Differential
regulation and properties of MAPKs. Oncogene. 26:3100–3112. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Carlino MS, Long GV, Kefford RF and Rizos
H: Targeting oncogenic BRAF and aberrant MAPK activation in the
treatment of cutaneous melanoma. Crit Rev Oncol Hematol.
96:385–398. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yuan TL and Cantley LC: PI3K pathway
alterations in cancer: Variations on a theme. Oncogene.
27:5497–5510. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Davies MA: The role of the PI3K-AKT
pathway in melanoma. Cancer J. 18:142–147. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Cohen C, Zavala-Pompa A, Sequeira JH,
Shoji M, Sexton DG, Cotsonis G, Cerimele F, Govindarajan B, Macaron
N and Arbiser JL: Mitogen-actived protein kinase activation is an
early event in melanoma progression. Clin Cancer Res. 8:3728–3733.
2002.PubMed/NCBI
|
|
56
|
Wang YF, Jiang CC, Kiejda KA, Gillespie S,
Zhang XD and Hersey P: Apoptosis induction in human melanoma cells
by inhibition of MEK is caspase-independent and mediated by the
Bcl-2 family members PUMA, Bim, and Mcl-1. Clin Cancer Res.
13:4934–4942. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lovly CM, Dahlman KB, Fohn LE, Su Z,
Dias-Santagata D, Hicks DJ, Hucks D, Berry E, Terry C, Duke M, et
al: Routine multiplex mutational profiling of melanomas enables
enrollment in genotype-driven therapeutic trials. PLoS One.
7:e353092012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Rubinstein JC, Sznol M, Pavlick AC, Ariyan
S, Cheng E, Bacchiocchi A, Kluger HM, Narayan D and Halaban R:
Incidence of the V600K mutation among melanoma patients with BRAF
mutations, and potential therapeutic response to the specific BRAF
inhibitor PLX4032. J Transl Med. 8:672010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Davies H, Bignell GR, Cox C, Stephens P,
Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W,
et al: Mutations of the BRAF gene in human cancer. Nature.
417:949–954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wan PTC, Garnett MJ, Roe SM, Lee S,
Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ,
Barford D and Marais R; Cancer Genome Project: Mechanism of
activation of the RAF-ERK signaling pathway by oncogenic mutations
of B-RAF. Cell. 116:855–867. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Richtig G, Hoeller C, Kashofer K,
Aigelsreiter A, Heinemann A, Kwong LN, Pichler M and Richtig E:
Beyond the BRAFV600E hotspot: Biology and clinical
implications of rare BRAF gene mutations in melanoma patients. Br J
Dermatol. 177:936–944. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jakob JA, Bassett RL Jr, Ng CS, Curry JL,
Joseph RW, Alvarado GC, Rohlfs ML, Richard J, Gershenwald JE, Kim
KB, et al: NRAS mutation status is an independent prognostic factor
in metastatic melanoma. Cancer. 118:4014–4023. 2012. View Article : Google Scholar :
|
|
63
|
Giehl K: Oncogenic Ras in tumour
progression and metastasis. Biol Chem. 386:193–205. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Fedorenko IV, Gibney GT and Smalley KS:
NRAS mutant melanoma: Biological behavior and future strategies for
therapeutic management. Oncogene. 32:3009–3018. 2013. View Article : Google Scholar
|
|
65
|
Maertens O, Johnson B, Hollstein P,
Frederick DT, Cooper ZA, Messiaen L, Bronson RT, McMahon M, Granter
S, Flaherty K, et al: Elucidating distinct roles for NF1 in
melanomagenesis. Cancer Discov. 3:338–349. 2013. View Article : Google Scholar :
|
|
66
|
Whittaker SR, Theurillat JP, Van Allen E,
Wagle N, Hsiao J, Cowley GS, Schadendorf D, Root DE and Garraway
LA: A genome-scale RNA interference screen implicates NF1 loss in
resistance to RAF inhibition. Cancer Discov. 3:350–362. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Nissan MH, Pratilas CA, Jones AM, Ramirez
R, Won H, Liu C, Tiwari S, Kong L, Hanrahan AJ, Yao Z, et al: Loss
of NF1 in cutaneous melanoma is associated with RAS activation and
MEK dependence. Cancer Res. 74:2340–2350. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Krauthammer M, Kong Y, Bacchiocchi A,
Evans P, Pornputtapong N, Wu C, McCusker JP, Ma S, Cheng E, Straub
R, et al: Exome sequencing identifies recurrent mutations in NF1
and RASopathy genes in sun-exposed melanomas. Nat Genet.
47:996–1002. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Gibney GT and Smalley KS: An unholy
alliance: Cooperation between BRAF and NF1 in melanoma development
and BRAF inhibitor resistance. Cancer Discov. 3:260–263. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Beadling C, Jacobson-Dunlop E, Hodi FS, Le
C, Warrick A, Patterson J, Town A, Harlow A, Cruz F III, Azar S, et
al: KIT gene mutations and copy number in melanoma subtypes. Clin
Cancer Res. 14:6821–6828. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Handolias D, Salemi R, Murray W, Tan A,
Liu W, Viros A, Dobrovic A, Kelly J and McArthur GA: Mutations in
KIT occur at low frequency in melanomas arising from anatomical
sites associated with chronic and intermittent sun exposure.
Pigment Cell Melanoma Res. 23:210–215. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Shain AH, Yeh I, Kovalyshyn I, Sriharan A,
Talevich E, Gagnon A, Dummer R, North J, Pincus L, Ruben B, et al:
The genetic evolution of melanoma from precursor lesions. N Engl J
Med. 373:1926–1936. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Castellano M, Pollock PM, Walters MK,
Sparrow LE, Down LM, Gabrielli BG, Parsons PG and Hayward NK:
CDKN2A/16 is inactivated in most melanoma cell lines. Cancer Res.
57:4868–4875. 1997.PubMed/NCBI
|
|
74
|
Sharpless E and Chin L: The INK4a/ARF
locus and melanoma. Oncogene. 22:3092–3098. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wu H, Goel V and Haluska FG: PTEN
signaling pathways in melanoma. Oncogene. 22:3113–3122. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Mirmohammadsadegh A, Marini A, Nambiar S,
Hassan M, Tannapfel A, Ruzicka T and Hengge UR: Epigenetic
silencing of the PTEN gene in melanoma. Cancer Res. 66:6546–6552.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Stahl JM, Cheung M, Sharma A, Trivedi NR,
Shanmugam S and Robertson GP: Loss of PTEN promotes tumor
development in malignant melanoma. Cancer Res. 63:2881–2890.
2003.PubMed/NCBI
|
|
78
|
Tsao H, Goel V, Wu H, Yang G and Haluska
FG: Genetic interaction between NRAS and BRAF mutations and
PTEN/MMAC1 inactivation in melanoma. J Invest Dermatol.
122:337–341. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Nogueira C, Kim KH, Sung H, Paraiso KH,
Dannenberg JH, Bosenberg M, Chin L and Kim M: Cooperative
interactions of PTEN deficiency and RAS activation in melanoma
metastasis. Oncogene. 29:6222–6232. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Shi H, Hugo W, Kong X, Hong A, Koya RC,
Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al:
Acquired resistance and clonal evolution in melanoma during BRAF
inhibitor therapy. Cancer Discov. 4:80–93. 2014. View Article : Google Scholar :
|
|
81
|
Damsky WE, Curley DP, Santhanakrishnan M,
Rosenbaum LE, Platt JT, Gould Rothberg BE, Taketo MM, Dankort D,
Rimm DL, McMahon M and Bosenberg M: β-catenin signaling controls
metastasis in Braf-activated Pten-deficient melanomas. Cancer Cell.
20:741–754. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Rimm DL, Caca K, Hu G, Harrison FB and
Fearon ER: Frequent nuclear/cytoplasmic localization of β-catenin
without exon 3 mutations in malignant melanoma. Am J Pathol.
154:325–329. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Ross MI and Gershenwald JE: Evidence-based
treatment of early-stage melanoma. J Surg Oncol. 104:341–353. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Luke JJ, Flaherty KT, Ribas A and Long GV:
Targeted agents and immunotherapies: Optimizing outcomes in
melanoma. Nat Rev Clin Oncol. 14:463–482. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Hodi FS, O'Day SJ, McDermott DF, Weber RW,
Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel
JC, et al: Improved survival with ipilimumab in patients with
metastatic melanoma. N Engl J Med. 363:711–723. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Hauschild A, Grob JJ, Demidov LV, Jouary
T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr,
Kaempgen E, et al: Dabrafenib in BRAF-mutated metastatic melanoma:
A multicentre, open-label, phase 3 randomised controlled trial.
Lancet. 380:358–365. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
McArthur GA, Chapman PB, Robert C, Larkin
J, Haanen JB, Dummer R, Ribas A, Hogg D, Hamid O, Ascierto PA, et
al: Safety and efficacy of vemurafenib in BRAF(V600E) and
BRAF(V600K) mutation-positive melanoma (BRIM-3): Extended follow-up
of a phase 3, randomised, open-label study. Lancet Oncol.
15:323–332. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Robert C, Long GV, Brady B, Dutriaux C,
Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C,
Kalinka-Warzocha E, et al: Nivolumab in previously untreated
melanoma without BRAF mutation. N Engl J Med. 372:320–330. 2015.
View Article : Google Scholar
|
|
89
|
Robert C, Karaszewska B, Schachter J,
Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R,
Grange F, Mortier L, et al: Improved overall survival in melanoma
with combined dabrafenib and trametinib. N Engl J Med. 372:30–39.
2015. View Article : Google Scholar
|
|
90
|
Long GV, Stroyakovskiy D, Gogas H,
Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A,
Grob JJ, et al: Dabrafenib and trametinib versus dabrafenib and
placebo for Val600 BRAF-mutant melanoma: A multicentre,
double-blind, phase 3 randomised controlled trial. Lancet.
386:444–451. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Ribas A, Puzanov I, Dummer R, Schadendorf
D, Hamid O, Robert C, Hodi FS, Schachter J, Pavlick AC, Lewis KD,
et al: Pembrolizumab versus investigator-choice chemotherapy for
ipilimumab-refractory melanoma (KEYNOTE-002): A randomised,
controlled, phase 2 trial. Lancet Oncol. 16:908–918. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Weber JS, D'Angelo SP, Minor D, Hodi FS,
Gutzmer R, Neyns B, Hoeller C, Khushalani I, Miller WH Jr, Lao CD,
et al: Nivolumab versus chemotherapy in patients with advanced
melanoma who progressed after anti-CTLA-4 treatment (CheckMate
037): A randomised, controlled, open-label, phase 3 trial. Lancet
Oncol. 16:375–384. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Ascierto PA, McArthur GA, Dréno B,
Atkinson V, Liszkay G, Di Giacomo AM, Mandalà M, Demidov L,
Stroyakovskiy D, Thomas L, et al: Cobimetinib combined with
vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM):
Updated efficacy results from a randomised, double-blind, phase 3
trial. Lancet Oncol. 17:1248–1260. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Malas S, Harrasser M, Lacy KE and
Karagiannis SN: Antibody therapies for melanoma: New and emerging
opportunities to activate immunity (Review). Oncol Rep. 32:875–886.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Zhang Y, Song Y and Gao Q: Increased
survival time of a patient with metastatic malignant melanoma
following immunotherapy: A case report and literature review. Oncol
Lett. 10:883–886. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Akiyama Y, Nonomura C, Kondou R, Miyata H,
Ashizawa T, Maeda C, Mitsuya K, Hayashi N, Nakasu Y and Yamaguchi
K: Immunological effects of the anti-programmed death-1 antibody on
human peripheral blood mononuclear cells. Int J Oncol.
49:1099–1107. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Larkin J, Lao CD, Urba WJ, McDermott DF,
Horak C, Jiang J and Wolchok JD: Efficacy and safety of nivolumab
in patients with BRAF V600 mutant and BRAF wild-type advanced
melanoma: A pooled analysis of 4 clinical trials. JAMA Oncol.
1:433–440. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Mangana J, Cheng PF, Schindler K, Weide B,
Held U, Frauchiger AL, Romano E, Kähler KC, Rozati S, Rechsteiner
M, et al: Analysis of BRAF and NRAS mutation status in advanced
melanoma patients treated with anti-CTLA 4 antibodies: Association
with overall survival. PLoS One. 10:e01394382015. View Article : Google Scholar
|
|
99
|
Robert C, Schachter J, Long GV, Arance A,
Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al
KEYNOTE-006 investigators: Pembrolizumab versus ipilimumab in
advanced melanoma. N Engl J Med. 372:2521–2532. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Simeone E and Ascierto PA:
Immunomodulating antibodies in the treatment of metastatic
melanoma: The experience with anti-CTLA-4, anti-CD137, and
anti-PD1. J Immunotoxicol. 9:241–247. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Zimmer L, Apuri S, Eroglu Z, Kottschade
LA, Forschner A, Gutzmer R, Schlaak M, Heinzerling L, Krackhardt
AM, Loquai C, et al: Ipilimumab alone or in combination with
nivolumab after progression on anti-PD-1 therapy in advanced
melanoma. Eur J Cancer. 75:47–55. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
McCubrey JA, Steelman LS, Kempf CR,
Chappell WH, Abrams SL, Stivala F, Malaponte G, Nicoletti F, Libra
M, Bäsecke J, et al: Therapeutic resistance resulting from
mutations in Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR signaling pathways.
J Cell Physiol. 226:2762–2781. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Steelman LS, Chappell WH, Abrams SL, Kempf
RC, Long J, Laidler P, Mijatovic S, Maksimovic-Ivanic D, Stivala F,
Mazzarino MC, et al: Roles of the Raf/MEK/ERK and
PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity
to therapy-implications for cancer and aging. Aging (Albany NY).
3:192–222. 2011. View Article : Google Scholar
|
|
104
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Franklin RA, Montalto G, Cervello M, Libra M, Candido S,
Malaponte G, et al: Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade
inhibitors: How mutations can result in therapy resistance and how
to overcome resistance. Oncotarget. 3:1068–1111. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Van Allen EM, Wagle N, Sucker A, Treacy
DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker
S, Kryukov GV, et al Dermatologic Cooperative Oncology Group of
Germany (DeCOG): The genetic landscape of clinical resistance to
RAF inhibition in metastatic melanoma. Cancer Discov. 4:94–109.
2014. View Article : Google Scholar :
|
|
106
|
Hugo W, Shi H, Sun L, Piva M, Song C, Kong
X, Moriceau G, Hong A, Dahlman KB, Johnson DB, et al: Non-genomic
and immune evolution of melanoma acquiring MAPKi resistance. Cell.
162:1271–1285. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Gray ES, Rizos H, Reid AL, Boyd SC,
Pereira MR, Lo J, Tembe V, Freeman J, Lee JH, Scolyer RA, et al:
Circulating tumor DNA to monitor treatment response and detect
acquired resistance in patients with metastatic melanoma.
Oncotarget. 6:42008–42018. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Santiago-Walker A, Gagnon R, Mazumdar J,
Casey M, Long GV, Schadendorf D, Flaherty K, Kefford R, Hauschild
A, Hwu P, et al: Correlation of BRAF Mutation status in
circulating-free dna and tumor and association with clinical
outcome across four BRAFi and MEKi clinical trials. Clin Cancer
Res. 22:567–574. 2016. View Article : Google Scholar
|
|
109
|
Girotti MR, Gremel G, Lee R, Galvani E,
Rothwell D, Viros A, Mandal AK, Lim KH, Saturno G, Furney SJ, et
al: Application of sequencing, liquid biopsies, and patient-derived
xenografts for personalized medicine in melanoma. Cancer Discov.
6:286–299. 2016. View Article : Google Scholar
|
|
110
|
Spranger S, Spaapen RM, Zha Y, Williams J,
Meng Y, Ha TT and Gajewski TF: Up-regulation of PD-L1, IDO, and
T(regs) in the melanoma tumor microenvironment is driven by CD8(+)
T cells. Sci Transl Med. 5:200ra1162013. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Donia M, Harbst K, van Buuren M, Kvistborg
P, Lindberg MF, Andersen R, Idorn M, Munir Ahmad S, Ellebæk E,
Mueller A, et al: Acquired immune resistance follows complete tumor
regression without loss of target antigens or IFNγ signaling.
Cancer Res. 77:4562–4566. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Spranger S, Bao R and Gajewski TF:
Melanoma-intrinsic β-catenin signalling prevents anti-tumour
immunity. Nature. 523:231–235. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Peng W, Chen JQ, Liu C, Malu S, Creasy C,
Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, et al: Loss of
PTEN promotes resistance to T cell-mediated immunotherapy. Cancer
Disco. 6:202–216. 2016. View Article : Google Scholar
|
|
114
|
Zaretsky JM, Garcia-Diaz A, Shin DS,
Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY,
Abril-Rodriguez G, Sandoval S, Barthly L, et al: Mutations
associated with acquired resistance to PD 1 blockade in melanoma. N
Engl J Med. 375:819–829. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Banna GL, Torino F, Marletta F, Santagati
M, Salemi R, Cannarozzo E, Falzone L, Ferraù F and Libra M:
Lactobacillus rhamnosus GG: An overview to explore the rationale of
its use in cancer. Front Pharmacol. 1(8): 6032017. View Article : Google Scholar
|
|
116
|
Gopalakrishnan V, Spencer CN, Nezi L,
Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman
K, Wei SC, et al: Gut microbiome modulates response to anti-PD-1
immunotherapy in melanoma patients. Science. 359:97–103. 2018.
View Article : Google Scholar
|
|
117
|
Routy B, Le Chatelier E, Derosa L, Duong
CPM, Alou MT, Daillère R, Fluckiger A, Messaoudene M, Rauber C,
Roberti MP, et al: Gut microbiome influences efficacy of PD-1-based
immunotherapy against epithelial tumors. Science. 359:91–97. 2018.
View Article : Google Scholar
|
|
118
|
Pappalardo F, Russo G, Candido S, Pennisi
M, Cavalieri S, Motta S, McCubrey JA, Nicoletti F and Libra M:
Computational modeling of PI3K/AKT and MAPK signaling pathways in
melanoma Cancer. PLoS One. 11:e01521042016. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Rambow F, Job B, Petit V, Gesbert F,
Delmas V, Seberg H, Meurice G, Van Otterloo E, Dessen P, Robert C,
et al: New functional signatures for understanding melanoma biology
from tumor cell lineage-specific analysis. Cell Reports.
13:840–853. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Pennisi M, Russo G, Di Salvatore V,
Candido S, Libra M and Pappalardo F: Computational modeling in
melanoma for novel drug discovery. Expert Opin Drug Discov.
11:609–621. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Chapman PB, Hauschild A, Robert C, Haanen
JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et
al BRIM-3 Study Group: Improved survival with vemurafenib in
melanoma with BRAF V600E mutation. N Engl J Med. 364:2507–2516.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Robert C, Thomas L, Bondarenko I, O'Day S,
Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, et al:
Ipilimumab plus dacarbazine for previously untreated metastatic
melanoma. N Engl J Med. 364:2517–2526. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Wolchok JD, Chiarion-Sileni V, Gonzalez R,
Rutkowski P, Grob JJ, Cowey CL, Lao CD, Wagstaff J, Schadendorf D,
Ferrucci PF, et al: Overall survival with combined nivolumab and
ipilimumab in advanced melanoma. N Engl J Med. 377:1345–1356. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Schachter J, Ribas A, Long GV, Arance A,
Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al:
Pembrolizumab versus ipilimumab for advanced melanoma: Final
overall survival results of a multicentre, randomised, open-label
phase 3 study (KEYNOTE-006). Lancet. 390:1853–1862. 2017.
View Article : Google Scholar : PubMed/NCBI
|