Introduction
Venous thromboembolism (VTE) is a common
complication in patients with multiple myeloma (MM), who can have
up to a 28-fold increase in risk of VTE (1,2). The
occurrence of thrombosis seriously impacts quality of life and
increases mortality of patients with MM (3). Notably, the incidence of VTE in
patients with MM treated with immunomodulatory drug (IMiD)
monotherapy is 2–6%, while dexamethasone (Dex)-containing regimens
dramatically increase the risk of VTE up to 17–26% in individual
clinical trials (4,5). Previous studies have reported that
patients with MM have several risk factors for VTE, including
increased levels of factor VIII and von Willebrand factor,
hypofibrinolysis and acquired activated protein C resistance
(6–9). However, these molecular mechanisms
are not sufficient to reflect the changes in plasma procoagulant
components and there is limited research studying the cellular
mechanism of VTE in patients with MM. In addition, the evidence to
guide optimal selection of thromboprophylaxis when managing
patients with MM is still lacking. Thus, it is important to
investigate the precise molecular and cellular mechanism of the
hypercoagulable state in these patients.
Phosphatidylserine (PS) is exposed by the action of
scramblase on the cell’s surface during biological processes such
as apoptosis and cell activation (10). Once exposed on the outer membrane,
PS acts as a catalytic surface for factor Xa and thrombin formation
in the coagulation cascade (11).
Our previous studies have demonstrated that PS expression on the
surface of circulating cells is associated with a risk of
developing venous thrombotic complications in various disorders,
including nephrotic syndrome and acute promyelocytic leukemia
(12,13). Previous studies have demonstrated
that peripheral blood cells and endothelial cells (ECs) can be
injured or activated by high levels of monoclonal paraprotein and
inflammatory cytokines circulating in the blood of patients with MM
(14–18). However, relatively little is known
about the extent to which PS is exposed on these cells, or whether
PS+ cells contribute to a hypercoagulable state in
patients with MM. Although other studies reported that IMiDs with
Dex increased PS exposure on monocyte and EC cell lines (19,20),
the inhibition assays were not performed to clarify whether PS was
responsible for cell-associated procoagulant activity (PCA).
Furthermore, it is unclear whether IMiD-based regimens will further
aggravate the amount of PS exposure on blood cells or ECs from
patients with MM treated with patient serum. Thus, lactadherin, a
superior PS probe (21), was used
to detect the exposure of PS on cells and define whether there is
an association between PS levels, cell-associated PCA and
development of the hypercoagulable state in patients with MM.
The main objective of the current study was to
evaluate the PCA of PS+ blood cells and ECs and to
assess a possible association with the development of venous
thrombosis. Furthermore, the effect of treatment with IMiDs with or
without Dex on PS exposure in blood cells and ECs was evaluated,
and their relevance to PCA of these cells. Additionally, inhibition
experiments were performed to evaluate the anticoagulant property
of lactadherin. The results suggested that PS exposure on blood
cells and ECs has a pivotal role in the hypercoagulability in
patients with MM.
Materials and methods
Study subjects
The study included 20 newly diagnosed patients with
MM (range 31–78, 45% males) according to the standards of the
International Myeloma Working Group who were admitted to the First
Affiliated Hospital of Harbin Medical University between October
2016 and February 2017, and 15 healthy volunteers (range 28–75,
46.67% males) (22). The exclusion
criteria were a recent (<6 months) thrombotic event, current
anticoagulant therapy, and associated disease, including
antiphospholipid syndrome, chronic renal disease, heart disease,
malignant or systemic disease, diabetes, acute infection,
immobilization, surgery, hereditary thrombophilia and
hyperviscosity, among other conditions. Harbin Medical University
Ethical Committee (Harbin, China) approved the study, and patients
provided written, informed consent.
Reagents
Thalidomide (Tha), lenalidomide (Len) and Dex were
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Human umbilical vein cells (HUVECs), ECs medium, and poly-L-lysine
were from ScienCell Research Laboratories, Inc. (San Diego, CA,
USA). RPMI-1640 medium, fetal bovine serum (FBS) and bovine serum
albumin (BSA) were obtained from Gibco (Thermo Fisher Scientific,
Inc., Waltham, MA, USA). Alexa Fluor 488 or Alexa Fluor
647-conjugated lactadherin were prepared in our laboratory. Human
factors (F) Va, VIIa, VIII, IXa, X, Xa, prothrombin, thrombin,
fluorescein EGR-chloromethylketone and biotinylated
EGR-chloromethylketone were all from Haematologic Technologies,
Inc. (Burlington, VT, USA). Mouse anti-fibrin II chain (cat. no.
NYBT2G1) was from Accurate Chemical & Scientific Corporation
(Westbury, NY, USA). Isotype control antibody (cat. no. X0931) was
from Dako (Agilent Technologies, Inc., Santa Clara, CA, USA).
Fluorescein-maleimide, fluorescein isothiocyanatephalloidin, DAPI,
Alexa 647-labeled isotype-matched control antibody (cat. no.
MA5-18168) were from Molecular Probes (Invitrogen; Thermo Fisher
Scientific, Inc.). Propidium iodide (PI) was obtained from Shanghai
Dobio Biotech Co., Ltd. (Shanghai, China). Chromogenic substrates
S-2765 and S-2238 were from Diapharma Group, Inc. (West Chester,
OH, USA). Percoll was from GE Healthcare Life Sciences (Uppsala,
Sweden). Tyrode’s buffer containing 1 mM HEPES was prepared in our
laboratory and filtered through a 0.22-mm syringe filter from EMD
Millipore (Billerica, MA, USA).
Protein purification and labeling
Lactadherin was purified from bovine milk and
labeled with Alexa Fluor 647 or Alexa Fluor 488 as described
previously (12). The ratio of
fluorescein to lactadherin was 1.2/1 or 1.1/1.
Preparation of blood cells
Blood was drawn prior to therapy with a 21-gauge
needle and was collected into an anticoagulant tube containing 3.2%
citrate (5 ml; BD Biosciences, San Jose, CA, USA). Blood cell
isolation was performed ≤30 min post-blood collection. Following
centrifuging at 200 × g for 15 min at 20°C, platelet-rich plasma
(PRP) was aspirated carefully from the upper layer and erythrocytes
were collected from the bottom layer of blood samples. Mixed
peripheral blood leukocytes were isolated from the blood of study
subjects using gradient centrifugation with 30% percoll and 68%
percoll, according to the manufacturer’s protocol. Erythrocytes,
PRP, and leukocytes were analyzed immediately by flow cytometry and
confocal microscopy following isolation. To prepare platelet-free
plasma (PFP), PRP was centrifuged for 20 min at 1,500 × g at room
temperature. For microparticle-depleted plasma (MDP) preparation,
600 μl of PFP was centrifuged for 30 min at 20,000 × g at
20°C. The supernatant (400 μl) was collected, snap-frozen in
liquid nitrogen, and then stored at −80°C prior to use.
Preparation of immunomodulatory drugs and
Dex
Tha and Len were dissolved in dimethyl sulfoxide
(DMSO; Sigma-Aldrich; Merck KGaA) and Dex was resuspended in 1X
PBS. The drugs were stored at −20°C prior to use. Tha or Len and
Dex were diluted in culture medium to reach the final
concentrations of 1.0 and 10 μM, respectively. The final
concentration of DMSO was 0.01% in all experiments as preliminary
experiments had demonstrated that DMSO concentrations of 0.01% had
no significant effect on PS exposure on cells (data not shown).
HUVEC culture and reconstitution
experiments
HUVECs were maintained in EC medium in
poly-L-lysine-coated cell culture flasks at 37°C and 5%
CO2 in a humid environment. HUVECs were incubated in
growth media containing 20% pooled serum obtained from patients
with MM or healthy subjects at room temperature for 24 h.
Serum-cultured ECs were then treated with DMSO (0.01%), Tal (1.0
μM), Len (1.0 μM), Tal (1.0 μM)/Dex (10
μM), or Len (1.0 μM)/Dex (10 μM) at room
temperature for 24 h. Cells in the logarithmic growth phase were
used for all experiments. All cell culture results presented are
based on at least three individual experiments.
Blood cells of incubation and
reconstitution experiments
Erythrocytes, platelets and leukocytes from patients
with MM were cultured separately with DMSO (0.01%), Tal (1.0
μM), Len (1.0 μM), Tal (1.0 μM)/Dex (10
μM), or Len (1.0 μM)/Dex (10 μM) in a 5%
CO2 atmosphere at 37°C. Erythrocytes (106/ml)
were incubated in vitro for 24 h at a hematocrit of 0.4% in
Ringer’s solution (23). Platelets
(106/ml) were suspended in Tyrode’s buffer for 1 h
(24). Leukocytes
(106/ml) were incubated for 24 h in RPMI-1640 medium
containing 10% FBS (25).
Erythrocytes, platelets and leukocytes were washed twice before
detection.
Flow cytometric analysis of PS exposure
on blood cells and ECs
To quantify PS exposure on blood cells and ECs, 5
μl of each cell suspension (0.5–1×106/ml) in
Tyrode’s buffer was incubated with 5 μl Alexa Fluor
488-conjugated lactadherin for 15 min at room temperature in the
dark. A total of 10,000 events per sample were acquired by flow
cytometry and analyzed with BD FACSDiva software 6.0 (BD
Biosciences).
Clotting time and inhibition assays
PCA of various cell types was evaluated using a
one-stage recalcification time assay in a KC4A-coagulometer
(Amelung; Labcon GmbH, Heppenheim, Germany). Cell suspensions [100
μl of erythrocyte (1×108), platelet
(1×107), leukocyte (1×106), or EC
(1×106)] were incubated with 100 μl MDP from
healthy volunteers at 37°C. After 3 min, 100 μl of warmed 25
mM CaCl2 was added to start the reaction, and the time
to subsequent fibrin strand formation was recorded. All clotting
assays were performed in triplicate. For inhibition assays, cells
were preincubated with lactadherin (final concentration 128 nM)
prior to the assay.
Factor Xa and prothrombinase formation
and inhibition assay
For the intrinsic Xa formation, a total of
104 platelets/leukocytes/ECs or 105
erythrocytes were incubated with 1 nM factor IXa, 5 nM factor VIII,
0.2 nM thrombin, 130 nM factor X, and 5 mM CaCl2 in
factor Xa buffer (TBS with 0.2% BSA) for 5 min at 20°C. The
reaction was stopped by the addition of EDTA (7 mM final
concentration). Immediately following the addition of 10 μl
S-2765 (0.8 mM) chromogenic substrate, factor Xa generation was
determined using a kinetic absorbance reading at 405 nm on a
SpectraMax M5 Microplate Reader (Molecular Devices, LLC, Sunnyvale,
CA, USA). The assay to measure formation of extrinsic factor Xa was
analogous to that for intrinsic factor Xa except that cells were
mixed with 1 nM factor VIIa, 130 nM factor X and 5 mM
CaCl2. For the prothrombinase assay, the samples were
incubated with 1 nM factor Va, 2 nM factor Xa, 1 μM
prothrombin and 5 mM CaCl2 in prothrombinase buffer (TBS
with 0.05% BSA) for 5 min at 25°C. Thrombin production was
evaluated after the addition of EDTA using the chromogenic
substrate S-2238 (0.8 mM). For the inhibition assays of
intrinsic/extrinsic factor Xa and prothrombinase formation, cells
suspensions were pre-incubated with lactadherin (final
concentration 128 nM) for 10 min at 25°C in Tyrode’s buffer. The
mixture was then incubated with the specified clotting factors
according to the above protocols. The quantity of thrombin or
factor Xa formation was assessed as previously described (12).
Fibrin generation assay
Fibrin formation was quantified by turbidity as
described (26). For assays in
normal human MDP, blood cells and cultured ECs were added to
recalcified (10 mM, final) normal human MDP (88% MDP, final) in the
absence or presence of lactadherin (final concentration 128 nM).
Fibrin production was measured by turbidity at 405 nm in a
SpectraMax 340PC plate reader (Molecular Devices).
Confocal microscopy
To locate PS, erythrocytes, platelets or leukocytes
were incubated with Alexa Fluor 488-lactadherin (final
concentration 4 nM) or PI, and ECs were incubated with the final
concentrations of 4 nM Alexa Fluor 488-lactadherin and Alexa Fluor
647-CD31 [purified CD31, (clone L133.1), BD Biosciences)] for 10
min at room temperature in the dark. Then, cells were washed to
remove unbound dye and imaged immediately. Observation of PS
exposure on cells by confocal microscopy was performed as
previously described (12). For
fibrin generation experiments in vitro, ECs were cultured on
1% gelatin-coated coverslips in media containing 20% normal or
patient serum for 24 h. Following rinsing with Tyrode’s buffer, the
ECs were then overlaid with prewarmed MDP (15%) in the presence of
3 mM calcium. Fibrin networks were imaged using laser confocal
microscopy in the presence of Alexa Fluor 647-conjugated
anti-fibrin (final concentration, 1 μg/ml; prepared in our
laboratory) at 37°C for 15 min. Background signal was calculated
using a similarly labeled isotype matched control antibody (final 1
μg/ml) at 37°C for 15 min. To observe the location of
coagulation factor binding sites, the ECs were co-stained with
factor Va-fluorescein-maleimide (final concentration, 2 nM) and
factor Xa-EGRck-biotin (final concentration, 2 nM, complexed to
Alexa Fluor 647-streptavidin) at 37°C for 10 min. Samples were
excited with 488 or 568 nm emission lines of a krypton-argon laser
and narrow bandpass filters were used for restricting emission
wavelength overlap. Images were obtained using the LSM 510 system
(Carl Zeiss AG, Oberkochen, Germany).
Statistical analysis
Results are presented as mean ± standard deviation
of at least triplicate measurements. Statistical analysis was
performed with Student’s t-test or one-way analysis of variance
followed by Fisher’s least significant difference post hoc tests
for multiple comparisons as appropriate. P<0.05 was considered
to indicate a statistically significant difference.
Results
Patient characteristics
The characteristics of the study participants (20
patients with MM and 15 healthy subjects) are detailed in Table I. Patients with MM had a high serum
concentration of M-component. Compared with healthy subjects,
patients with MM had significantly higher levels of
β2-microglobulin, fibrinogen, D-dimer, von Willebrand factor and
factor VIII and shortened prothrombin time. However, patients with
MM had lower levels of erythrocytes and hemoglobin than healthy
subjects.
 | Table IClinical characteristics of healthy
subjects and patients with multiple myeloma. |
Table I
Clinical characteristics of healthy
subjects and patients with multiple myeloma.
| Characteristic | Healthy
subjects | MM |
|---|
| Total n | 15 | 20 |
| Age (years) | 57±7 | 60±11 |
| Male, n (%) | 7 (46.67%) | 9 (45%) |
| International
Staging System stage I/II/III, n (%) | NA | 4/7/9
(20%/35%/45%) |
| M-protein class, n
(%) | | |
| IgG | NA | 14 (70%) |
| IgA | NA | 6 (30%) |
| Serum M protein
(g/l) | NA | 25±12 |
| β2-microglobulin
(mg/l) | 1.65±0.55 | 6.88±5.08a |
| Erythrocytes
(×1012/l) | 4.10±0.44 | 3.31±1.01a |
| Platelet
(×109/l) | 228±25 | 188±82 |
| Leukocytes
(×109/l) | 5.79±0.63 | 5.31±1.38 |
| Albumin (g/l) | 42.4±3.6 | 37.6±7.4 |
| Hemoglobin
(g/l) | 124±8 | 101±29a |
| Prothrombin time
(sec) | 13.48±0.76 | 12.52±1.04a |
| Activated partial
thromboplastin time (sec) | 33.9±2.0 | 31.6±4.5 |
| Fibrinogen
(g/l) | 2.61±0.32 | 3.07±0.96 |
| D-dimer (mg/l) | 0.20±0.07 | 1.81±1.26a |
| Von Willebrand
factor (U/ml) | 1.06±0.26 | 2.23±0.71a |
| Factor VIII
(U/ml) | 0.87±0.18 | 2.10±0.62a |
PS exposure of blood cells from healthy
subjects and patients with MM patients, and in cultured ECs
As lactadherin binds to PS with high affinity
(21), fluorescence-labeled was
used lactadherin to detect the level of PS exposure on various
blood cells using a flow cytometer. Patients with MM had a
significantly higher percentage of PS+ blood cells than
healthy subjects (RBC, 1.12±0.58 vs. 0.54±0.29%; PLT, 10.24±3.37
vs. 2.35±1.10%; WBC, 19.26±2.55 vs. 5.46±1.41%; P<0.05 for all
cell types; Fig. 1A). The
percentage of PS+ ECs after treatment with patient serum
was 4.30-fold higher than after treatment with healthy plasma
(P<0.05; Fig. 1B). The absolute
number of PS+ blood cells was also significantly higher
in patients with MM compared with healthy subjects (P<0.05, for
all; Table II). To observe PS
binding on the outer membrane of cells, erythrocytes, platelets, or
leukocytes were incubated with Alexa Fluor 488-lactadherin and PI,
and imaged using confocal microscopy. Alexa Fluor 488-lactadherin
staining was not observed on the membranes of platelets,
erythrocytes and leukocytes from healthy subjects (Fig. 1C), whereas light green fluorescence
was observed on platelets, leukocytes and erythrocytes from
patients with MM (Fig. 1D). These
results further confirmed that there is increased PS exposure on
blood cells in patients with MM compared with healthy subjects.
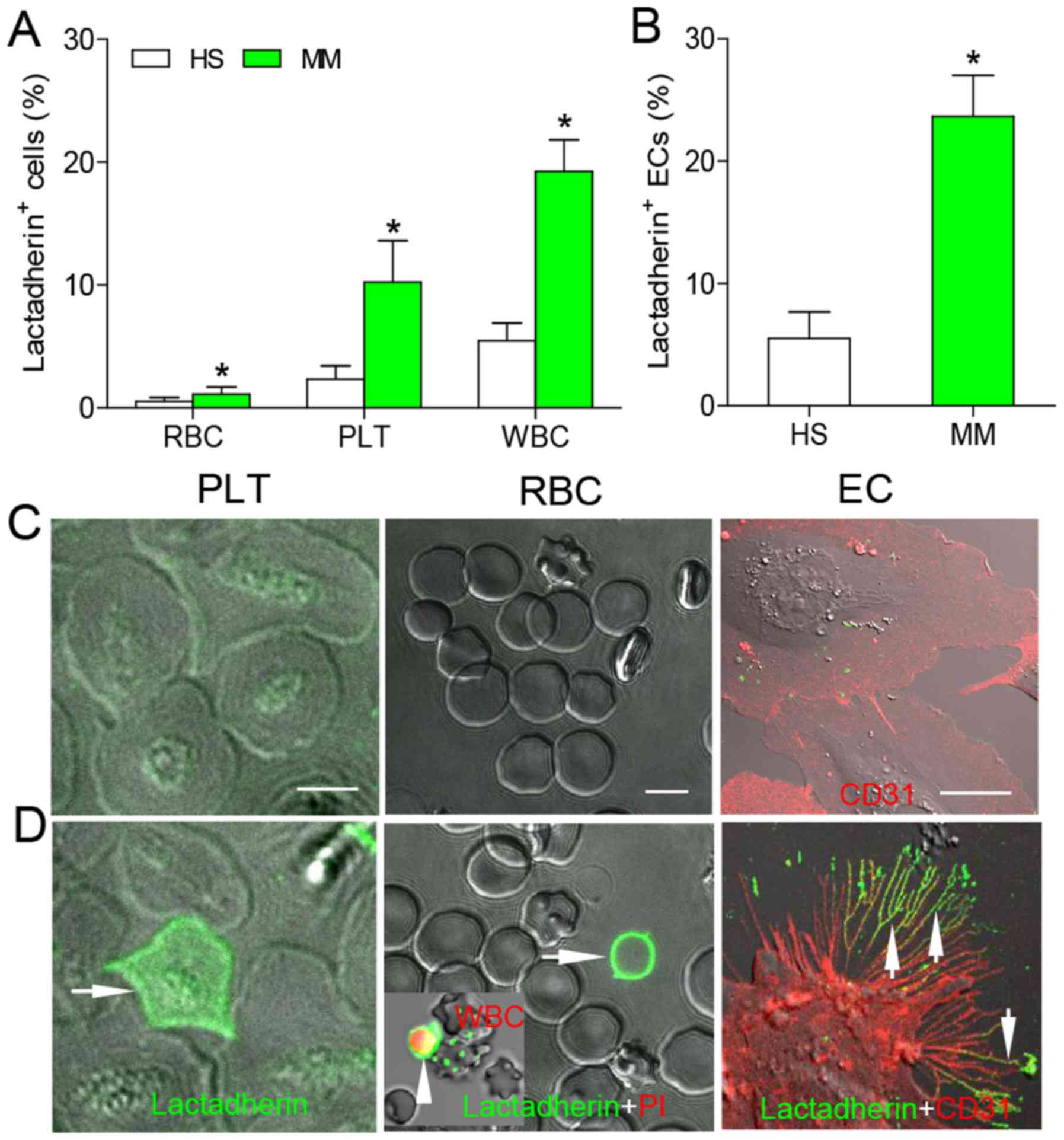 | Figure 1PS exposure on blood cells and HUVECs
was analyzed by flow cytometry and confocal microscopy. Blood cells
of (A) HS and patients with MM and (B) HUVECs treated with HS or MM
serum were incubated with Alexa 488-conjugated lactadherin and
assessed by flow cytometry. Data are presented as the mean ±
standard deviation, *P<0.05 vs. HS. Alexa Fluor
488-lactadherin staining was performed on blood samples from (C) HS
and (D) patients with MM; almost no staining was observed on cells
from HS; WBCs were identified using PI staining (red fluorescence,
inset). ECs with (C) HS or (D) patient serum were stained with
CD31-Alexa Fluor 647 and PS exposure was detected using
lactadherin-Alexa Fluor 488 at 24-h incubation; almost no
lactadherin staining was observed on normal HUVECs. Treatment of
HUVECs with patient serum led to retraction of cell margins,
extension of filopods, and lactadherin (green) binding on filopods
(arrows); scale bar, 5 μm. PS, phosphatidylserine; HUVEC,
human umbilical vein endothelial cells; HS, healthy subjects; MM,
multiple myeloma; PLT, platelets; RBCs, red blood cells
(erythrocytes); EC, endothelial cell; WBCs, white blood cells
(leukocytes); PI, propidium iodide. |
| Table IIAbsolute number of PS-positive blood
cells in study subjects. |
Table II
Absolute number of PS-positive blood
cells in study subjects.
| PS+
blood cells | Healthy
subjects | Multiple
myeloma |
|---|
| Erythrocyte
(×1010/l) | 2.20±1.30 | 3.71±5.93a |
| Platelet
(×109/l) | 5.33±2.96 | 19.24±9.69a |
| Leukocytes
(×108/l) | 3.16±0.93 | 10.23±3.34a |
ECs incubated for 24 h with serum from patients with
MM or healthy control were stained with CD31-Alexa Fluor 647 and PS
exposure was detected using lactadherin-Alexa Fluor 488. Confocal
laser-scanning microscopy demonstrated that there was limited
lactadherin staining on ECs cultured with normal serum (Fig. 1C), while large amounts of
lactadherin, exhibited by green fluorescence, was observed on ECs
treated with patient serum (Fig.
1D). Notably, the percentage of PS+ ECs was higher
than that of erythrocytes, platelets and leukocytes in patients
with MM.
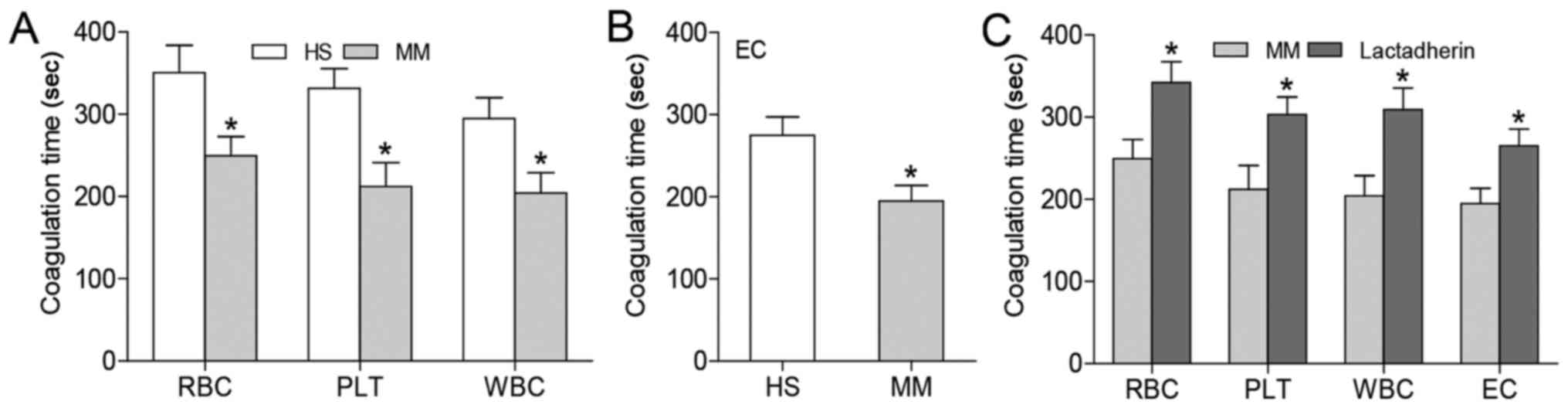
Coagulation time and inhibition
assay
To examine whether the increase in PS+
cells contributes to enhanced PCA, recalcification-time assays were
performed using a KC4A-coagulometer. Compared with samples from
healthy subjects, patient suspensions of erythrocytes, platelets
and leukocytes (cell number controlled) exhibited significantly
shorter coagulation time (P<0.05; Fig. 2A). ECs cultured in serum from
patients with MM induced a shorter coagulation time when cultured
in serum from healthy subjects (P<0.05; Fig. 2B). To confirm whether this
increased PCA was attributed to membrane PS exposure, inhibition
assays were performed where cells were incubated with 128 nM
lactadherin prior to coagulation testing. Lactadherin prolonged the
coagulation time of PS+ cells to similar values as
healthy subjects (P<0.05; Fig.
2C).
Role of PS in formation of procoagulant
enzyme complexes
In order to further evaluate the capacity of blood
cells and ECs to support the formation of procoagulant enzyme
complexes, intrinsic/extrinsic factor Xa and thrombin formation
assays were performed using purified coagulation factors. The
production of all three procoagulant enzyme complexes was increased
in MM groups compared with healthy subjects (P<0.05; Fig. 3A–D). These findings suggest that
PS+ blood cells contribute to coagulation, and that the
relative number of these blood cells may increase thrombosis risk.
In inhibition assays for all cell types, lactadherin (at 128 nM)
blocked production of the three procoagulant enzyme complexes by
~75% (Fig. 3E). PS blockade almost
entirely inhibited the formation of these complexes, suggesting
that PS independently increases PCA in patients with MM.
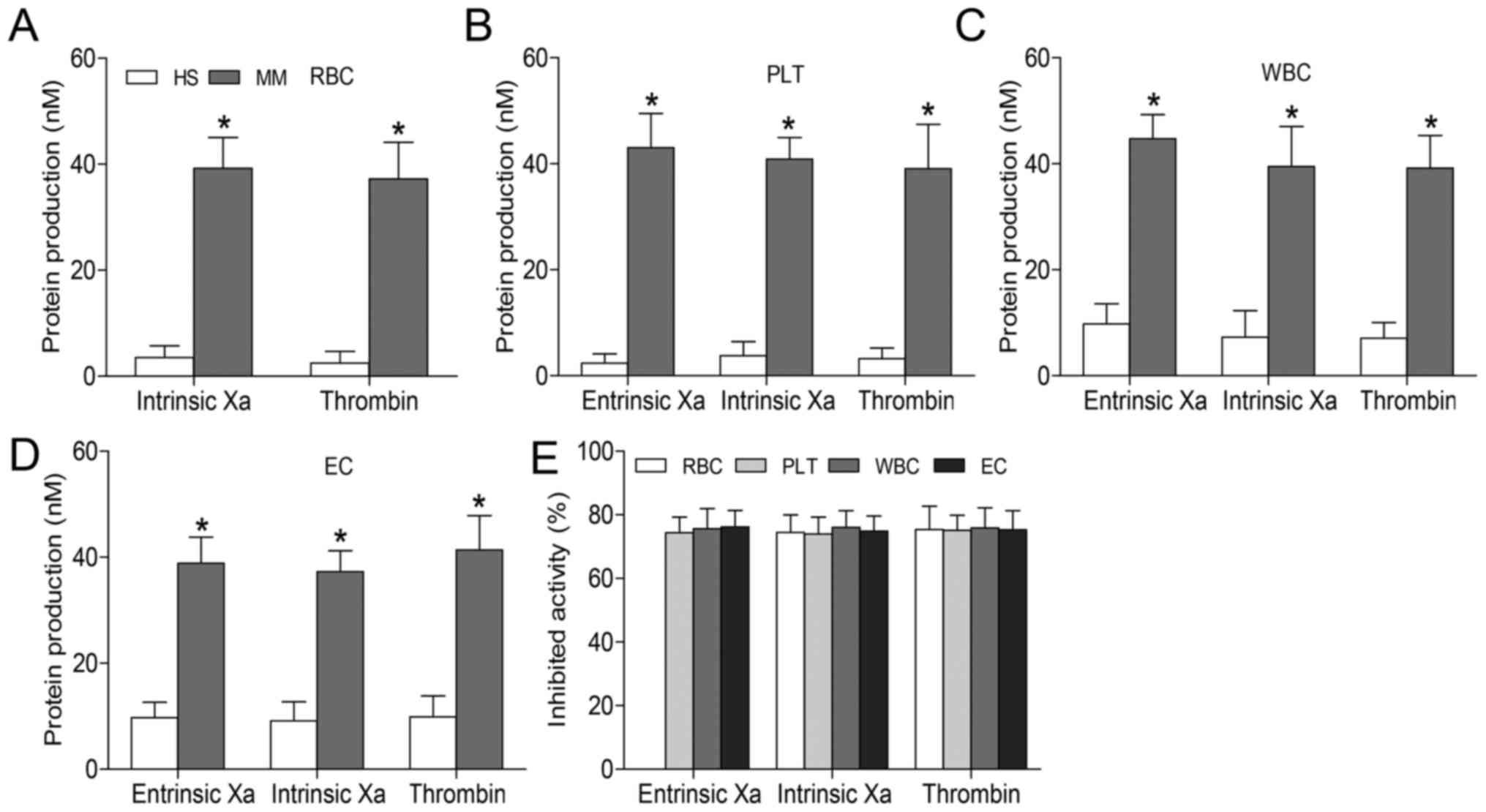 | Figure 3Formation and inhibition assays of
procoagulant enzyme complexes. Xa and thrombin production in the
presence of (A) 2×105 RBCs, (B) 2×104 PLTs or
(C) 2×104 WBCs from healthy subjects and patients with
MM. Experiments were also performed using (D) 2×104 ECs
cultured with normal or patient serum for 24 h. Intrinsic Xa
formation was measured in the presence of intrinsic Xa, FVIII and
thrombin. Extrinsic Xa production was assessed in the presence of
FVIIa. Thrombin generation was investigated in the presence of Xa
and FVa. (E) Capacity of lactadherin (128 nM) to block procoagulant
enzyme complexes on cells from patients with MM was evaluated; in
each cell type, lactadherin decreased activity of the procoagulant
enzyme complexes by ~75%. Results displayed as the mean ± standard
deviation, *P<0.05 vs. HS. FVIII, factor VIII; FVa,
factor Va; HS, healthy subjects; MM, multiple myeloma; RBCs, red
blood cells (erythrocytes); Xa, factor Xa; PLT, platelets; WBCs,
white blood cells (leukocytes); EC, human umbilical vein
endothelial cells. |
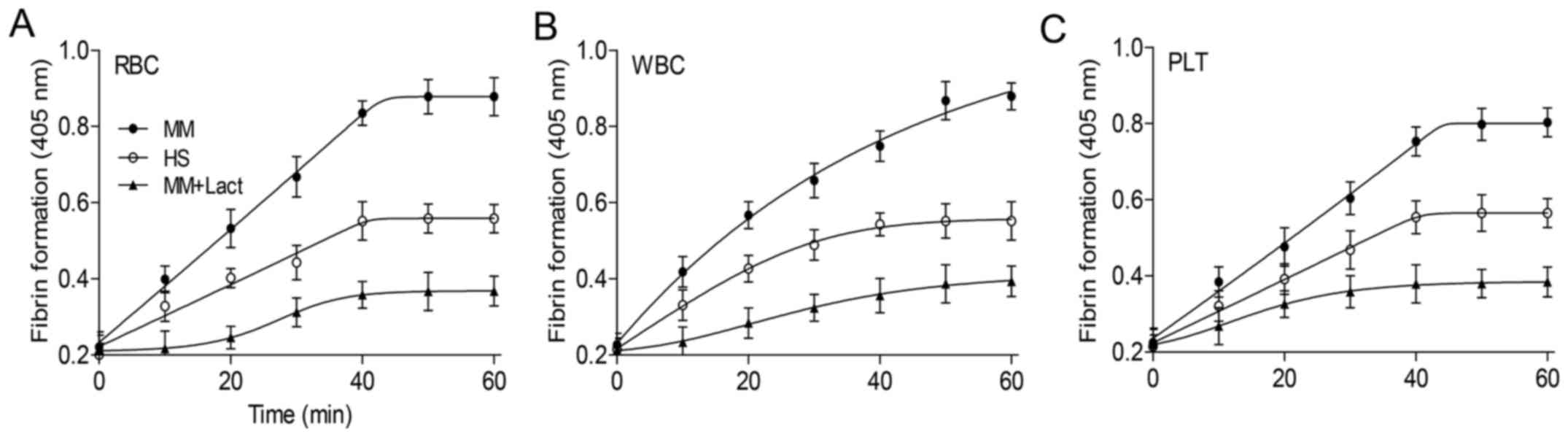
Role of PS in fibrin formation on blood
cells
Fibrin constitutes the primary structural protein of
blood clots and intravascular thrombi and its formation involves
the concerted action of coagulation factors and blood cells.
Therefore, fibrin formation was evaluated in normal MDP. More
fibrin was deposited on erythrocytes, platelets and leukocytes from
patients with MM than those from healthy subjects (P<0.05;
Fig. 4). The inhibition assays
demonstrated that lactadherin reduced fibrin formation of all cells
to a level lower than healthy subjects.
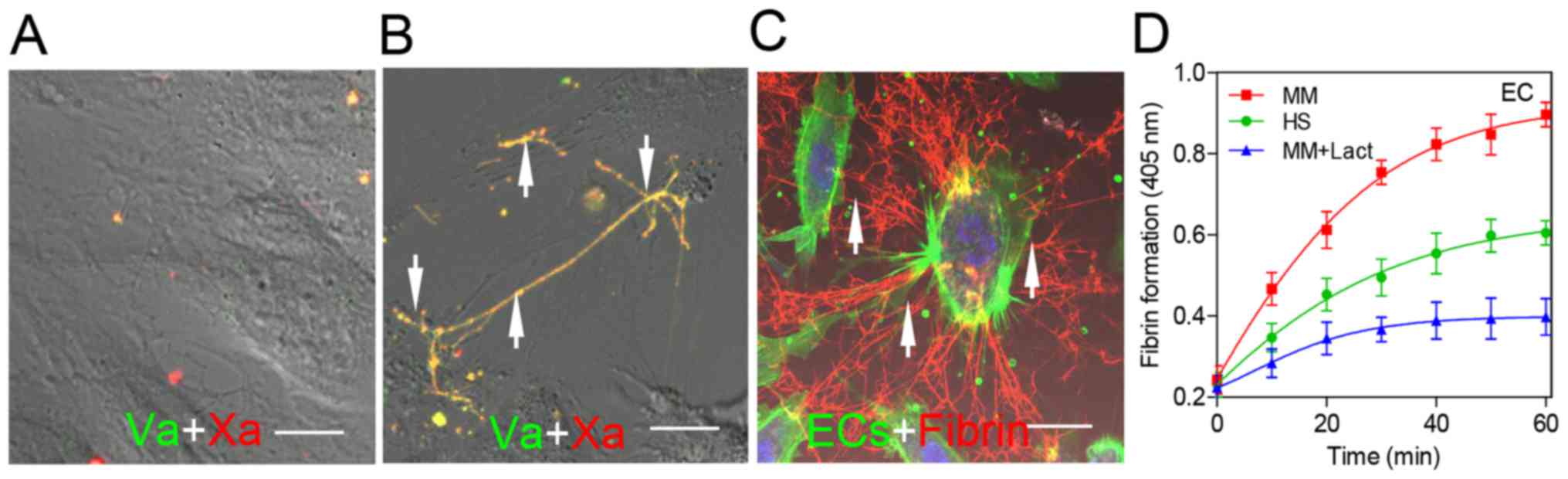
Factor Xa/factor Va binding and fibrin
deposition around the PS on ECs
Confocal microscopy provided more evidence that PS
on ECs has an important role in fibrin clot formation. Limited
factor Va (red) or factor Xa (green) binding was observed on ECs
treated with serum from healthy subjects (Fig. 5A, arrow). When ECs were cultured
with the serum from patients with MM, factor Va and factor Xa were
observed to be bound to the PS-enriched areas of the cell membrane
(Fig. 5B, arrow). ECs were
incubated with plasma in the presence of calcium and Alexa
647-conjugated anti-fibrin. Considerable fibrin fibrils (red) were
spread along the filopodia of ECs visualized using actin and DAPI
staining (Fig. 5C, arrow). The
patient serum resulted in a 1.48-fold higher fibrin generation in
ECs than the serum from healthy subjects (P<0.05; Fig. 5D).
Effects of IMiDs with or without Dex to
PS exposure on blood cells and ECs
The effects of the IMiDs (Tha and Len, with or
without Dex treatment) on PS exposure of blood cells in
vitro were determined. Erythrocytes (24 h), platelets (1 h),
and leukocytes (24 h) from patient with MM were cultured with Tha
(1.0 μM), Len (1.0 μM), Tha (1.0 μM)/Dex (10
μM), or Len (1.0 μM)/Dex (10 μM) at 37°C. Flow
cytometry analysis demonstrated that, compared with control or
either IMiD alone, Tha or Len plus Dex increased the level of PS
exposure on erythrocytes (P<0.05; Fig. 6A). IMiD treatment alone did not
significantly increase PS exposure on erythrocytes compared with
controls. However, Tha and Len significantly increased PS exposure
on platelets and leukocytes compared with controls (P<0.05;
Fig. 6B and C). Treatment of
platelets and leukocytes with an IMiD in combination with Dex
resulted in higher PS exposure compared with IMiD treatment alone.
The effects of IMiDs and Dex, either alone or in combination, on PS
exposure on ECs were determined by flow cytometry. Incubation of
ECs with an IMiD alone resulted in increased PS exposure compared
with controls (P<0.05; Fig.
6D). Furthermore, an increased percentage of PS+ ECs
was induced by the combination of an IMiD and Dex, as compared with
Tha or Len alone. The results indicate that IMiDs and Dex used in
combination can further increase PS exposure on blood cells from MM
patients and ECs in vitro.
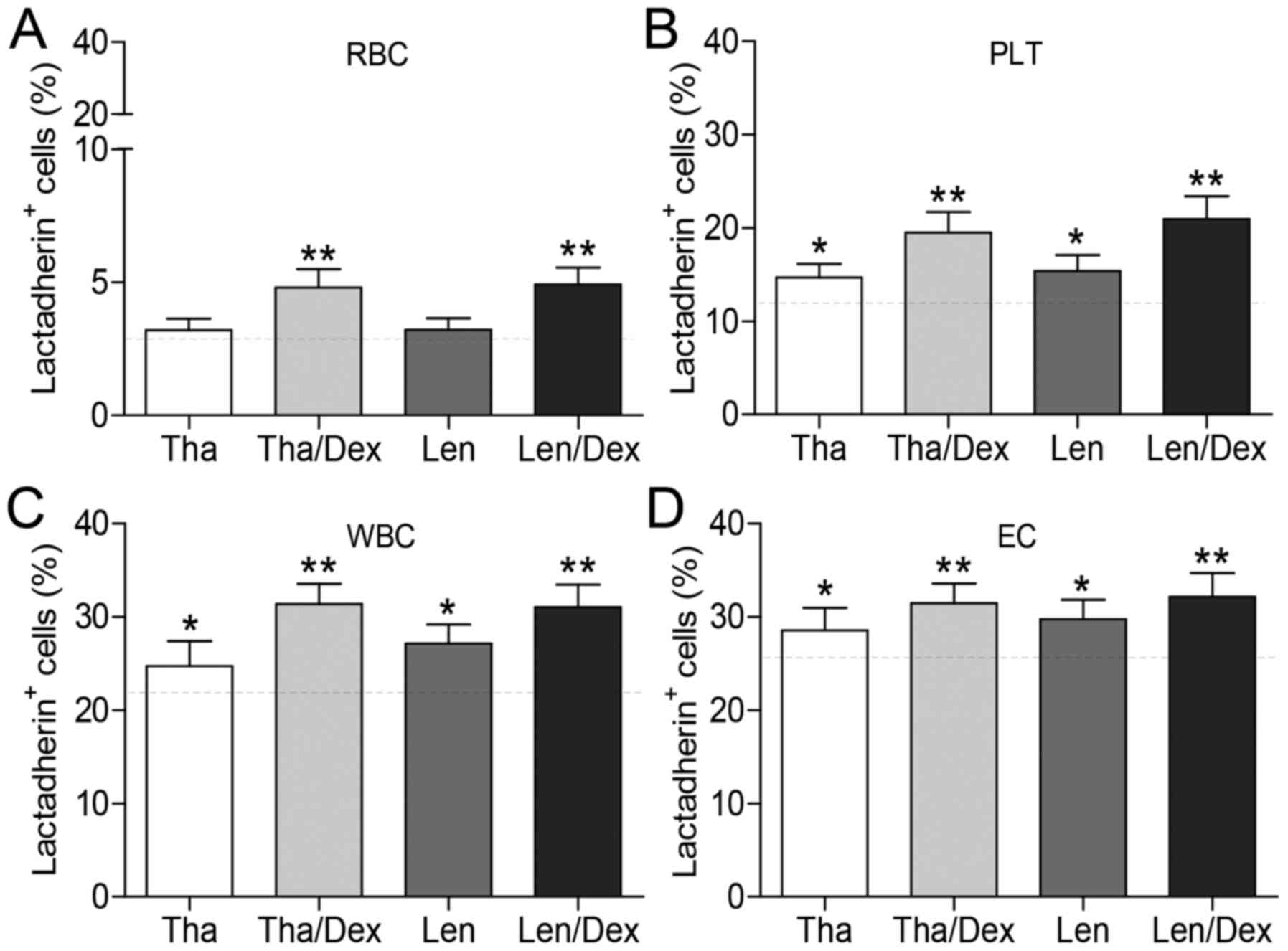 | Figure 6Effect of immunomodulatory
drugs-based treatments on PS exposure of blood cells and ECs. (A)
RBCs (24 h), (B) PLTs (1 h), (C) WBCs (24 h) from patients with MM
were incubated with dimethyl sulfoxide (0.01%), Tha (1.0
μM), Len (1.0 μM), Tal (1.0 μM)/Dex (10
μM), or Len (1.0 μM)/Dex (10 μM) at 37°C. (D)
ECs were incubated in five groups containing 20% pooled serum
obtained from patients with MM at room temperature for 24 h. The
gray dashed line represents PS exposure with no drug treatment as
control. PS exposure was measured as the % of cells that were
positive for Alexa Fluor 488-lactadherin using flow cytometry. Data
are presented as the mean ± standard deviation,
*P<0.05 vs. control, **P<0.05 vs. Tha
or Len. PS, phosphatidylserine; MM, multiple myeloma; RBC, red
blood cells (erythrocytes); Tha, thalidomide; Len, lenalidomide;
Dex, dexamethasone; PLT, platelets; WBC, WBCs, white blood cells
(leukocytes); EC, human umbilical vein endothelial cells. |
Discussion
The findings of the current study demonstrated that
PS+ blood cells may potentially have a role in the
induction of a hypercoagulable state in patients with MM, and that
patient serum components can induce PS exposure in cultured ECs.
Compared with healthy subjects, patients with MM had markedly
higher levels of PS+ blood cells, and serum from
patients with MM increased PS+ cultured ECs compared
with healthy serum. In addition, the percentage of PS+
ECs was higher than that of erythrocytes, platelets and leukocytes
in patients with MM. Exposed PS contributes to tenase and
prothrombinase complex formation, leading to a shorter coagulation
time and greater intrinsic/extrinsic factor Xa, thrombin and fibrin
generation. In addition, combined treatment with IMiDs plus Dex
induced greater PS exposure on platelets, leukocytes and
erythrocytes than treatment with IMiDs alone. Finally, lactadherin
inhibited production of all three procoagulant enzyme complexes by
≥75%, further confirming the procoagulant role of PS in patients
with MM.
Although platelet dysfunction often occurs in MM
(27), it remains unclear whether
platelets contribute to thrombosis in patients with MM. To the best
of our knowledge, this is the first study evaluating the role of
PS+ platelets in the prothrombotic state in patients
with MM. The percentage of PS+ platelets was
significantly higher in patients with MM than in healthy subjects,
which indicates abnormal activation or apoptosis of platelets. In a
previous study, the serum level of platelet factor 4 was
significantly elevated in patients with MM (15), supporting this finding. Notably,
PS+ platelets had the capacity to increase tenase and
prothrombinase complex formation, leading to greater factor Xa,
thrombin and fibrin generation. Activated platelets release
thrombin leading to platelet aggregation and three-dimensional clot
formation (28). Thus, we
hypothesize that PS+ platelets may recruit more
circulating platelets by producing thrombin. On this basis,
platelet PS exposure is an important mediator of the
hypercoagulable state in patients with MM.
Clinical studies have reported that activated
leukocytes are associated with venous thrombosis (29). However, the pathogenic pathways
linking leukocyte abnormalities to thrombosis in patients with MM
remain unclear. The results of the current study indicated that
injured or activated leukocytes increased PCA through PS exposure.
This process increases the serum concentration of three
procoagulant enzyme complexes by PS+ leukocyte-derived
PCA and potentially forms the nidus upon which the thrombus
develops. Typically, erythrocytes are considered as passive
participants in coagulation, merely providing bulk material for the
obstructive clot. The observations in the current study provide
evidence that erythrocytes may be actively involved in the
hypercoagulable state of patients with MM. PS exposure on
erythrocytes provides a catalytic surface to support the assembly
of blood coagulation factors, thus promoting the coagulation
cascade activation and thrombin generation (10). Previous studies reported that
PS+ erythrocytes are also more adhesive to endothelial
cells and prone to form erythrocyte aggregates in chronic uremia
and obesity, supporting the conclusions of the present study
(30,31).
To investigate the role of vascular endothelium
dysfunction in MM-associated thrombosis, cultured ECs were
incubated with patient serum, which resulted in a significant
increase in PS exposure compared with incubation in serum from
healthy subjects. A previous study has indicated patients with MM
exhibit pathologically enhanced von Willebrand factor, a marker of
ECs activation, which is consistent with the findings of the
present study (7). In addition to
promoting the formation of thrombin, the results of the current
study demonstrate that patient serum-treated ECs highly expressed
PS, which supported binding of factor Va and factor Xa, and fibrin
deposition. Following activation, imbalance or alteration of
‘differential expression of procoagulants and anticoagulants in the
endothelium’ may modulate endothelial thromboresistance from an
anticoagulant state into a procoagulant one (32,33).
Thus, it seems reasonable to hypothesize that PS has a pivotal role
in the localized procoagulant phenotype of ECs by bringing clotting
factors together, resulting in fibrin formation.
It was previously reported that patients with MM
have higher levels of endogenous thrombin potential in a global
assay of thrombin generation (34). The data of the current study
demonstrated that PS+ blood cells and ECs from patients
with MM supported the formation of intrinsic/extrinsic factor Xase
and prothrombinase, essential components of the coagulation cascade
that lead to increased thrombin generation. In addition,
PS+ cells from patients with MM have a shorter
coagulation time and higher production of fibrin than samples from
healthy subjects. Blockade of PS with lactadherin prolonged
coagulation time and decreased fibrin formation to control levels
and inhibited ~75% of the procoagulant enzyme production. By
contrast, in a previous study, anti-tissue factor antibody had a
negligible effect on PCA of cells from patients with MM; this may
be explained by the fact that plasma exposed tissue factor is
generally quiescent, with little or no detectable PCA, unless it
resides in a membrane containing PS (35,36).
The results indicate that increased PS contributes to the
hypercoagulable state in patients with MM.
The exact mechanism of the increased risk of VTE in
patients with MM treated with IMiDs-based regimens is not yet fully
understood. The results of the current study have demonstrated that
treatment with IMiDs plus Dex induces higher levels of
PS+ blood cells and ECs than controls or IMiDs alone,
in vitro. IMiDs with Dex has been previously reported to
increase P-selectin expression on platelets and induce EC injury,
supporting the findings of the present study (37,38).
In addition, IMiDs stimulate T-cell production and activation of
natural killer cells (39). Thus,
we hypothesize that treatment with Dex may aggravate the abnormal
activation or apoptosis of blood cells in patients with MM using
IMiDs-based regimens. Considering the high cost of treatment
(40), the number of patients that
receive IMiDs-based regimens is low in China. The in vivo
effect of IMiDs-based regimens on PCA of blood cells and ECs from
patients with MM will be investigated in future studies.
In summary, PS+ blood cells and
PS-associated PCA were increased in the circulation of patients
with MM and patient serum induced PS exposure on cultured ECs. In
addition, IMiDs-based treatment increased PS exposure on blood
cells and ECs in vitro. Increasing levels of PS+
cells were associated with a hypercoagulable state in patients with
MM. Furthermore, PS inhibition assays using lactadherin suggest
that blockage of PS may constitute a novel therapy for preventing
thrombosis. Future clinical trials are required to investigate this
hypothesis.
Acknowledgments
Not applicable.
Funding
This study was supported by grants from the National
Natural Science Foundation of China (cat. no. 81470301 and
81670128).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors’ contributions
Study concept and design (LG, JS); data acquisition,
clinical data and sample collection, statistical analysis (LG, DT,
MY, CW, PZ, JJ, BL, YL, RL); data interpretation and manuscript
drafting (LG, JS); critical revision of the manuscript (LG, VN, DT,
MY, YZ, TL, JS); YZ, TL, ZD, YT, JZ, YB and JK provided valuable
advice for this study. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Harbin Medical University Ethical Committee (Harbin,
China) approved the study, and patients provided written, informed
consent.
Consent for publication
Not applicable.
Competing interests
JS has a patent for the use of lactadherin as a
probe for phosphatidylserine. Other authors declare that they have
no competing interests.
References
|
1
|
Kristinsson SY, Pfeiffer RM, Björkholm M,
Goldin LR, Schulman S, Blimark C, Mellqvist UH, Wahlin A, Turesson
I and Landgren O: Arterial and venous thrombosis in monoclonal
gammopathy of undetermined significance and multiple myeloma: A
population-based study. Blood. 115:4991–4998. 2010. View Article : Google Scholar
|
|
2
|
Blom JW, Doggen CJ, Osanto S and Rosendaal
FR: Malignancies, prothrombotic mutations, and the risk of venous
thrombosis. JAMA. 293:715–722. 2005. View Article : Google Scholar
|
|
3
|
Kristinsson SY, Pfeiffer RM, Björkholm M,
Schulman S and Landgren O: Thrombosis is associated with inferior
survival in multiple myeloma. Haematologica. 97:1603–1607. 2012.
View Article : Google Scholar
|
|
4
|
Palumbo A, Rajkumar SV, Dimopoulos MA,
Richardson PG, San Miguel J, Barlogie B, Harousseau J, Zonder JA,
Cavo M, Zangari M, et al International Myeloma Working Group:
Prevention of thalidomide- and lenalidomide-associated thrombosis
in myeloma. Leukemia. 22:414–423. 2008. View Article : Google Scholar
|
|
5
|
De Stefano V, Za T and Rossi E: Venous
thromboembolism in multiple myeloma. Semin Thromb Hemost.
40:338–347. 2014. View Article : Google Scholar
|
|
6
|
Auwerda JJ, Sonneveld P, de Maat MP and
Leebeek FW: Prothrombotic coagulation abnormalities in patients
with newly diagnosed multiple myeloma. Haematologica. 92:279–280.
2007. View Article : Google Scholar
|
|
7
|
Minnema MC, Fijnheer R, De Groot PG and
Lokhorst HM: Extremely high levels of von Willebrand factor antigen
and of procoagulant factor VIII found in multiple myeloma patients
are associated with activity status but not with thalidomide
treatment. J Thromb Haemost. 1:445–449. 2003. View Article : Google Scholar
|
|
8
|
Elice F, Fink L, Tricot G, Barlogie B and
Zangari M: Acquired resistance to activated protein C (aAPCR) in
multiple myeloma is a transitory abnormality associated with an
increased risk of venous thromboembolism. Br J Haematol.
134:399–405. 2006. View Article : Google Scholar
|
|
9
|
van Marion AM, Auwerda JJ, Minnema MC, van
Oosterom R, Adelmeijer J, de Groot PG, Leebeek FW, Sonneveld P,
Lokhorst HM and Lisman T: Hypofibrinolysis during induction
treatment of multiple myeloma may increase the risk of venous
thrombosis. Thromb Haemost. 94:1341–1343. 2005.
|
|
10
|
Zwaal RF and Schroit AJ: Pathophysiologic
implications of membrane phospholipid asymmetry in blood cells.
Blood. 89:1121–1132. 1997.
|
|
11
|
Vance JE and Steenbergen R: Metabolism and
functions of phosphatidylserine. Prog Lipid Res. 44:207–234. 2005.
View Article : Google Scholar
|
|
12
|
Gao C, Xie R, Yu C, Wang Q, Shi F, Yao C,
Xie R, Zhou J, Gilbert GE and Shi J: Procoagulant activity of
erythrocytes and platelets through phosphatidylserine exposure and
microparticles release in patients with nephrotic syndrome. Thromb
Haemost. 107:681–689. 2012. View Article : Google Scholar
|
|
13
|
Zhou J, Shi J, Hou J, Cao F, Zhang Y,
Rasmussen JT, Heegaard CW and Gilbert GE: Phosphatidylserine
exposure and procoagulant activity in acute promyelocytic leukemia.
J Thromb Haemost. 8:773–782. 2010. View Article : Google Scholar
|
|
14
|
Jurczyszyn A, Czepiel J, Biesiada G,
Gdula-Argasińska J, Cibor D, Owczarek D, Perucki W and Skotnicki
AB: HGF, sIL-6R and TGF-β1 play a significant role in the
progression of multiple myeloma. J Cancer. 5:518–524. 2014.
View Article : Google Scholar
|
|
15
|
Fritz E, Ludwig H, Scheithauer W and
Sinzinger H: Shortened platelet half-life in multiple myeloma.
Blood. 68:514–520. 1986.
|
|
16
|
Lee H, Kong SY, Sohn JY, Shim H, Youn HS,
Lee S, Kim HJ and Eom HS: Elevated red blood cell distribution
width as a simple prognostic factor in patients with symptomatic
multiple myeloma. Biomed Res Int. 2014:1456192014.
|
|
17
|
Kerr R, Stirling D and Ludlam CA:
Interleukin 6 and haemostasis. Br J Haematol. 115:3–12. 2001.
View Article : Google Scholar
|
|
18
|
Mechtcheriakova D, Schabbauer G, Lucerna
M, Clauss M and De Martin R, Binder BR, Hofer E and De Martin R:
Specificity, diversity, and convergence in VEGF and TNF-alpha
signaling events leading to tissue factor up-regulation via EGR-1
in endothelial cells. FASEB J. 15:230–242. 2001. View Article : Google Scholar
|
|
19
|
Hoshi A, Matsumoto A, Chung J, Isozumi Y
and Koyama T: Activation of coagulation by a thalidomide-based
regimen. Blood Coagul Fibrinolysis. 22:532–540. 2011. View Article : Google Scholar
|
|
20
|
Isozumi Y, Arai R, Fujimoto K and Koyama
T: Activation of coagulation by lenalidomide-based regimens for the
treatment of multiple myeloma. PLoS One. 8:e643692013. View Article : Google Scholar
|
|
21
|
Shi J, Shi Y, Waehrens LN, Rasmussen JT,
Heegaard CW and Gilbert GE: Lactadherin detects early
phosphatidylserine exposure on immortalized leukemia cells
undergoing programmed cell death. Cytometry A. 69:1193–1201. 2006.
View Article : Google Scholar
|
|
22
|
Rajkumar SV, Dimopoulos MA, Palumbo A,
Blade J, Merlini G, Mateos MV, Kumar S, Hillengass J, Kastritis E,
Richardson P, et al: International Myeloma Working Group updated
criteria for the diagnosis of multiple myeloma. Lancet Oncol.
15:e538–e548. 2014. View Article : Google Scholar
|
|
23
|
Lang E, Gatidis S, Freise NF, Bock H,
Kubitz R, Lauermann C, Orth HM, Klindt C, Schuier M, Keitel V, et
al: Conjugated bilirubin triggers anemia by inducing erythrocyte
death. Hepatology. 61:275–284. 2015. View Article : Google Scholar
|
|
24
|
NaveenKumar SK, Thushara RM, Sundaram MS,
Hemshekhar M, Paul M, Thirunavukkarasu C, Basappa, Nagaraju G,
Raghavan SC, Girish KS, et al: Unconjugated bilirubin exerts
pro-apoptotic effect on platelets via p38-MAPK activation. Sci Rep.
5:150452015. View Article : Google Scholar
|
|
25
|
Khan NM and Poduval TB: Immunomodulatory
and immu-notoxic effects of bilirubin: Molecular mechanisms. J
Leukoc Biol. 90:997–1015. 2011. View Article : Google Scholar
|
|
26
|
Campbell RA, Overmyer KA, Selzman CH,
Sheridan BC and Wolberg AS: Contributions of extravascular and
intravascular cells to fibrin network formation, structure, and
stability. Blood. 114:4886–4896. 2009. View Article : Google Scholar
|
|
27
|
Eby C: Pathogenesis and management of
bleeding and thrombosis in plasma cell dyscrasias. Br J Haematol.
145:151–163. 2009. View Article : Google Scholar
|
|
28
|
Koupenova M, Kehrel BE, Corkrey HA and
Freedman JE: Thrombosis and platelets: An update. Eur Heart J.
38:785–791. 2017.
|
|
29
|
Swystun LL and Liaw PC: The role of
leukocytes in thrombosis. Blood. 128:753–762. 2016. View Article : Google Scholar
|
|
30
|
Bonomini M, Sirolli V, Gizzi F, Di Stante
S, Grilli A and Felaco M: Enhanced adherence of human uremic
erythrocytes to vascular endothelium: Role of phosphatidylserine
exposure. Kidney Int. 62:1358–1363. 2002. View Article : Google Scholar
|
|
31
|
Solá E, Vayá A, Martínez M, Moscardó A,
Corella D, Santaolaria ML, España F and Hernández-Mijares A:
Erythrocyte membrane phosphatidylserine exposure in obesity.
Obesity (Silver Spring). 17:318–322. 2009. View Article : Google Scholar
|
|
32
|
Aird WC: Phenotypic heterogeneity of the
endothelium: II. Representative vascular beds. Circ Res.
100:174–190. 2007. View Article : Google Scholar
|
|
33
|
Levi M, Nieuwdorp M, van der Poll T and
Stroes E: Metabolic modulation of inflammation-induced activation
of coagulation. Semin Thromb Hemost. 34:26–32. 2008. View Article : Google Scholar
|
|
34
|
Petropoulou AD, Gerotziafas GT, Samama MM,
Hatmi M, Rendu F and Elalamy I: In vitro study of the
hypercoagulable state in multiple myeloma patients treated or not
with thalidomide. Thromb Res. 121:493–497. 2008. View Article : Google Scholar
|
|
35
|
Cimmino G, Ciccarelli G and Golino P: Role
of tissue factor in the coagulation network. Semin Thromb Hemost.
41:708–717. 2015. View Article : Google Scholar
|
|
36
|
Chen VM and Hogg PJ: Encryption and
decryption of tissue factor. J Thromb Haemost. 11(Suppl 1):
277–284. 2013. View Article : Google Scholar
|
|
37
|
Dunkley S and Gaudry L: Thalidomide causes
platelet activation, which can be abrogated by aspirin. J Thromb
Haemost. 5:1323–1325. 2007. View Article : Google Scholar
|
|
38
|
Rosovsky R, Hong F, Tocco D, Connell B,
Mitsiades C, Schlossman R, Ghobrial I, Lockridge L, Warren D,
Bradwin G, et al: Endothelial stress products and coagulation
markers in patients with multiple myeloma treated with lenalidomide
plus dexamethasone: An observational study. Br J Haematol.
160:351–358. 2013. View Article : Google Scholar
|
|
39
|
Quach H, Ritchie D, Stewart AK, Neeson P,
Harrison S, Smyth MJ and Prince HM: Mechanism of action of
immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia.
24:22–32. 2010. View Article : Google Scholar
|
|
40
|
Wang J, Guo H and Zhou X: Clinical utility
and patient consideration in the use of lenalidomide for multiple
myeloma in Chinese patients. Onco Targets Ther. 8:1277–1284.
2015.
|