Introduction
Osteosarcoma is the most common form of primary
malignant bone tumour (1). The
annual incidence estimated at 2–5/100,000 individuals is
distributed in two peaks regarding the frequency, emerging around
the second and seventh decade of life (2). Typical symptoms are swelling, pain
and hyperthermia at the affected locus, while in advanced stages
pathological fractures occur (3).
Here, predominantly the metaphyses of long bones, especially at the
knee joints in distal femur and proximal tibia are affected
(4). Currently, modern imaging
techniques including computer tomography, magnet resonance and
positron emission tomography are used for diagnosis and staging.
Invasive biopsy and subsequent histological analyses are used for
confirming the diagnosis (5).
Approximately 85% of tumours are classified as 'highly malignant'
and predominantly show early hematogenous pulmonary metastases
(1,6). Still, 80–90% are thought to present
with occult metastases, which cannot be detected with current
technology.
The therapeutic strategy for osteosarcoma consists
of neoadjuvant systemic polychemotherapy with e.g. doxorubicin,
cisplatin, ifosfamide and methotrexate for 10 weeks before surgical
treatment (7). Systemic therapy is
maintained for up to 30 weeks after surgery. Thereafter, ~70% of
patients are cured (8). The
sensitivity of osteosarcoma for radiation therapy is rather
limited; however it still remains a treatment option for palliative
care (9). In Europe, the
immunomodulator mifamurtide or liposomal muramyl tripeptide
phosphatidyl ethanolamine (L-MTP-PE) (Mepact®) is
available for the treatment of high-grade, non-metastatic
osteosarcoma, while it does not display significant effects in the
case that the tumour has already spread (10,11).
Nevertheless, systemic cancer therapy is a great burden to the
body. Apart from osteosarcoma itself, side effects of chemotherapy
such as myelosuppression or kidney failure, which can be induced by
e.g. cisplatin/ifosfamide, or cardiac failure caused by
anthracyclines e.g. doxorubicin, account for the second most common
cause of death (12,13). Thus, there is a need for
alternative or additive therapeutic strategies, which are less
harmful to the body.
Statins are administered for cholesterol lowering
and in cardiovascular disease due to inhibition of the HMG-CoA
reductase and the mevalonate pathway. However, they have been
reported to exert anti-invasive, proapoptotic and
antiprolifera-tive properties in numerous in vivo and in
vitro studies (14). A recent
meta-analysis with 7,813 cases demonstrated that statin therapy
after cancer diagnosis reduced the hazard of death by 21% (15). Another meta-analysis with more than
one million patients demonstrated that among post diagnosis, statin
users had a significantly higher relapse-free survival (16). In numerous human cancer entities
e.g. lung, prostate, colon, breast, liver or renal cancer,
lipophilic simvastatin caused therapeutic benefits (14,17–20).
An impact on small G proteins and/or suppressed prenylation of key
proteins, which regulate the cell cycle and apoptosis (e.g. Rho
kinases) are discussed as possible underlying mechanism of the mode
of action of statins (20,21). However, the molecular mechanism of
simvastatin on osteosarcoma progression has not yet been
clarified.
Given the widespread use of statins and the limited
systemic treatment options for osteosarcoma, the aim of this study
was to evaluate the osteosarcoma cell growth and apoptosis, and
underlying mechanisms, as well as the involved regulation of small
G proteins in optional simvastatin therapy.
Materials and methods
Cell lines and cell cultures
Human osteosarcoma cell lines SaOS-2 and U2OS were
purchased from Cell Line Services (Eppenheim, Germany). Tumour
cells were cultured at 37°C under 5% CO2 in RPMI-1640
medium (Seromed, Berlin, Germany) supplemented with 10%
heat-inactivated fetal calf serum (FCS), 100 IU/ml penicillin and
100 µg/ml streptomycin (Gibco, Karlsruhe, Germany) and 20 mM
HEPES buffer (Sigma, Steinheim, Germany). The culture media were
changed every 2 or 3 days.
Drug treatment
Simvastatin was obtained from Calbiochem (Darmstadt,
Germany) and activated prior to the experiments by alkaline
hydrolysis of the lactone moiety according to the manufacturer's
protocol. Tumour cells were treated for 48 and 72 h with
simvastatin (4–66 µM) or with vehicle (ctrl) with fresh
changes of culture medium and simvastatin after 48 h. Additionally,
mevalonate (160 µM), farnesyl pyrophosphate (FPP, 16
µM) and geranylgeranyl pyrophosphate (GGPP, 16 µM,
all from Sigma) were added to the medium containing simvastatin to
address simvastatin's site of action along the mevalonate
pathway.
Tumour cell growth
MTT kit I
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Roche Diagnostics, Penzberg, Germany) was used to evaluate
MTT-reducing activity of the cellular mitochondria. SaOS-2 and U2OS
cells (100 µl, 1×104/ml) were seeded onto 96-well
tissue culture plates and incubated with simvastatin or vehicle as
described above to monitor time- and dose-response. Twenty-four
hours before the evaluation of each time-point, MTT (0.5 mg/ml) was
added to each well, and the cells were incubated for 4 h at 37°C.
Thereafter, cells were lysed in a solubilisation solution
containing 10% sodium dodecyl sulphate (SDS) in 0.01 M hydrogen
chloride (HCl) and incubated overnight at 37°C in 5%
CO2. The following day, the absorbance of each well was
measured with a multimode microplate reader (Infinite M200; Tecan,
Crailsheim, Germany) at 550 nm. Each experiment was performed in
triplicate. After subtracting the background absorbance, the
results were expressed as mean cell number. For better
comparability of cell growth, the control group was set to 100% and
other groups were measured in relation to the control.
Cell cycle analysis
SaOS-2 and U2OS cells were grown to 70% confluency,
and then treated with simvastatin or vehicle (ctrl). Cell cycle
analyses were carried out after 48 or 72 h. Tumour cells were
stained with propidium iodide (PI) using a Cycle Test Plus DNA
Reagent kit and then subjected to flow cytometry with a FACScan
flow cytometer (both from BD Biosciences, Heidelberg, Germany).
From each sample, 10,000 cells were measured. Data acquisition was
carried out using CellQuest software and cell cycle distribution
calculated using ModFit software (BD Biosciences). The number of
gated cells in G1, G2/M or S phase are presented as a percentage
(%). Each experiment was performed in triplicate.
Assessment of apoptosis
After treatment with simvastatin or vehicle (ctrl),
SaOS-2 and U2OS cells were incubated with PI and Annexin
V-conjugated fluorescein isothiocyanate (FITC) from an Annexin
V-FITC Apoptosis Detection kit 1 (BD Biosciences) according to the
manufacturer's instructions. Annexin V-FITC/PI binding was
evaluated by flow cytometry with a FACScan flow cytometer
(excitation wavelength 488 nm, emission wavelength 530 nm; BD
Biosciences). The population of PI-negative/Annexin V-positive
cells was related to early apoptosis, and that of
PI-positive/Annexin V-positive cells was related to late
apoptosis/secondary necrosis.
Ribonucleic acid (RNA) isolation,
quantitative reverse-transcription-polymerase chain reaction
(RT-PCR)
Total RNA was isolated using the RNeasy system
(Qiagen, Hilden, Germany) according to the manufacturer's
instructions. The quality as well as the amount of the isolated RNA
were determined photometrically using the NanoDrop ND-1000 device
(NanoDrop Technologies, Wilmington, DE, USA). Afterwards, RNA was
used for qRT-PCR. Here, RNA was reversely transcribed using the
Affinity Script qPCR-cDNA synthesis kit (Stratagene, La Jolla, CA,
USA) following the manufacturer's instructions. To determine the
mRNA expression of TP53, Cdk1 (CDC 2), CDKN1A, Bax
and Bcl-2, qRT-PCR was carried out on a Stratagene Mx3005p qPCR
system (Stratagene) using gene-specific primers for human TP53 (NM
000546, UniGene#: Hs.437460), human Cdk1 (NM 001786, UniGene#:
Hs.732435), human CDKN1A (NM 000389, UniGene#: Hs.370771), human
Bax (NM 004324, UniGene#: Hs.624291) and human Bcl-2 (NM 000633,
UniGene#: Hs.150749) purchased from SABiosciences (SuperArray,
Frederick, MD, USA). The expression of GAPDH with human
GAPDH (NM 002046, UniGene#: Hs.592355; SABiosciences) was measured
as a reference gene. qRT-PCR reaction was setup with 1X
RT2 SYBR-Green/Rox qPCR Master Mix (SABiosciences) in a
25 µl volume according to the manufacturer's instructions. A
two-step amplification protocol consisting of initial denaturation
at 95°C for 10 min followed by 40 cycles with a 15-sec denaturation
at 95°C and a 60-sec annealing/extension at 60°C was chosen. The
amount of target mRNA in each sample was normalized to the amount
of GAPDH mRNA. The relative mRNA expression of target genes
after normalization to GAPDH is shown.
Western blot analysis
Total content of cleaved caspase-3 and cleaved PARP,
p-JNK, p-c-Jun and c-Jun in SaOS-2 and U2OS cells was evaluated by
western blot analysis using anti-cleaved caspase-3 (clone Asp175,
5A1E), anti-cleaved PARP (clone Asp214, D64E10), anti-p-JNK (clone
Thr183/Tyr185), anti-human p-c-Jun (clone Ser63, 54B3), anti-human
Jun (clone 60A8) (Cell Signaling Technologies, Cambridge, UK).
Tumour cell lysates (50 µg protein) were separated by
electrophoresis on 12% polyacrylamide SDS gels and transferred to
nitrocellulose membranes (Amersham-Buchler, Braunschweig, Germany).
Determination of β-actin with anti-β-actin antibody (clone AC-15;
Sigma, Taufkirchen, Germany) served as the loading control. Blots
were blocked (10% non-fat dry milk in 1 mM Tris, 150 mM NaCl, pH
7.4) for 1 h, and then incubated for 1 h at room temperature (RT)
with the primary antibody (diluted according to the manufacturer's
instructions in blocking buffer with 0.5% Tween-20 and 0.5% bovine
serum albumin), and then incubated for another hour with
horseradish peroxidase-conjugated secondary antibody
(HRP-conjugated goat anti-mouse or goat anti-rabbit IgG; cat. no.
12-349 or cat. no. 12-348; Upstate Biotechnology, Lake Placid, NY,
USA) diluted 1:5,000 in blocking buffer with supplements at RT.
Proteins were detected with ECL™ western blot detection reagents
(GE Healthcare, Munich, Germany), visualized, digitized, and the
integrated density of individual bands was determined using the
software Multianalyst (Bio-Rad Laboratories, Inc., Munich,
Germany). By densitometric measurements using the same software the
amount of protein expression was normalized to β-actin.
Statistical analysis
All experiments were performed 3–6 times.
Differences between groups were determined by Wilcoxon-Mann-Whitney
U-test. A P-value of <0.05 was considered significant. Data are
expressed as mean ± standard error of the mean (SEM).
Results
Dose-response analysis of simvastatin in
response to U2OS and SaOS-2 cells
Simvastatin was applied in doses of 0, 4, 8, 16, 33
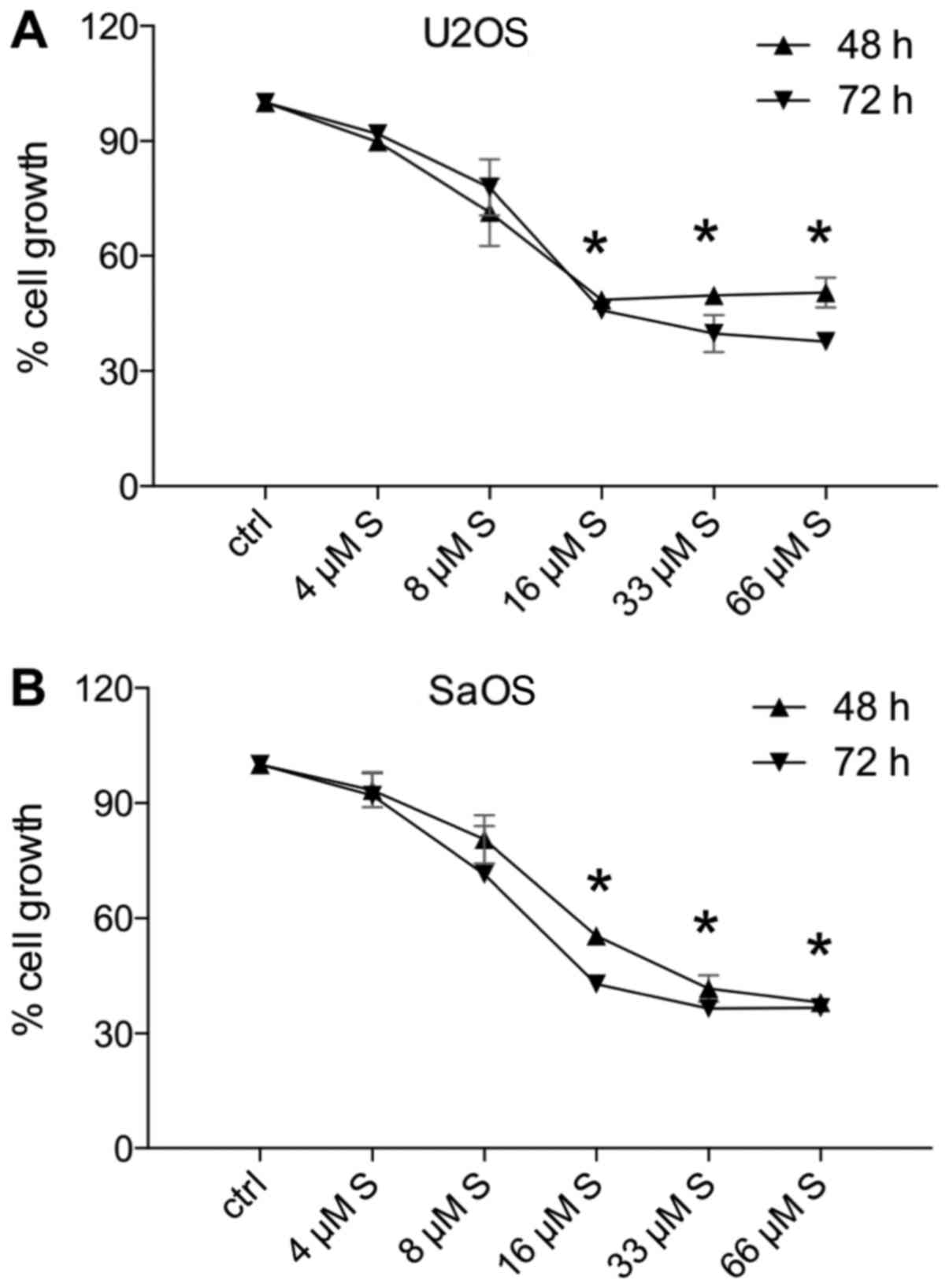
and 66 µM for 48 or 72 h. Both cell lines U2OS (Fig. 1A) and SaOS-2 (Fig. 1B) responded to low-dose simvastatin
of 8 µM; however, this growth reduction was not significant.
Fig. 1 reveals that U2OS (Fig. 1A) and SaOS-2 (Fig. 1B) showed a significant reduction in
cell growth following treatment with 16, 33 or 66 µM
simvastatin for either 48 or 72 h when compared to the control
cells (P<0.05 vs. 100% control) (Fig. 1). Treatment for 48 or 72 h,
respectively, evoked similar results with regard to cell growth.
Therefore, for the following analyses, a dose of 0, 16 or 33
µM simvastatin and both incubation periods (48 and 72 h,
respectively) were applied.
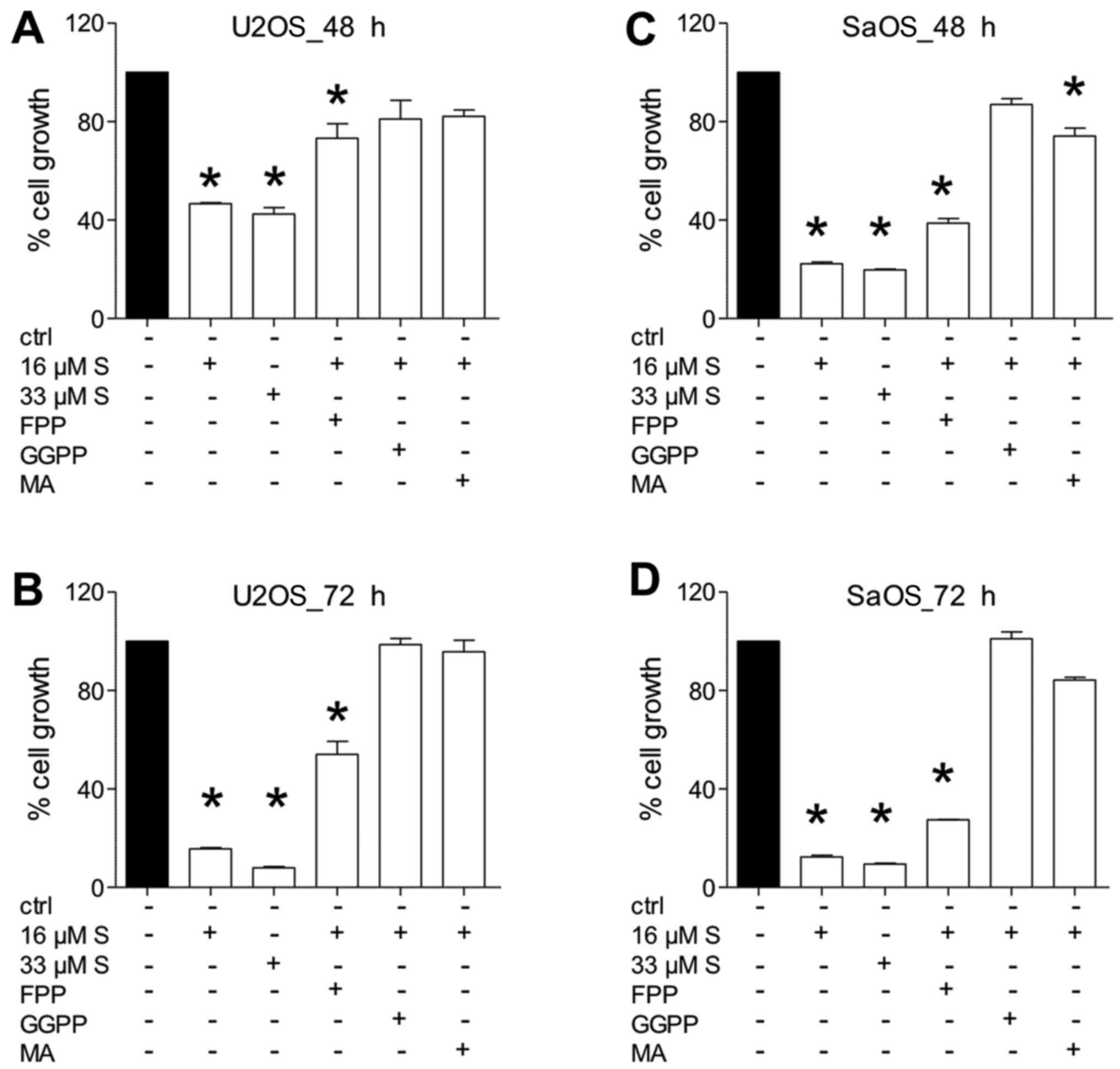
Cell growth inhibition potential of
simvastatin
Treatment of U2OS (Fig.
2A and B) or SaOS-2 cells (Fig. 2C
and D) with either 16 or 33 µM simvastatin confirmed the
growth-suppressive data, which are shown in Fig. 1. Treatment with doses of 16 or 33
µM reduced cell growth of U2OS cells to below 50%, and in
SaOS-2 to below 30% compared to 100% of controls (P<0.05)
(Fig. 2). After 72 h, viable cells
exposed to a high dose of simvastatin reached a growth of 7% in
U2OS and 10% in SaOS-2 cells (P<0.05) (Fig. 2B and D).
Substituting FFP to simvastatin-treated cells showed
a trend of increasing cell growth in all experiments. However, the
growth of U2OS (Fig. 2A and B) as
well as SaOS-2 cells (Fig. 2C and
D) was still significantly reduced when compared to the
untreated controls (P<0.05) (Fig.
2).
Addition of GGPP to simvastatin-treated cells
resulted in increased cell growth in all experiments, which was not
significantly different compared to the untreated controls either
after 48 or 72 h, respectively (P<0.05) (Fig. 2).
Addition of MA to simvastatin-treated cells showed
increased cell growth in U2OS cells, which was comparable to the
untreated controls (Fig. 2A and
B). Treatment of SaOS-2 cells with MA reduced cell growth when
compared to the untreated controls (Fig. 2C and D); however this difference
was significant only after 48 h of treatment (P<0.05) (Fig. 2C).
Nonetheless, the cell growth was consistently
increased in all add-back experiments as compared with cells
treated with simvastatin alone.
Simvastatin induces cell cycle
arrest
To elucidate the underlying mechanism of
simvastatin-reduced tumour cell growth, the impact of simvastatin
on cell cycle distribution was analysed by flow cytometry (Fig. 3A–D). Additionally, the gene
expression of relevant cell cycle proteins TP53, CDKN1A and CDC2
(CDK1) (Fig. 3E–G) was examined
via qRT-PCR.
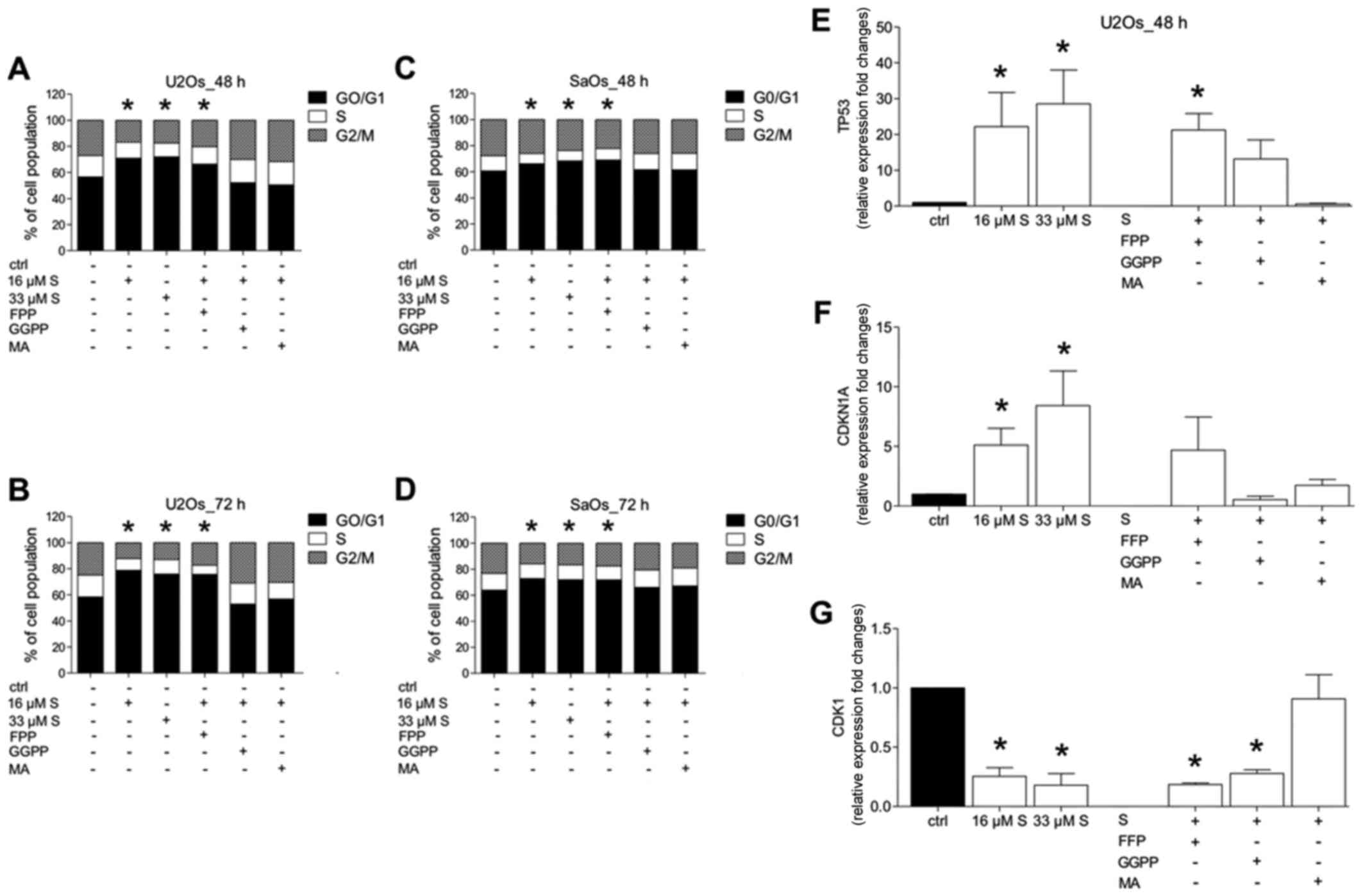 | Figure 3Effects of the mevalonate pathway on
the cell cycle of osteosarcoma cells. U2OS (A and B) and SaOS-2 (C
and D) cells were treated with simvastatin (S, 16 or 33 µM)
or vehicle (ctrl) for 48 or 72 h, and add-back experiments with
farnesyl pyrophosphate (FPP, 16 µM), geranylgeranyl
pyrophosphate (GGPP, 16 µM) and mevalonate (MA, 160
µM) were carried out. Distribution of cell cycle fractions
(G0/G1, cell cycle arrest; S, synthesis phase; and G2/M,
proliferation or mitosis phase) was determined using propidium
iodide staining. Percentages of populations in each cell cycle
phase were averaged from three independent experiments (G0/G1 cell
cycle arrest, *P<0.05 vs. ctrl). (E–G) U2OS cells
were treated as indicated. Gene expression of TP53 (E), CDKN1A (F)
and CDK1 (G) after 48 h was assessed. Data are expressed as mean ±
SEM; *P<0.05 vs. ctrl. |
Following treatment of U2OS and SaOS-2 cells with
simvastatin at each dose, a significant G0/G1 phase arrest was
induced compared to the untreated control cells after 48 as well as
72 h, respectively (P<0.05) (Fig.
3A–D). Simvastatin reduced markedly both, the S phase as well
as the G2/M phase concomitant to G0/G1 phase induction (Fig. 3A–D).
Supplementation of FFP to simvastatin-treated cells
did not markedly influence the cell cycle distribution as compared
to that noted in the simvastatin-treated only cells, and was
significant compared to the untreated controls (P<0.05)
(Fig. 3A–D).
However, addition of GGPP or MA to
simvastatin-treated cells resulted in a cell cycle distribution,
which did not significantly differ from that of the untreated
controls (Fig. 3A–D).
To confirm the G0/G1 phase arrest, gene expression
of tumour-suppressor protein TP53, which is known to induce cell
cycle arrest or apoptosis, the cyclin-dependent kinase inhibitor
CDKN1A, which is known to function as a regulator of cell cycle
progression at G1, and cyclin-dependent kinase CDK1, which is
responsible for cell cycle progression, were assessed. The gene
expression in untreated control cells was set as a reference.
Simvastatin induced a significant increase in TP53
gene expression of up to 20-fold compared to that observed in the
untreated control cells (P<0.05) (Fig. 3E). In cells treated with FFP and
simvastatin, TP53 was significantly increased compared to controls
and to levels that were observed in the simvastatin-treated only
cells (P<0.05). Substitution with GGPP or MA, respectively, did
not induce significant changes in TP53 gene expression compared to
the untreated controls (Fig.
3E).
Both doses of simvastatin, 16 and 33 µM,
induced a significant increase in CDKN1A gene expression compared
to that noted in the untreated control cells (P<0.05) (Fig. 3F). Cells treated with either FFP,
GGPP or MA and simvastatin did not induce significant changes in
CDKN1A gene expression compared to that observed in the untreated
controls (Fig. 3F).
Simvastatin significantly decreased CDK1 gene
expression compared to that noted in the untreated control cells
(P<0.05) (Fig. 3G). In cells
treated with FFP or GGPP and simvastatin, respectively, CDK1 was
significantly decreased compared to the controls, and similar to
levels that were observed in the simvastatin-treated only cells
(P<0.05) (Fig. 3G).
Substitution with MA did not significantly change CDK1 gene
expression compared to the untreated controls (Fig. 3G).
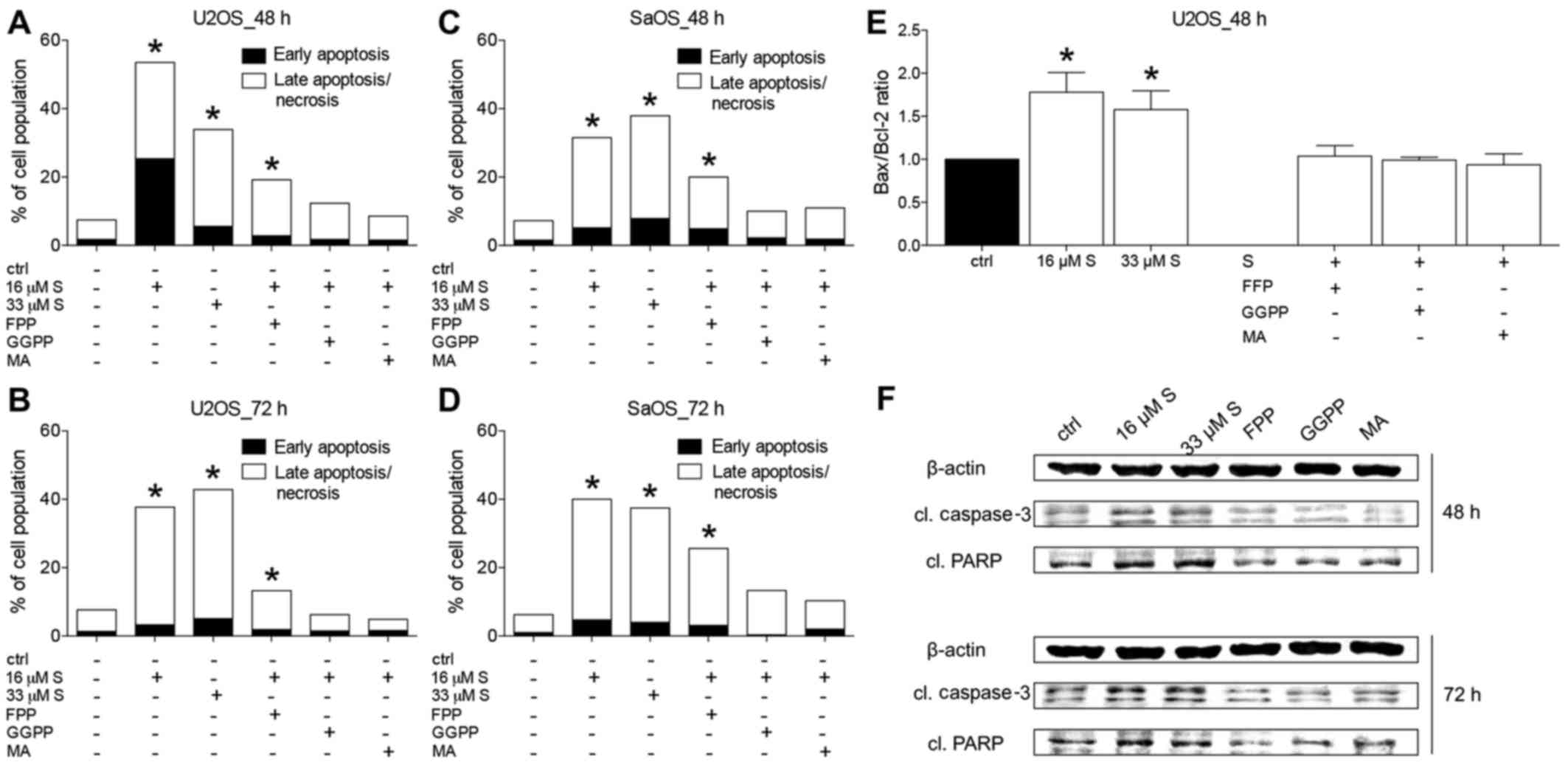
Induction of apoptosis in U2OS and SaO2
cells by simvastatin
To study the impact of simvastatin on the survival
of U2OS and SaOS-2 cells, respectively, flow cytometric analyses,
gene expression analyses of pro-apoptotic Bax and anti-apoptotic
Bcl-2 as well as western blot analysis for apoptosis inducers
cleaved caspase-3 (cl. caspase-3) or cleaved
poly(ADP-ribose)-Polymerase (cl. PARP) were performed.
In U2OS and SaOS-2 cells, early and late
apoptosis/necrosis were significantly increased by simvastatin
treatment (16 and 33 µM, respectively) at 48 or 72 h,
compared to the untreated controls (P<0.05) (Fig. 4A–D). Similarly, in all experiments,
substituting FFP significantly increased apoptosis compared to that
noted in the untreated controls (P<0.05). Treatment with GGPP or
MA did not significantly alter the apoptosis rates when compared to
the untreated controls (Fig.
4A–D).
The ratio of the gene expression of pro-apoptotic
Bax to anti-apoptotic Bcl-2 has been established as a reliable
marker for the regulation of cell survival. Examination of the
Bax/Bcl-2 ratio demonstrated U2OS cells to have a significantly
increased Bax/Bcl-2 ratio after their exposure to simvastatin for
48 h compared to the controls (P<0.05) (Fig. 4E). This effect was not
significantly altered by supplementing FFP, GGPP or MA to
simvastatin (Fig. 4E).
Caspase-3 and PARP are crucial to p53-mediated
apoptosis. As a zymogen, caspase-3 becomes activated by its
cleavage to cleaved caspase-3, and can in turn process PARP to
cleaved PARP. Cleaved PARP cannot fulfill its function on DNA
repair and is thus inactive. Both act as surrogate markers for
apoptosis. Treating U2OS cells with simvastatin for either 48 or 72
h induced a strong cleavage of both, caspase-3 as well as PARP
compared to the control (Fig. 4F).
Addition of either FFP, GGPP or MA to simvastatin-treated cells
reduced the cleavage of caspase-3 and PARP, respectively, and the
protein levels were comparable to the untreated controls (Fig. 4F).
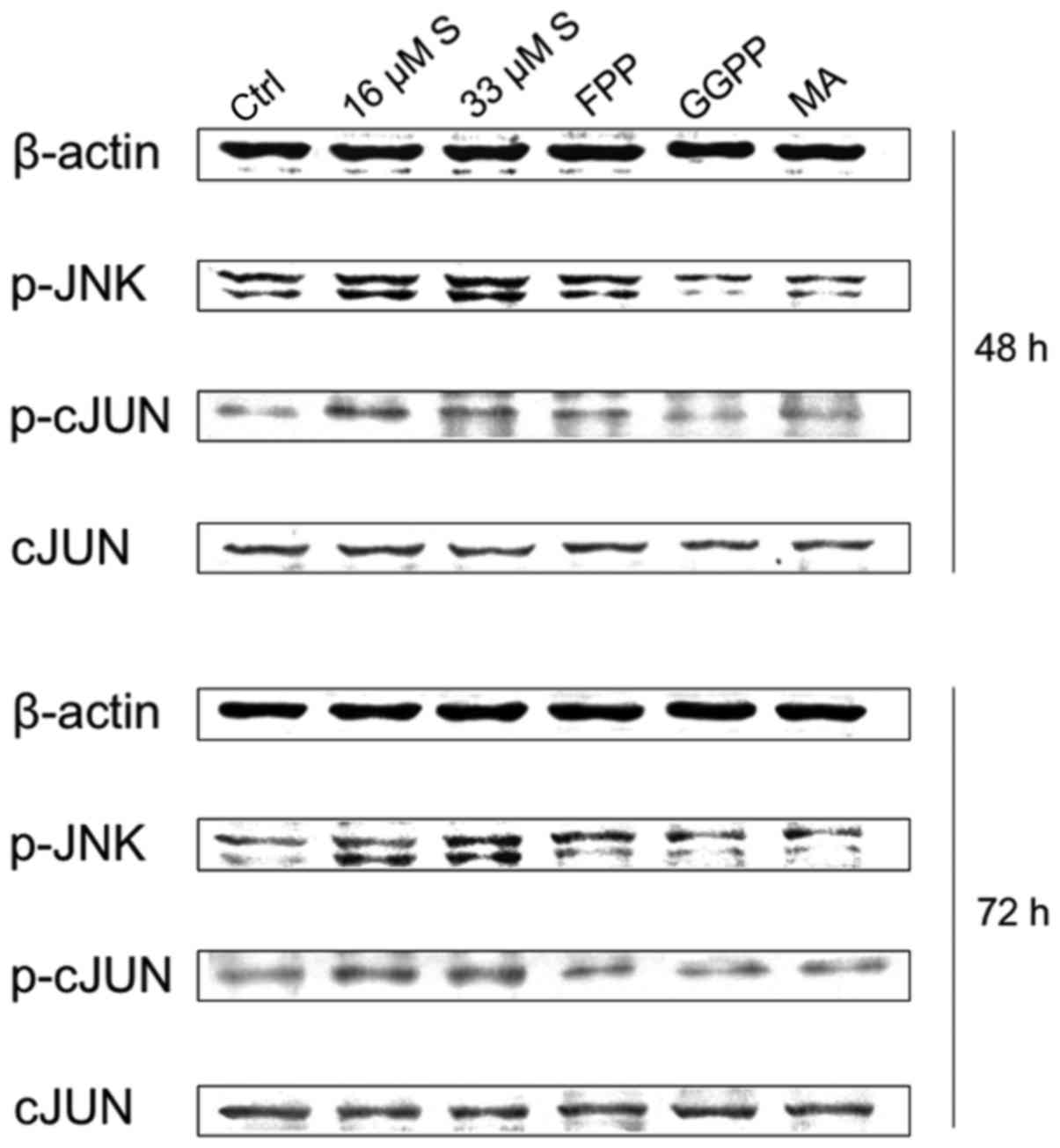
Activation of JNK and c-Jun
To further examine the mechanisms of the anticancer
mode of action of simvastatin, protein expression levels of
activated/phosphorylated JNK (p-JNK) and c-Jun as well as
activated/phosphorylated c-Jun (p-c-Jun) were analysed by western
blot analysis. Simvastatin treatment at both doses induced a strong
increase in the expression of p-JNK and p-c-Jun compared to levels
in the untreated control groups (representative figure of U2OS is
shown) (Fig. 5). This effect could
be partly reversed by addition of FFP, and nearly completely
abolished by substitution of either GGPP or MA after 48 as well as
72 h. In Fig. 6. we demonstrate a
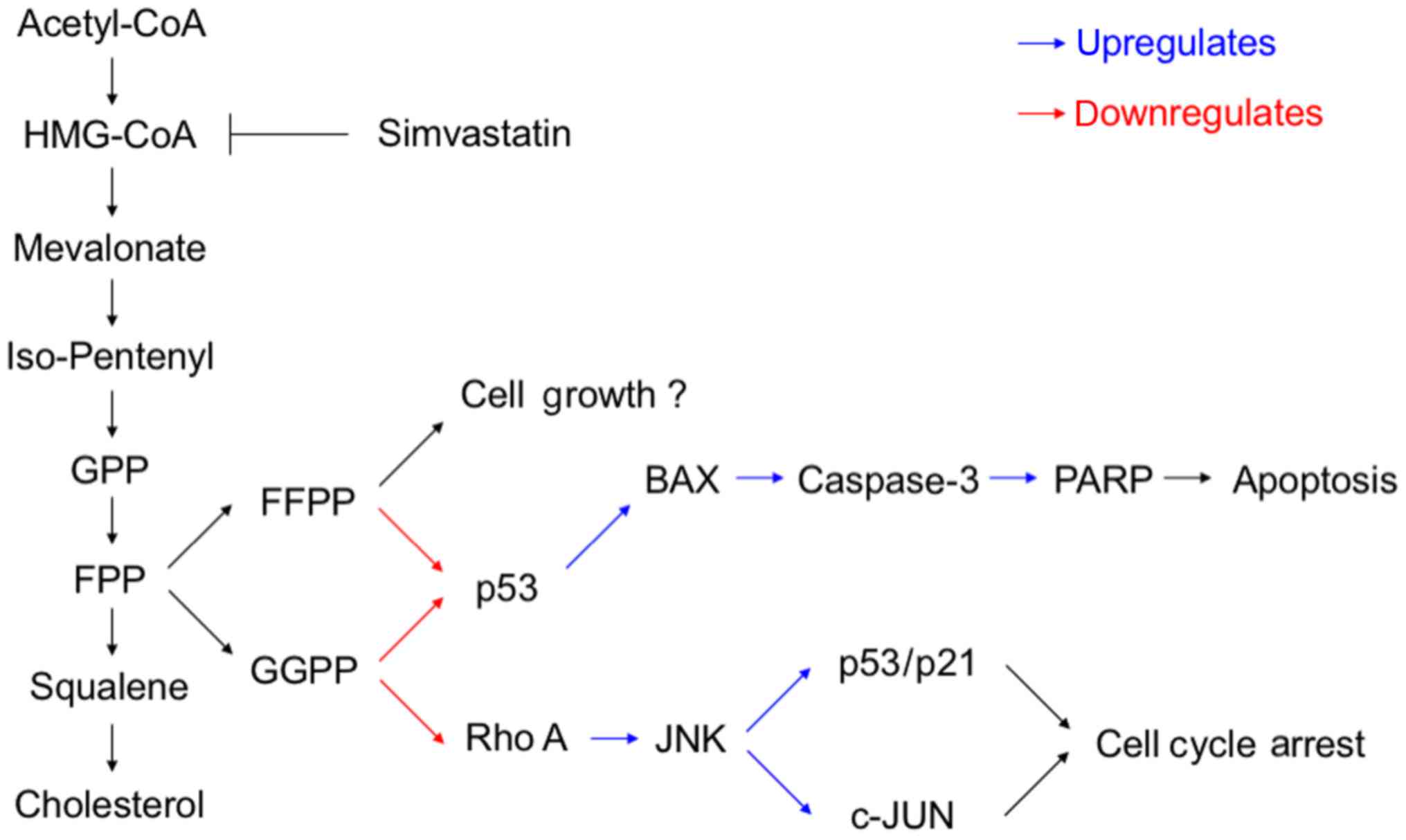
possible molecular mechanism.
Discussion
The present study shows that simvastatin, a potent
HMG-CoA reductase inhibitor, has the potential to dose- and
time-dependently inhibit growth and induce apoptosis in
osteosarcoma cells. The proposed underlying mechanism of
simvastatin's mode of action is via JNK and c-Jun.
Statins, including simvastatin, inhibit HMG-CoA
reductase via the mevalonate pathway, and are commonly used for
cholesterol-lowering in cardiovascular disease (22,23).
They exert numerous pleiotropic effects including modulation of
inflammation, angiogenesis, neurodegeneration and cancer
metastasis, as shown in vitro and in vivo (20). With regard to the mevalonate
pathway, the downstream products FPP and GGPP are essential for the
prenylation of proteins (24).
Many oncogenes from the Ras family, such as Rho A, are partially
dependent on post-translational prenylation to be active in cell
signaling (24). Cancer cells with
their upregulated metabolism imply prenylation of Ras proteins for
cell growth, cell cycle and cell signaling. Therefore, inhibiting
the mevalonate pathway effectively reduced cancer growth in
numerous malignancies including melanoma, glioma, hepatocellular
carcinoma and breast cancer among many others (14,20).
The proliferative ability of malignant cells is a
major property of sarcoma cells and imperative for cancer
assessment (25). The potential of
simvastatin to inhibit cell growth of osteosarcoma cells has been
controversially discussed. While simvastatin's protective effects
for human osteosarcoma cells by reducing oxidative stress and
apoptosis have been reported (26), Fromigué et al demonstrated
the apoptosis-inducing effects in human osteosarcoma cells as well
as suppression of proliferation (27). The latter did, however, not
investigate the cell cycle or underlying mechanism addressing the
relevant downstream substrates of the mevalonate pathway nor the
cell cycle involvement as analysed in the present study. Another
important disparity between both studies is the applied
concentration of simvastatin. While Zhao et al (26) used 0.001–0.1 µM to
demonstrate the 'protective' properties, Fromigué et al used
10 µM. Sandoval-Usme et al reported that simvastatin
inhibited the cell proliferation of osteosarcoma cells at a
concentration higher than 10 µM (28). Inhibition of osteosarcoma cell
proliferation by simvastatin (10 µM) was also shown by a
Japanese group (29). Our findings
demonstrated significantly inhibited osteosarcoma cell growth with
16 to 33 µM simvastatin, emphasizing the need for higher
concentrations for effective cancer treatment with simvastatin.
However, increasing the dose of simvastatin raises a question about
its clinical safety. Regarding the clinical application of
simvastatin, this drug is administered for long-term conditions.
The therapeutical range of simvastatin is at blood levels of
simvastatin between 1 and 15 nmol/l (30). Yet, the potential side effects of
simvastatin have been reported in large trials such as the Heart
Protection Study (HPS) and Scandinavian Simvastatin Study (4S)
(31,32). HPS included over 20,500 patients
who received a daily dose of 40 mg simvastatin for a period of up
to 5 years. Significant side-effects were myalgia (0.1% of
patients) and elevation of transaminases (n=21 or 0.21%) and CK.
The 4S study with 4,444 patients and a 5-year follow-up is a famous
study that shows the myalgia-inducing effect of simvastatin.
However, clinical trials to measure the maximum dose that is still
tolerable in patients are sparse. A Dutch group treated 28 patients
with myeloma or lymphoma with a 7-day period of escalating
simvastatin dose. Tolerated doses were up to 15 mg/kg body
weight/day and most reported side effects were fatigue and
neutropenic fever (33). This
indicates a potential role of a far higher simvastatin dose in
oncologic patients than the statin therapy for cardiovascular
disease, that is probably due to shorter application periods. The
dose that was chosen in the present study was meant to clarify the
molecular mechanism, and the possible use of simvastatin as an
add-on drug to current chemotherapy. Additionally, we expect that a
short-term exposure as a synergistic drug to chemotherapy will
presumably have no significant side-effects.
Successful rescue from simvastatin's effects with
FFP, GGPP and MA, implies the importance of the mevalonate pathway
for malignant cells. Next to cell proliferation/growth, cancer
cells elude the process of programmed cell death in order to grow
exponentially beyond their physiological life span (34). In fact, the ability to evade
apoptosis is a major criterion for cells to be considered malignant
(35). Among other mechanisms, the
release of proteins of the Bcl-2 family that mediate mitochondrial
permeability can induce apoptosis (36). The balance between pro-apoptotic
proteins (e.g. Bax, Bad and Bid) and anti-apoptotic proteins
(Bcl-2, Bcl-W, Bfl-1) is the key to cytochrome c release,
which forms the apoptosome (with Apf-1 and caspase-9), that cleaves
and thereby activates caspase-3 (36). Caspase-3 is the effective protease
that cleaves DNA and e.g. PARP-1, hereby inactivating PARP-1 in its
function to repair broken DNA strands, and stimulating further cell
death (37,38). Consequently, the reduction in Bcl-2
gene expression constitutes one target for activating apoptosis in
cancer cells. Statin-induced apoptosis, which has been associated
with Bcl-2 suppression, has been observed in breast cancer, human
glioma, human cardiac myocytes and adenocarcinoma cells (39–41).
A common feature in these studies is the concentrations (1–20
µM) needed to cause apoptosis. While statin's effect on
protein prenylation and mevalonate pathway is explained via HMG-CoA
reductase inhibition, the exact mechanism underlying the
suppression of Bcl-2 remains unknown. The present study showed a
significant increase in apoptotic cells after simvastatin therapy.
This was reversed by adding GGPP but not FPP, which implies that
GGPP-Ras and not FFP-Ras may dominantly activate certain pathways.
In summary, simvastatin treatment shifted the Bax/Bcl-2 ratio at
the mRNA level to a more pro-apoptotic ratio, an effect that was
again reversible with downstream mediators of the mevalonate
pathway. Considering that laryngeal cancer patients with increased
Bax/Bcl-2 mRNA ratio have longer disease-free and overall survival
(42), possibly, in osteosarcoma
patients, this could lead to benefits as well. To verify our
findings, we also investigated the protein expression of cleaved,
and thus activated caspase-3 and cleaved, and thus inactivated
PARP. As malignant cells are dependent on DNA damage repair by PARP
as discussed above, an inhibition of the named protein constitutes
a target for new therapies in breast cancer for example (43). Both cleaved proteins (caspase-3 and
PARP) were upregulated by simvastatin therapy, and indicate their
key role in the apoptosis of osteosarcoma cells. Comparable
findings were reported by Woschek et al in regards to renal
cancer cells by using 16 or 33 µM simvastatin (19). Taken together, our study showed an
induction of apoptosis mediators and subsequently apoptosis in
malignant osteosarcoma cells by simvastatin in vitro.
Apart from eluding programmed cell death, deficient
cell cycle regulation is another trait of cancer cells and their
growth, which has been discussed above. The cell cycle is regulated
by cyclin-dependent kinases (CDKs) and CDK inhibitors, that
associate with cyclin proteins (44).
However, many cancer entities display mutations in
cell cycle regulators, and thus exert an increased cell cycle rate
(45). Therefore, the strategy of
slowing down or arresting the cell cycle constitutes another
important target for cancer therapies (45). Simvastatin was found to induce cell
cycle arrest in pancreatic and endometrial cancer (46,47).
Similarly, simvastatin (16–64 µM) induced cell cycle arrest
via inhibition of CDKs in hepatic and renal cancer cells (18,19).
Yet, as the cell cycle is of upmost importance to cancer genesis it
certainly has to be taken into account for the underlying
mode-of-action of simvastatin in osteosarcoma cells, an approach
that has not been addressed previously. In the present study,
simvastatin treatment led to a significantly higher percentage of
osteosarcoma cells in the G0/G1 phase, and therefore, in cell cycle
arrest compared with the controls. Only GGPP and MA abolished these
effects, findings that again indicate a mevalonate pathway
dependency. However, data from the literature as well as our report
suggest that further studies are needed to verify the proposed
underlying mechanisms for simvastatin's mode-of-action.
Furthermore, in previous studies with osteosarcoma samples obtained
during surgery, alterations of TP53 were found in tumour tissue but
not in normal tissue, thus indicating the importance of TP53 in
tumour genesis (48). In more
recent pathological studies of human tumour tissue, TP53 gene
mutation was linked to susceptibility to osteosarcoma in childhood,
resistance to chemotherapy as well as poor overall prognosis
(49–51). In the present study, we found that
simvastatin is a potent enhancer of TP53. Similar data were found
for CDKN1A, which codes for p21 that in turn regulates cell cycle
proteins including CDK1 and CDK2. After 48 h, CDKN1A expression was
significantly and dose-dependently reduced. Notably, CDC2 is
responsible for cell cycle progression and thus a natural target
for cell cycle arrest (52).
Expression of CDC2 was markedly reduced under simvastatin therapy.
In summary, malignant osteosarcoma cells treated with simvastatin
are promoted to the G0/G1 phase with upregulation of TP53 and
CDKN1A while CDC2 is downregulated. This is a novel finding for
osteosarcoma cells, and may be a possible treatment target, which
should be investigated in further in vivo studies.
One pathway more exclusively induced by GGPP-Ras is
JNK (53). However, it has not
been investigated for osteosarcoma in terms of statin treatment
yet. Many effects of JNK are mediated by transcription factors of
the activator protein-1 (AP-1) family, of which c-Jun is the most
commonly known. Once these are activated, they form dimers and bind
to enhancer or promoter regions of DNA. Research findings have
indicated that AP-1 complexes do play a role in the expression of
p53, and are able to enhance expression of p21 in osteosarcoma
(54). Subsequently, c-Jun and JNK
pathway not only mediate apoptosis but also the cell cycle arrest
(55). The present study
demonstrated a considerable increase in p-JNK and p-c-Jun
expression, which are the phosphorylated and active forms of those
proteins. However, there are groups, which have reported a
downregulation of c-Jun after simvastatin therapy in U2OS cells
(56). Certainly, JNK and c-Jun
are activated by Rho kinases, so the question remains why these can
be upregulated by simvastatin. A possible explanation for this
phenomenon has been offered by others showing that statins may
inhibit prenylation of Rho kinases, but some Rho proteins (RhoA,
Rac1 and Cdc42) seem to be upregulated (53,57,58).
They may have reached their functionality without prenylation, and
may subsequently activate JNK via Map kinases. This is supported by
a recent study by Kamel et al, who demonstrated an impaired
Rho efficacy and an enhanced MAPK pathway in an osteosarcoma
simvastatin model (29).
Limitations of the study exist. We did not address
an optional additive therapy option to cytostatic agents using
simvastatin in combination with those. Optionally, a dose-reduction
of simvastatin may be reached by this approach. Further notably
detailed mechanistical studies are required to evaluate the
underlying pathways as well as the efficacy of the proposed
treatment regime.
In conclusion, simvastatin has distinct effects on
apoptosis and viability of osteosarcoma cells via JNK, and has to
be considered and evaluated for the treatment of this disease in
future studies.
Acknowledgments
We thank Katrin Jurida for outstanding technical
assistance.
Notes
[1]
Funding
The present study was funded by institutional
means.
[2] Availability
of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
[3] Authors'
contributions
BR designed the study, performed the statistical
analyses and revised the manuscript. SK and MW carried out analyses
and made the first draft of the manuscript. NK and RS carried out
analyses. MK and MH contributed intellectually to the completion of
the study.
[4] Ethics
approval and consent to participate
Not applicable.
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Ottaviani G and Jaffe N: The etiology of
osteosarcoma. Cancer Treat Res. 152:15–32. 2009. View Article : Google Scholar
|
|
2
|
Berner K, Hall KS, Monge OR, Weedon-Fekjær
H, Zaikova O and Bruland OS: Prognostic factors and treatment
results of high-grade osteosarcoma in norway: A scope beyond the
'classical' patient. Sarcoma. 2015:5168432015. View Article : Google Scholar
|
|
3
|
Li J, Yang Z, Li Y, Xia J, Li D, Li H, Ren
M, Liao Y, Yu S, Chen Y, et al: Cell apoptosis, autophagy and
necroptosis in osteosarcoma treatment. Oncotarget. 7:44763–44778.
2016.PubMed/NCBI
|
|
4
|
Bielack SS, Kempf-Bielack B, Delling G,
Exner GU, Flege S, Helmke K, Kotz R, Salzer-Kuntschik M, Werner M,
Winkelmann W, et al: Prognostic factors in high-grade osteosarcoma
of the extremities or trunk: An analysis of 1,702 patients treated
on neoadjuvant cooperative osteosarcoma study group protocols. J
Clin Oncol. 20:776–790. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Eftekhari F: Imaging assessment of
osteosarcoma in childhood and adolescence: Diagnosis, staging, and
evaluating response to chemotherapy. Cancer Treat Res. 152:33–62.
2009. View Article : Google Scholar
|
|
6
|
Salah S, Ahmad R, Sultan I, Yaser S and
Shehadeh A: Osteosarcoma with metastasis at initial diagnosis:
Current outcomes and prognostic factors in the context of a
comprehensive cancer center. Mol Clin Oncol. 2:811–816. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Luetke A, Meyers PA, Lewis I and Juergens
H: Osteosarcoma treatment - where do we stand? A state of the art
review. Cancer Treat Rev. 40:523–532. 2014. View Article : Google Scholar
|
|
8
|
Errani C, Longhi A, Rossi G, Rimondi E,
Biazzo A, Toscano A, Alì N, Ruggieri P, Alberghini M, Picci P, et
al: Palliative therapy for osteosarcoma. Expert Rev Anticancer
Ther. 11:217–227. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schwarz R, Bruland O, Cassoni A, Schomberg
P and Bielack S: The role of radiotherapy in oseosarcoma. Cancer
Treat Res. 152:147–164. 2009. View Article : Google Scholar
|
|
10
|
Meyers PA, Schwartz CL, Krailo MD, Healey
JH, Bernstein ML, Betcher D, Ferguson WS, Gebhardt MC, Goorin AM,
Harris M, et al Children's Oncology Group: Osteosarcoma: The
addition of muramyl tripeptide to chemotherapy improves overall
survival: A report from the Children's Oncology Group. J Clin
Oncol. 26:633–638. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chou AJ, Kleinerman ES, Krailo MD, Chen Z,
Betcher DL, Healey JH, Conrad EU III, Nieder ML, Weiner MA, Wells
RJ, et al Children's Oncology Group: Addition of muramyl tripeptide
to chemotherapy for patients with newly diagnosed metastatic
osteosarcoma: A report from the Children's Oncology Group. Cancer.
115:5339–5348. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Skinner R, Cotterill SJ and Stevens MC;
United Kingdom Children's Cancer Study Group: Risk factors for
nephrotoxicity after ifosfamide treatment in children: A UKCCSG
Late Effects Group study. Br J Cancer. 82:1636–1645.
2000.PubMed/NCBI
|
|
13
|
Arunkumar PA, Viswanatha GL, Radheshyam N,
Mukund H and Belliyappa MS: Science behind cisplatin-induced
nephrotoxicity in humans: A clinical study. Asian Pac J Trop
Biomed. 2:640–644. 2012. View Article : Google Scholar
|
|
14
|
Davies JT, Delfino SF, Feinberg CE,
Johnson MF, Nappi VL, Olinger JT, Schwab AP and Swanson HI: Current
and emerging uses of statins in clinical therapeutics: A Review.
Lipid Insights. 9:13–29. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jeon CY, Pandol SJ, Wu B, Cook-Wiens G,
Gottlieb RA, Merz CN and Goodman MT: The association of statin use
after cancer diagnosis with survival in pancreatic cancer patients:
A SEER-medicare analysis. PLoS One. 10:e01217832015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mei Z, Liang M, Li L, Zhang Y, Wang Q and
Yang W: Effects of statins on cancer mortality and progression: A
systematic review and meta-analysis of 95 cohorts including
1,111,407 individuals. Int J Cancer. 140:1068–1081. 2017.
View Article : Google Scholar
|
|
17
|
Relja B, Meder F, Wang M, Blaheta R,
Henrich D, Marzi I and Lehnert M: Simvastatin modulates the
adhesion and growth of hepatocellular carcinoma cells via decrease
of integrin expression and ROCK. Int J Oncol. 38:879–885. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Relja B, Meder F, Wilhelm K, Henrich D,
Marzi I and Lehnert M: Simvastatin inhibits cell growth and induces
apoptosis and G0/G1 cell cycle arrest in hepatic cancer cells. Int
J Mol Med. 26:735–741. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Woschek M, Kneip N, Jurida K, Marzi I and
Relja B: Simvastatin reduces cancerogenic potential of renal cancer
cells via heranylgeranyl pyrophosphate and mevalonate pathway. Nutr
Cancer. 68:420–427. 2016. View Article : Google Scholar
|
|
20
|
Mullen PJ, Yu R, Longo J, Archer MC and
Penn LZ: The interplay between cell signalling and the mevalonate
pathway in cancer. Nat Rev Cancer. 16:718–731. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou Q and Liao JK: Statins and
cardiovascular diseases: From cholesterol lowering to pleiotropy.
Curr Pharm Des. 15:467–478. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Klein MS, Koffarnus RL, Minze MG and Ochoa
P: Achieving cholesterol goals with low-cost
3-hydroxy-3-methylglutaryl coenzyme-A (HMG Co-A) reductase
inhibitors. Am J Health Syst Pharm. 73(Suppl 1): S63–S68. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tricarico PM, Crovella S and Celsi F:
Mevalonate pathway blockade, mitochondrial dysfunction and
autophagy: A possible link. Int J Mol Sci. 16:16067–16084. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Garay T, Kenessey I, Molnár E, Juhász É,
Réti A, László V, Rózsás A, Dobos J, Döme B, Berger W, et al:
Prenylation inhibition-induced cell death in melanoma: Reduced
sensitivity in BRAF mutant/PTEN wild-type melanoma cells. PLoS One.
10:e01170212015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wood CE, Hukkanen RR, Sura R,
Jacobson-Kram D, Nolte T, Odin M and Cohen SM: Scientific and
Regulatory Policy Committee (SRPC) Review: Interpretation and use
of cell proliferation data in cancer risk assessment. Toxicol
Pathol. 43:760–775. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao XH, Xu ZR, Zhang Q and Yang YM:
Simvastatin protects human osteosarcoma cells from oxidative
stress-induced apoptosis through mitochondrial-mediated signaling.
Mol Med Rep. 5:483–488. 2012.
|
|
27
|
Fromigué O, Haÿ E, Modrowski D, Bouvet S,
Jacquel A, Auberger P and Marie PJ: RhoA GTPase inactivation by
statins induces osteosarcoma cell apoptosis by inhibiting
p42/p44-MAPKs-Bcl-2 signaling independently of BMP-2 and cell
differentiation. Cell Death Differ. 13:1845–1856. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sandoval-Usme MC, Umaña-Pérez A, Guerra B,
Hernández-Perera O, García-Castellano JM, Fernández-Pérez L and
Sánchez-Gómez M: Simvastatin impairs growth hormone-activated
signal transducer and activator of transcription (STAT) signaling
pathway in UMR-106 osteosarcoma cells. PLoS One. 9:e877692014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kamel WA, Sugihara E, Nobusue H,
Yamaguchi-Iwai S, Onishi N, Maki K, Fukuchi Y, Matsuo K, Muto A,
Saya H, et al: Simvastatin-induced apoptosis in osteosarcoma cells:
A Key role of RhoA-AMPK/p38 MAPK signaling in antitumor activity.
Mol Cancer Ther. 16:182–192. 2017. View Article : Google Scholar
|
|
30
|
Björkhem-Bergman L, Lindh JD and Bergman
P: What is a relevant statin concentration in cell experiments
claiming pleiotropic effects? Br J Clin Pharmacol. 72:164–165.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
No authors listed. Randomised trial of
cholesterol lowering in 4444 patients with coronary heart disease:
The Scandinavian Simvastatin Survival Study (4S). Lancet.
344:1383–1389. 1994.PubMed/NCBI
|
|
32
|
Heart Protection Study Collaborative
Group: MRC/BHF Heart Protection Study of cholesterol lowering with
simvastatin in 20,536 high-risk individuals: A randomised
placebo-controlled trial. Lancet. 360:7–22. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
van der Spek E, Bloem AC, van de Donk NW,
Bogers LH, van der Griend R, Kramer MH, de Weerdt O, Wittebol S and
Lokhorst HM: Dose-finding study of high-dose simvastatin combined
with standard chemotherapy in patients with relapsed or refractory
myeloma or lymphoma. Haematologica. 91:542–545. 2006.PubMed/NCBI
|
|
34
|
Fulda S: The mechanism of necroptosis in
normal and cancer cells. Cancer Biol Ther. 14:999–1004. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fulda S: Targeting apoptosis for
anticancer therapy. Semin Cancer Biol. 31:84–88. 2015. View Article : Google Scholar
|
|
36
|
Jendrossek V: The intrinsic apoptosis
pathways as a target in anticancer therapy. Curr Pharm Biotechnol.
13:1426–1438. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW,
Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, et al:
Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad
Sci USA. 103:18308–18313. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kroemer G, Galluzzi L and Brenner C:
Mitochondrial membrane permeabilization in cell death. Physiol Rev.
87:99–163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Spampanato C, De Maria S, Sarnataro M,
Giordano E, Zanfardino M, Baiano S, Cartenì M and Morelli F:
Simvastatin inhibits cancer cell growth by inducing apoptosis
correlated to activation of Bax and downregulation of BCL-2 gene
expression. Int J Oncol. 40:935–941. 2012. View Article : Google Scholar
|
|
40
|
Konturek PC, Burnat G and Hahn EG:
Inhibition of Barret's adenocarcinoma cell growth by simvastatin:
Involvement of COX-2 and apoptosis-related proteins. J Physiol
Pharmacol. 58(Suppl 3): 141–148. 2007.PubMed/NCBI
|
|
41
|
Lee HY, Kim IK, Lee HI, Mo JY, Yeo CD,
Kang HH, Moon HS and Lee SH: The apoptotic effect of simvastatin
via the upregulation of BIM in nonsmall cell lung cancer cells. Exp
Lung Res. 42:14–23. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Giotakis AI, Kontos CK, Manolopoulos LD,
Sismanis A, Konstadoulakis MM and Scorilas A: High BAX/BCL2 mRNA
ratio predicts favorable prognosis in laryngeal squamous cell
carcinoma, particularly in patients with negative lymph nodes at
the time of diagnosis. Clin Biochem. 49:890–896. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
O'Connor MJ: Targeting the DNA damage
response in cancer. Mol Cell. 60:547–560. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bendris N, Lemmers B and Blanchard JM:
Cell cycle, cyto-skeleton dynamics and beyond: The many functions
of cyclins and CDK inhibitors. Cell Cycle. 14:1786–1798. 2015.
View Article : Google Scholar :
|
|
45
|
Dey N, Williams C, Leyland-Jones B and De
P: Mutation matters in precision medicine: A future to believe in.
Cancer Treat Rev. 55:136–149. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Babcook MA, Sramkoski RM, Fujioka H,
Daneshgari F, Almasan A, Shukla S, Nanavaty RR and Gupta S:
Combination simvastatin and metformin induces G1-phase cell cycle
arrest and Ripk1- and Ripk3-dependent necrosis in C4-2B osseous
metastatic castration-resistant prostate cancer cells. Cell Death
Dis. 5:e15362014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Schointuch MN, Gilliam TP, Stine JE, Han
X, Zhou C, Gehrig PA, Kim K and Bae-Jump VL: Simvastatin, an
HMG-CoA reductase inhibitor, exhibits anti-metastatic and
anti-tumorigenic effects in endometrial cancer. Gynecol Oncol.
134:346–355. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Masuda H, Miller C, Koeffler HP, Battifora
H and Cline MJ: Rearrangement of the p53 gene in human osteogenic
sarcomas. Proc Natl Acad Sci USA. 84:7716–7719. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mirabello L, Yeager M, Mai PL,
Gastier-Foster JM, Gorlick R, Khanna C, Patiño-Garcia A,
Sierrasesúmaga L, Lecanda F, Andrulis IL, et al: Germline TP53
variants and susceptibility to osteosarcoma. J Natl Cancer Inst.
107:1072015. View Article : Google Scholar
|
|
50
|
Goto A, Kanda H, Ishikawa Y, Matsumoto S,
Kawaguchi N, Machinami R, Kato Y and Kitagawa T: Association of
loss of heterozygosity at the p53 locus with chemoresistance in
osteosarcomas. Jpn J Cancer Res. 89:539–547. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chen Z, Guo J, Zhang K and Guo Y: TP53
mutations and survival in osteosarcoma patients: A meta-analysis of
published data. Dis Markers. 2016:46395752016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Williams GH and Stoeber K: The cell cycle
and cancer. J Pathol. 226:352–364. 2012. View Article : Google Scholar
|
|
53
|
Liang SL, Liu H and Zhou A:
Lovastatin-induced apoptosis in macrophages through the
Rac1/Cdc42/JNK pathway. J Immunol. 177:651–656. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ogawa E, Okuyama R, Egawa T, Nagoshi H,
Obinata M, Tagami H, Ikawa S and Aiba S: p63/p51-induced onset of
keratinocyte differentiation via the c-Jun N-terminal kinase
pathway is counteracted by keratinocyte growth factor. J Biol Chem.
283:34241–34249. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Dhanasekaran DN and Reddy EP: JNK
signaling in apoptosis. Oncogene. 27:6245–6251. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fromigué O, Hamidouche Z and Marie PJ:
Blockade of the RhoA-JNK-c-Jun-MMP2 cascade by atorvastatin reduces
osteosarcoma cell invasion. J Biol Chem. 283:30549–30556. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhu Y, Casey PJ, Kumar AP and Pervaiz S:
Deciphering the signaling networks underlying simvastatin-induced
apoptosis in human cancer cells: Evidence for non-canonical
activation of RhoA and Rac1 GTPases. Cell Death Dis. 4:e5682013.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Sassano A, Katsoulidis E, Antico G, Altman
JK, Redig AJ, Minucci S, Tallman MS and Platanias LC: Suppressive
effects of statins on acute promyelocytic leukemia cells. Cancer
Res. 67:4524–4532. 2007. View Article : Google Scholar : PubMed/NCBI
|