Introduction
Vascular endothelial growth factor (VEGF) and
VEGF-receptor (VEGF-R) family members are the central regulators of
tumor angiogenesis (1). Therefore,
several angiogenesis inhibitors targeting the VEGF/VEGF-R pathway
have been developed and have become an important option for the
management of a number of human malignancies, including colorectal
cancer (1–4). However, there is increasing evidence
that the clinical results of VEGF/VEGF-R-targeted therapies are
very modest, resulting in a moderate improvement in overall
survival (5–8). Pre-clinical studies have shown that
VEGF/VEGF-R-targeting agents accelerate invasion and metastasis
(9,10). Additionally, the clinical outcome
is associated with the development of resistance to the
VEGF/VEGF-R-targeting agents and the increased risk of invasion and
metastasis (11–14). Therefore, acquired and evasive
resistance to VEGF/VEGF-R inhibitors is a growing concern in the
clinic.
Several mechanisms by which tumor cells acquire
resistance to VEGF/VEGF-R pathway targeting therapies and escape
from aggravated microenvironments have been elucidated (1–4,11).
VEGF/VEGF-R blockers primarily target vascular endothelial cells
and inhibit tumor angiogenesis, leading to hypoxia within the
tumor. Therefore, hypoxic stress is considered to be a central
mechanism for the aggressive malignant progression of tumor cells
(15–17). Several preclinical studies have
shown that VEGF pathway inhibitors facilitate tumor cell invasion
and metastasis, which require hypoxic conditions and
hypoxia-inducible factors (9–11).
Tumor cells are important targets of VEGF/VEGF-R
inhibitors, as several types of cancer cells express functional
VEGF-Rs and utilize VEGF as an autocrine survival factor (18–22).
Therefore, it is possible that VEGF/VEGF-R blockers could directly
act on tumor cells and elicit an adaptive and evasive response,
resulting in more aggressive phenotypes. Notably, several studies,
including one by the present research group, have shown that the
chronic treatment of colon cancer cells with anti-VEGF-A antibody
alters their phenotype, for example, by increasing cell motility,
apoptosis resistance under hypoxic conditions and spheroid
formation (19–22).
VEGF/VEGF-R-targeting agents are classified into the
following two groups: Those that target VEGF ligands [VEGF-A,
VEGF-B and placental growth factor (PlGF)], such as bevacizumab and
aflibercept, and those that target VEGF-Rs (VEGF-R1, -R2 and -R3),
including sunitinib, sorafenib, regorafenib, pazopanib, axitinib,
vandetanib, dovitinib, nintedanib, lenvatinib, foretinib and
cabozantinib (5–7). The aim of the present study was to
elucidate whether bevacizumab (a VEGF-A inhibitor) and sunitinib (a
blocker of all VEGF-Rs) directly affect the evasive adaptation of
tumor cells. The different mechanisms of evasive activation of the
two drugs were also investigated. For this aim, VEGF/VEGF-R
inhibitor-adapted cells were developed via the prolonged (3 months)
exposure of human colon cancer cell lines to bevacizumab
(bevacizumab-adapted cells) or sunitinib (sunitinib-adapted cells).
This experiment was based on the observation of tumor invasiveness
occurring within a few months of the initiation of treatment with
VEGF/VEGF-R-targeting agents in several preclinical models.
Notably, preclinical studies have demonstrated that extensive
treatment (1–3 months) with sunitinib accelerates local invasion
and distant metastasis and leads to a shortening of overall
survival (9,10).
Materials and methods
Reagents
Sunitinib (a VEGF-R tyrosine kinase inhibitor),
foretinib [a tyrosine-protein kinase Met (cMet) and VEGF-R tyrosine
kinase inhibitor], capmatinib (a selective inhibitor of cMet),
GW788388 [a selective inhibitor of transforming growth factor
(TGF)β-RI and -RII], SSR128129E [an allosteric fibroblast growth
factor receptor (FGF-R) 1–3 inhibitor], CP-673451 [a selective
inhibitor of platelet-derived growth factor receptor (PDGF-R)-α/β]
and V1/A7R [a neuropilin-1 (NRP1)-binding heptapeptide, ATWLPPR,
that specifically inhibits NRP1) were obtained from Selleck
Chemicals (Cosmo Bio Co., Ltd., Tokyo, Japan). Neutralizing
antibodies against human hepato-cyte growth factor (HGF; MAB294),
human NRP1 (AF3870), control non-immune sheep IgG (5-001-A) and
control non-immune mouse IgG (MAB002) were from R&D Systems,
Inc. (Minneapolis, MN, USA). Specific antagonistic inhibitors for
VEGF-R1 and VEGF-R3 were purchased from Genscript Japan, Inc.
(Tokyo, Japan).
Establishment of cell models adapted to
bevacizumab and sunitinib
Human colon cancer cell lines (HCT116 and RKO) were
obtained from American Type Culture Collection (Manassas, VA, USA)
and maintained in RPMI-1640 medium (Nacalai Tesque, Inc., Tokyo,
Japan) with 10% fetal bovine serum (FBS; SAFS Biosciences; Merck
KGaA, Darmstadt, Germany), 100 U/ml penicillin and 0.1 mg/ml
streptomycin (both from Nacalai Tesque, Inc.) at 37°C under 5%
CO2 and 95% air. To establish the bevacizumab-adapted
cells (HCT/bev and RKO/bev, respectively), cells were chronically
exposed to bevacizumab (Chugai Pharmaceuticals Co., Ltd., Tokyo,
Japan) for 3 months at a clinically relevant dose (100
µg/ml), based on the US Food and Drug
Administration-approved bevacizumab dose (5 mg/kg) corresponding to
a concentration of 100 µg/ml in cell culture experiments
(20). To develop the
sunitinib-adapted cells (HCT/suni and RKO/suni, respectively) or
foretinib-adapted cells (HCT/fore and RKO/fore, respectively),
cells were chronically treated with a pharmacologically relevant
concentration of sunitinib (0.1 µM) or foretinib (5 nM) for
3 months (23,24). The vehicle-treated control cells
(HCT/ctl and RKO/ctl, respectively) were obtained by chronic
treatment with DMSO (0.005%) for 3 months.
The bevacizumab-adapted cells were pretreated with
DMSO (0.005%), VEGF-R1 inhibitor (50 µM) or VEGF-R3
inhibitor (75 µM) for 1 h at 37°C, and then their migration
and invasion abilities were determined. The control and
sunitinib-adapted cells were pretreated with vehicle (DMSO,
0.005%), capmatinib (2 nM), GW788388 (0.35 µM), SSR128129E
(0.2 µM), CP-673451 (0.05 µM), NRP1 antagonist V1/A7R
(3 µM), anti-NRP1 neutralizing antibody (5 µg/ml),
anti-HGF neutralizing antibody (1 µg/ml), a control
non-immune sheep IgG (5 µg/ml; as a control for anti-NRP1
neutralizing antibody) or a control non-immune mouse IgG (1
µg/ml; as a control for anti-HGF neutralizing antibody) for
1 h at 37°C, and then their migration and invasion activities were
evaluated. Parental HCT116 and RKO cells were pretreated with
vehicle (DMSO, 0.005%), sunitinib (0.1 µM) or foretinib (10
nM) for 48 h at 37°C, then their migration and invasion activities
were examined.
Cell migration and invasion assay
Equal numbers of cells (50,000 cells) were suspended
in 0.25 ml RPMI-1640 containing 1% FBS with various treatment
agents, as described in the section entitled 'establishment of cell
models adapted to bevacizumab and sunitinib', and then placed in
the top compartment of an uncoated 8-µm pore membrane
chamber (BD Biosciences, Franklin Lakes, NJ, USA); 0.75 ml
RPMI-1640 containing 4% FBS for the migration assay or 10% FBS for
the invasion assay was added to the bottom compartment. Following
24–48 h incubation under standard conditions (37°C/5%
CO2), non-migrating cells were scraped from the top
compartment, and cells that had migrated to the bottom compartment
were fixed with 100% methanol at 25°C for 30 sec, and stained at
25°C for 30 sec using Hemacolor Rapid staining of blood smear
(Merck KGaA). Membranes were excised and mounted on a standard
microscope slide. The numbers of migrated cells were determined
from 4–6 random high-power fields visualized at ×20
magnification.
Invasion assays were performed using a protocol
similar to that of the migration assay with minor modifications.
The inserts used in the invasion assays were coated with Matrigel
(BD Biosciences) and prehydrated with 1% FBS-supplemented medium
for 2 h prior to the addition of the cell suspension. Following
48-h incubation under standard conditions (37°C/5% CO2),
the numbers of invading cells were quantified as described
above.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The extraction of total RNA was carried out using an
RNeasy mini kit (Qiagen, Tokyo, Japan), and total RNA (1 µg)
was used to synthesize cDNA using the PrimeScript RT reagent kit
(Takara Bio, Inc., Tokyo, Japan) according to the following steps:
37°C for 10 min, 85°C for 30 sec, and cooling down to 4°C. The
levels of transcripts for human NRP1 and human GAPDH
in the cells were measured by RT-qPCR using the following specific
primer sets: NRP1, 5′-CCCTGAGAATGGGTGGACT-3′ (forward) and
5′-CGTGACAAAGCGCAGAAG-3′ (reverse); GAPDH,
5′-GCTAGGGACGGCCTGAAG-3′ (forward) and 5′-GCCCAATACGACCAAATCC-3′
(reverse). qPCR analysis was performed using a SYBR-Green Master
Mix (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) according to the following steps: 95°C for 30 sec (1
cycle), 60°C for 30 sec and 95°C for 5 sec (40 cycles).
Amplification and quantification of the PCR products were performed
using the Applied Biosystems 7500 System (Applied Biosystems;
Thermo Fisher Scientific, Inc.). Standards were run in the same
plate and the relative standard curve quantification method was
used to calculate the relative mRNA expression (25). RNA quantities were normalized
against the GAPDH mRNA levels.
Western blot analysis
Total cell lysates were prepared using a lysis
buffer containing 100 mM Tris-HCl (pH 6.8), 300 mM NaCl, 2 mM EDTA
and 4% (v/v) sodium dodecylsulfate (SDS). Protein concentrations
were determined using a bicinchoninic acid protein assay (Pierce;
Thermo Fisher Scientific, Inc.). The cell extracts (50 µg
protein/lane) were subjected to 10% SDS-polyacrylamide gel
electrophoresis (PAGE) and transferred to a polyvinylidene
difluoride membrane (EMD Millipore, Bedford, MA, USA). The membrane
was blocked with 4% skimmed milk for 1 h at 25°C, and then
incubated for 15 h at 4°C in phosphate-buffered saline (PBS)
containing 0.05% Tween-20 with the primary antibody according to
the instructions provided by the manufacturer; a rabbit monoclonal
anti-human NRP1 antibody (D62C6) at 1:3,000 dilution, a rabbit
monoclonal anti-human cMet antibody (D1C2) at 1:2,000 dilution, a
rabbit monoclonal anti-human phospho-cMet (Tyr1234/Tyr1235)
antibody (D26) at 1:3,000 dilution, a rabbit monoclonal anti-human
p130Cas antibody (E1L9G) at 1:2,000 dilution, a rabbit monoclonal
anti-human phospho-p130Cas (Tyr410) antibody (#4011) at a 1:1,000
dilution, a rabbit monoclonal anti-human Slug antibody (C19G7) at a
1:1,000 dilution, a rabbit monoclonal anti-human N-cadherin
antibody (D4R1H) at a 1:2,000 dilution and a mouse monoclonal
anti-human β-actin antibody (8H10D10) at a 1:10,000 dilution. All
primary antibodies were acquired from Cell Signaling Technology,
Inc. (Danvers, MA, USA). The membrane was probed for 1 h at 25°C
with secondary antibodies; anti-rabbit IgG horseradish peroxidase
(HRP)-linked antibody at a 1:10,000 dilution (#7074) and anti-mouse
IgG HRP-linked antibody at a 1:10,000 dilution (#7076) (both from
Cell Signaling Technology, Inc.). The membranes were developed
using ECL western blot detection reagents (GE Healthcare Life
Sciences, Little Chalfont, UK).
Co-immunoprecipitation analysis
For NRP1/cMet co-immunoprecipitation analysis, cells
were crosslinked with dithiobis(succinimidyl propionate) (Pierce;
Thermo Fisher Scientific, Inc.) prior to cell lysis. Cells were
harvested in ice-cold lysis buffer (50 mM HEPES, pH 7.0, 150 mM
NaCl, 10 mM EDTA, 1.5 mM MgCl2, 1% Nonidet P-40, 1%
Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 1 mM sodium
orthovanadate, 0.5% sodium deoxycholate, 5 mg/ml aprotinin, 5 mg/ml
leupeptin, 20 mM sodium fluoride and 20 mM sodium pyrophosphate),
homogenized through a 23-gauge needle 10 times, then centrifuged at
10,000 × g for 20 min at 4°C. The clarified lysate (1 mg) was
incubated with a rabbit monoclonal anti-human NRP1 antibody (D62C6)
overnight at 4°C with constant gentle rocking, followed by the
addition of Protein G magnet Dynabeads (Thermo Fisher Scientific,
Inc.) for 4 h at 4°C. The immunoprecipitates were washed, eluted
with SDS-PAGE sample buffer, and subjected to SDS-PAGE using 4–20%
polyacrylamide gradient gels. They were transferred to a
polyvinylidene difluoride membrane (EMD Millipore). The membrane
was blocked with 4% skimmed milk for 1 h at 25°C, and then
incubated for 15 h at 4°C in PBS containing 0.05% Tween-20 with the
primary antibody; a rabbit monoclonal anti-human cMet antibody
(D1C2) at 1:2,000 dilution, a rabbit monoclonal anti-human
phospho-cMet (Tyr1234/Tyr1235) antibody (D26) at 1:3,000 dilution,
and rabbit monoclonal anti-human NRP1 antibody (D62C6) at 1:3,000
dilution. All primary antibodies were acquired from Cell Signaling
Technology, Inc. The membrane was probed for 1 h at 25°C with
anti-rabbit IgG HRP-linked antibody at a 1:10,000 dilution (#7074;
Cell Signaling Technology, Inc.). The membranes were developed
using ECL western blot detection reagents (GE Healthcare Life
Sciences).
Cell survival assay
Cell survival was assessed using a CellTiter
96® AQueous One Solution Cell Proliferation Assay
(Promega Corporation, Madison, WI, USA), according to the
manufacturer's protocol. Cells were plated (1×104
cells/well) in a 96-well flat-bottom plate for 24 h, then they were
treated with sunitinib (0.1, 1 or 10 µM) or with bevacizumab
(10 or 100 µg/ml) for 3 days. Following treatment, 100
µl RPMI-1640 medium containing the
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxy-methoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
reagent (20 µl/well) was added to each well and the cells
were incubated for 1 h at 37°C. The levels of blue formazan were
measured spectrophotometrically at 490 nm immediately using a
microplate reader (Wallac 1420 ARVO MX; PerkinElmer, Inc., Waltham,
MA, USA).
ELISA
Secreted VEGF ligands (VEGF-A, PlGF and VEGF-C) in
the cell culture supernatants were measured using a VEGF
isotype-specific ELISA kit (DVE00, DPG00 and DVEC00; R&D
Systems, Inc.) according to the manufacturer's protocol. Notably,
an ELISA that measures VEGF-B was not available from R&D
Systems, Inc. when the study was conducted. Total and
phosphorylated VEGF-Rs (R1 and R3) in cell lysates were measured
using the respective human VEGF-R DuoSet IC ELISA kit and the
respective human Phospho-VEGF-R DuoSet IC ELISA kit (both from
R&D Systems, Inc.) according to the manufacturer's
protocol.
Statistical analysis
Results are expressed as the mean ± standard
deviation. Differences between groups were analyzed via analysis of
variance and Scheffe's test using SPSS software (release 6.1; SPSS
Japan, Tokyo, Japan). P<0.01 was considered to indicate a
statistically significant difference.
Results
Development and characterization of
sunitinib- and bevacizumab-adapted cells
The human colon cancer cell lines HCT116 and RKO
were selected because they express several VEGF/VEGF-R family
members and utilize autocrine VEGF signals for their survival; this
has been demonstrated by several groups, including the present
research team (20–22). To investigate the direct effects of
sunitinib and bevacizumab, two cell models (sunitinib- and
bevacizumab-adapted cells) were established by continuously
culturing the cells in the presence of sunitinib or bevacizumab for
3 months; a similar procedure has been used in previous studies
(21,22).
To characterize the resistance of the
sunitinib-adapted cells (HCT/suni and RKO/suni) to sunitinib, the
cells were treated with sunitinib at several concentrations and
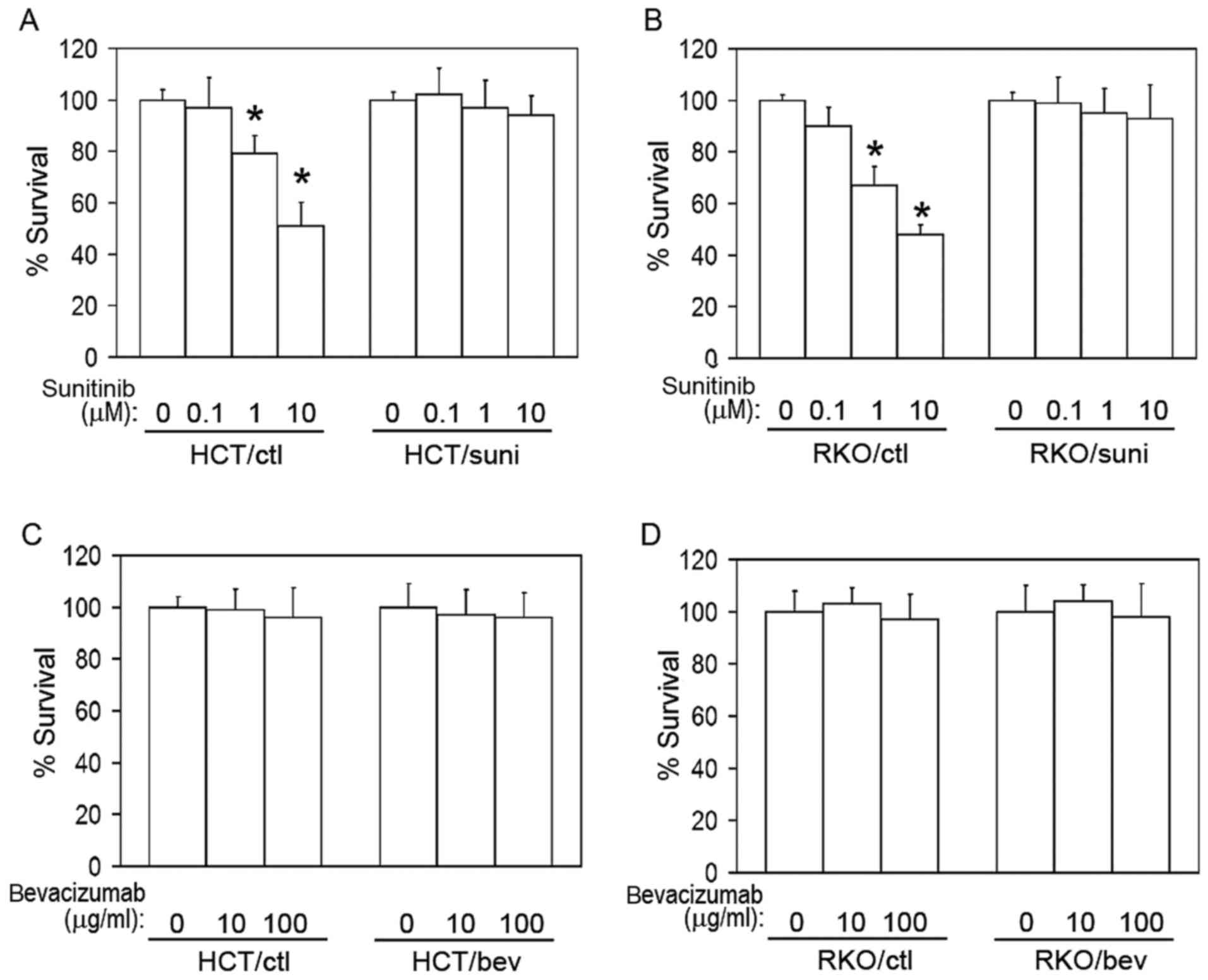
analyzed using a cell survival assay (Fig. 1A and B). A marked resistance to
sunitinib was observed in the sunitinib-adapted cells compared with
the respective vehicle-treated control cells (Fig. 1A and B). However, the
bevacizumab-adapted cells (HCT/bev and RKO/bev) exhibited similar
growth rates to their respective control cells when treated with
bevacizumab (Fig. 1C and D).
Cell migration and invasion activities of
the bevacizumab- and sunitinib-adapted cells
It has been reported that bevacizumab-adapted cells
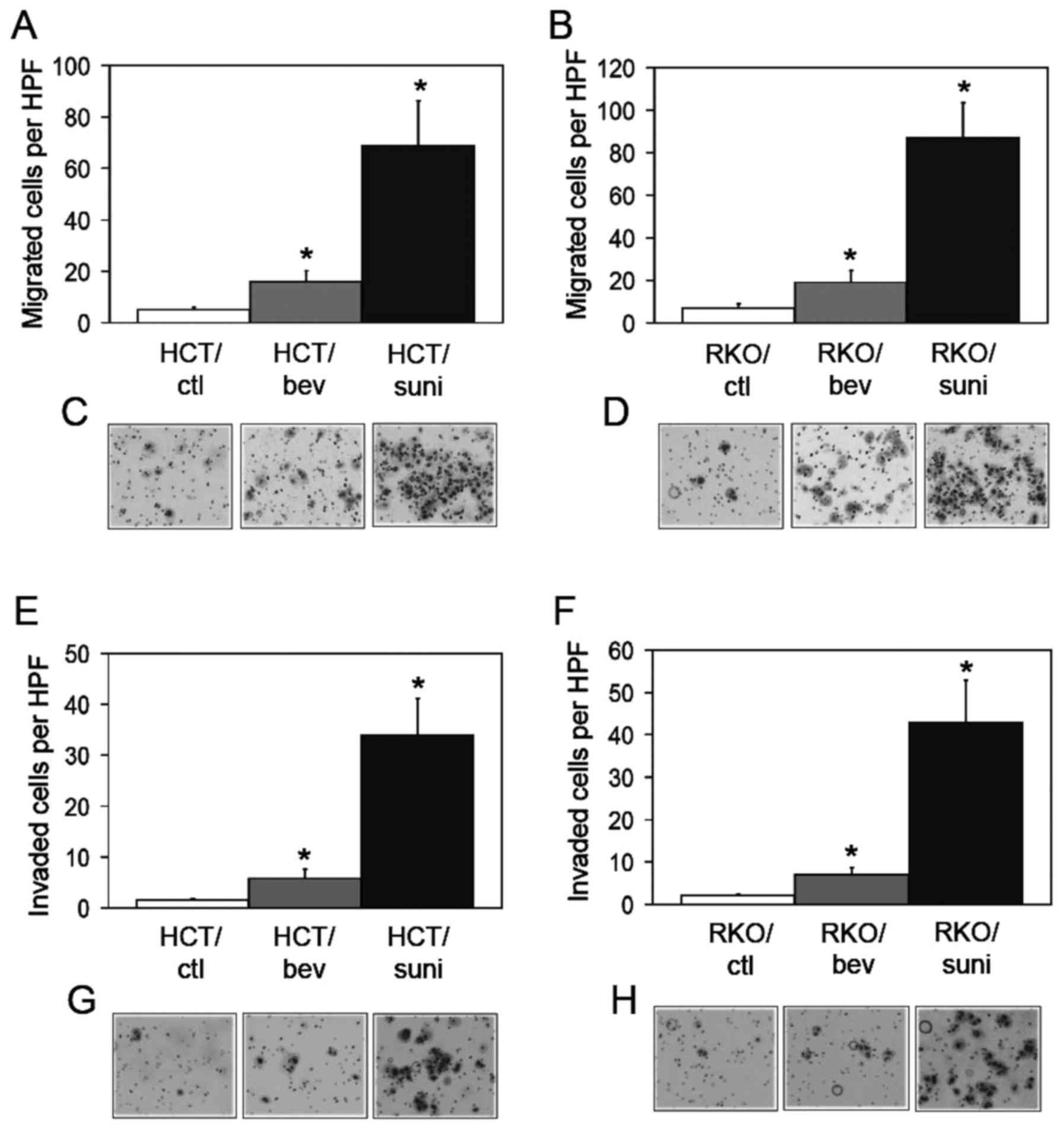
exhibit increased motility and invasive activities (21). Thus, these activities were compared
in the bevacizumab- and sunitinib-adapted cells in the present
study. A modified Boyden chamber assay demonstrated that the
migration activities of the bevacizumab-adapted cells were
significantly increased compared with those of the control cells
(Fig. 2A–D). The sunitinib-adapted
cells showed significant increases in migration activities compared
with those of the control and of the bevacizumab-adapted cells
(Fig. 2A–D). Cell invasion was
also examined using a Matrigel invasion assay. Consistent with the
migration activity, the invasion activities of the
bevacizumab-adapted cells were significantly increased compared
with those of the control cells Fig.
2E–H). The sunitinib-adapted cells exhibited significant
increases in invasion activities compared with the control and with
the bevacizumab-adapted cells (Fig.
2E–H). These findings indicate that the evasive phenotype was
more potently activated by the inhibition of all VEGF-Rs compared
with inhibition of the VEGF-A ligand alone and suggest that the
evasive activation was induced by a different mechanism under the
VEGF-R-inhibited conditions compared with the VEGF-A-inhibited
conditions.
Different mechanisms of evasive
adaptation in the bevacizumab- and sunitinib-adapted cells
Whether the mechanism of evasive adaptation differed
between the bevacizumab- and sunitinib-adapted cells was
investigated. It was hypothesized that the bevacizumab-adapted
cells may redundantly depend on VEGF/VEGF-R family members aside
from VEGF-A whereas the sunitinib-adapted cells may be independent
of these proteins because all VEGF-Rs are inhibited in the cells.
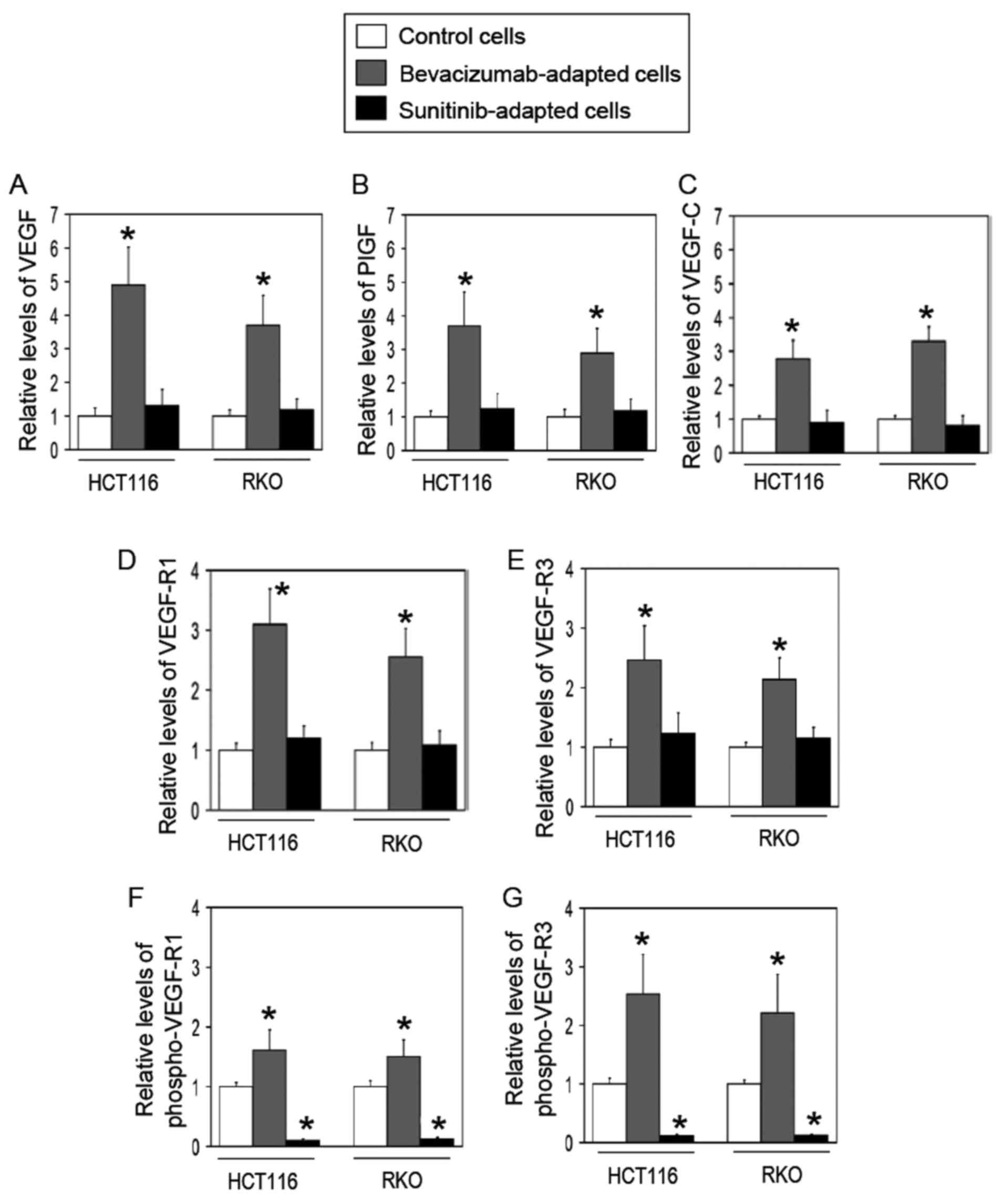
To test this hypothesis, the expression profiles of VEGF ligands
and their receptors at the protein levels were compared in the two
cell models using ELISAs. Consistent with the aforementioned
hypothesis, the bevacizumab-adapted cells exhibited a significant
increase in the expression levels of VEGF-A, PlGF, VEGF-C, VEGF-R1
and VEGF-R3 compared with the control cells among the VEGF family
members tested (Fig. 3A–E).
Additionally, the phosphorylated levels of VEGF-R1 and VEGF-R3 were
significantly elevated in the bevacizumab-adapted cells compared
with the control cells (Fig. 3F and
G), suggesting that the bevacizumab-adapted cells utilize
VEGF-R1 and VEGF-R3 autocrine systems. By contrast, the
sunitinib-adapted cells did not demonstrate any significant
difference in the expression of VEGF family members when compared
with the control cells (Fig.
3).
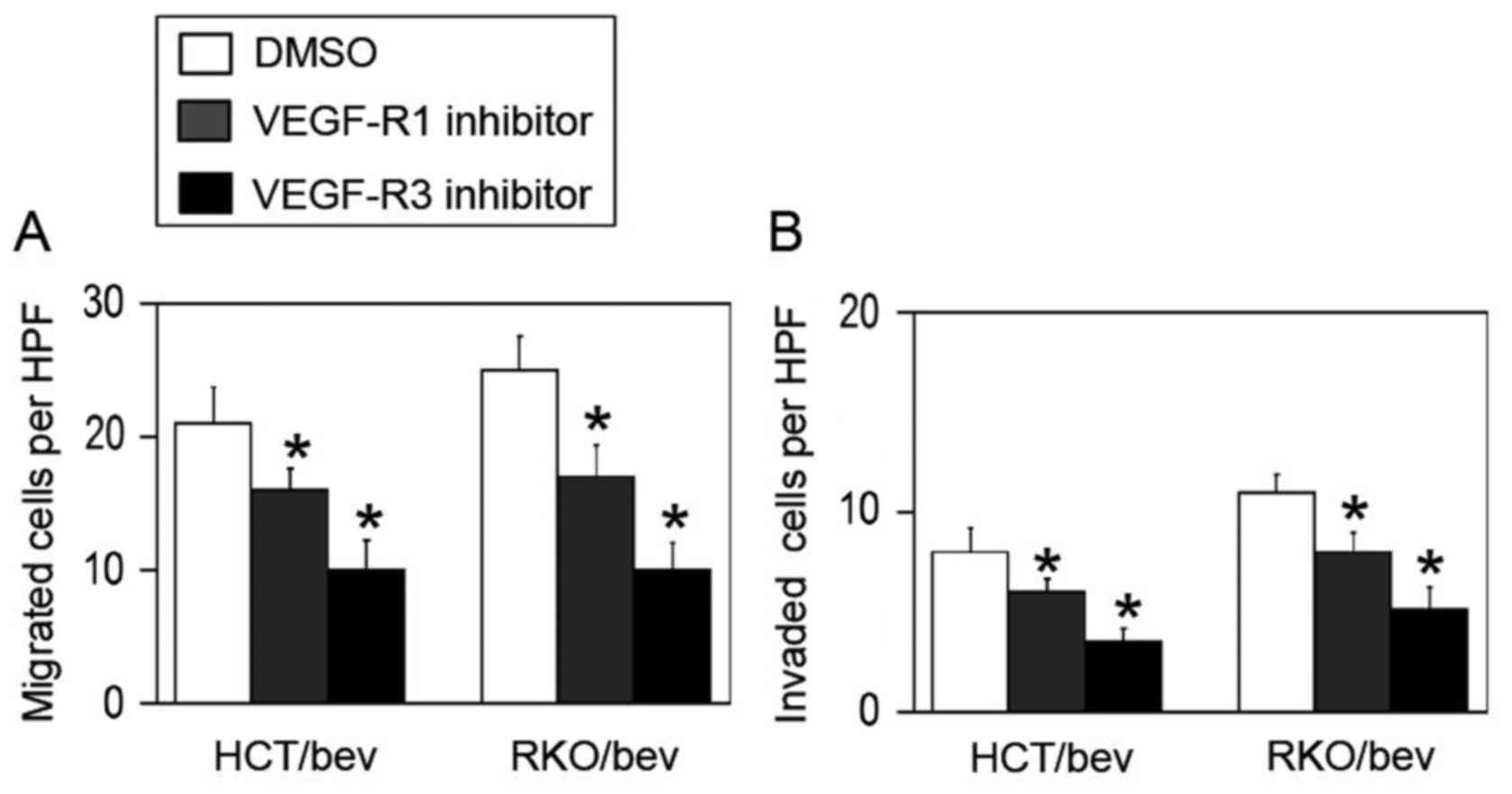
To investigate whether the bevacizumab-adapted cells
utilized VEGF-R1 and VEGF-R3 systems for their evasive adaptation,
specific antagonistic inhibitors for VEGF-R1 (26) or VEGF-R3 (27) were used. Treatment with the VEGF-R1
inhibitor significantly decreased the migration and invasion
activities of the HCT/bev and RKO/bev cells compared with the
DMSO-treated controls (Fig. 4).
When VEGF-R3 was blocked, the evasive activities of the HCT/bev and
RKP/bev cells were also significantly inhibited compared with the
DMSO-treated controls (Fig. 4).
These results support the hypothesis that the bevacizumab-adapted
cells were redundantly dependent on VEGF family members.
To clarify the mechanism in the sunitinib-adapted
cells, the present study focused on NRP1 as it is a multifunctional
co-receptor involved in migration and invasion in several cancer
cells (28–30). Thus, the expression levels of NRP1
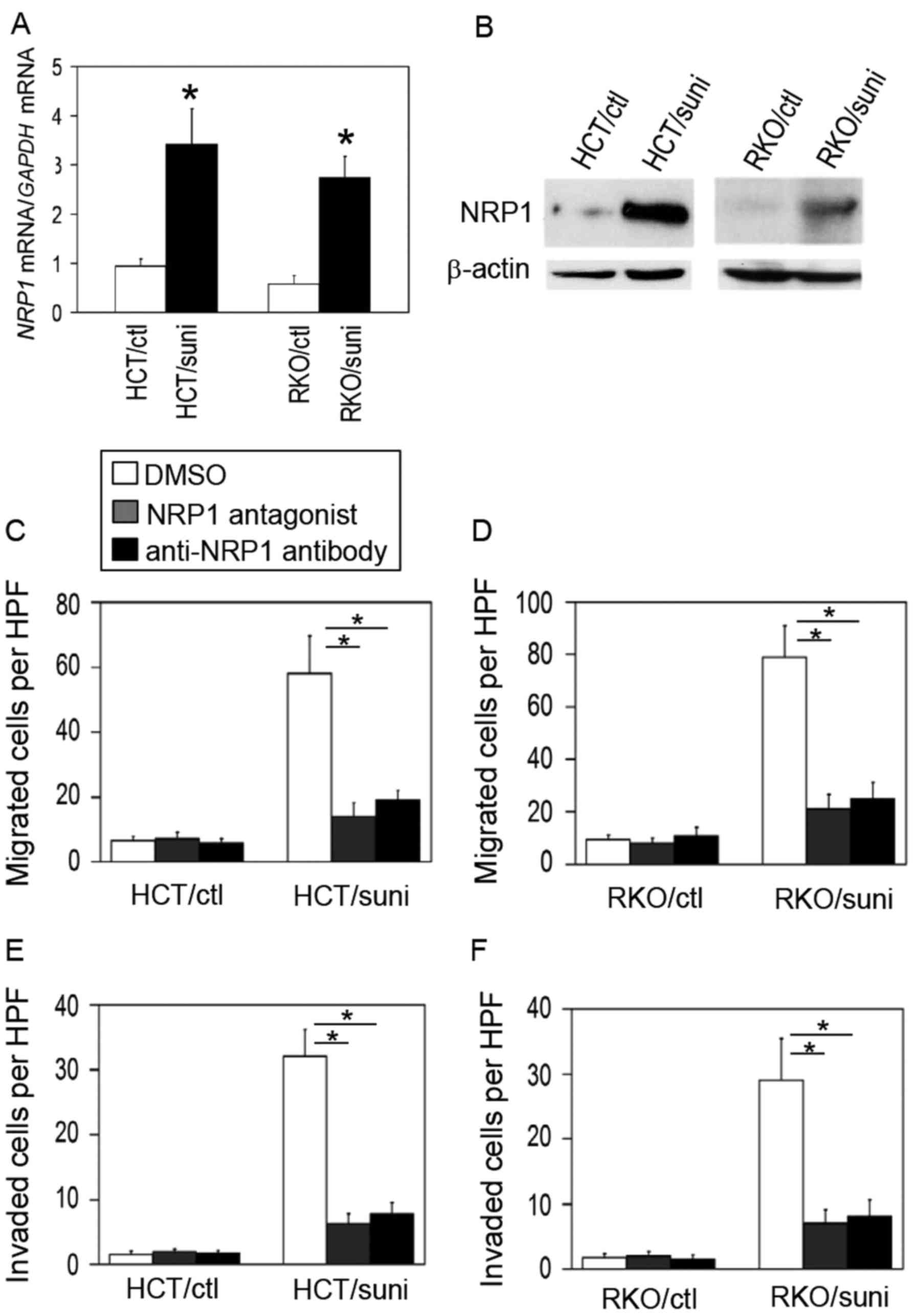
were evaluated in the sunitinib-adapted cells. As shown in Fig. 5A and B, NRP1 expression was
increased at the mRNA and protein levels in the sunitinib-adapted
cells compared with the control cells.
To block NRP1 function, two different types of
inhibitors [an NRP1 antagonistic peptide, which binds to the NRP1
extracellular domain (31), and a
neutralizing anti-NRP1 antibody] were used. Treatment with the NRP1
blockers significantly decreased the migration and invasion
activities of the sunitinib-adapted cells but not the control cells
when compared with the respective DMSO-treated controls (Fig. 5C–F). These results indicate that
the sunitinib-adapted cells switched from a VEGF-R-dependent
phenotype to an NRP1-dependent one.
Identification of the NRP1 partner
receptor in the sunitinib-adapted cells
The NRP1-dependent evasive mechanism in the
sunitinib-adapted cells was investigated. NRP1 functions as a
co-receptor for several growth factor receptors, including cMet,
TGFβ-R, FGF-R and PDGF-R (28–30).
To explore the partner receptor of NRP1, receptor kinase inhibitors
specific for cMet (capmatinib), TGFβ-R (GW788388), FGF-R
(SSR128129E) and PDGF-R (CP-673451) were used. As shown in Fig. 6A and B, the inhibition of cMet, but
not of TGFβ-R, FGF-R and PDGF-R, significantly reduced the
migration activity of the sunitinib-adapted cells compared with
that of the DMSO-treated control. By contrast, blocking cMet did
not affect the control cells.
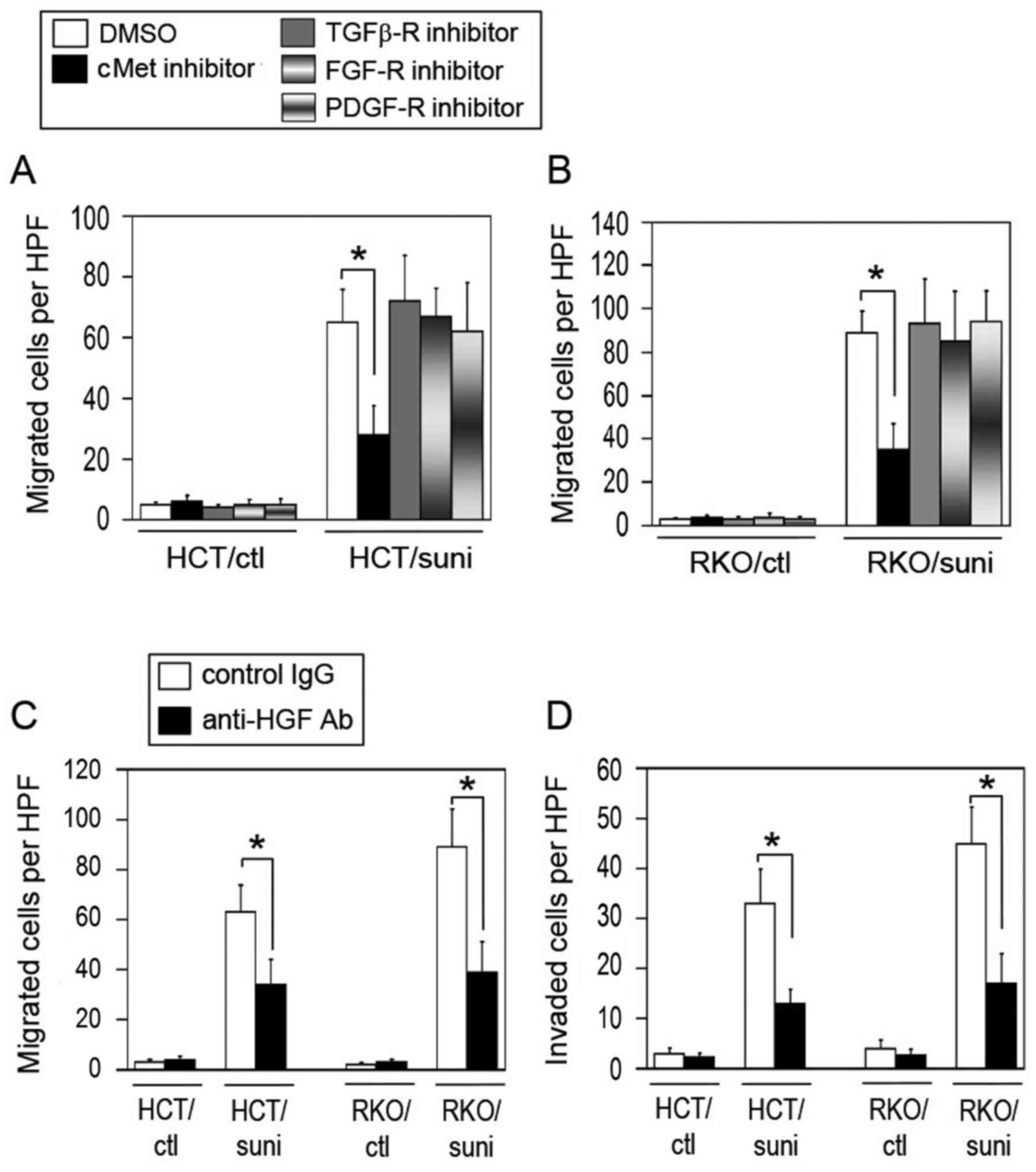 | Figure 6cMet participates in the evasive
activation of the sunitinib-adapted cells. Effect of the inhibition
of cMet, TGFβ-R, FGF-R or PDGF-R on the migration activity in the
sunitinib-adapted (A) HCT116 and (B) RKO cells. The control and
sunitinib-adapted cells were pretreated for 24 h with vehicle
(DMSO), cMet inhibitor (capmatinib), TGFβ-R inhibitor (GW788388),
FGF-R inhibitor (SSR128129E) or PDGF-R inhibitor (CP-673451), and
the migration activity of the cells was analyzed (n=4–5; mean ±
standard deviation). *P<0.01 vs. the respective
DMSO-treated control cells. (C and D) Blockade of tumor
cell-derived HGF suppresses the migration and invasion activities
of the sunitinib-adapted cells. The control and sunitinib-adapted
cells were pretreated for 24 h with a control non-immune IgG or
anti-HGF neutralizing antibody, then their (C) migration and (D)
invasion activities were evaluated (n=4–5; mean ± standard
deviation). *P<0.01 vs. the respective control
IgG-treated cells. cMet, tyrosine-protein kinase Met; TGFβ-R,
transforming growth factor β receptor; FGF-R, fibroblast growth
factor receptor; PDGF-R, platelet-derived growth factor receptor;
DMSO, dimethylsulfoxide; HGF, hepatocyte growth factor; IgG,
immunoglobulin G; Ab, antibody; HCT/ctl, control HCT116 cells;
RKO/ctl, control RKO cells; HCT/suni, sunitinib-adapted HCT116
cells; RKO/suni, sunitinib-adapted RKO cells; HPF, high power
field. |
To further confirm the involvement of cMet in the
evasive activation of the sunitinib-adapted cells, the effect of
blocking the HGF derived from the cells was examined. As shown in
Fig. 6C and D, the blockade of HGF
with an anti-HGF neutralizing antibody significantly reduced the
migration and invasion activities of the sunitinib-adapted cells
but not the control cells compared with the respective IgG-treated
control cells. These results are similar to those for cMet
inhibition (Fig. 6A and B).
NRP1-dependent cMet activation in the
sunitinib-adapted cells
The results observed in Figs. 5 and 6 suggest that NRP1 participated in cMet
activation in the sunitinib-adapted cells. To test this, the cMet
phosphorylation levels were evaluated by immunoblot analysis. As
shown in Fig. 7A, the levels of
phospho-cMet were increased in the sunitinib-adapted cells compared
with the control cells. Whether NRP1 was required for cMet
phosphorylation in the sunitinib-adapted cells was then
investigated. Blocking NRP1 markedly reduced the levels of
phosphorylated cMet (Fig. 7B) but
did not affect the expression levels of total cMet in the
sunitinib-adapted cells, indicating that cMet activation is
dependent on NRP1.
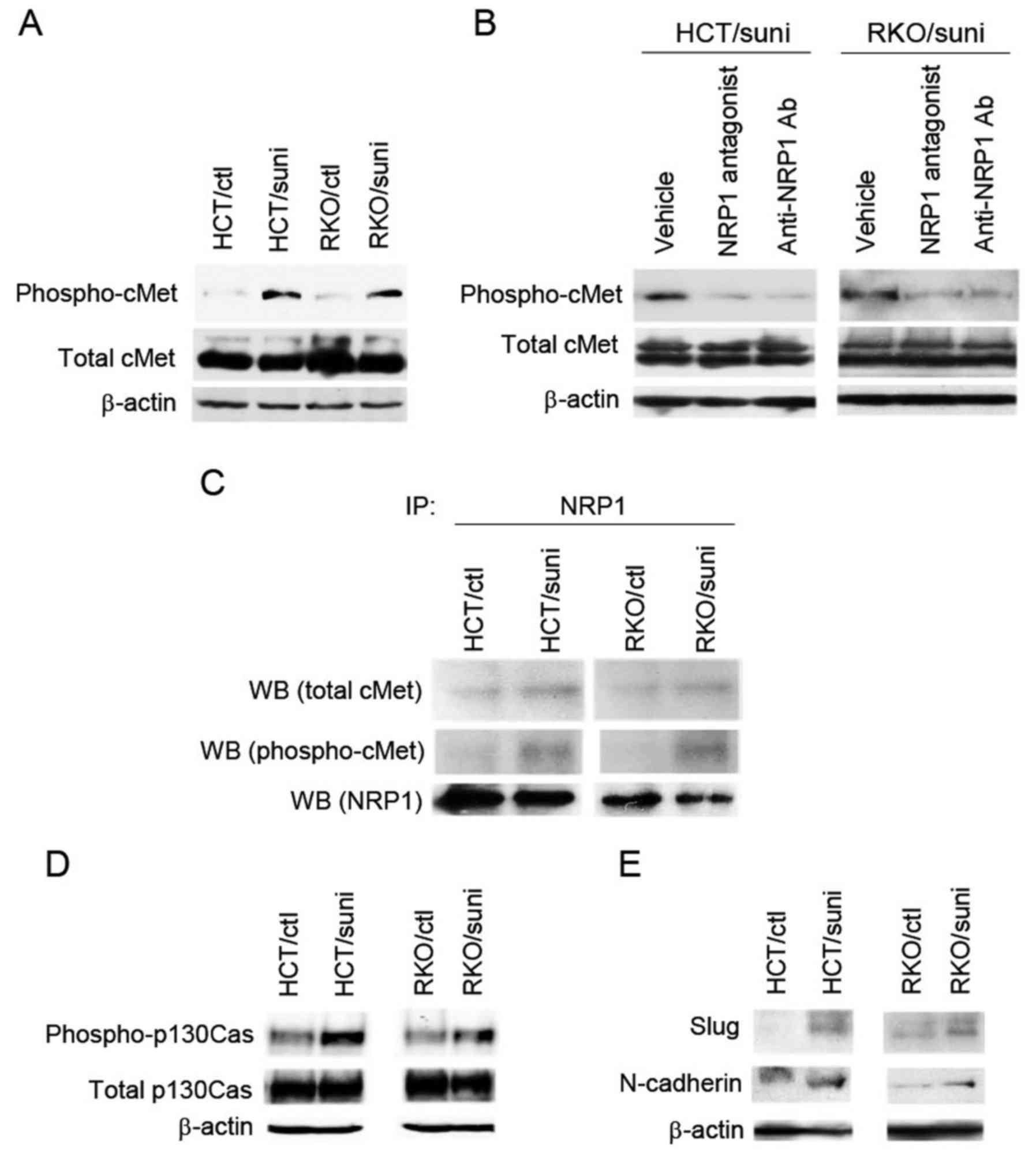 | Figure 7NRP1-dependent cMet activation in the
sunitinib-adapted cells. (A) Expression levels of phosphorylated
cMet and total cMet were measured by western blot analysis. (B)
NRP1-dependent cMet phosphorylation in the sunitinib-adapted cells.
The sunitinib-adapted cells were pretreated for 2 h with vehicle
(DMSO), NRP1 antagonist or anti-NRP1 neutralizing antibody, and
then analyzed by western blotting using antibodies against
phospho-cMet and total cMet, respectively. (C) Interaction of NRP1
with phosphorylated cMet in the sunitinib-adapted cells. NRP1 was
immunoprecipitated with anti-NRP1 antibody using cell lysates from
the control and the sunitinib-adapted cells. Immunoblotting was
performed using antibodies against phospho-cMet, total cMet and
NRP1. Western blot analyses were also conducted to determine the
expression levels of (D) phosphorylated p130Cas and total p130Cas
and (E) Slug and N-cadherin. (A, B, D and E) The levels of β-actin
are shown as a loading control. NRP1, neuropilin-1; cMet,
tyrosine-protein kinase Met; DMSO, dimethylsulfoxide; Ab, antibody;
HCT/ctl, control HCT116 cells; RKO/ctl, control RKO cells;
HCT/suni, sunitinib-adapted HCT116 cells; RKO/suni,
sunitinib-adapted RKO cells; IP, immunoprecipitation; WB, western
blot. |
To further investigate whether NRP1 physically
interacted with cMet and was associated with its activation in the
sunitinib-adapted cells, immunoprecipitation in combination with
immunoblot analysis was performed. NRP1 protein was
immunoprecipitated, and then the levels of co-precipitated cMet and
its phosphorylation were measured by immunoblot analysis. The
association of NRP1 with cMet was increased in the
sunitinib-adapted cells compared with the control cells (Fig. 7C, upper panel). Importantly, the
co-precipitation of phosphorylated cMet was observed only in the
sunitinib-adapted cells (Fig. 7C,
middle panel). These results indicate that NRP1 interacted with and
was involved in cMet activation in the sunitinib-adapted cells.
The activation of the downstream effector of NRP1,
p130Cas, which is an important NRP1 signaling molecule that
activates cell motility (32) was
also investigated. The levels of phospho-p130Cas were elevated in
the sunitinib-adapted cells compared with the control cells
(Fig. 7D, upper panel), while the
levels of total p130Cas did not differ between the control and
sunitinib-adapted cells (Fig. 7D,
middle panel). As previous studies demonstrated that the activation
of p130Cas is associated with the induction of the mesenchymal
markers Slug and N-cadherin (33,34),
the levels of these markers were evaluated in the sunitinib-adapted
cells. Consistent with the activation of p130Cas, Slug and
N-cadherin expression levels were increased in the
sunitinib-adapted cells compared with the control cells (Fig. 7E).
Effect of dual blockade of VEGF-R and
cMet on the evasive activation
Based on the aforementioned findings, it was
hypothesized that the dual blockade of VEGF-R and cMet would
inhibit the NRP1/HGF/cMet adaptive pathway under VEGF-R inhibition
conditions. To test this, foretinib was used; this compound
inhibits VEGF-R and cMet activities with nanomolar potency
(24). Short-term treatment with
foretinib for 48 h did not activate the migration and invasion
activities of HCT116 and RKO cells (Fig. 8A and B). By contrast, treatment
with sunitinib for 48 h significantly elevated these activities
compared with those of the DMSO-treated control (Fig. 8A and B).
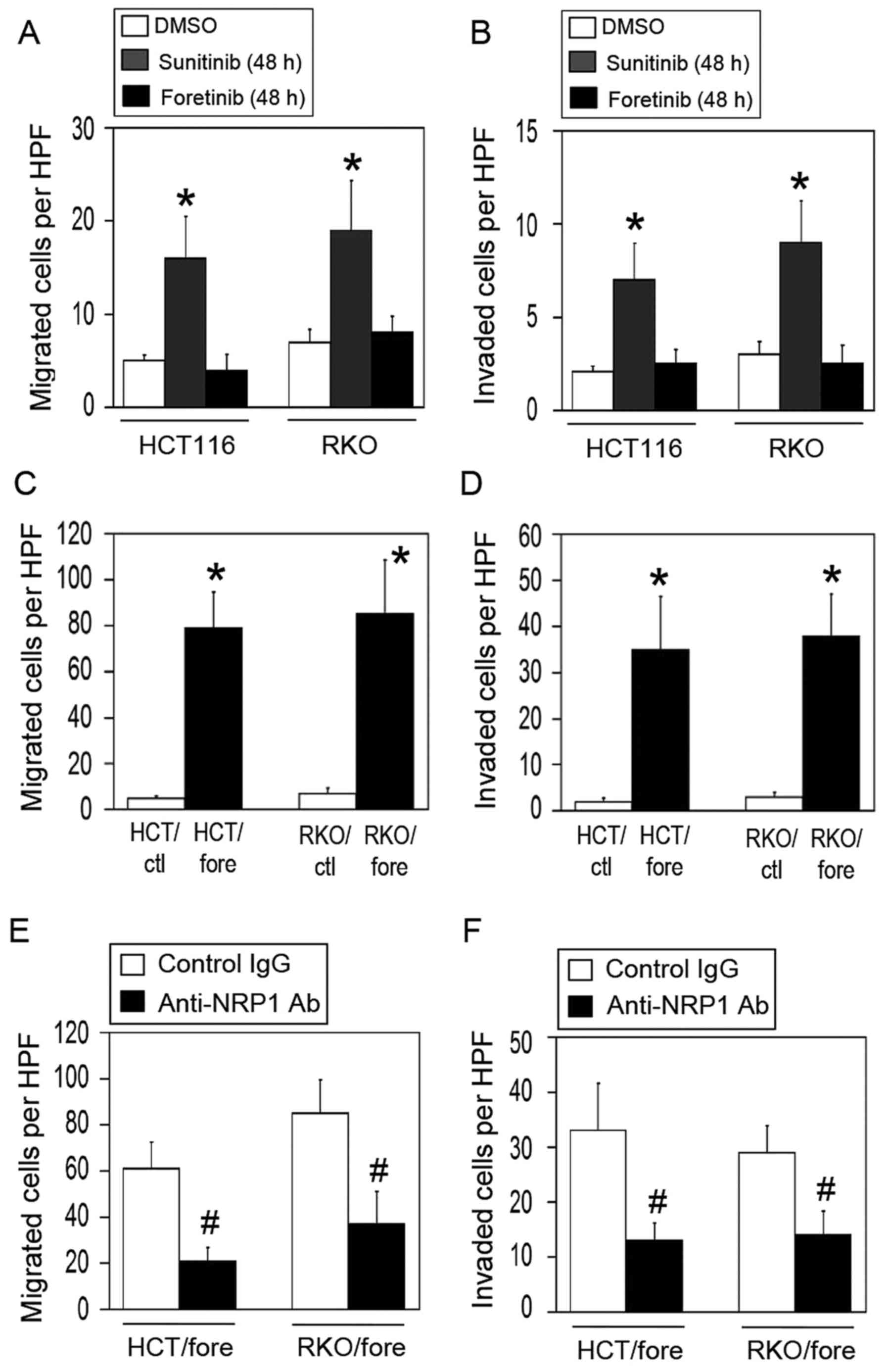 | Figure 8Effect of dual blockade of VEGF-R and
cMet on the evasive activation of the colon cancer cells. (A and B)
Effect of short-term treatment with sunitinib or foretinib on the
migration and invasion activities of colon cancer cells. Following
the exposure of parental HCT116 and RKO cells to sunitinib or
foretinib for 48 h, the (A) migration and (B) invasion activities
of the cells were evaluated (n=4–6; mean ± standard deviation).
*P<0.01 vs. the respective DMSO-treated control
cells. (C and D) Effect of long-term treatment with foretinib on
the migration and invasion activities of colon cancer cells.
Parental HCT116 and RKO cells were chronically exposed to foretinib
for 3 months, and the (C) migration and (D) invasion activities of
the cells were evaluated (n=4–6; mean ± standard deviation). HPF,
high power field. *P<0.01 vs. the respective
DMSO-treated control cells. (E and F) Effect of NRP1 blockade on
the evasive activation of the foretinib-adapted cells. The
foretinib-adapted cells were pretreated for 24 h with control
(non-immune) IgG or anti-NRP1 neutralizing antibody, and the (E)
migration and (F) invasion activities of the cells were evaluated
(n=4–6; mean ± standard deviation). #P<0.01 vs. the
respective control IgG-treated cells. VEGF-R, vascular endothelial
growth factor receptor; cMet, tyrosine-protein kinase Met; NRP1,
neuropilin-1; IgG, immunoglobulin G; DMSO, dimethylsulfoxide;
HCT/ctl, control HCT116 cells; RKO/ctl, control RKO cells;
HCT/fore, foretinib-adapted HCT116 cells; RKO/fore,
foretinib-adapted RKO cells; HPF, high power field. |
The effect of chronic foretinib exposure for 3
months on the evasive activities of the cells was then evaluated. A
foretinib-adapted cell model (HCT/fore and RKO/fore) similar to the
sunitinib cell model was established. However, the
foretinib-adapted cells exhibited a marked activation of migration
and invasion, similar to the sunitinib-adapted cells (Fig. 8C and D).
Finally, whether blocking NRP1 was effective in
reducing the migration and invasion of the foretinib-adapted cells
was examined. Notably, the blockade of NRP1 significantly decreased
the migration and invasion activities in these cells compared with
the IgG-treated control (Fig. 8E and
F), suggesting that the foretinib-adapted cells retained a
dependence on NRP1.
Discussion
In the present study, the direct effects of two
different VEGF pathway-targeting drugs, sunitinib and bevacizumab,
on the evasive adaptation of colon cancer cells were compared. The
results demonstrated that sunitinib activated the migration and
invasion activities of the cells more strongly than did
bevacizumab, and by a distinct mechanism. Under the bevacizumab
VEGF-A ligand-blocking conditions, cancer cells remained dependent
upon the VEGF-R1 and -R3 pathways, as their evasive abilities were
significantly decreased by the specific inhibitors of VEGF-R1 and
-R3 (Fig. 3F and G). This result
is supported by a previous study (21), in which a pan-VEGF-R inhibitor,
SU5416, suppresses the motility of bevacizumab-adapted cells. By
contrast, when all VEGF-Rs were inhibited by sunitinib, cancer
cells became dependent on NRP1 and switched from a VEGF-R-dependent
pathway to an alternative adaptive one (the NRP1/HGF/cMet pathway)
(Figs. 6 and 7).
The present study revealed that NRP1 was required
for cMet activation under conditions where all VEGF-Rs were
inhibited by sunitinib. Previous studies have demonstrated that
cMet is a critical receptor for the induction of evasive resistance
to VEGF/VEGF-R inhibitors in several cancer models (35–37);
however, NRP1 did not appear participate in the evasive mechanisms.
In a pancreatic neuroendocrine tumor model treated with sunitinib,
tumor cell local invasion was accelerated by cMet activation
directly induced by hypoxic stress, but not by the direct action of
sunitinib on cancer cells (36).
In addition, VEGF-R2, but not VEGF-R1, usually interacts with and
inactivates cMet in mouse models of glioblastoma multiforme, and
blocking VEGF-R2 induces the dissociation of the two receptors and
thus activates cMet, leading to increased invasive and metastatic
activities in vitro and in vivo (35). Notably, NRP1 is not involved in the
VEGF-R2/cMet system (35).
NRP1 has been suggested to serve critical roles in
cancer progression, since NRP1 overexpression increased the
migration and invasion activities of gastric and esophageal
squamous cell carcinoma cell lines (38–40).
Additionally, NRP1 is preferentially expressed in metastatic cells.
For example, NRP1 expression has been detected in MDA-MB-231
metastatic breast cancer and MBA-MB-435 melanoma cells, but not in
the MDA-MB-453 non-metastatic cell line and certain non-metastatic
tumors (41,42).
The possible importance of NRP1 in the malignant
phenotypes of tumor cells is supported by several clinical studies,
including those regarding tumor progression and the poor survival
of patients (43–47). In advanced colorectal carcinomas,
patients with tumors expressing high levels of NRP1 exhibited a
significantly higher incidence of lymph node or liver metastasis
than did those with tumors expressing low levels of NRP1 (44). Increased NRP1 expression occurs in
gastrointestinal tumors, and this upregulation appears to parallel
the invasive behavior of the tumor (43). The survival time of patients with
tumors expressing high NRP1 levels is significantly shorter than
that of patients with low NRP1 levels (45–47).
Therefore, NRP1 is suggested to be a prognostic marker in several
cancers and an important target for cancer therapy.
During anti-angiogenic therapy using VEGF/VEGF-R
inhibitors, antitumor effects are mainly caused by the reduction of
tumor microvessel density and the resulting hypoxic conditions. It
is widely accepted that hypoxic stress selects a sub-population of
tumor cells and activates several phenotypic changes by which tumor
cells are able to survive and progress their malignancy under the
hypoxic conditions. However, studies have demonstrated that
VEGF/VEGF-R inhibitors directly act on tumor cells and accelerate
their malignant phenotypes, independent of hypoxia (18–22).
Notably, Han et al (48)
demonstrated that simple hypoxic stress is not sufficient to
trigger evasive resistance whereas VEGF-R inhibition induces
resistance in renal cell carcinoma cells. Thus, malignant
phenotypes may be accelerated additively or synergistically by the
direct effects of VEGF/VEGF-R inhibitors and hypoxic stress in the
tumor microenvironment in vivo.
In the present study, treatment with sunitinib was
demonstrated to markedly accelerate the evasive activities of the
cells compared with bevacizumab treatment. This is in agreement
with previous studies documenting that anti-VEGF antibody therapy
does not affect metastasis in mouse tumor models, whereas VEGF-R
tyrosine kinase inhibitors, such as sunitinib, promote metastasis
(49,50). Additionally, head-to-head
comparisons between bevacizumab and sunitinib treatments have been
conducted. In a randomized phase III clinical trial in patients
with advanced breast cancer, bevacizumab was clinically superior to
sunitinib (51). In a randomized
phase II trial in patients with advanced renal-cell carcinoma,
treatment with bevacizumab produced better efficacy results than
were obtained with sunitinib (52).
Based on the findings of the present study and
evidence from several reports indicating that activation of the
cMet pathway is critical for evasive resistance to VEGF-R-targeting
therapy (35–37), it appears possible that a dual
inhibitor of VEGF-R and cMet, such as foretinib, may overcome the
resistance to VEGF-R inhibitors. Indeed, short-term treatment with
foretinib completely suppressed the evasive activation of colon
cancer cells in comparison with sunitinib treatment (Fig. 8A and B). However, chronic exposure
to foretinib induced evasive activation similar to sunitinib
treatment (Fig. 8C and D).
Notably, blocking NRP1 suppressed this activation (Fig. 8E and F). These findings suggest
that under conditions involving the chronic inhibition of cMet and
VEGF-R, cancer cells may switch from the NRP1/cMet pathway to
another pathway that remains to be dependent upon NRP1. Therefore,
the findings of the present study suggest that targeting NRP1 may
represent a promising approach for the treatment of cancer with
drugs targeting VEGF-R and cMet. Further studies are required to
understand the exact mechanism and molecular interactions of NRP1
under chronic cMet and VEGF-R inhibition conditions.
In summary, it is concluded that
VEGF/VEGF-R-targeting drugs directly induced evasive adaptation in
colon cancer cells independently of hypoxia. The present study
demonstrated that sunitinib markedly activated an evasive phenotype
through an alternative NRP1/HGF/cMet axis, while bevacizumab
accomplished this through redundant VEGF/VEGF-R1 and VEGF-R3
pathways.
Acknowledgments
Not applicable.
Notes
[1]
Funding
This study was supported in part by grants from
Grants-in-Aid for JSPS fellows (grant no. 16J04163 to CT),
Grants-in-Aid for Scientific Research (grant no. 15H04931 to STK)
from JSPS, and the Princess Takamatsu Cancer Research Fund (grant
no. 14-24611 to STK).
[2] Availability
of data and materials
The datasets generated during and/or analyzed during
the current study are available from the corresponding author on
reasonable request.
[3] Authors'
contributions
CT carried out the cellular, biochemical and
molecular biological studies and drafted the manuscript. NY, HN and
TU performed cellular studies. AO, KH and TN contributed to
experimental design and helped to draft the manuscript. STK
designed and directed the study, and helped to draft the
manuscript. All authors read and approved the final manuscript.
[4] Ethics
approval and consent to participate
Not applicable.
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Bergers G and Hanahan D: Modes of
resistance to anti-angiogenic therapy. Nat Rev Cancer. 8:592–603.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ellis LM and Reardon DA: Is there really a
yin and yang to VEGF-targeted therapies? Lancet Oncol. 11:809–811.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ferrara N: Pathways mediating
VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev.
21:21–26. 2010. View Article : Google Scholar
|
|
4
|
Ribatti D: Tumor refractoriness to
anti-VEGF therapy. Oncotarget. 19:46668–46677. 2016.
|
|
5
|
Jayson GC, Kerbel R, Ellis LM and Harris
AL: Antiangiogenic therapy in oncology: Current status and future
directions. Lancet. 388:518–529. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jayson GC, Hicklin DJ and Ellis LM:
Antiangiogenic therapyevolving view based on clinical trial
results. Nat Rev Clin Oncol. 9:297–303. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Meadows KL and Hurwitz HI: Anti-VEGF
therapies in the clinic. Cold Spring Harb Perspect Med.
2:a0065772012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bottsford-Miller JN, Coleman RL and Sood
AK: Resistance and escape from antiangiogenesis therapy: Clinical
implications and future strategies. J Clin Oncol. 30:4026–4034.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason
GA, Christensen JG and Kerbel RS: Accelerated metastasis after
short-term treatment with a potent inhibitor of tumor angiogenesis.
Cancer Cell. 15:232–239. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pàez-Ribes M, Allen E, Hudock J, Takeda T,
Okuyama H, Viñals F, Inoue M, Bergers G, Hanahan D and Casanovas O:
Antiangiogenic therapy elicits malignant progression of tumors to
increased local invasion and distant metastasis. Cancer Cell.
15:220–231. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ebos JM and Kerbel RS: Antiangiogenic
therapy: Impact on invasion, disease progression, and metastasis.
Nat Rev Clin Oncol. 8:210–221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kieran MW, Kalluri R and Cho YJ: The VEGF
pathway in cancer and disease: Responses, resistance, and the path
forward. Cold Spring Harb Perspect Med. 2:a0065932012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Moserle L, Jiménez-Valerio G and Casanovas
O: Antiangiogenic therapies: Going beyond their limits. Cancer
Discov. 4:31–41. 2014. View Article : Google Scholar
|
|
14
|
Piao Y, Liang J, Holmes L, Henry V, Sulman
E and de Groot JF: Acquired resistance to anti-VEGF therapy in
glioblastoma is associated with a mesenchymal transition. Clin
Cancer Res. 19:4392–4403. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Graeber TG, Osmanian C, Jacks T, Housman
DE, Koch CJ, Lowe SW and Giaccia AJ: Hypoxia-mediated selection of
cells with diminished apoptotic potential in solid tumours. Nature.
379:88–91. 1996. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Semenza GL: Hypoxia-inducible factors:
Mediators of cancer progression and targets for cancer therapy.
Trends Pharmacol Sci. 33:207–214. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Semenza GL: Oxygen sensing,
hypoxia-inducible factors, and disease pathophysiology. Annu Rev
Pathol. 9:47–71. 2014. View Article : Google Scholar
|
|
18
|
Goel HL and Mercurio AM: VEGF targets the
tumour cell. Nat Rev Cancer. 13:871–882. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Simon T, Gagliano T and Giamas G: Direct
effects of anti-angiogenic therapies on tumor cells: VEGF
signaling. Trends Mol Med. 23:282–292. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Videira PA, Piteira AR, Cabral MG, Martins
C, Correia M, Severino P, Gouveia H, Carrascal M, Almeida JF,
Trindade H, et al: Effects of bevacizumab on autocrine VEGF
stimulation in bladder cancer cell lines. Urol Int. 86:95–101.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fan F, Samuel S, Gaur P, Lu J, Dallas NA,
Xia L, Bose D, Ramachandran V and Ellis LM: Chronic exposure of
colorectal cancer cells to anti-VEGF mAb promotes compensatory
pathways that mediate tumor cell migration. Br J Cancer.
104:1270–1277. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yamagishi N, Kondo TS, Masuda K, Nishida
K, Kuwano Y, Dang DT, Dang LH, Nikawa T and Rokutan K: Chronic
inhibition of tumor cell-derived VEGF enhances the malignant
phenotype of colorectal cancer cells. BMC Cancer. 13:2292013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang D, Ding Y, Li Y, Luo WM, Zhang ZF,
Snider J, Vandenbeldt K, Qian CN and Teh BT: Sunitinib acts
primarily on tumor endothelium rather than tumor cells to inhibit
the growth of renal cell carcinoma. Cancer Res. 70:1053–1062. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shah MA, Wainberg ZA, Catenacci DV,
Hochster HS, Ford J, Kunz P, Lee FC, Kallender H, Cecchi F, Rabe
DC, et al: Phase II study evaluating 2 dosing schedules of oral
foretinib (GSK1363089), cMet/VEGFR2 inhibitor, in patients with
metastatic gastric cancer. PLoS One. 8:e540142013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pfaffl MW: Relative quantification.
Real-time PCR. Dorak MT: 1st edition. Taylor & Francis; London:
2006
|
|
26
|
Bae DG, Kim TD, Li G, Yoon WH and Chae CB:
Anti-flt1 peptide, a vascular endothelial growth factor receptor
1-specific hexapeptide, inhibits tumor growth and metastasis. Clin
Cancer Res. 11:2651–2661. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chang YW, Su CM, Su YH, Ho YS, Lai HH,
Chen HA, Kuo ML, Hung WC, Chen YW, Wu CH, et al: Novel peptides
suppress VEGFR-3 activity and antagonize VEGFR-3-mediated oncogenic
effects. Oncotarget. 5:3823–3835. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Parker MW, Guo HF, Li X, Linkugel AD and
Vander Kooi CW: Function of members of the neuropilin family as
essential pleiotropic cell surface receptors. Biochemistry.
51:9437–9446. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Prud'homme GJ and Glinka Y: Neuropilins
are multifunctional coreceptors involved in tumor initiation,
growth, metastasis and immunity. Oncotarget. 3:921–939. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rizzolio S and Tamagnone L: Multifaceted
role of neuropilins in cancer. Curr Med Chem. 18:3563–3575. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Binétruy-Tournaire R, Demangel C, Malavaud
B, Vassy R, Rouyre S, Kraemer M, Plouët J, Derbin C, Perret G and
Mazié JC: Identification of a peptide blocking vascular endothelial
growth factor (VEGF)-mediated angiogenesis. EMBO J. 19:1525–1533.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Evans IM, Yamaji M, Britton G, Pellet-Many
C, Lockie C, Zachary IC and Frankel P: Neuropilin-1 signaling
through p130Cas tyrosine phosphorylation is essential for growth
factor-dependent migration of glioma and endothelial cells. Mol
Cell Biol. 31:1174–1185. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bisaro B, Montani M, Konstantinidou G,
Marchini C, Pietrella L, Iezzi M, Galiè M, Orso F, Camporeale A,
Colombo SM, et al: p130Cas/cyclooxygenase-2 axis in the control of
mesenchymal plasticity of breast cancer cells. Breast Cancer Res.
14:R1372012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mui KL, Bae YH, Gao L, Liu SL, Xu T,
Radice GL, Chen CS and Assoian RK: N-cadherin induction by ECM
stiffness and FAK overrides the spreading requirement for
proliferation of vascular smooth muscle cells. Cell Rep.
10:1477–1486. 2015. View Article : Google Scholar
|
|
35
|
Lu KV, Chang JP, Parachoniak MM, Aghi MK,
Meyronet D, Isachenko N, Fouse SD, Philips JJ, Cheresh DA, Park M,
et al: VEGF inhibits tumor cell invasion and mesenchymal transition
through a MET/VEGFR2 complex. Cancer Cell. 22:21–35. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sennino B, Ishiguro-Oonuma T, Wei Y,
Naylor RM, Williamson CW, Bhagwandin V, Tabruyn SP, You WK, Chapman
HA, Christensen JG, et al: Suppression of tumor invasion and
metastasis by concurrent inhibition of c-Met and VEGF signaling in
pancreatic neuroendocrine tumors. Cancer Discov. 2:270–287. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mezquita B, Pineda E, Mezquita J, Mezquita
P, Pau M, Codony-Servat J, Martínez-Balibrea E, Mora C, Maurel J
and Mezquita C: LoVo colon cancer cells resistant to oxaliplatin
overexpress c-MET and VEGFR-1 and respond to VEGF with
dephosphorylation of c-MET. Mol Carcinog. 55:411–419. 2016.
View Article : Google Scholar
|
|
38
|
Peng Y, Liu YM, Li LC, Wang LL and Wu XL:
MicroRNA-338 inhibits growth, invasion and metastasis of gastric
cancer by targeting NRP1 expression. PLoS One. 9:e944222014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shi F, Shang L, Pan BQ, Wang XM, Jiang YY,
Hao JJ, Zhang Y, Cai Y, Xu X, Zhan QM, et al: Calreticulin promotes
migration and invasion of esophageal cancer cells by upregulating
neuropilin-1 expression via STAT5A. Clin Cancer Res. 20:6153–6162.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hong TM, Chen YL, Wu YY, Yuan A, Chao YC,
Chung YC, Wu MH, Yang SC, Pan SH, Shih JY, et al: Targeting
neuropilin 1 as an antitumor strategy in lung cancer. Clin Cancer
Res. 13:4759–4768. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bachelder RE, Crago A, Chung J, Wendt MA,
Shaw LM, Robinson G and Mercurio AM: Vascular endothelial growth
factor is an autocrine survival factor for neuropilin-expressing
breast carcinoma cells. Cancer Res. 61:5736–5740. 2001.PubMed/NCBI
|
|
42
|
Soker S, Takashima S, Miao HQ, Neufeld G
and Klagsbrun M: Neuropilin-1 is expressed by endothelial and tumor
cells as an isoform-specific receptor for vascular endothelial
growth factor. Cell. 92:735–745. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hansel DE, Wilentz RE, Yeo CJ, Schulick
RD, Montgomery E and Maitra A: Expression of neuropilin-1 in
high-grade dysplasia, invasive cancer, and metastases of the human
gastrointestinal tract. Am J Surg Pathol. 28:347–356. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ochiumi T, Kitadai Y, Tanaka S, Akagi M,
Yoshihara M and Chayama K: Neuropilin-1 is involved in regulation
of apoptosis and migration of human colon cancer. Int J Oncol.
29:105–116. 2006.PubMed/NCBI
|
|
45
|
Ben Q, Zheng J, Fei J, An W, Li P, Li Z
and Yuan Y: High neuropilin 1 expression was associated with
angiogenesis and poor overall survival in resected pancreatic
ductal adenocarcinoma. Pancreas. 43:744–749. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lee SW, Lee JE, Yoo CY, Ko MS, Park CS and
Yang SH: NRP-1 expression is strongly associated with the
progression of pituitary adenomas. Oncol Rep. 32:1537–1542. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhu H, Cai H, Tang M and Tang J:
Neuropilin-1 is overexpressed in osteosarcoma and contributes to
tumor progression and poor prognosis. Clin Transl Oncol.
16:732–738. 2014. View Article : Google Scholar
|
|
48
|
Han KS, Raven PA, Frees S, Gust K, Fazli
L, Ettinger S, Hong SJ, Kollmannsberger C, Gleave ME and So AI:
Cellular adaptation to VEGF-targeted antiangiogenic therapy induces
evasive resistance by overproduction of alternative endothelial
cell growth factors in renal cell carcinoma. Neoplasia. 17:805–816.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Singh M, Couto SS, Forrest WF, Lima A,
Cheng JH, Molina R, Long JE, Hamilton P, McNutt A, Kasman I, et al:
Anti-VEGF antibody therapy does not promote metastasis in
genetically engineered mouse tumour models. J Pathol. 227:417–430.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chung AS, Kowanetz M, Wu X, Zhuang G, Ngu
H, Finkle D, Komuves L, Peale F and Ferrara N: Differential drug
class-specific metastatic effects following treatment with a panel
of angiogenesis inhibitors. J Pathol. 227:404–416. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Robert NJ, Saleh MN, Paul D, Generali D,
Gressot L, Copur MS, Brufsky AM, Minton SE, Giguere JK, Smith JW
II, et al: Sunitinib plus paclitaxel versus bevacizumab plus
paclitaxel for first-line treatment of patients with advanced
breast cancer: A phase III, randomized, open-label trial. Clin
Breast Cancer. 11:82–92. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Négrier S, Gravis G, Pérol D, Chevreau C,
Delva R, Bay JO, Blanc E, Ferlay C, Geoffrois L, Rolland F, et al:
Temsirolimus and bevacizumab, or sunitinib, or interferon alfa and
bevacizumab for patients with advanced renal cell carcinoma
(TORAVA): A randomised phase 2 trial. Lancet Oncol. 12:673–680.
2011. View Article : Google Scholar : PubMed/NCBI
|