Introduction
Bladder cancer (BCa) is one of the most prevalent
urinary tract malignancies worldwide (1,2).
Effective diagnostic and therapeutic methods are available for
patients with superficial disease, and the majority of these
patients respond well to treatment; however, this type of cancer
frequently recurs. Invasive and metastatic BCa remains a lethal
malignancy (3,4). Therefore, novel treatment strategies
based on new molecular networks are urgently needed to improve the
poor prognosis of patients with BCa. Programmed cell death protein
4 (PDCD4), a tumor suppressor, localizes to the nucleus and is
associated with tumor progression and prognosis in a number of
human cancers (5,6).
Overexpression of PDCD4 has been shown to inhibit
cell proliferation, and downregulation of PDCD4 has been identified
in several cancerous tissues compared with adjacent normal tissues.
PDCD4 plays a major role in a number of biological processes that
may lead to tumor development. A variety of apoptosis inducers
upregulate the expression of PDCD4, thus affecting multiple
signaling pathways (7–9). c-Jun N-terminal kinase (JNK) is a
member of the mitogen-activated protein kinase (MAPK) family, and
its activation is a requirement for the development of various
cancers (10,11). More recently, the JNK signaling
pathway was found to play an important role in the proliferation
and apoptosis of human BCa cells (12–14).
Previous studies (15–17) have demonstrated that PDCD4 gene
knockdown may increase the phosphorylation of c-Jun; hence, the
transformation-suppressor function of PDCD4 may be due to the
inhibition of c-Jun activity. Overexpression of miR-21
significantly promotes cell migration and invasion by targeting
PDCD4 and activating its downstream c-Jun N-terminal kinase
signaling pathway. These data indicate that PDCD4-related signal
transduction through the JNK pathway may be a novel therapeutic
target. However, the number of studies on PDCD4 and JNK signaling
in BCa is limited, and the specific mechanism of action requires
further elucidation. The aim of the present study was to evaluate
the effects of PDCD4 on cell proliferation and apoptosis, and
determine whether the mechanism of tumor cell inhibition by PDCD4
partially involves activation of the JNK/c-Jun pathway.
Materials and methods
Cell transfection
In the present study, EJ cells were used, which have
been found to be contaminated by T24 cells, with the resultant cell
line being a mixed BCa type. A large number of studies still use
these cells for BCa research, and the contamination does not affect
the outcomes of the experiments (18,19).
The BCa cells were purchased from the Shanghai Cell Bank of Chinese
Academy of Sciences (Shanghai, China), and were maintained in
RPMI-1640 (HyClone; GE Healthcare Life Sciences, Logan, UT, USA)
supplemented with 10% (v/v) heat-inactivated fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific Inc., Waltham, MA, USA)
containing 100 μ/ml penicillin and 100 mg/ml streptomycin in
a humidified atmosphere of 5% CO2 at 37°C. Cells were
harvested following a brief incubation in 0.02% (w/v) EDTA in
phosphate-buffered saline (PBS). The cells were transfected with
plasmids (pDsRed2-N1 and pDsRed2-N1-PDCD4) and assigned to three
groups for the experiments as follows: BCa, empty vector (mock) and
PDCD4 transfection groups.
PDCD4 expression detection
The expression of PDCD4 was analyzed by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis and western blotting. Briefly, at 48 h after transfection,
total RNA was extracted from cells using the Qiagen RNeasy kit
(Qiagen, Basel, Switzerland), and reverse-transcribed with random
primers for complementary DNA (cDNA) synthesis using the Reverse
Transcription System (Promega, Madison, WI, USA). Primers were
designed and synthesied as previously reported (20) and β-actin was used as a positive
control. Each experiment was performed at least three times.
Total protein extracts were harvested using protein
extraction buffer. Protein concentrations were assessed using a
bicinchoninic acid protein assay kit (Santa Cruz Biotechnology,
Inc., Dallas, TX, USA). Total protein (10 μg) was separated
by electrophoresis on 4–20% SDS-PAGE gels and transferred to
nitrocellulose membranes; the membranes were blocked with 5%
skimmed milk-Tris-buffered saline plus Tween-20 solution for 1 h,
followed by incubation at 4°C overnight with specific primary
antibodies against PDCD4 [1:1,000, (D29C6) XP rabbit mAb, cat. no.
9535P] or human β-actin antibody [1:1,000, (D6A8) rabbit mAb, cat.
no. 8457s] (both from Cell Signaling Technology, Inc., Danvers, MA,
USA). Following incubation with peroxidase-conjugated AffiniPure
goat anti-rabbit IgG (1:5,000; cat. no. ZB-2301; ZSGB-Bio Co.,
Ltd., Beijing, China), the bound antibodies were visualized using
an enhanced chemiluminescence reagent (Millipore, Billerica, MA,
USA) followed by Bio-Rad ImageLab™ (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Data are expressed as the relative density of
the protein normalized to β-actin. The percentages of the increase
or decrease of protein were estimated by comparison to vehicle
control (100%).
Cell growth assay
Cells stably transfected with PDCD4 were plated in
48-well plates at a density of 5×103 cells/well. After
6, 12, 24 and 48 h, the cells were collected, resuspended in PBS
and treated with 2 μg/ml cisplatin for 24 h. The cells were
divided into the following five groups: BCa cells; empty vector
(mock); mock + cisplatin; PDCD4; and PDCD4 + cisplatin. Cell growth
and viability were evaluated by the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. The controls were treated with an equal volume of the drug
vehicle dimethyl sulfoxide, but the applied concentration did not
affect cell growth. The optical density of each culture was read by
a Universal Microplate Reader at a wavelength of 570 nm. The
absorbance of the cells and growth inhibition ratio were
calculated.
Wound healing assay
The cells were plated onto 24-well plates at a
density of 5×105/per well, and were grown for 48 h to a
confluence of >90%. BCa cells stably transfected with PDCD4 at
0, 12 and 24 h were treated with or without 2 μg/ml
cisplatin and incubated for 24 h at 37°C under 5% CO2. A
wound (cell-free area) was created by manually scraping the cell
monolayer with a 10-μl plastic pipette tip. Cell debris was
removed and the cells were then cultured in RPMI-1640 containing 1%
FBS. Images (magnification, ×40) were captured using a Nikon
Eclipse 90i microscope (Nikon Corporation, Tokyo, Japan). The
experiments were repeated three times.
Cell migration assay
The migratory function of mixed BCa cells was
evaluated using a modified Boyden chamber (Transwell; Corning Life
Sciences, Inc., Tewksbury, MA, USA) assay with a polycarbonate
filter with 8-μm pores placed between the upper and lower
chambers. In brief, at 0 and 24 h following transfection, cells
were treated with or without 2 μg/ml cisplatin containing 1%
FBS and added (1×106 cells/100 μl) to the upper
chamber. The lower chamber was filled with complete medium in the
presence of 10% FBS. After a 48-h incubation at 37°C under 5%
CO2, cells that had not migrated were removed, whereas
migrated cells were fixed in 4% paraformaldehyde for 10 min at room
temperature and stained with the Crystal Violet Staining Solution
kit (Solarbio, Beijing Solarbio Science & Technology Co., Ltd.,
Beijing, China). The number of migrated cells was counted using a
Nikon Eclipse 90i microscope.
Flow cytometry and apoptosis assay
The Annexin V-FITC/PI apoptosis detection kit was
used according to the manufacturer’s instructions, as previously
reported (21). Briefly, cells
were transfected at 12 and 24 h, then treated with or without 2
μg/ml cisplatin and incubated for a further 24 h at 37°C
under 5% CO2 in 6-well plates. The cells
(1×106) were collected and suspended in 500 μl
binding buffer, and 5 μl Annexin V-FITC and 5 μl PI
were added to each sample and incubated for 15 min in the dark. The
cell surface phosphatidylserine was quantitatively estimated in
apoptotic cells by using the Annexin V/FITC apoptosis detection kit
according to the manufacturer’s instructions (Roche Diagnostics,
Indianapolis, IN, USA). Data were detected using FACScan flow
cytometry (FACS LSRFortessa; BD Biosciences, Franklin Lakes, NJ,
USA). Triplicate experiments with triplicate samples were
performed.
Epithelial-to-mesenchymal transition
(EMT) markers and immunofluorescence staining analysis
E-cadherin, vimentin and N-cadherin were detected by
cell immunofluorescence techniques to investigate the EMT signaling
pathway-related markers. Briefly, cells in each group under
different conditions were seeded on coverslips in 6-well plates
(1×105/well) for 24 h, fixed in 4% paraformaldehyde for
30 min and washed three times with PBS. The slips were
permeabilized for 10 min with 0.1% Triton X-100 and washed three
times with PBS, and the cells were then blocked with 10% normal
goat serum for 30 min. The cells were incubated overnight at 4°C
with primary antibodies against E-cadherin (1:50 dilution;
sc-7870), vimentin (1:200, ab92547; Abcam) and N-cadherin (1:50,
sc-7939), followed by incubation for 1 h in the dark with
fluorescein isothiocyanate-conjugated and tetraethyl rhodamine
isothiocyanate goat anti-rabbit secondary antibody (ZSGB-Bio Co.,
Beijing, China). Fluorescent images were captured with a Nikon
Eclipse 90i microscope. The staining results were analyzed with the
Image-Pro Plus 6 image-analyzing system (Media Cybernetics, Inc.,
Rockville, MD, USA).
Western blotting
To further investigate the potential role of PDCD4
in BCa cells and the possible underlying mechanism, western
blotting was performed to analyze the protein expression of JNK
signaling pathway and EMT markers. Blots were probed with specific
primary antibodies: c-Jun (1:1,000, no. 9165), p-JNK (1:1,000, no.
4668) and p(Ser-73)-c-Jun (1:1,000, no. 9261/no. 3270) (Cell
Signaling Technology, Beverly, MA, USA), E-cadherin, vimentin,
N-cadherin and matrix metalloproteinase-9 (MMP-9). The
peroxidase-conjugated AffiniPure goat anti-rabbit IgG secondary
antibody was added, followed by incubation at 37°C for 1 h. The
bound antibodies were visualized using an enhanced
chemiluminescence reagent (Millipore) followed by analysis with
Bio-Rad Image Lab™. Data are expressed as the relative density of
the protein normalized to β-actin. The percentages of increase or
decrease of the protein level were estimated by comparison to
vehicle control (100%).
Statistical analysis
Statistical analyses were performed with one-way
analysis of variance followed by the Bonferroni test or t-test when
appropriate, using the SPSS statistical software, version 10.0
(SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to
indicate statistically significant differences. Data of continuous
variables are presented as the mean ± standard deviation.
Results
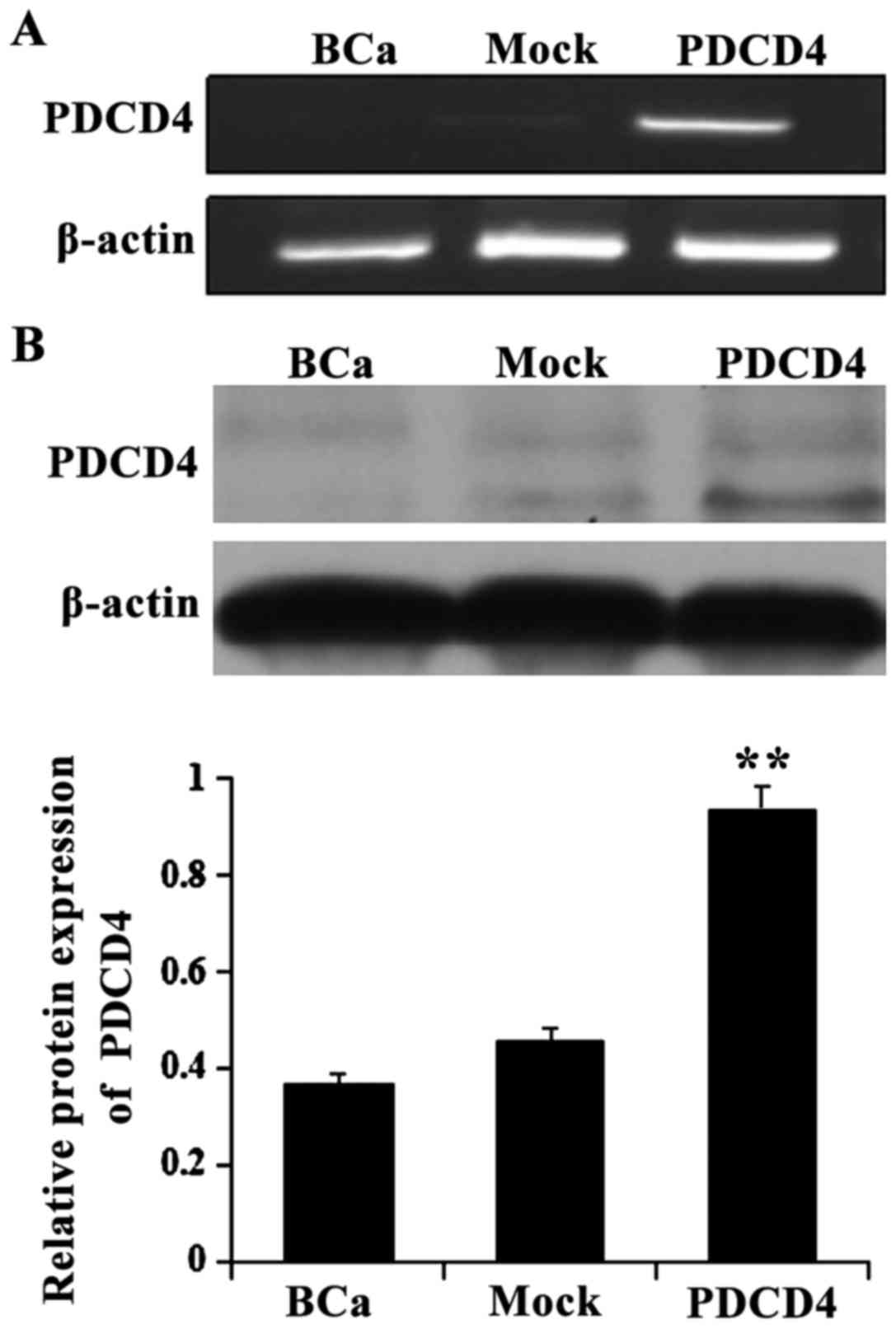
Overexpression of PDCD4 in BCa cells
To provide direct evidence that PDCD4 inhibits the
proliferation of BCa cells, we introduced recombinant pDsRed2-N1
plasmids that carry the full-length PDCD4 cDNA, as well as parental
vector controls into the cells, which express only low levels of
endogenous PDCD4. RT-PCR and western blot analysis revealed that
cells transfected with the PDCD4 expression plasmid exhibited
stronger PDCD4 expression compared with the control cells
(P<0.05; Fig. 1). These results
indicate that the recombinant pDsRed2-N1-PDCD4 plasmid promoted
PDCD4 expression in BCa cells.
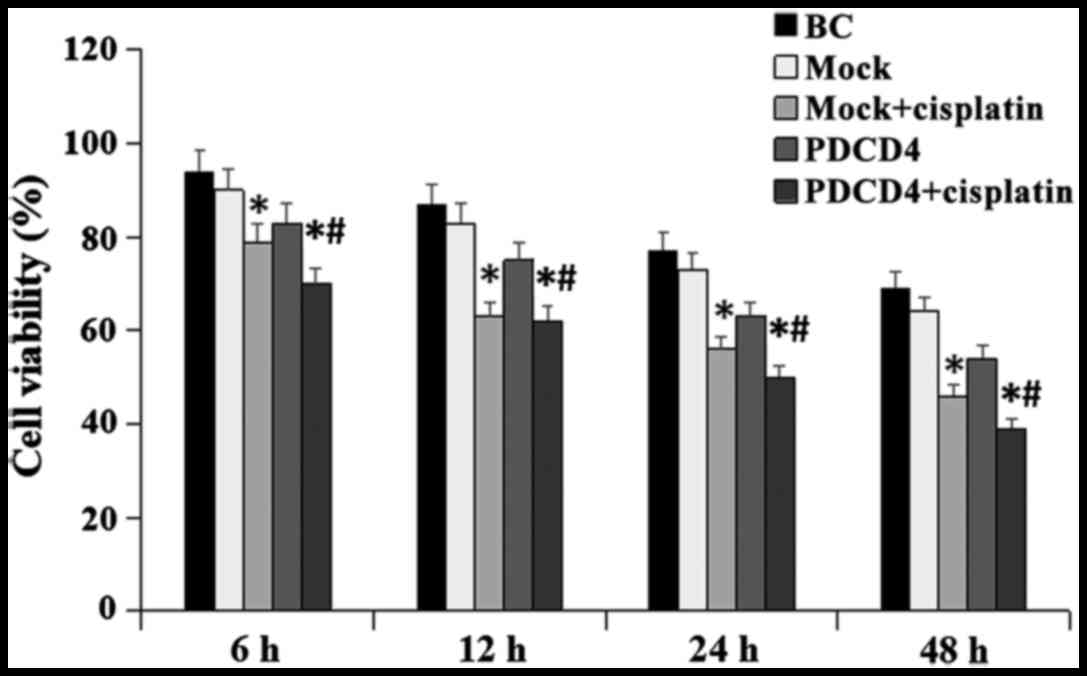
PDCD4 overexpression inhibits cell
viability and improves sensitivity to cisplatin
Following transfection, the effect of PDCD4 on cell
growth was further investigated. The results revealed that, when
the cells were treated with 2 μg/ml cisplatin for 24 h,
overexpression of PDCD4 significantly inhibited the viability of
BCa cells compared with cells without treatment (P<0.05;
Fig. 2). Our results indicated
that enforced expression of PDCD4 was associated with cisplatin
sensitivity in BCa cells.
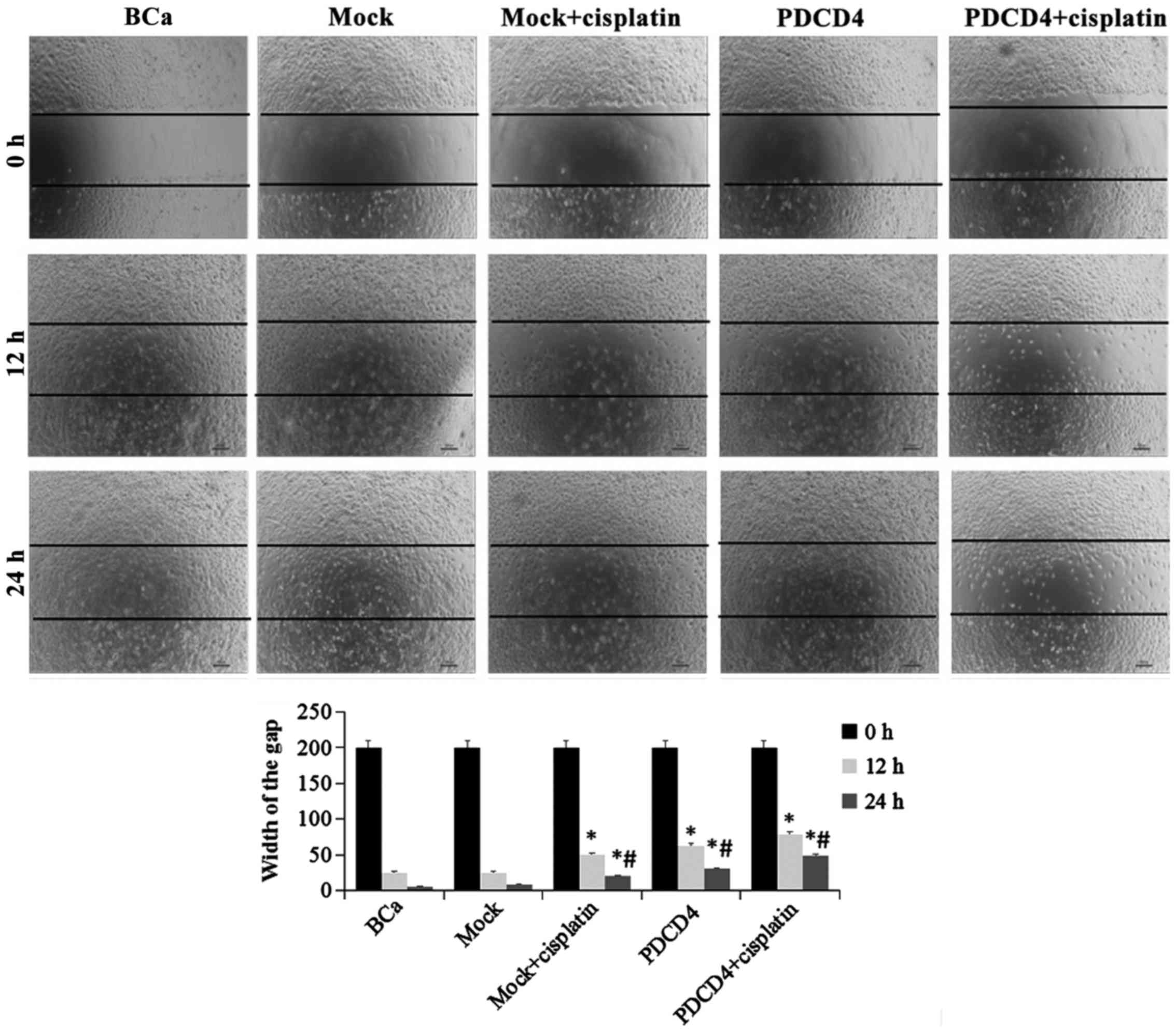
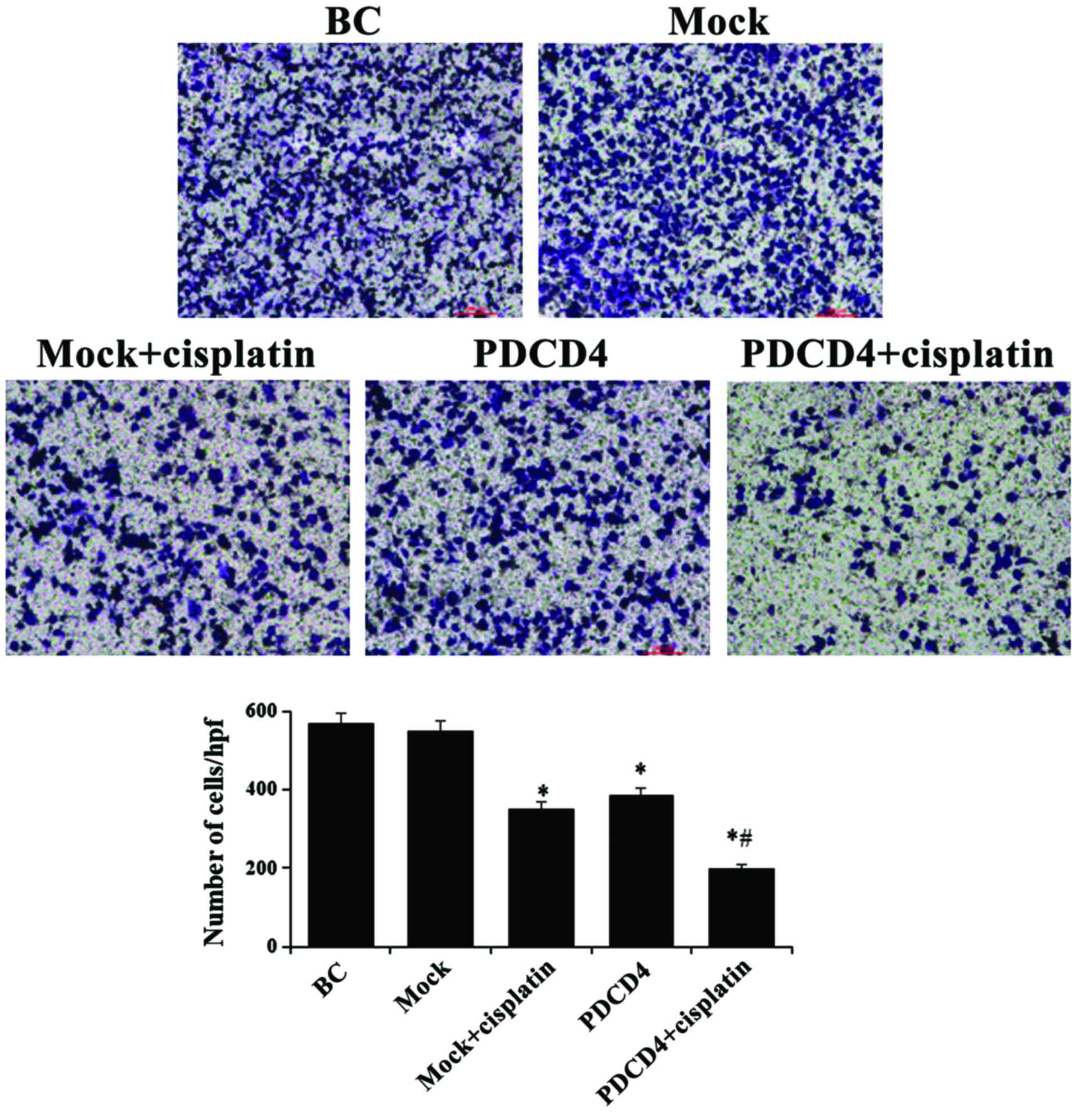
PDCD4 overexpression inhibits cell
migration and invasion
The migration and invasion properties of cells in
each group under different conditions were evaluated. As
demonstrated in Fig. 3, the
migration of cells was significantly inhibited in the PDCD4 group
compared with the mock group at 12 and 24 h, as measured by the
wound healing assay. Similarly, invasion, as assessed by the
Transwell assay, was also markedly suppressed in the PDCD4 group
compared with the mock group at 24 h (P<0.05; Fig. 4). Furthermore, after
PDCD4-transfected cells were treated with cisplatin, their
migration and invasion ability were clearly lower compared with
those in the mock + cisplatin and PDCD4 groups. These results
indicated that, in addition to its cytotoxic effect, PDCD4
overexpression increased the sensitivity to cisplatin in BCa
cells.
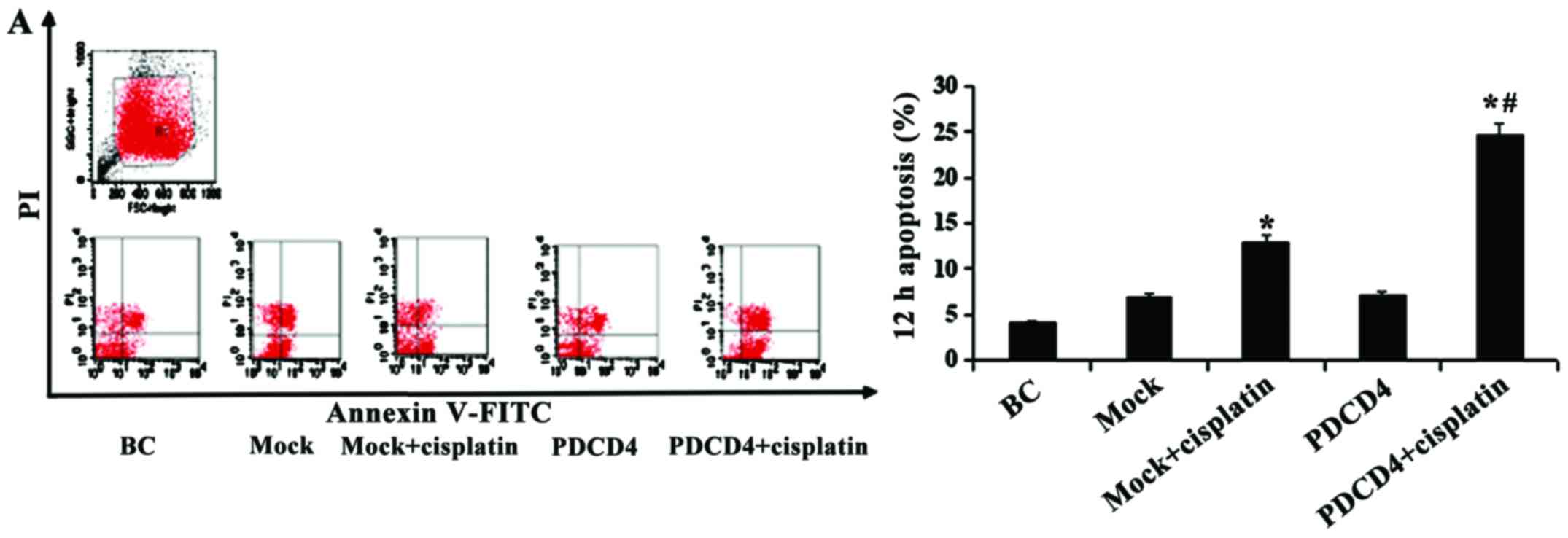
PDCD4 overexpression induces BCa cell
apoptosis
To evaluate the effect of PDCD4 overexpression on
BCa cell apoptosis, the cells in each group under different
conditions were stained with Annexin V/FITC and analyzed by flow
cytometry. As shown in Fig. 5, the
percentage of apoptotic cells gradually increased after 12 and 24 h
of treatment with cisplatin (P<0.05). Of note, there was a more
prominent elevation of the positive ratio in PDCD4 cells compared
with mock cells when they were treated with cisplatin
(P<0.05).
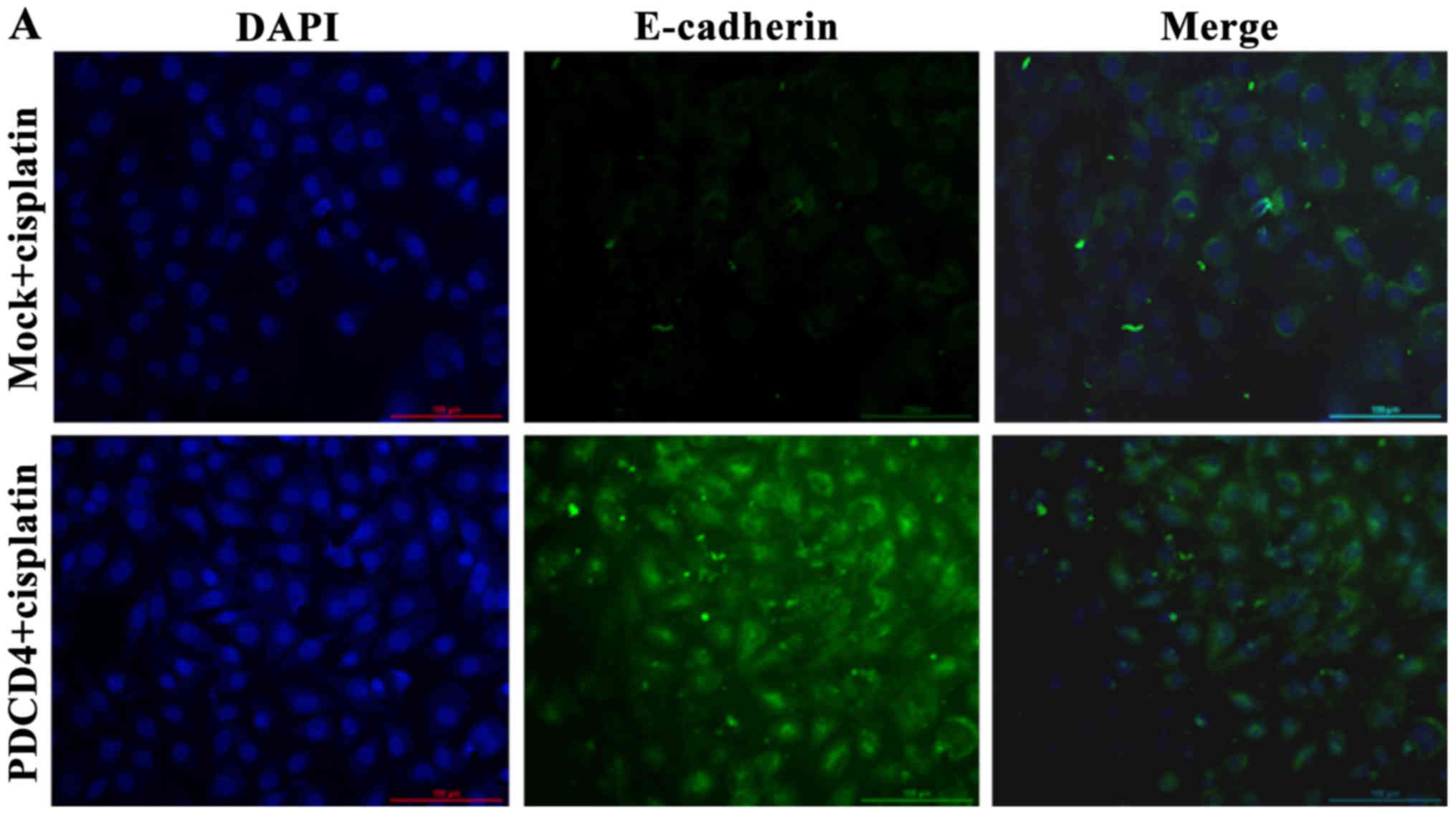
PDCD4 overexpression inhibits the
expression of EMT markers
In order to investigate the mechanism underlying the
inhibitory effect of PDCD4 on BCa cells, the EMT transition signal
pathway-related markers were detected by cell immunofluorescence
techniques and western blotting.
As shown in Fig. 6,
the immunofluorescence techniques revealed that the expression of
E-cadherin was significantly decreased, whereas that of vimentin
and N-cadherin was significantly increased when the cells were
treated with cisplatin for 24 h in the PDCD4 + cisplatin group
compared with the mock + cisplatin group.
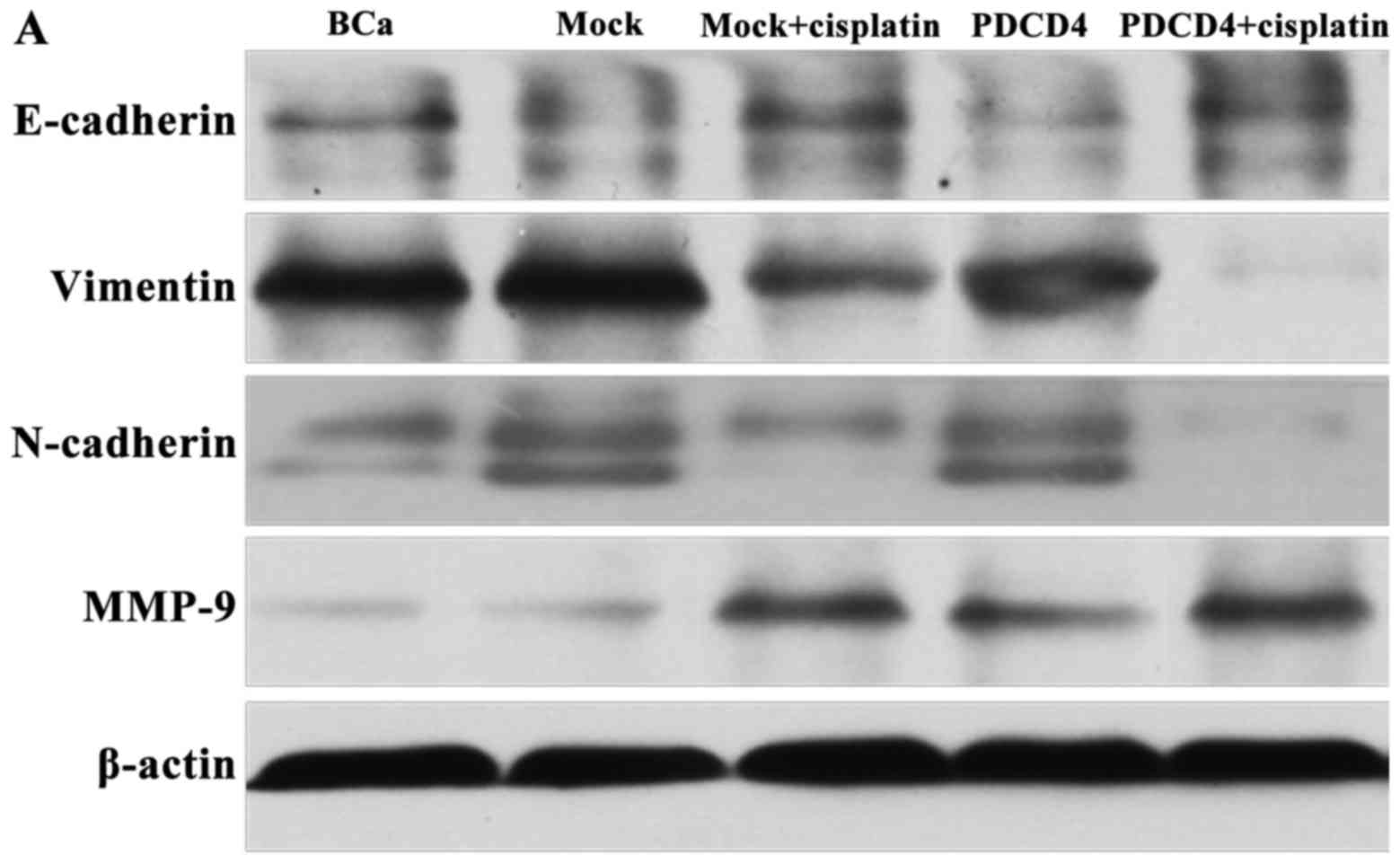
The western blotting results also demonstrated that
the protein expression of E-cadherin and the EMT-associated protein
MMP-9 was upregulated, but vimentin and N-cadherin were
downregulated when the cells were treated with cisplatin for 24 h
in the PDCD4 + cisplatin group compared with the mock + cisplatin
group (P<0.05; Fig. 7).
These results revealed that overexpression of PDCD4
enhances the efficiency of EMT inhibition when the cells are
treated with cisplatin.
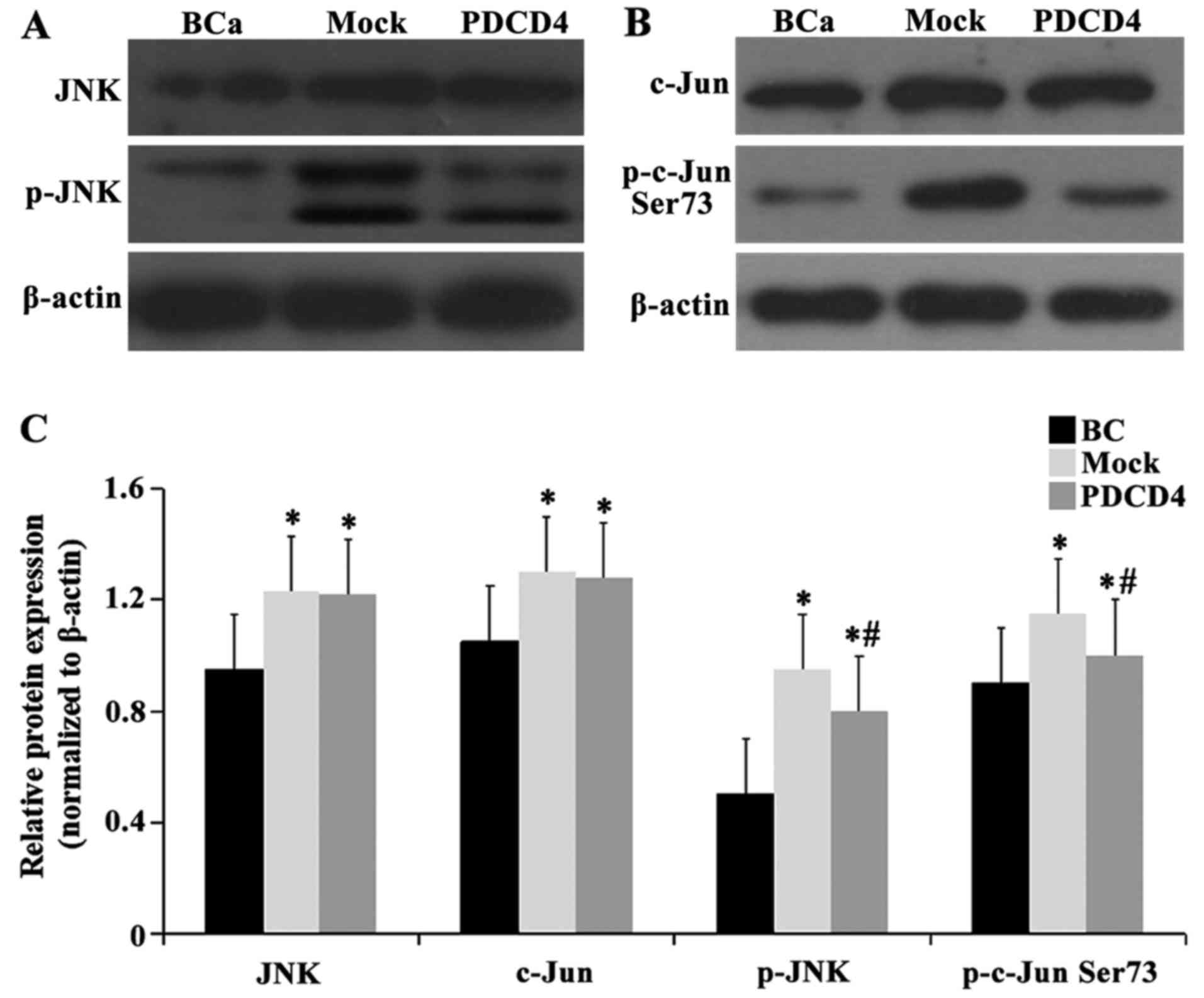
Effect of PDCD4 overexpression on protein
levels of the JNK signaling pathway
We analyzed the protein expression levels of JNK,
c-Jun, p-JNK and p-c-Jun Ser73 by western blotting. As demonstrated
in Fig. 8, the protein levels of
JNK and c-Jun were increased in the mock and PDCD4 groups compared
with the BCa group (P<0.05), with no significant difference
(P>0.05) between the mock and PDCD4 groups. The expression of
p-JNK and p-c-Jun Ser73 was obviously increased in the mock and
PDCD4 groups compared with the BCa group (P<0.05); moreover, the
expression of p-JNK and p-c-Jun Ser73 was significantly decreased
in the PDCD4 group compared with the mock group (P<0.05). Taken
together, these findings demonstrated that the enhancement in the
sensitivity of cells to cisplatin is partially mediated via
regulation of the JNK/c-Jun pathway.
Discussion
To the best of our knowledge, the present study is
the first to demonstrate that PDCD4 overexpression in BCa can
inhibit cancer cell proliferation and invasion and tumor growth,
and enhance sensitivity to cisplatin, possibly through activation
of the JNK/c-Jun signaling pathway, further suppressing the EMT
process.
PDCD4 is a tumor suppressor gene that is involved in
cell apoptosis, transformation and invasion, as well as tumor
progression (22). Targeted
inhibition of PDCD4 significantly enhanced cancer cell migration
and invasion in hepatocellular carcinoma (23). More recently, PDCD4 has been shown
to suppress BCa progression, and increased PDCD4 expression
efficiently sensitized muscle-invasive BCa cells to cisplatin
chemotherapy and suppressed cell invasiveness (24). However, the mechanisms underlying
PDCD4-induced apoptosis have yet to be fully elucidated. Modulation
of the JNK1/2 signaling pathway by PDCD4, particularly
c-Jun/AP-1-dependent transcription through the downstream
MAP4K1/JNK/AP-1 signaling pathway, suggests that this is a novel
upstream target of this signaling cascade (25). Increased c-Jun phosphorylation was
positively correlated with clinical grade in BCa tissues, and
deregulation of c-Jun has been reported in several different types
of cancer, including its overexpression in BCa (26). Mitochondria-related apoptosis in
human BCa cells is also associated with activation of the JNK
signaling pathway (27). In the
present study, we found that cell proliferation, invasion and tumor
growth were significantly inhibited, whereas cell apoptosis was
enhanced in the PDCD4 transfection group compared with the mock
group in BCa cells when they were treated with cisplatin. More
importantly, we found that PDCD4 overexpression reduced the protein
levels of p-JNK and p-c-Jun. Collectively, the findings of our
study suggest that PDCD4 potentiates the antitumor activity of
cisplatin against human BCa cells by inhibiting cell proliferation
and invasion and promoting apoptosis, partly via activation of the
JNK/c-Jun pathway.
EMT is a complex and reversible process
characterized by the loss of intercellular cohesion and epithelial
makers. Recent studies reported that EMT promotes cell migration
and invasion in multiple types of cancer, and it is crucial for the
invasiveness and metastasis of BCa (17,28).
EMT is characterized by the acquisition of a mesenchymal phenotype
and loss of epithelial cell polarity, includes conversion of
epithelial cells to a fibroblastoid phenotype, and involves
downregulation of the epithelial-specific protein E-cadherin and
upregulation of the mesenchymal-specific proteins N-cadherin and
vimentin, increasing migration through the extracellular matrix
(29). This switch leads to loss
of the affinity for neighboring epithelial cells and development of
affinity for mesenchymal cells, resulting in increased migration
and invasion (9). It has been
demonstrated that the association between the MMP family and EMT
plays an important role in different types of cancer, such as lung,
ovarian, liver and breast cancer (30–33),
and knockdown of PDCD4 may lead to EMT, changes in adhesion, and
promotion of migration and metastasis. These data indicated that
both PDCD4 and EMT play important roles in the progression of
cancer; however, the mechanisms underlying the interaction between
PDCD4 and EMT signaling pathways has not been fully elucidated.
Recent studies also demonstrated that the development and
progression of BCa are associated with the involvement of both EMT
and MMPs (34). MMP-2 and MMP-9
are involved in the metastatic spread of various tumors, including
transitional cell carcinoma and BCa (35,36).
We herein demonstrated that PDCD4 + cisplatin significantly
downregulated the protein expression levels of epithelial marker
E-cadherin and MMP-9, while upregulating the mesenchymal markers
vimentin and N-cadherin compared with the mock + cisplatin group.
Based on these results, it was hypothesized that PDCD4 may enhance
the sensitivity of BCa cells to cisplatin via inhibition of the
MMP-related EMT process. Suppression of cell migration, invasion
and adhesive ability caused by EMT may be mediated via inhibition
of JNK-related pathways (37,38).
JNKs increase circulating tumor cell survival and tissue homing,
promote cell attachment to the endothelium, stimulate EMT to
facilitate tumor cell extravasation, and enhance the secretion of
endothelial barrier disrupters (39). Reversing EMT to inhibit gastric
cancer cell metastasis and suppressing the expression of MMP-9 and
MMP-2 may be mediated via the JNK and extracellular-regulated
kinase signaling pathways (40).
Of note, the present study also demonstrated that PDCD4
overexpression markedly reduced the protein levels of p-JNK and
p-c-Jun. Taken together, our data suggest that PDCD4 increases the
antitumor activity of cisplatin against human BCa cells through
activation of the JNK/c-Jun pathway and further suppression of the
EMT process.
Modulating immune inhibitory pathways has been a
major recent breakthrough in cancer treatment. Checkpoint blockade
antibodies targeting cytotoxic T-lymphocyte antigen 4 and programed
cell-death protein 1 (PD-1) have demonstrated acceptable toxicity
(41). The discovery of PD-1 and
its ligand 1 (PD-L1) has introduced a modern era of cancer
immunotherapy. Blocking the PD-1 pathway using monoclonal
antibodies against PD-1 or PD-L1 may therefore revamp the immune
response against tumor cells (42). Several anti-PD agents have been
approved for BCa therapy. Our current data may be able to be
incorporated and used together with these novel agents, and the
results of the present study may help design new checkpoint
blockade therapies targeted at PDCD4 in the future.
In summary, our results suggest that PDCD4, a
nuclear/cytoplasmic shuttling protein with multiple functions,
plays an important role in the development and progression of human
BCa. The mechanism underlying the increased sensitivity of BCa
cells to cisplatin may be mediated via the JNK/c-Jun signaling
pathway, further reversing the MMP-related EMT process.
Acknowledgments
The authors are grateful to Central Research
Laboratory of the Second Hospital of Shandong University for the
technical assistance and generous support.
Notes
[1]
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81500042), the
Natural Science Foundation of Shandong Province (grant no.
ZR2016HM67), the Science and Technology Development Project of
Shandong Province (grant nos. 2016GSF201044 and 2016GSF201203), and
the Medical and Health Science Technology Development Plan Project
of Shandong Province (grant no. 2015WS0307).
[2] Availability
of data and materials
All data generated or analyzed during this study are
included in this published article.
[3] Authors’
contributions
YL designed the research and oversaw the writing of
the manuscript; JL, RZ, JW, QX and XX performed the experiments and
wrote the manuscript, and FK, JZ analyzed the data. All authors
have read and approved the manuscript.
[4] Ethics
approval and consent to participate
Not applicable.
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Wu CL, Ho JY, Chou SC and Yu DS: miR-429
reverses epithelial-mesenchymal transition by restoring E-cadherin
expression in bladder cancer. Oncotarget. 7:26593–26603.
2016.PubMed/NCBI
|
|
2
|
Rao Q, Chen Y, Yeh CR, Ding J, Li L, Chang
C and Yeh S: Recruited mast cells in the tumor microenvironment
enhance bladder cancer metastasis via modulation of ERβ/CCL2/CCR2
EMT/MMP9 signals. Oncotarget. 7:7842–7855. 2016. View Article : Google Scholar
|
|
3
|
Wu CT, Chang YH, Lin P, Chen WC and Chen
MF: Thrombomodulin expression regulates tumorigenesis in bladder
cancer. BMC Cancer. 14:3752014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shirodkar SP and Lokeshwar VB: Potential
new urinary markers in the early detection of bladder cancer. Curr
Opin Urol. 19:488–493. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Asangani IA, Rasheed SA, Nikolova DA,
Leupold JH, Colburn NH, Post S and Allgayer H: MicroRNA-21 (miR-21)
post-transcriptionally downregulates tumor suppressor Pdcd4 and
stimulates invasion, intravasation and metastasis in colorectal
cancer. Oncogene. 27:2128–2136. 2008. View Article : Google Scholar
|
|
6
|
Yu H, Zeng J, Liang X, Wang W, Zhou Y, Sun
Y, Liu S, Li W, Chen C and Jia J: Helicobacter pylori promotes
epithelial-mesenchymal transition in gastric cancer by
downregulating programmed cell death protein 4 (PDCD4). PLoS One.
9:e1053062014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ferraro A, Kontos CK, Boni T, Bantounas I,
Siakouli D, Kosmidou V, Vlassi M, Spyridakis Y, Tsipras I, Zografos
G, et al: Epigenetic regulation of miR-21 in colorectal cancer:
ITGB4 as a novel miR-21 target and a three-gene network
(miR-21-ITGβ4-PDCD4) as predictor of metastatic tumor potential.
Epigenetics. 9:129–141. 2014. View Article : Google Scholar
|
|
8
|
Brønnum H, Andersen DC, Schneider M,
Sandberg MB, Eskildsen T, Nielsen SB, Kalluri R and Sheikh SP:
miR-21 promotes fibrogenic epithelial-to-mesenchymal transition of
epicardial mesothelial cells involving Programmed Cell Death 4 and
Sprouty-1. PLoS One. 8:e562802013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Q, Zhu J, Zhang Y, Sun Z, Guo X, Wang
X, Lee E, Bakthavatchalu V, Yang Q and Yang HS: Down-regulation of
programmed cell death 4 leads to epithelial to mesenchymal
transition and promotes metastasis in mice. Eur J Cancer.
49:1761–1770. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang X, Wu H, Lakdawala VS, Hu F, Hanson
ND and Miller AH: Inhibition of Jun N-terminal kinase (JNK)
enhances glucocorticoid receptor-mediated function in mouse
hippocampal HT22 cells. Neuropsychopharmacology. 30:242–249. 2005.
View Article : Google Scholar
|
|
11
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fang Y, Wang Y, Wang Y, Meng Y, Zhu J, Jin
H, Li J, Zhang D, Yu Y, Wu XR, et al: A new tumour suppression
mechanism by p27Kip1: EGFR down-regulation mediated by JNK/c-Jun
pathway inhibition. Biochem J. 463:383–392. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gong Y, Qiu W, Ning X, Yang X, Liu L, Wang
Z, Lin J, Li X and Guo Y: CCDC34 is up-regulated in bladder cancer
and regulates bladder cancer cell proliferation, apoptosis and
migration. Oncotarget. 6:25856–25867. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shen KH, Li CF, Chien LH, Huang CH, Su CC,
Liao AC and Wu TF: Role of galectin-1 in urinary bladder urothelial
carcinoma cell invasion through the JNK pathway. Cancer Sci.
107:1390–1398. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Subedi A, Kim MJ, Nepal S, Lee ES, Kim JA,
Sohn DH, Song K, Lee SH, Park WS, Jeong BS, et al: Globular
adiponectin modulates expression of programmed cell death 4 and
miR-21 in RAW 264.7 macrophages through the MAPK/NF-κB pathway.
FEBS Lett. 587:1556–1561. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang Q, Zhang Y and Yang HS: Pdcd4
knockdown up-regulates MAP4K1 expression and activation of AP-1
dependent transcription through c-Myc. Biochim Biophys Acta.
1823:1807–1814. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Echevarría-Vargas IM, Valiyeva F and
Vivas-Mejía PE: Upregulation of miR-21 in cisplatin resistant
ovarian cancer via JNK-1/c-Jun pathway. PLoS One. 9:e970942014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Song Y, Zhang P, Sun Y, Li X, Chen L, Xiao
Y and Xing Y: AMPK activation-dependent autophagy compromises
oleanolic acid-induced cytotoxicity in human bladder cancer cells.
Oncotarget. 8:67942–67954. 2017.PubMed/NCBI
|
|
19
|
Huang B, Zhang J, Zhang X, Huang C, Hu G,
Li S, Xie T, Liu M and Xu Y: Suppression of LETM1 by siRNA inhibits
cell proliferation and invasion of bladder cancer cells. Oncol Rep.
38:2935–2940. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wei ZT, Zhang X, Wang XY, Gao F, Zhou CJ,
Zhu FL, Wang Q, Gao Q, Ma CH, Sun WS, et al: PDCD4 inhibits the
malignant phenotype of ovarian cancer cells. Cancer Sci.
100:1408–1413. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Luan Y, Liu J, Liu X, Xue X, Kong F, Sun
C, Wang J, Liu L and Jia H: Tetramethypyrazine inhibits renal cell
carcinoma cells through inhibition of NKG2D signaling pathways. Int
J Oncol. 49:1704–1712. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wei X, Wang W, Wang L, Zhang Y, Zhang X,
Chen M, Wang F, Yu J, Ma Y and Sun G: MicroRNA-21 induces
5-fluorouracil resistance in human pancreatic cancer cells by
regulating PTEN and PDCD4. Cancer Med. 5:693–702. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Selaru FM, Olaru AV, Kan T, David S, Cheng
Y, Mori Y, Yang J, Paun B, Jin Z, Agarwal R, et al: MicroRNA-21 is
overexpressed in human cholangiocarcinoma and regulates programmed
cell death 4 and tissue inhibitor of metalloproteinase 3.
Hepatology. 49:1595–1601. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lei Y, Hu X, Li B, Peng M, Tong S, Zu X,
Wang Z, Qi L and Chen M: miR-150 modulates cisplatin
chemosensitivity and invasiveness of muscle-invasive bladder cancer
cells via targeting PDCD4 in vitro. Med Sci Monit. 20:1850–1857.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ferreira DM, Afonso MB, Rodrigues PM,
Simão AL, Pereira DM, Borralho PM, Rodrigues CM and Castro RE:
c-Jun N-terminal kinase 1/c-Jun activation of the p53/microRNA
34a/sirtuin 1 pathway contributes to apoptosis induced by
deoxycholic acid in rat liver. Mol Cell Biol. 34:1100–1120. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yuan F, Xu Z, Yang M, Wei Q, Zhang Y, Yu
J, Zhi Y, Liu Y, Chen Z and Yang J: Overexpressed DNA polymerase
iota regulated by JNK/c-Jun contributes to hypermutagenesis in
bladder cancer. PLoS One. 8:e693172013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Duan F, Yu Y, Guan R, Xu Z, Liang H and
Hong L: Vitamin K2 induces mitochondria-related apoptosis in human
bladder cancer cells via ROS and JNK/p38 MAPK Signal Pathways. PLoS
One. 11:e01618862016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Roth B, Jayaratna I, Sundi D, Cheng T,
Melquist J, Choi W, Porten S, Nitti G, Navai N, Wszolek M, et al:
Employing an orthotopic model to study the role of
epithelial-mesenchymal transition in bladder cancer metastasis.
Oncotarget. 8:34205–34222. 2017. View Article : Google Scholar :
|
|
29
|
Chaffer CL, Brennan JP, Slavin JL, Blick
T, Thompson EW and Williams ED: Mesenchymal-to-epithelial
transition facilitates bladder cancer metastasis: Role of
fibroblast growth factor receptor-2. Cancer Res. 66:11271–11278.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hulit J, Suyama K, Chung S, Keren R,
Agiostratidou G, Shan W, Dong X, Williams TM, Lisanti MP, Knudsen
K, et al: N-cadherin signaling potentiates mammary tumor metastasis
via enhanced extracellular signal-regulated kinase activation.
Cancer Res. 67:3106–3116. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Baek SH, Ko JH, Lee JH, Kim C, Lee H, Nam
D, Lee J, Lee SG, Yang WM, Um JY, et al: Ginkgolic acid inhibits
invasion and migration and TGF-β-induced EMT of lung cancer cells
through PI3K/Akt/mTOR inactivation. J Cell Physiol. 232:346–354.
2017. View Article : Google Scholar
|
|
32
|
Vos MC, Hollemans E, Ezendam N, Feijen H,
Boll D, Pijlman B, van der Putten H, Klinkhamer P, van Kuppevelt
TH, van der Wurff AA, et al: MMP-14 and CD44 in
epithelial-to-mesenchymal transition (EMT) in ovarian cancer. J
Ovarian Res. 9:532016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zeng Y, Yao X, Chen L, Yan Z, Liu J, Zhang
Y, Feng T, Wu J and Liu X: Sphingosine-1-phosphate induced
epithelial-mesenchymal transition of hepatocellular carcinoma via
an MMP-7/syndecan-1/TGF-β autocrine loop. Oncotarget.
7:63324–63337. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wu CL, Ho JY, Chou SC and Yu DS: miR-429
reverses epithelial-mesenchymal transition by restoring E-cadherin
expression in bladder cancer. Oncotarget. 7:26593–26603.
2016.PubMed/NCBI
|
|
35
|
Wang R, Ke ZF, Wang F, Zhang WH, Wang YF,
Li SH and Wang LT: GOLPH3 overexpression is closely correlated with
poor prognosis in human non-small cell lung cancer and mediates its
metastasis through upregulating MMP-2 and MMP-9. Cell Physiol
Biochem. 35:969–982. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Song H, Pan D, Sun W, Gu C, Zhang Y, Zhao
P, Qi Z and Zhao S: SiRNA directed against annexin II receptor
inhibits angiogenesis via suppressing MMP2 and MMP9 expression.
Cell Physiol Biochem. 35:875–884. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cheng HL, Lin CW, Yang JS, Hsieh MJ, Yang
SF and Lu KH: Zoledronate blocks geranylgeranylation not
farnesylation to suppress human osteosarcoma U2OS cells metastasis
by EMT via Rho A activation and FAK-inhibited JNK and p38 pathways.
Oncotarget. 7:9742–9758. 2016.PubMed/NCBI
|
|
38
|
Lee YS, Kim SY, Song SJ, Hong HK, Lee Y,
Oh BY, Lee WY and Cho YB: Crosstalk between CCL7 and CCR3 promotes
metastasis of colon cancer cells via ERK-JNK signaling pathways.
Oncotarget. 7:36842–36853. 2016.PubMed/NCBI
|
|
39
|
Ebelt ND, Cantrell MA and Van Den Berg CL:
c-Jun N-terminal kinases mediate a wide range of targets in the
metastatic cascade. Genes Cancer. 4:378–387. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ji J, Jia S, Jia Y, Ji K, Hargest R and
Jiang WG: WISP-2 in human gastric cancer and its potential
metastatic suppressor role in gastric cancer cells mediated by JNK
and PLC-γ pathways. Br J Cancer. 113:921–933. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ma W, Gilligan BM, Yuan J and Li T:
Current status and perspectives in translational biomarker research
for PD-1/PD-L1 immune checkpoint blockade therapy. J Hematol Oncol.
9:472016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu B, Song Y and Liu D: Recent
development in clinical applications of PD-1 and PD-L1 antibodies
for cancer immunotherapy. J Hematol Oncol. 10:1742017. View Article : Google Scholar : PubMed/NCBI
|