Introduction
Ovarian cancer is the most lethal gynecologic
malignancy and the fifth leading cause for cancer mortality among
women in the United States (1). A
reason for this high mortality is chemotherapeutic resistance, as
recurrent and metastatic ovarian cancer usually exhibits resistant
to platinum-based chemotherapy (2). Thus, there is an urgent need to
identify novel drugs for use in ovarian cancer therapy.
Metformin (1,1-dimethylbiguanide), an oral biguanide
first introduced in the 1950s, has been used as the first-line
medication for the treatment of type II diabetes mellitus for
decades. Recently metformin has gained renewed interest as a
potential cancer chemotherapy adjuvant in adjuvant or neoadjuvant
setting (3,4). Numerous studies have demonstrated
antitumor effects of metformin in vitro and in vivo
using models using different types of cancer cell lines, including
breast, endometrial and ovarian cancer cell lines (5–7).
Furthermore, clinical observational studies demonstrated that
patients with ovarian cancer exposed to metformin had reduced
disease recurrences and cancer-specific mortalities compared with
unexposed women (8–10). A phase II clinical trial to assess
whether the addition of metformin to standard chemotherapy improves
survival in non-diabetic ovarian cancer patients is ongoing
(NCT02122185; clinicaltrials.gov).
Although metformin has been used as an anti-diabetic
agent for over half a century, its molecular mechanisms are still
not fully understood. The most described mechanism of metformin is
inhibition of mitochondrial respiratory chain complex I, leading to
reduced ATP production and activation of AMP-activated protein
kinase (AMPK), which in turn leads to increased glucose uptake by
muscle, decreased glucose production in liver, and reduced blood
glucose (11). In cancer cells,
AMPK is considered a tumor-suppressing pathway, which inhibits the
mammalian target of the rapamycin signaling, suppresses cell
proliferation, and promotes apoptosis and cell-cycle arrest
(12,13). AMPK-independent effects of
metformin in cancer, such as the inhibition of protein kinase
phosphorylation in breast and lung cancers, have been also reported
(14). However, whether metformin
inhibits ovarian cancer via the AMPK pathway and underlying
downstream molecular mechanisms remains elusive.
Epigenetic modifications, including DNA methylation
of the CpG sites, and covalent modifications of the N-terminal tail
of the core histones, have critical roles in tumor development and
progression (15). Histone H3
lysine 27 trimethylation (H3K27me3) is recognized as an epigenetic
marker in malignancies, because H3K27me3 reprograms epigenetic
landscape and gene expression, which is associated with various
pathways to drive tumorigenesis (16). In ovarian cancer, H3K27me3
contributes to the formation of the tumor microenvironment
(17), the development of
resistance to cisplatin and tumor progression (18). H3K27me3 is catalyzed by
histone-lysine N-methyltransferase EZH2 that interacts with
polycomb protein SUZ12 and polycomb protein EED as polycomb
repressive complex 2 (PRC2) and mediates gene silencing through
promoter methylation and chromatin remodeling (19). EZH2 depletion and H3K27me3
inhibition in ovarian cancer cells could inhibit tumor growth,
migration, and invasion, and enhance sensitized tumor cells to
cisplatin in vitro and in vivo (20,21).
Notably, EZH2 and H3K27me3 have been reported to contribute to the
development of renal injury and chronic inflammation in type 2
diabetes (22,23), demonstrating the role of H3K27me3
in energy stress.
In the present study, it was aimed to investigate
whether metformin inhibits ovarian cancer through repressing
H3K27me3. Given that metformin is predominantly used to treat
patients with type II diabetes and high glucose concentration
inhibits the activation of AMPK (24), the role of glucose concentration in
the effects of metformin in ovarian cancer cells was also
examined.
Materials and methods
Chemical, reagents and antibodies
The following chemicals were used in the current
study. Metformin (1,1-dimethylbiguanide,) was purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany; cat. no. 150959).
2-Deoxy-D-glucose (2-DG; cat. no. S4701) and dorsomorphin 2HCl
(Compound C) (cat. no. S7306) were purchased from Selleck Chemicals
(Houston, TX, USA). Antibodies against phospho-AMPKα (p-AMPKα,
Thr172; cat. no. 2535), AMPKα (cat. no. 2603), EZH2 (cat. no.
5246), SUZ12 (cat. no. 3737) were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Histone H3 (cat. no. 2348),
H3K27me3 (cat. no. 2363), EED (cat. no. 5371) antibodies were
purchased from ABclonal Biotech Co., Ltd. (Woburn, MA, USA).
β-actin antibody (cat. no. 66009-1-Ig) was purchased from
Proteintech, Inc. (Chicago, IL, USA). Horseradish
peroxidase-conjugated (HRP) anti-mouse antibody (cat. no.
074-1806-1) and HRP anti-rabbit antibody (cat. no. 074-1506-1) were
purchased from KPL, Inc. (Gaithersburg, MD, USA).
Cell lines and culture conditions
Human epithelial ovarian cancer cell lines SKOV3
(ovarian adenocarcinoma), ES2 (ovarian clear cell carcinoma), and
A2780 (ovarian carcinoma) were purchased from China Center for Type
Culture Collection (Wuhan University, Wuhan, China) and were
cultured in Dulbecco's modified Eagle's medium (DMEM) (HyClone; GE
Healthcare Life Sciences, Logan, UT, USA) containing 25 mM glucose
(mimicking hyperglycemia) or 5.5 mM glucose (mimicking
normoglycemia) supplemented with 10% fetal bovine serum (v/v;
Hangzhou Sijiqing Biological Engineering Materials Co., Ltd.,
Hangzhou, China) at 37°C in 95% air and 5% CO2.
For metformin treatment, ovarian cancer cells were
seeded at a density of 4×105 per well on 6-well plates
and incubated for 24 h. Then they were treated with metformin of
different concentrations (0, 2.5, 5 or 10 mM) for 24 h in
triplicate. 2-DG, an AMPK activator, was used to induce AMPK
phosphorylation in tumor cells, whereby cells were treated with 25
mM 2-DG for 24 h. To inhibit the metformin-mediated AMPK
activation, cells were pretreated with 20 µM Compound C for
2 h followed by treatment with 5 mM metformin for 24 h.
Transduction in vitro
A recombinant lentivirus harboring EZH2 DNA
(NM_004456; GeneChem Co., Ltd., Shanghai, China) was used to
upregulate the EZH2 level in the SKOV3 and ES2 cells. An empty
vector (Con307; GeneChem Co., Ltd.) was used as control. Stable
transduction was conducted following the manufacturers'
instructions. Transduction cells were selected with 25 µg/ml
puromycin and monoclonal cells were used for subsequent
experiments. The transfection efficiency was confirmed by western
blot analysis.
Proliferation assay
5-Ethynyl-20-deoxyuridine (EdU) DNA cell
proliferation kits were purchased from Guangzhou RiboBio Co., Ltd.
(Guangzhou, China). Cell proliferation activity was evaluated
according to the manufacturer's instructions. Briefly, ovarian
cancer cells were seeded in triplicate at a density of
5×103 cells/well in 96-well plates. After24 h, cells
were washed with PBS twice and fixed with 4% formaldehyde at room
temperature for 30 min. Subsequently, the cells were treated with
0.5% Triton X-100 for 10 min at room temperature for
permeabilization, followed by incubation with EdU at room
temperature for 1 h. Finally, the cells were counter-stained with 5
µg/ml Hoechst 33342 at room temperature for 30 min. The
proportion of EdU-incorporated cells was defined as the
proliferation rate. The assay was performed in three biological
replicates.
Analysis of cell apoptosis via flow
cytometry
The effects of metformin on ovarian cancer cell
apoptosis were assessed by flow cytometry using Annexin
V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) staining
kit (BD Pharmingen; BD Biosciences, San Jose, CA, USA). As cells
transduced with lenti-virus carried green fluorescence, their
apoptosis rates were assessed by using Annexin V-phycoerythrin
(PE)/7-aminoactinomycin D (7-AAD) staining kit (BD Pharmingen; BD
Biosciences). After 24 h of exposure to metformin, cells were
washed twice with ice-cold PBS, re-suspended in 500 µl
binding buffer, and stained with 5 µl FITC-conjugated 5
µl Annexin V or 5 µl Annexin V-PE conjugated and 7
µl 7-AAD, following gentle mixing, cells were incubated at
room temperature shielded from light for 15 min. Following washing
with binding buffer, flow cytometry analysis was performed using a
flow cytometry sorting system (MoFlo XDP) with Summit 6.2 software
(both from Beckman Coulter, Inc., Brea, CA, USA). Each assay was
run at least three times.
Scratch wound healing assay
Cells were seeded into 6-well plates at a destiny of
6×105 cells/well. Following incubation overnight, a
scratch wound was introduced on the cell mono-layer using a 100
µl pipette tip. PBS was used to wash away the floating cells
and then cultured in serum-free growth media. The migration of
cells into the wound area was observed at 0 h and 24 h. For
quantification, the wound gap was imaged using a light microscope
(Olympus Corporation, Tokyo, Japan), then measured using Image-Pro
Plus 6.0 (Media Cybernetics, Inc., Rockville, MD, USA) and the mean
recovery gaps were to normalized 0 h. Each assay was performed at
least three times.
Transwell migration assay
Transwell chambers (6.5-mm diameter; 24-well) were
used for migration assays (Costar; Corning Incorporated, Corning,
NY, USA). SKOV3 (5×104 cells/ml), ES2 (5×104
cells/ml), A2780 (8×104 cells/ml) in 100 µl
serum-free DMEM were seeded in each Transwell insert. The lower
chamber was contained 650 µl DMEM medium supplemented with
20% fetal bovine serum. After incubation at 37°C for 24 h,
non-migrated cells on the upper membrane surface were wiped with a
cotton swab. Cells that had migrated to the lower surfaces of each
filter were fixed with 4% paraformaldehyde for 30 min and stained
with 0.1% crystal violet at room temperature for 1 h. Then, cells
were counted in five randomly selected visual fields under a light
microscope (Olympus Corporation). Each assay was performed at least
three times.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated and extracted using
TRIzol® reagent (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) according to the manufacturer's instructions, and
1 µg total RNA in a final volume of 20 µl was used
for RT with PrimeScript RT Master Mix Kit (Takara Bio, Inc., Otsu,
Japan). Reverse transcription reaction was performed under the
following conditions: 37°C for 15 min, 85°C for 5 sec, 4°C for 5
min. PCR reaction system included 10 µl Eva Green Mix, 1
µl cDNA, 10 µM forward primer, 10 µM reverse
primer, with ddH2O up to 20 µl. The mRNA was
quantitated using a TaqMan One Step Gold RT-PCR kit (Applied
Biosystems; Thermo Fisher Scientific, Inc.). qPCR was performed
under the following conditions: 40 cycles at 95°C for 5 sec and
60°C for 30 sec (EZH2, SUZ12 and β-actin); 40 cycles at 95°C for 5
sec and 52°C for 30 sec (EED). Primer sequences are shown in
Table I. The relative expression
levels of EZH2, SUZ12 and EED to β-actin were calculated using the
2−ΔΔCq method (25).
All reactions were in triplicate and at least three biological
replicated were assessed.
 | Table IPrimer sequences used for reverse
transcription-quantitative polymerase chain reaction. |
Table I
Primer sequences used for reverse
transcription-quantitative polymerase chain reaction.
| Primer | Forward
(5′-3′) | Reverse
(5′-3′) |
|---|
| EZH2 |
TTGTTGGCGGAAGCGTGTAAAATC |
TCCCTAGTCCCGCGCAATGAGC |
| SUZ12 |
GCATTGCCCTTGGTGTACTC |
TGGTCCGTTGCGACTAAAA |
| EED |
ATGCTGTCAGTATTGAGAGTGGC |
GAGGCTGTTCACACATTTGAAAG |
| β-actin |
GCCAACACAGTGCTGTCTGG |
GCTCAGGAGGAGCAATGATCTTG |
Western blot analysis
Ovarian cancer cells were homogenized in cold NP40
buffer (Beyotime Institute of Biotechnology), protease inhibitor
cocktail (Complete™, EDTA-free) and phosphatase inhibitor cocktail
(PhosStop) (both from Roche Applied Science, Penzberg, Germany) and
sonicated on ice, and the lysates were centrifuged at 13,684 x g
for 10 min at 4°C. Protein was quantified using a bicinchoninic
acid assay (Beyotime Institute of Biotechnology). The protein
samples were boiled in protein sample buffer for 8 min. Then, 30
µg protein per well was resolved on 10% SDS-polyacrylamide
gels and transferred onto polyvinylidene fluoride membranes (GE
Healthcare Life Sciences, Little Chalfont, UK). The membranes were
probed with H3K27me3 antibody (1:1,000), H3 antibody (1:5,000),
EZH2 antibody (1:2,000), SUZ12 antibody (1:1,000), EED antibody
(1:2,000), AMPKα antibody (1:1,000), p-AMPKα antibody (1:1,000),
and β-actin antibody (1:5,000) for 24 h in 4°C following blocking
with 5% skim milk in Tris-buffered saline and Tween-20 (TBST).
Following washing in TBST the membrane was incubated with HRP
anti-mouse antibody (1:5,000) or HRP anti-rabbit antibody (1:5,000)
at room temperature for 1 h. Protein bands were measured via Novex
ECL HRP chemiluminescent substrate reagent (Thermo Fisher
Scientific, Inc.) and scanned using Image Lab Software in Molecular
Imager® ChemiDoc™ XRS+ (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) and normalized to internal control
β-actin. All the experiments were repeated at least three
times.
Statistical analysis
Data are presented as the mean ± standard deviation.
One-way analysis of variance with Tukey's post hoc tests were used
for multiple comparisons between groups. P<0.05 was considered
to indicate a statistically significant difference.
Results
Metformin inhibits ovarian cancer growth
and migration in vitro
To examine the effect of metformin on the aggressive
progression of ovarian cancer, SKOV3, A2780 and ES2 cells cultured
in hyperglycemic medium or normoglycemic medium were treated with
10 mM metformin for 24 h, and cell proliferation, apoptosis and
migration ability were then determined. Untreated cells served as
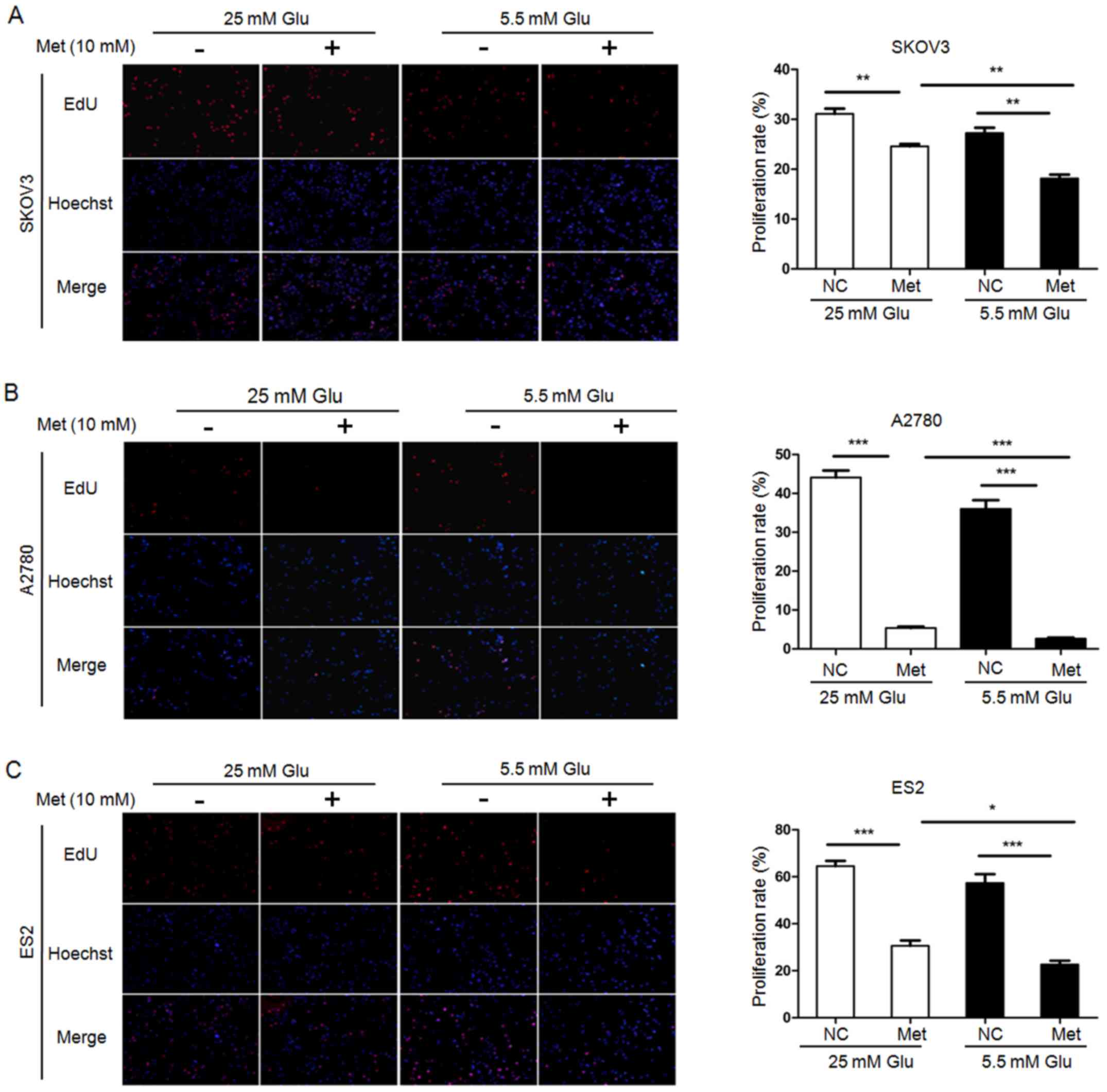
control. EdU assays demonstrated that the cell proliferation was
significantly reduced by metformin under both hyperglycemic and
normoglycemic conditions. Compared with the control cells, the
proliferation rates in SKOV3, A2780 and ES2 cells were decreased by
6.55, 38.72 and 33.96% following metformin treatment in
hyperglycemic medium, respectively. In normoglycemic medium, the
proliferation rates in SKOV3, A2780 and ES2 were decreased by 9.1,
33.37 and 34.7%, respectively. Intriguingly, the cell proliferation
rates in the normoglycemic cells were significantly lower than
those in the hyperglycemic cells exposure to metformin (Fig. 1).
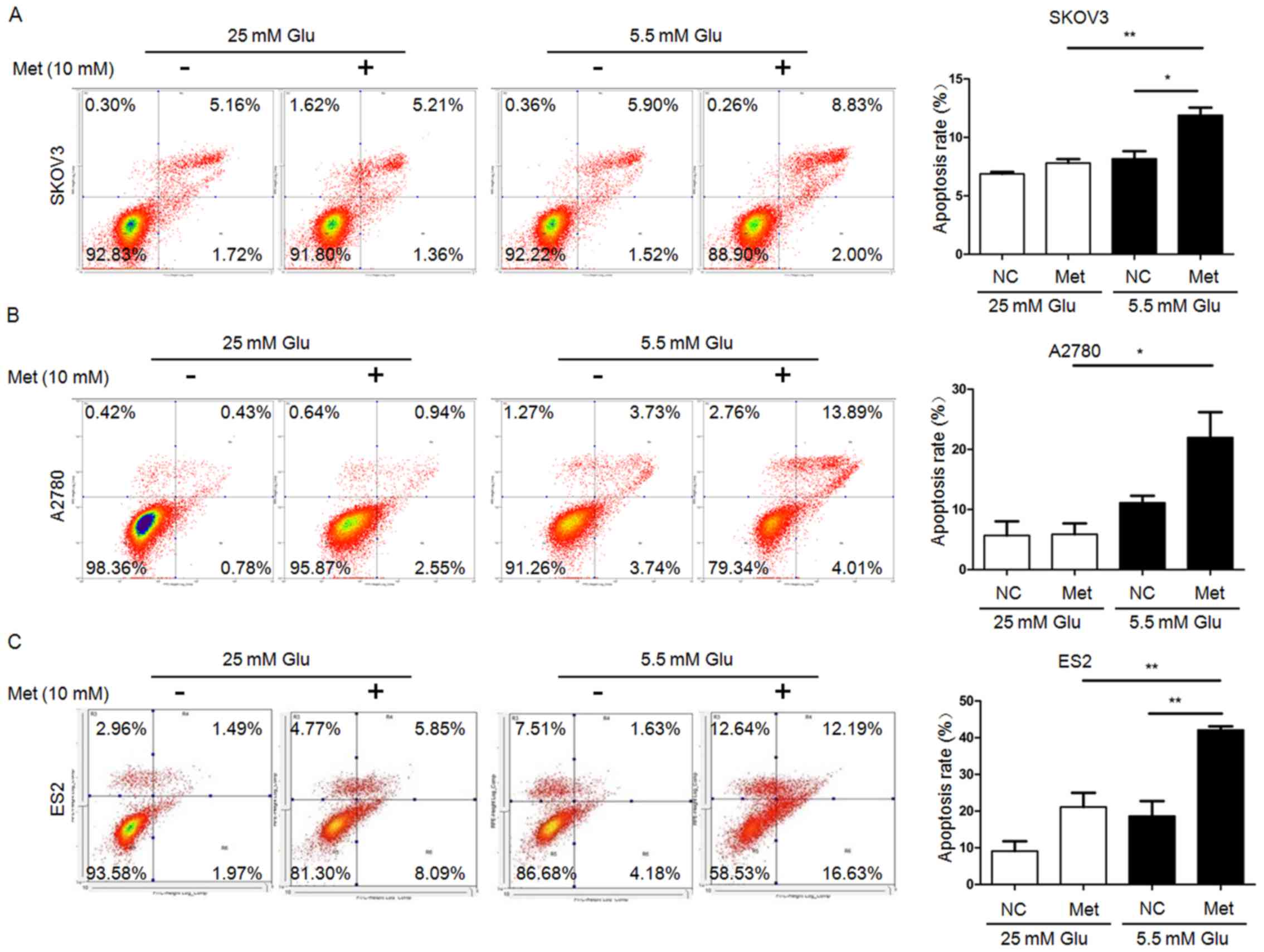
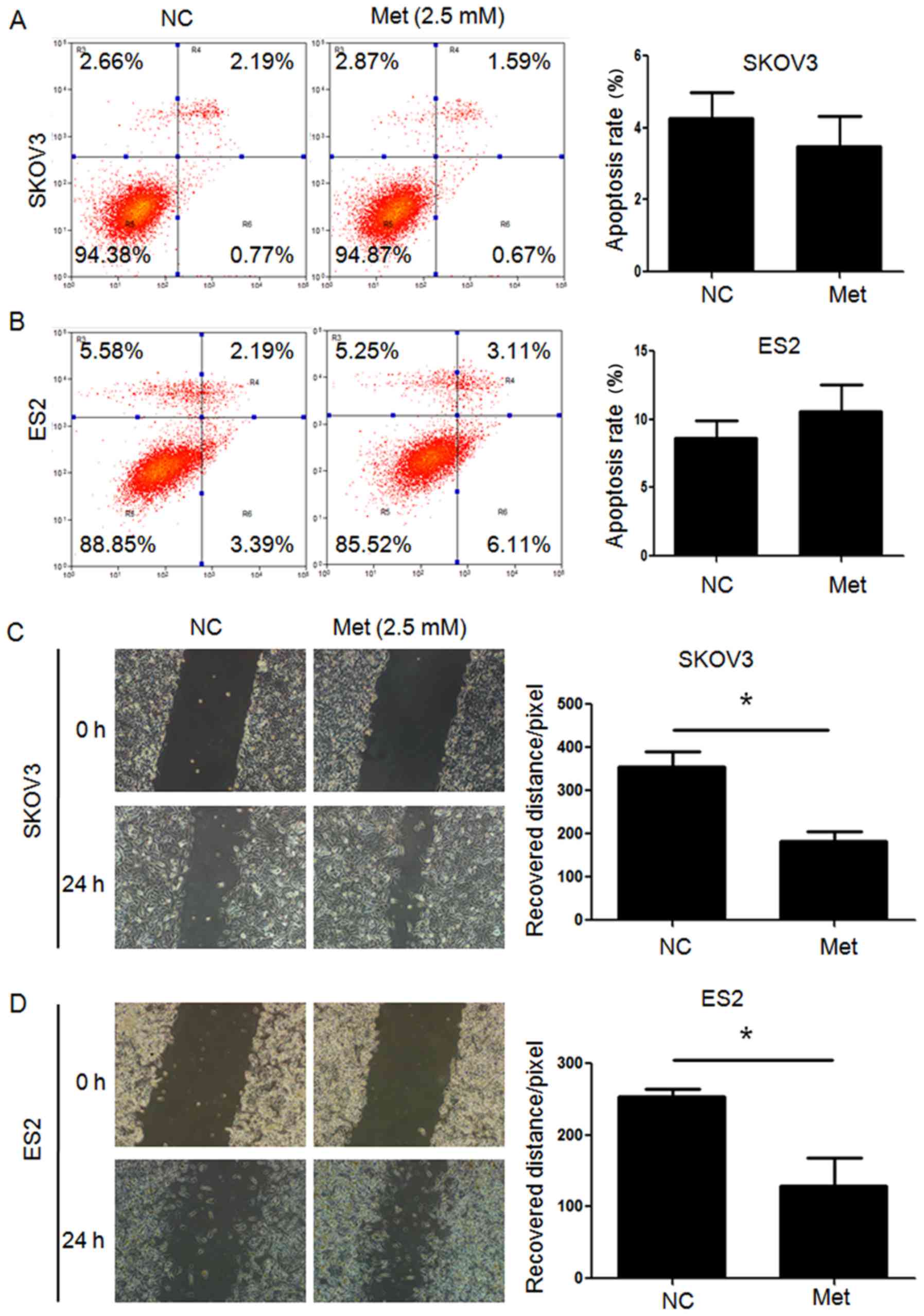
FITC/PI flow cytometry assays demonstrated that the
apoptosis rates in SKOV3 and ES2 cells were significantly increased
following metformin treatment under normoglycemia, but not under
hyperglycemia. Furthermore, all three ovarian cancer cell lines
undergoing metformin treatment in normoglycemic condition exhibited
a higher apoptosis rate compared with those treated with metformin
in hyperglycemic condition (Fig.
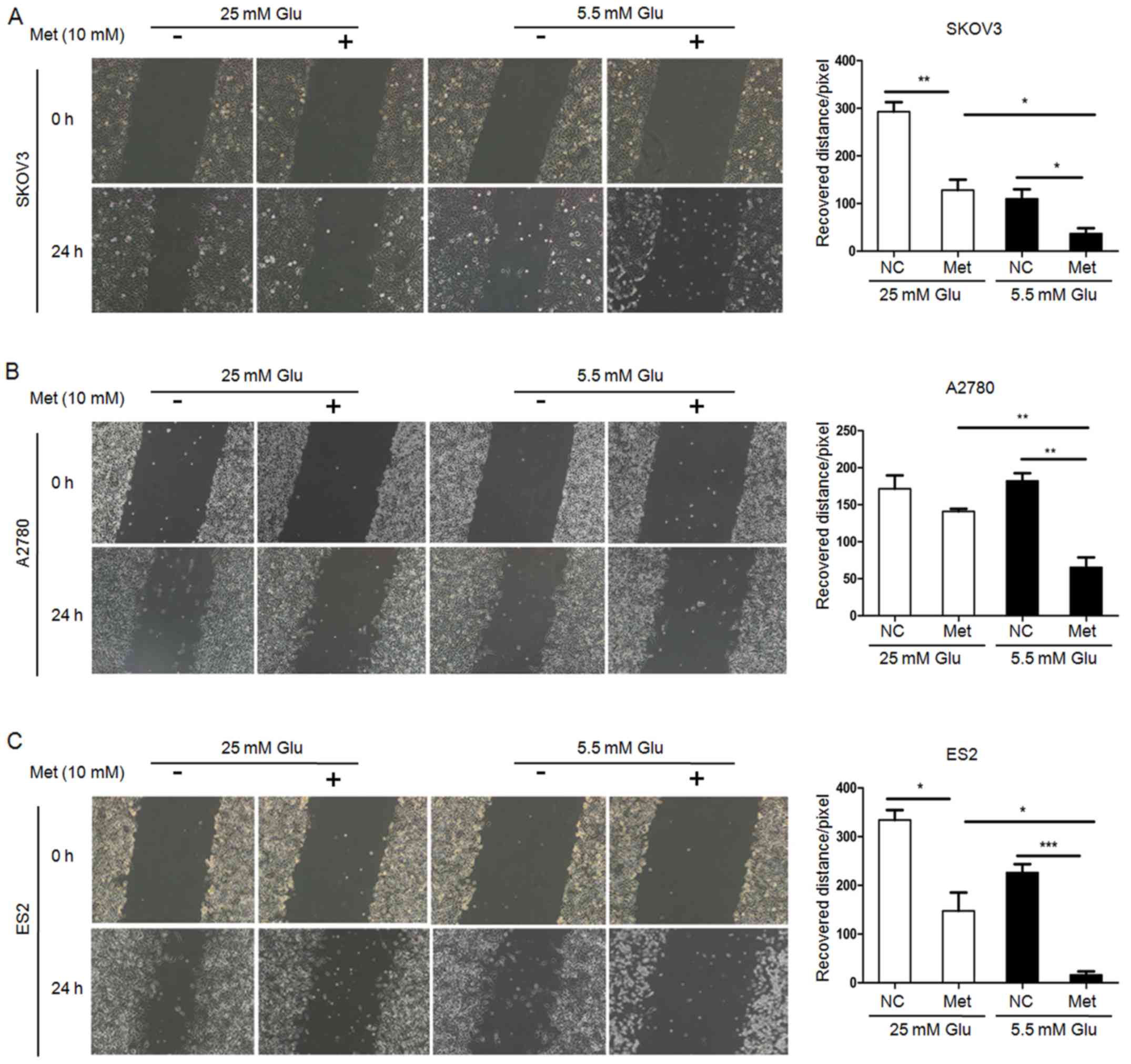
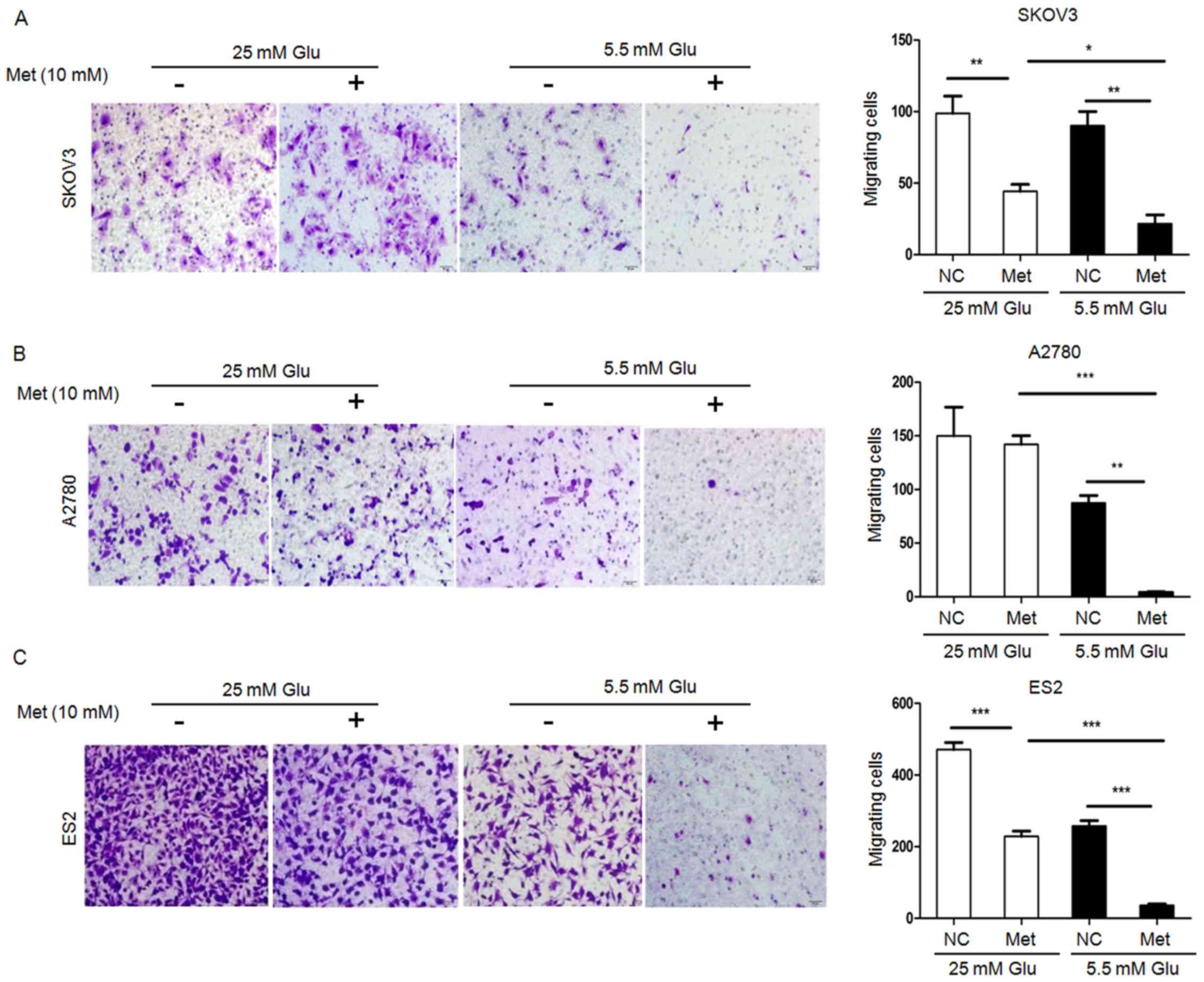
2). Scratch wound healing assays and Transwell migration assays
revealed that the migration capacity of ovarian cancer cells under
hyperglycemic and normoglycemic conditions were inhibited by
metformin, with more intensive inhibition under normoglycemic
condition (Figs. 3 and 4). As 10 mM metformin increases apoptosis
under hypoglycaemia, it may impact the migration results. To
exclude the influence of increased apoptosis on H3K27me3 and
migration, ovarian cancer cells were exposed to a low metformin
concentration (2.5 mM) under normoglycemic condition. As shown in
Fig. 5A and B, 2.5 mM metformin
did not enhance the cell apoptosis, while the migration and
H3K27me3 level were decreased (Figs.
5C and 6A). These results
suggest that metformin inhibits cell proliferation and migration,
and promotes apoptosis, and that reduction of glucose concentration
enhances the sensitivity of ovarian cancer cells to metformin.
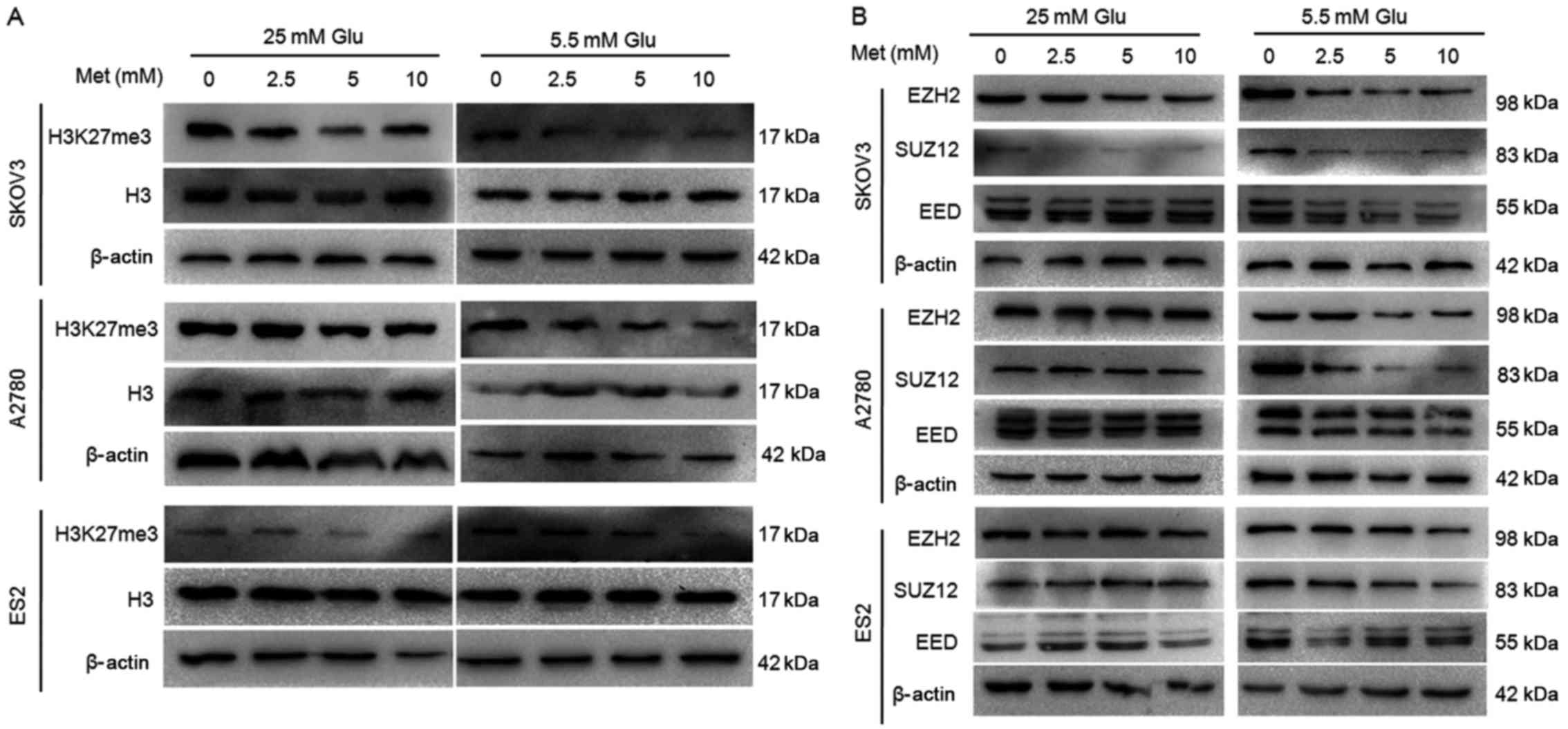
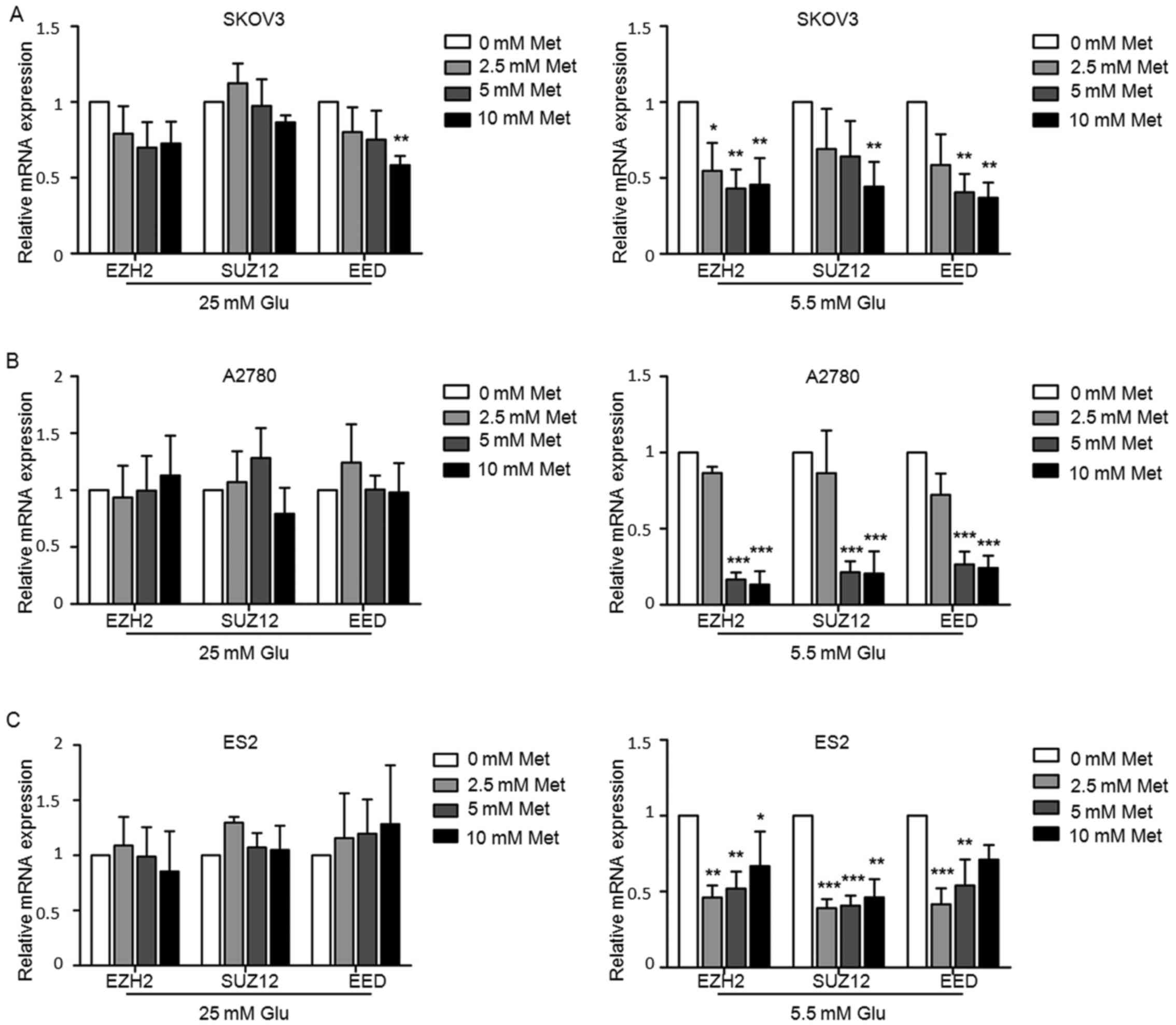
Metformin inhibits H3K27me3 in ovarian
cancer cells
To investigate whether H3K27me3 has a role in the
mechanism of metformin, the alterations in H3K27me3 level and the
expression of three main components of the PRC2 (i.e. EZH2, SUZ12
and EED) following metformin treatment were assessed. Ovarian
cancer cells were treated with various concentrations of metformin
(0, 2.5, 5 and 10 mM) in medium containing 5.5 or 25 mM glucose for
24 h. Western blot analysis demonstrated that metformin suppressed
H3K27me3 level in ovarian cancer cells in a dose-dependent manner,
particularly in medium with lower glucose concentration (Fig. 6A). The protein and mRNA expression
of EZH2, SUZ12 and EED were decreased to varying extents following
exposure to metformin under normoglycemic condition in all three
cell lines, while the suppression was rather modest in the cells
under hyperglycemic conditions (Figs.
6B and 7). Under hyperglycemic
conditions, the protein level of SUZ12 was reduced in SKOV3
(Fig. 6B), but there was no
significant inhibition in mRNA levels (Fig. 7), which may be due to
post-transcriptional modifications.
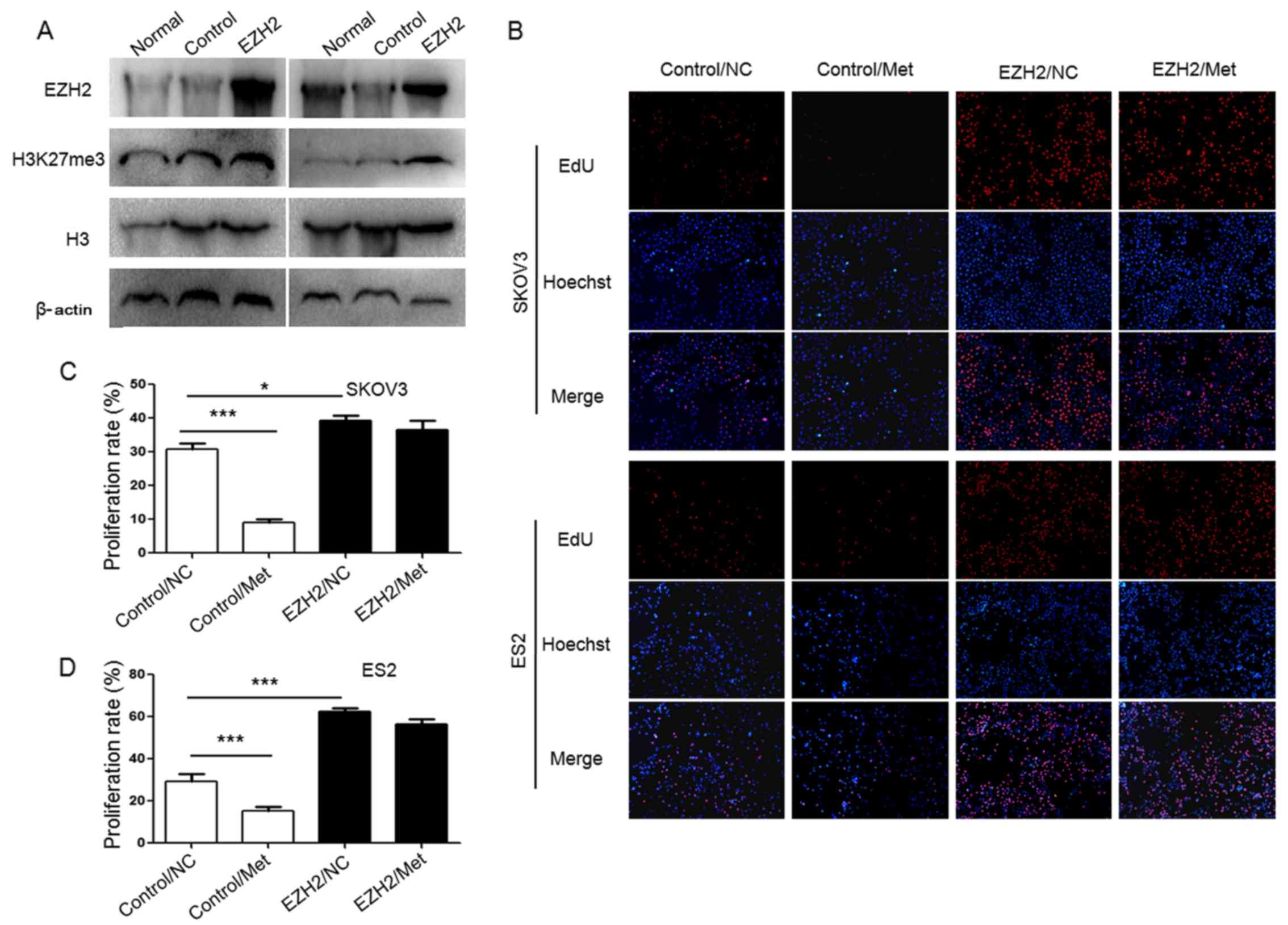
Metformin inhibits ovarian cancer through
decreased-H3K27me3
To demonstrate the role of H3K27me3 in the
metformin-mediated antitumor effect, H3K27me3 level was upregulated
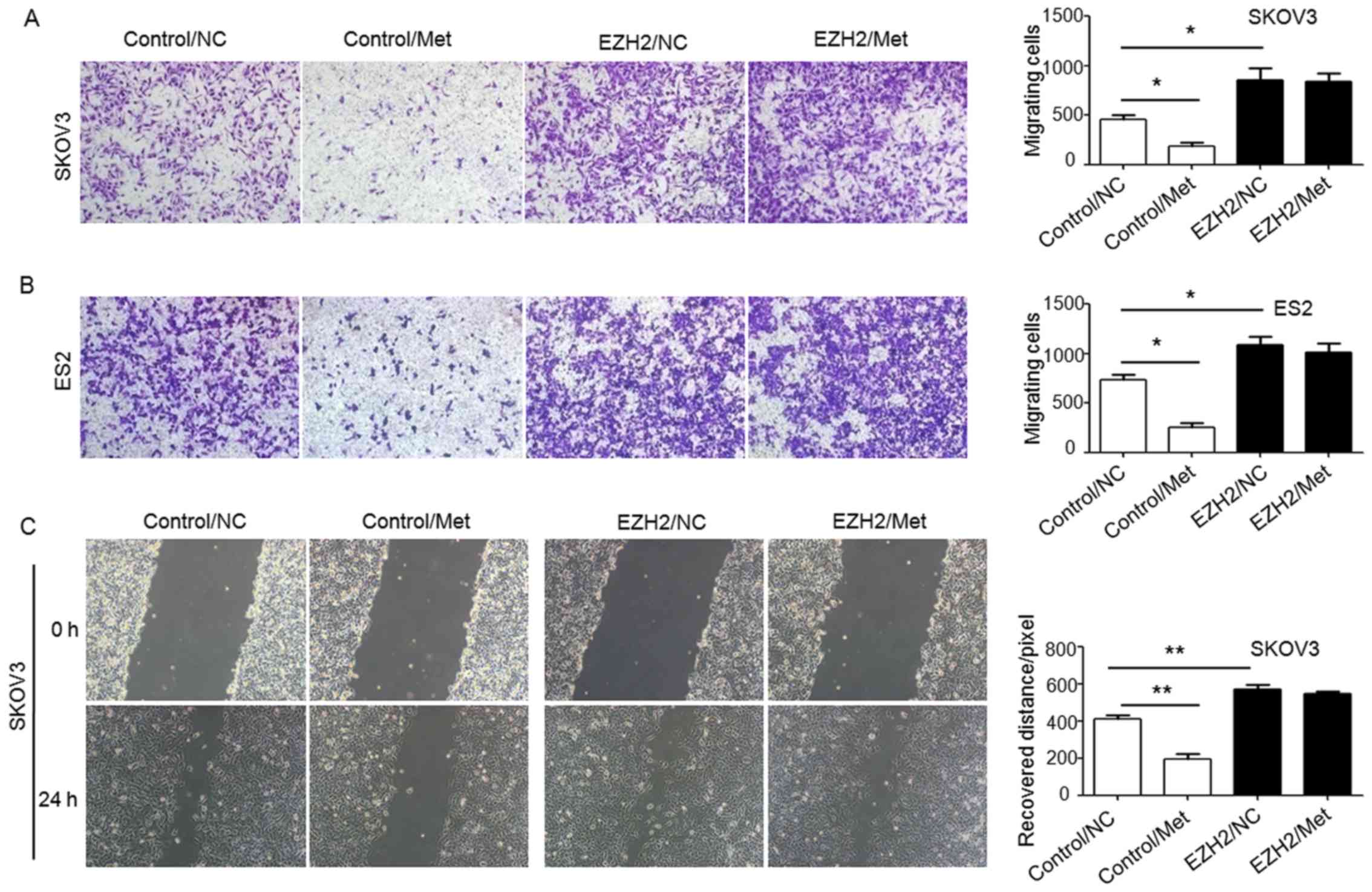
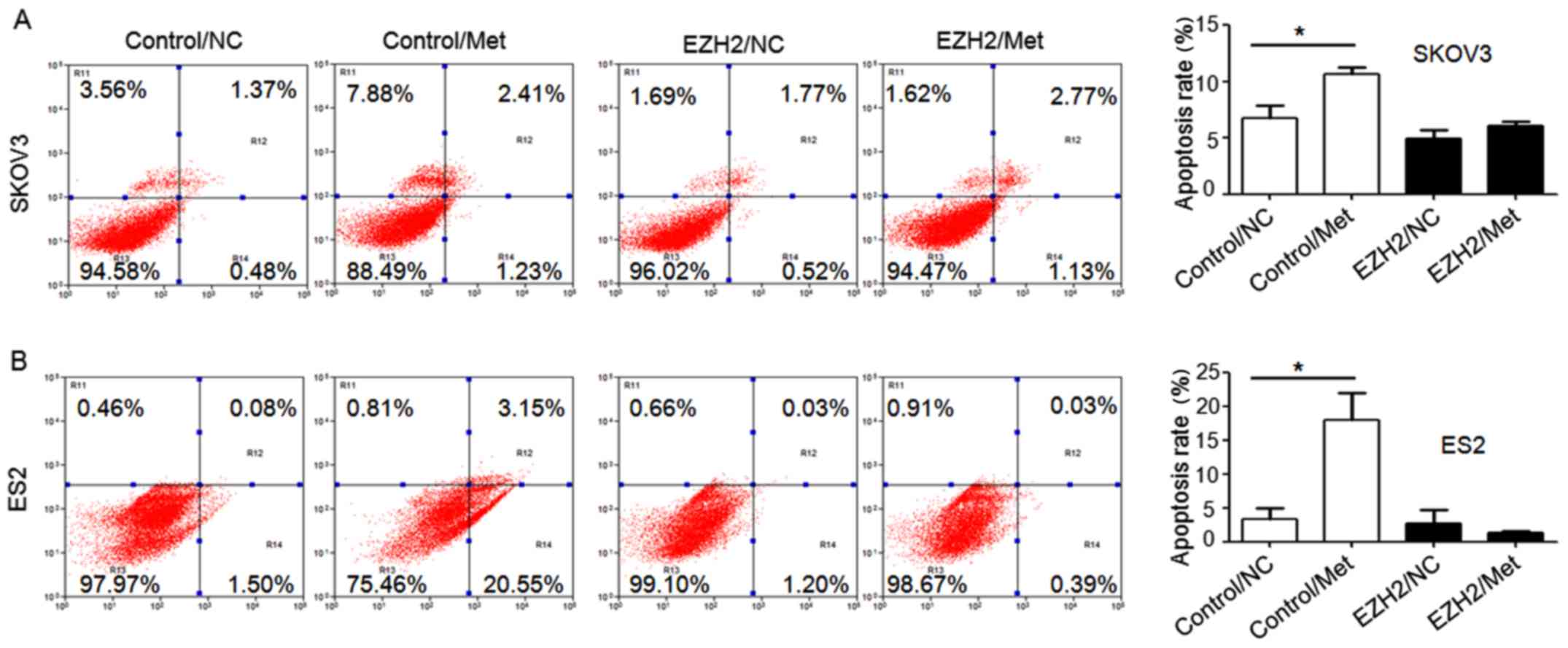
in SKOV3 and ES2 cells using an EZH2 DNA lentivirus (Fig. 8A), and cell proliferation (Fig. 8B–D), migration abilities (Fig. 9) and apoptosis (Fig. 10) were assessed. Ovarian cancer
cells were treated with 10 mM metformin in medium containing 5.5 mM
glucose under normoglycemic condition for 24 h. Cells transfected
with empty lentivirus served as the negative control. Upregulation
of H3K27me3 enhanced the proliferation and migration of ovarian
cancer cells (Figs. 8 and 9). Compared with the cells transfected
with empty lentivirus, metformin did not significantly affect the
cell proliferation, apoptosis or migration of SKOV3 and ES2 cells
transfected with EZH2 lentivirus (Figs. 8Figure 9–10). These results indicate that
decreased H3K27me3 is essential for the antitumor effect of
metformin in ovarian cancer cells.
Metformin inhibits H3K27me3 through AMPK
pathway
Given that metformin is an AMPK activator, whether
metformin suppresses PRC2 through the AMPK pathway was
investigated. AMPKα and p-AMPKα levels were assessed in cancer
cells treated with 0, 2.5, 5 and 10 mM metformin. As shown in
Fig. 11A, metformin induced
phosphorylation of AMPKα at Thr-172 in a dose-dependent manner, the
ratio of p-AMPKα/AMPKα was dramatically increased in the ovarian
cancer cells cultured in normoglycemic medium, while the increase
was slight in the cells in hyperglycemic medium. These results
confirmed that metformin induced the activation of AMPK in ovarian
cancer cells. Another AMPK activator, 2-DG, and an AMPK inhibitor,
Compound C, were used to evaluate the effects of AMPK activation on
H3K27me3 and PRC2. Similar to metformin, 2-DG increased the
p-AMPK/AMPK ratio, and repressed the expression of H3K27me3, EZH2,
SUZ12 and EED (Fig. 11B). In the
cells pretreated with Compound C, metformin was not able to induce
AMPK phosphorylation or inhibitEZH2, SUZ12, EED or H3K27me3
(Fig. 11C), suggesting AMPK
activation is required for metformin-induced PRC2 inhibition.
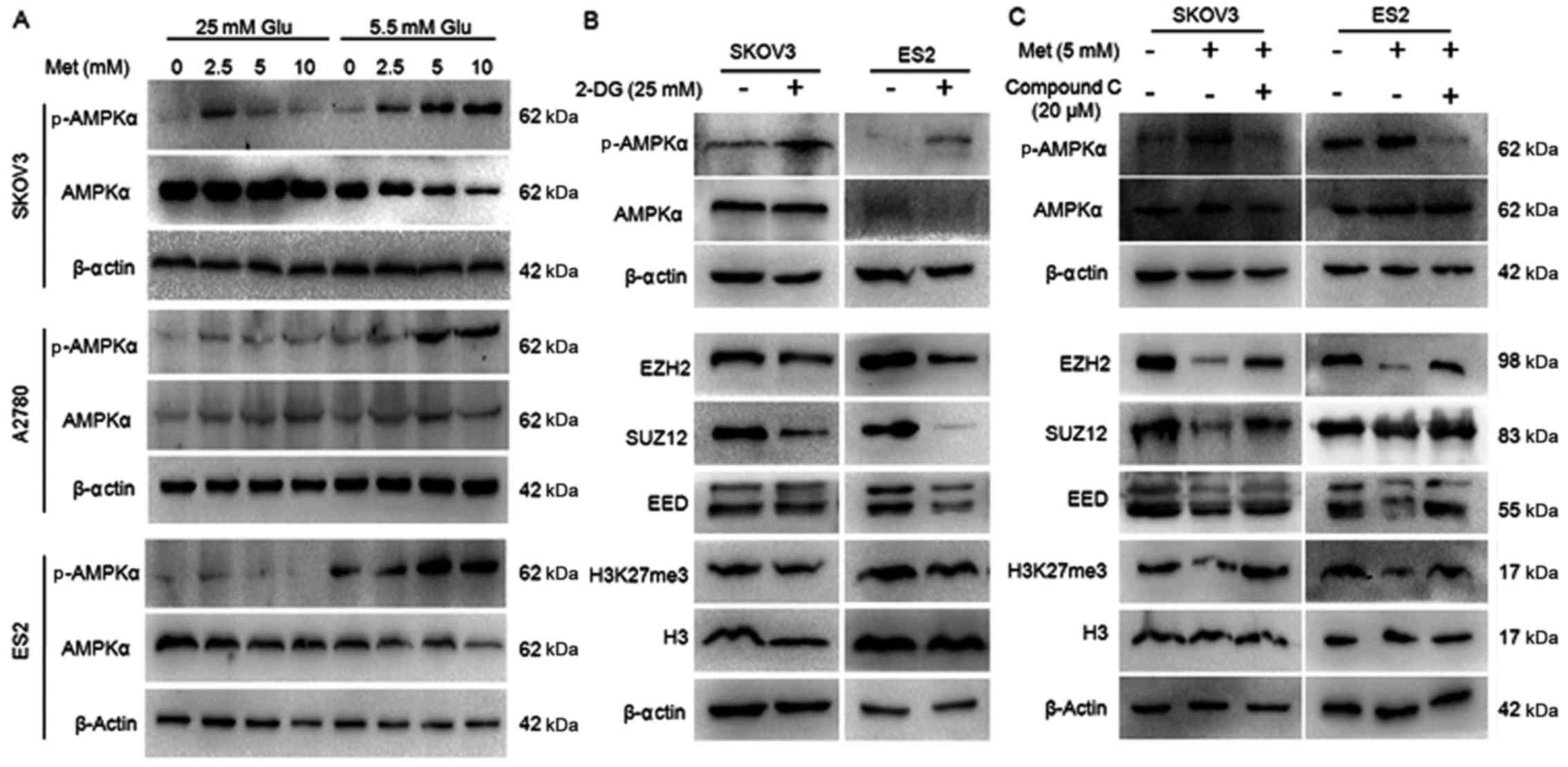 | Figure 11Metformin inhibits H3K27me3 through
the AMPK pathway. (A) Metformin activates AMPK of ovarian cancer
cells in normoglycemic condition. (B) 2-DG activates AMPK and
suppresses H3K27me3, PRC2 of ovarian cancer cells in normoglycemic
condition. (C) Compound C inhibits the effect of metformin on PRC2
and H3K27me3 when ovarian cancer cells were cultured in
normoglycemic condition. The protein expression of p-AMPKα, AMPKα,
H3K27me3, EZH2, SUZ12 and EED in A2780, SKOV3 and ES2 cells were
assayed by western blot analysis. The expression was normalized to
β-actin. Met, metformin; Glu, glucose; p-AMPKα, phospho-AMPKα;
AMPKα, AMP-activated protein kinase; 2-DG, 2-deoxy-D-glucose; EZH2,
histone-lysine N-methyltransferase EZH2;SUZ12, polycomb protein
SUZ12; EED, polycomb protein EED; 2-DG, dorsomorphin 2HCl;
H3K27me3, histone H3 lysine 27 trimethylation. |
Discussion
Multiple meta-analyses have now reported that
metformin may reduce the incidence of overall cancer by 10–40%,
with lower risks of cancer-specific mortality (26–29).
In patients with ovarian cancer, as revealed by a case-control
study, metformin intake is significantly associated with improved
5-year survival (73 vs. 44%, P=0.002) (10). In mouse models, metformin inhibited
the growth of ovarian cancer xenografts (5). The present study produced in
vitro evidence supporting the anti-tumor effects of metformin
in ovarian cancer and reported that H3K27me3 was abrogated by
metformin through AMPK activation, which sheds new light on the
underlying mechanism of metformin as an antitumor drug.
Metformin promoted cell apoptosis and repressed cell
proliferation and migration in all three epithelial ovarian cancer
cell lines used in the study (A2780, ES2 and SKOV3). Similar
results have been reported in other epithelial ovarian cancer cell
lines, including PA-1, OVCAR-3 and HO8910-PM (30,31).
It is well established that tumor cells can be starved by the
metformin-mediated ATP depletion that confers susceptibility to
cell death. In addition to regulating energy homeostasis, metformin
was previously reported to alter folate metabolism, disturbing
nucleotide synthesis and inhibiting cell proliferation (32). It is notable that the antitumor
effects of metformin were more robust in the low glucose medium,
which provides preliminary evidence supporting the use of metformin
for the treatment of ovarian cancer in non-diabetic patients. In
line with the findings of the current study, low glucose was
previously reported to enhance the cytotoxicity of metformin in
breast cancer and thyroid cancer cells (33,34).
Zhuang et al (35) reported
that lowering glucose potentiated metformin mediated ATP depletion
and cell death by reducing metformin-stimulated glycolysis in
ovarian and breast cancer cells, which may partially explain the
influences of glucose on metformin actions. Consistently, in the
current study, AMPK was more strongly activated by metformin in low
glucose medium, compared with high glucose medium.
The clarification of the molecular mechanisms under
the antitumor effect of metformin will be of important clinical
relevance regarding predicting the response to metformin therapy
and selecting appropriate patients for the treatment. In the
present study, metformin treatment resulted in decreased-H3K27me3
accompanied by repressed EZH2, SUZ12 and EED expression in ovarian
cancer cells. The inhibition of EZH2 transcription by metformin has
been previously observed in breast cancer and pancreatic cancer
cells, which may be associated with re-expression of the microRNAs
(miRs) targeting EZH2, including miR-26a and miR-101 (36,37).
In prostate cancer cells, metformin reduced the level of
histone-lysine N-methyltransferase SUV39H1, a histone
methyltransferase of H3 Lys9, and inhibited SUV39H1-mediated cell
migration (38). These findings
suggest that reduced specific histone methylation marks may mediate
the antitumor effects of metformin. Further research may be
warranted to investigate the presence of these histone marks on
gene promoters and the consequent alterations in gene transcription
in tumor cells.
AMPK is an important metabolic sensor that regulates
energy homeostasis; activated AMPK induces energy sparing pathways
to maintain ATP levels in response to hypoxia, fuel deprivation and
low glucose level (39). As
expected, in the present study, metformin-stimulated AMPK
activation was dependent on the content of glucose in the
environment. Notably, the metformin-induced PRC2 inhibition was
rescued by pretreatment with an AMPK inhibitor (Compound C),
suggesting that AMPK activation was required for the inhibitory
effect of metformin on H3K27me3. Recently, Demoinet et al
(40) demonstrated that AMPK is a
pivotal molecular link to couple the chromatin environment and
consequent adaptive changes in gene expression, and resolution of
epigenetic modifications in Caenorhabditis elegans.
Furthermore, AMPK inactivation was reported to be associated with
enrichment of H3K27me3 in gut epithelial cells (41). All these findings suggest
cross-talking between energy stress and the methylation of histones
in tumor cells.
In addition, the findings of the current study
demonstrated that the decrease in H3K27me3 is required for the
anti-tumor effect mediated by metformin in ovarian cancer cells.
H3K27me3 contributes to CpG methylation in gene promoter regions
and chromatin configuration, which in turn reduces the
accessibility of transcription factors and the transcriptional
activity of nearby genes, resulting in transcriptional repression.
H3K27me3 has been reported to regulate the expression of genes
involved in ovarian cancer cell proliferation, apoptosis and
metastasis, such asp57, Harakiri, Aplasia Ras homolog member I and
tissue inhibitor of metalloproteinase 2 (20,42–44).
Notably, Zhao et al (45)
demonstrated that enhancing H3K27me3 occupancy at the E3
ubiquitin-protein ligase NEDD4-likepromoter increased transforming
growth factor-β signal transduction in ovarian cancer cells, which
has a critical role in tumor progression (46). These downstream mechanisms of
H3K27me3 may partially explain the antitumor effects of
metformin.
Taken together, the findings suggest that the
antitumor effect of metformin in ovarian cancer cells may be
partially attributed to the metformin-mediated AMPK activation and
the consequent reduction of H3K27me3, particularly under
normoglycemia condition. Several small molecular inhibitors of EZH2
are being assessed in clinical trials to treat patients with
malignancies. For example, CPI-1205 and Tazemetostat are current
being used in patients with B-cell lymphoma and synovial sarcoma in
phase I clinical trials (NCT02395601 and NCT02601937; clinicaltrials.gov), respectively. However, it usually
takes >10 years to take a novel drug from the laboratory to
clinical use. By contrast, repurposing of metformin as an antitumor
agent is a shortcut, as above all, its biological safety has been
well documented. The findings of the current study indicate that
metformin maybe an epigenetic regulator targeting PRC2 and support
the use of metformin in treatment of patients with epithelial
ovarian cancer without diabetes.
Acknowledgments
Not applicable.
Funding
This study was supported by the National Science
Foundation of China (grant nos. 81672573, 81472443 and
81372807).
Availability of data and materials
The data sets generated during the current study are
available from the corresponding author on reasonable request.
Authors' contributions
ZW, JG and JC contributed to the conception and
design; GT, YZ and TL contributed to acquisition of data; GT, ZH
and LY contributed to analysis and interpretation of data for the
work; all authors drafted the work and revised it critically for
important intellectual content; all authors approved the final
manuscript to be published; all authors agree to be accountable for
all aspects of the work in ensuring that questions related to the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Davis A, Tinker AV and Friedlander M:
'Platinum resistant' ovarian cancer: What is it, who to treat and
how to measure benefit? Gynecol Oncol. 133:624–631. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Coyle C, Cafferty FH, Vale C and Langley
RE: Metformin as an adjuvant treatment for cancer: A systematic
review and meta-analysis. Ann Oncol. 27:2184–2195. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dowling RJ, Niraula S, Chang MC, Done SJ,
Ennis M, McCready DR, Leong WL, Escallon JM, Reedijk M, Goodwin PJ,
et al: Changes in insulin receptor signaling underlie neoadjuvant
metformin administration in breast cancer: A prospective window of
opportunity neoadjuvant study. Breast Cancer Res. 17:322015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rattan R, Graham RP, Maguire JL, Giri S
and Shridhar V: Metformin suppresses ovarian cancer growth and
metastasis with enhancement of cisplatin cytotoxicity in vivo.
Neoplasia. 13:483–491. 2011. View Article : Google Scholar :
|
|
6
|
Blandino G, Valerio M, Cioce M, Mori F,
Casadei L, Pulito C, Sacconi A, Biagioni F, Cortese G, Galanti S,
et al: Metformin elicits anticancer effects through the sequential
modulation of DICER and c-MYC. Nat Commun. 3:8652012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sivalingam VN, Kitson S, McVey R, Roberts
C, Pemberton P, Gilmour K, Ali S, Renehan AG, Kitchener HC and
Crosbie EJ: Measuring the biological effect of presurgical
metformin treatment in endometrial cancer. Br J Cancer.
114:281–289. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Currie CJ, Poole CD, Jenkins-Jones S, Gale
EAM, Johnson JA and Morgan CL: Mortality after incident cancer in
people with and without type 2 diabetes: Impact of metformin on
survival. Diabetes Care. 35:299–304. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Romero IL, McCormick A, McEwen KA, Park S,
Karrison T, Yamada SD, Pannain S and Lengyel E: Relationship of
type II diabetes and metformin use to ovarian cancer progression,
survival, and chemosensitivity. Obstet Gynecol. 119:61–67. 2012.
View Article : Google Scholar
|
|
10
|
Kumar S, Meuter A, Thapa P, Langstraat C,
Giri S, Chien J, Rattan R, Cliby W and Shridhar V: Metformin intake
is associated with better survival in ovarian cancer: A
case-control study. Cancer. 119:555–562. 2013. View Article : Google Scholar
|
|
11
|
Jenkins Y, Sun TQ, Markovtsov V, Foretz M,
Li W, Nguyen H, Li Y, Pan A, Uy G, Gross L, et al: AMPK activation
through mitochondrial regulation results in increased substrate
oxidation and improved metabolic parameters in models of diabetes.
PLoS One. 8:e818702013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Han G, Gong H, Wang Y, Guo S and Liu K:
AMPK/mTOR-mediated inhibition of survivin partly contributes to
metformin-induced apoptosis in human gastric cancer cell. Cancer
Biol Ther. 16:77–87. 2015. View Article : Google Scholar :
|
|
13
|
Ochoa-Gonzalez F, Cervantes-Villagrana AR,
Fernandez-Ruiz JC, Nava-Ramirez HS, Hernandez-Correa AC,
Enciso-Moreno JA and Castañeda-Delgado JE: Metformin induces cell
cycle arrest, reduced proliferation, wound healing impairment in
vivo and is associated to clinical outcomes in diabetic foot ulcer
patients. PLoS One. 11:e01509002016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pollak M: The insulin and insulin-like
growth factor receptor family in neoplasia: An update. Nat Rev
Cancer. 12:159–169. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Portela A and Esteller M: Epigenetic
modifications and human disease. Nat Biotechnol. 28:1057–1068.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chan KM, Fang D, Gan H, Hashizume R, Yu C,
Schroeder M, Gupta N, Mueller S, James CD, Jenkins R, et al: The
histone H3.3K27M mutation in pediatric glioma reprograms H3K27
methylation and gene expression. Genes Dev. 27:985–990. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Peng D, Kryczek I, Nagarsheth N, Zhao L,
Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W, et al: Epigenetic
silencing of TH1-type chemokines shapes tumour immunity and
immunotherapy. Nature. 527:249–253. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chapman-Rothe N, Curry E, Zeller C, Liber
D, Stronach E, Gabra H, Ghaem-Maghami S and Brown R: Chromatin
H3K27me3/H3K4me3 histone marks define gene sets in high-grade
serous ovarian cancer that distinguish malignant, tumour-sustaining
and chemo-resistant ovarian tumour cells. Oncogene. 32:4586–4592.
2013. View Article : Google Scholar
|
|
19
|
Cao R and Zhang Y: The functions of
E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr
Opin Genet Dev. 14:155–164. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yi X, Guo J, Guo J, Sun S, Yang P, Wang J,
Li Y, Xie L, Cai J and Wang Z: EZH2-mediated epigenetic silencing
of TIMP2 promotes ovarian cancer migration and invasion. Sci Rep.
7:35682017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hu S, Yu L, Li Z, Shen Y, Wang J, Cai J,
Xiao L and Wang Z: Overexpression of EZH2 contributes to acquired
cisplatin resistance in ovarian cancer cells in vitro and in vivo.
Cancer Biol Ther. 10:788–795. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Siddiqi FS, Majumder S, Thai K, Abdalla M,
Hu P, Advani SL, White KE, Bowskill BB, Guarna G, Dos Santos CC, et
al: The histone methyltransferase enzyme enhancer of zeste homolog
2 protects against podocyte oxidative stress and renal injury in
diabetes. J Am Soc Nephrol. 27:2021–2034. 2016. View Article : Google Scholar :
|
|
23
|
Gallagher KA, Joshi A, Carson WF, Schaller
M, Allen R, Mukerjee S, Kittan N, Feldman EL, Henke PK, Hogaboam C,
et al: Epigenetic changes in bone marrow progenitor cells influence
the inflammatory phenotype and alter wound healing in type 2
diabetes. Diabetes. 64:1420–1430. 2015. View Article : Google Scholar :
|
|
24
|
Litchfield LM, Mukherjee A, Eckert MA,
Johnson A, Mills KA, Pan S, Shridhar V, Lengyel E and Romero IL:
Hyperglycemia-induced metabolic compensation inhibits metformin
sensitivity in ovarian cancer. Oncotarget. 6:23548–23560. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
26
|
Wu L, Zhu J, Prokop LJ and Murad MH:
Pharmacologic therapy of diabetes and overall cancer risk and
mortality: A meta-analysis of 265 studies. Sci Rep. 5:101472015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gandini S, Puntoni M, Heckman-Stoddard BM,
Dunn BK, Ford L, DeCensi A and Szabo E: Metformin and cancer risk
and mortality: A systematic review and meta-analysis taking into
account biases and confounders. Cancer Prev Res (Phila). 7:867–885.
2014. View Article : Google Scholar
|
|
28
|
Zhang P, Li H, Tan X, Chen L and Wang S:
Association of metformin use with cancer incidence and mortality: A
meta-analysis. Cancer Epidemiol. 37:207–218. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang ZJ and Li S: The prognostic value of
metformin for cancer patients with concurrent diabetes: A
systematic review and meta-analysis. Diabetes Obes Metab.
16:707–710. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Moon HS, Kim B, Gwak H, Suh DH and Song
YS: Autophagy and protein kinase RNA-like endoplasmic reticulum
kinase (PERK)/eukaryotic initiation factor 2 alpha kinase (eIF2α)
pathway protect ovarian cancer cells from metformin-induced
apoptosis. Mol Carcinog. 55:346–356. 2016. View Article : Google Scholar
|
|
31
|
Milewicz T, Kiałka M, Mrozińska S, Ociepka
A and Krzysiek J: Metformin - new treatment strategies for
gynecologic neoplasms. Przegl Lek. 70:81–84. 2013.In Polish.
|
|
32
|
Jara JA and López-Muñoz R: Metformin and
cancer: Between the bioenergetic disturbances and the antifolate
activity. Pharmacol Res. 101:102–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Menendez JA, Oliveras-Ferraros C, Cufí S,
Corominas-Faja B, Joven J, Martin-Castillo B and Vazquez-Martin A:
Metformin is synthetically lethal with glucose withdrawal in cancer
cells. Cell Cycle. 11:2782–2792. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bikas A, Jensen K, Patel A, Costello J Jr,
McDaniel D, Klubo-Gwiezdzinska J, Larin O, Hoperia V, Burman KD,
Boyle L, et al: Glucose-deprivation increases thyroid cancer cells
sensitivity to metformin. Endocr Relat Cancer. 22:919–932. 2015.
View Article : Google Scholar
|
|
35
|
Zhuang Y, Chan DK, Haugrud AB and
Miskimins WK: Mechanisms by which low glucose enhances the
cytotoxicity of metformin to cancer cells both in vitro and in
vivo. PLoS One. 9:e1084442014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bao B, Wang Z, Ali S, Ahmad A, Azmi AS,
Sarkar SH, Banerjee S, Kong D, Li Y, Thakur S, et al: Metformin
inhibits cell proliferation, migration and invasion by attenuating
CSC function mediated by deregulating miRNAs in pancreatic cancer
cells. Cancer Prev Res (Phila). 5:355–364. 2012. View Article : Google Scholar
|
|
37
|
Cabello P, Pineda B, Tormo E, Lluch A and
Eroles P: The antitumor effect of metformin is mediated by miR-26a
in breast cancer. Int J Mol Sci. 17:12982016. View Article : Google Scholar :
|
|
38
|
Yu T, Wang C, Yang J, Guo Y, Wu Y and Li
X: Metformin inhibits SUV39H1-mediated migration of prostate cancer
cells. Oncogenesis. 6:e3242017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bonini MG and Gantner BN: The multifaceted
activities of AMPK in tumor progression - why the 'one size fits
all' definition does not fit at all? IUBMB Life. 65:889–896. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Demoinet E, Li S and Roy R: AMPK blocks
starvation-inducible transgenerational defects in Caenorhabditis
elegans. Proc Natl Acad Sci USA. 114:E2689–E2698. 2017. View Article : Google Scholar :
|
|
41
|
Sun X, Yang Q, Rogers CJ, Du M and Zhu MJ:
AMPK improves gut epithelial differentiation and barrier function
via regulating Cdx2 expression. Cell Death Differ. 24:819–831.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fu Y, Chen J, Pang B, Li C, Zhao J and
Shen K: EZH2-induced H3K27me3 is associated with epigenetic
repression of the ARHI tumor-suppressor gene in ovarian cancer.
Cell Biochem Biophys. 71:105–112. 2015. View Article : Google Scholar
|
|
43
|
Li H, Cai Q, Wu H, Vathipadiekal V, Dobbin
ZC, Li T, Hua X, Landen CN, Birrer MJ, Sánchez-Beato M, et al:
SUZ12 promotes human epithelial ovarian cancer by suppressing
apoptosis via silencing HRK. Mol Cancer Res. 10:1462–1472. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Guo J, Cai J, Yu L, Tang H, Chen C and
Wang Z: EZH2 regulates expression of p57 and contributes to
progression of ovarian cancer in vitro and in vivo. Cancer Sci.
102:530–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhao R, Cui T, Han C, Zhang X, He J,
Srivastava AK, Yu J, Wani AA and Wang QE: DDB2 modulates TGF-β
signal transduction in human ovarian cancer cells by downregulating
NEDD4L. Nucleic Acids Res. 43:7838–7849. 2015. View Article : Google Scholar :
|
|
46
|
Principe DR, Doll JA, Bauer J, Jung B,
Munshi HG, Bartholin L, Pasche B, Lee C and Grippo PJ: TGF-β:
Duality of function between tumor prevention and carcinogenesis. J
Natl Cancer Inst. 106:djt3692014. View Article : Google Scholar
|