Introduction
Pancreatic ductal adenocarcinoma (PDAC) accounts for
>90% of pancreatic cancers, and it has one of the highest
mortality rates of all human cancers worldwide; most patients
succumb within 1 year following diagnosis (1). It is hypothesized that of the ~53,670
people estimated to be diagnosed with this malignancy in the United
States 43,090 of them will succumb in 2017 (2). Less than 15% of patients with PDAC
are eligible for surgical resection, and the 5-year survival rate
remains low, 20–25% (3). Owing to
the absence of specific symptoms of PDAC and a lack of early
detection and screening techniques, the initial diagnosis of
pancreatic cancer often occurs at the advanced and metastatic
stages, at which surgical resection is unfeasible (4).
Over the past several decades, most
chemotherapeutic, targeted or immune-based therapeutic approaches
have only yielded limited clinical outcomes (5). Since 1996, the deoxycytidine analog
gemcitabine has been the most commonly used frontline therapeutic
agent for patients with advanced pancreatic cancer; however, it
only provides a median overall survival rate of 5.7 months
(6). A previous study examined the
effects of a combination therapy comprising several monotherapeutic
agents, including 5-fluorouracil, leucovorin, irinotecan and
oxaliplatin (collectively termed FOLFIRINOX), with gemcitabine in
patients with advanced pancreatic cancer and demonstrated that
FOLFIRINOX may be an effective agent; however, the median overall
survival is <1 year (7). There
has been an increased effort in the development of more potent
treatment regimens.
The etiology of pancreatic cancer remains unknown,
but may involve the dysregulation of multiple signaling effectors
or cytokines, including interleukin (IL)-8, signal transducer and
activator of transcription (STAT)-3, RAC-α serine/threonine-protein
kinase (AKT), extracellular signal-regulated kinase (ERK) and
others (8,9). An increasing number of studies have
revealed that IL-8 and its receptors, C-X-C chemokine receptor
(CXCR)-1 and CXCR2, serve crucial roles in orchestrating the
initiation and progression phases of various tumors (10,11),
including breast cancer (12),
melanoma (13), colorectal cancer
(14) and pancreatic cancers
(8). The CXCR1/2 signaling cascade
may be inhibited by a number of strategies, such as through the
administration of small-molecule inhibitors, and preclinical
studies have demonstrated their promising effects on reducing the
progression of human cancers (15,16).
Reparixin was originally developed to prevent IL-8-induced
reperfusion injury (17–19), but was later revealed to be an
effective small-molecule inhibitor that blocks CXCR1/2, and was
used in the clinic to delay the progression and advancement of
breast cancer (20,21). Another small-molecule inhibitor,
SCH527123, also exhibits antitumoral effects in mouse models and
has been demonstrated to be effective against melanoma (22), breast cancer (23) and colorectal cancers (24). Clinical trials revealed that
neither reparixin nor SCH527123 induced noticeable cytotoxicity
upon treating breast cancer, ischemia-reperfusion injury (IRI) or
inflammatory diseases such as asthma (19,20,25).
However, the efficacy of reparixin or SCH527123 on PDAC remains
unknown.
The present study is the first, to the best of our
knowledge, to investigate the efficacy of reparixin and SCH527123
on pancreatic cancer cell lines in vitro. The antitumoral
effects of these CXCR1/2 antagonists on pancreatic cancer were
determined by investigating viability, proliferation, colony
formation and migration of PDAC cells treated with these agents.
The results demonstrated that reparixin and SCH527123 not only
blocked the mitogenic effects triggered by IL-8, but also inhibited
overall cell survival, proliferation and migration. Molecular
investigation further uncovered the underlying mechanism involved
with the inhibition of downstream effectors, as demonstrated by the
reduced phosphorylation levels of AKT, ERK, STAT3 and ribosomal
protein S6 (S6). Results from the present study suggested that the
CXCR1/2 antagonists, reparixin and SCH-527123, may be novel
therapeutic candidates in treating pancreatic cancer, in part by
disrupting the IL-8/CXCR1/2 signaling cascade.
Materials and methods
Cell culture and reagents
Human pancreatic cancer cell lines HPAC, MIA PaCa-2,
Capan-1, AsPC-1 and BxPC-3 were purchased from The American Type
Culture Collection (Manassas, VA, USA). All cell lines were
routinely cultured in Dulbecco's modified Eagle's medium containing
4.5 g/l glucose, l-glutamine and sodium pyruvate
(DMEM; cat. no. 10013 CV; Mediatech, Inc. A Corning Subsidiary;
Manassas, VA, USA) supplemented with 10% fetal bovine serum (FBS;
cat. no. S11150; Atlanta Biologicals, Flowery Branch, GA, USA) and
1% penicillin/streptomycin (cat. no. P0781; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) in a humidified 37°C incubator with 5%
CO2. Cells were routinely assessed microscopically for
the expected morphologies.
Reagents used in the present study were as follows:
Recombinant human IL-8 (cat. no. 8921SF; Cell Signaling Technology,
Inc., Danvers, MA, USA),
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
cat. no. M5655; Sigma-Aldrich; Merck KGaA), dimethyl sulfoxide
(DMSO; cat. no. D2650; Sigma-Aldrich; Merck KGaA),
N,N-dimethylformamide (DMF; cat. no. D119-4; Fisher Scientific;
Thermo Fisher Scientific, Inc., Waltham, MA, USA), crystal violet
(cat. no. C6158; Sigma-Aldrich; Merck KGaA), Bromodeoxyuridine
(BrdU) Cell Proliferation assay kit (cat. no. 6813S; Cell Signaling
Technology, Inc.) and lysis buffer (cat. no. 9803; Cell Signaling
Technology, Inc.). The IL-8 powder was dissolved in sterile PBS to
constitute a stock solution (250 ng/μl), which was stored in
aliquots at −20°C. Reparixin was purchased from INDOFINE Chemical
Co., Inc. (cat. no. 1106143; Hillsborough Township, NJ, USA), and
SCH527123 was purchased from AdooQ Bioscience (cat. no. A11555;
Irvine, CA, USA); both were dissolved in sterile DMSO to make a
stock reagent (20 mM) and stored in aliquots at −20°C.
MTT cell viability assay
AsPC-1, BxPC-3, HPAC, Capan-1 and MIA PaCa-2 cells
were seeded (3,000 cells/well) in 96-well microtiter plates and
cultured overnight at 37°C in 100 μl DMEM containing 10%
FBS. The next day, when the cells reached ~20% confluency, the
culture medium in each well was changed with 100 μl DMEM +
10% FBS supplemented with various concentrations of reparixin (10,
20, 40, 60 and 80 μM), SCH527123 (20, 40, 60, 80 and 100
μM) or with DMSO vehicle control under the same conditions
(DMEM + 10% FBS) at 37°C. Following 72-h incubation, MTT solution
(25 μl) was added to each well and the cells were incubated
for 4 h. Subsequently, DMF solubilization solution (150 μl)
was added to dissolve the formazan crystals, and the cells were
incubated overnight at room temperature in a sealed moistened
chamber protected from the light. Cell viability was assessed by
measuring the absorbance at 595 nm in each well (which reflected
the amount of MTT taken up by the metabolically active cells),
comparing with the absorbance in the mock control wells and
quantifying. Half-maximal inhibitory concentrations
(IC50) were determined using Sigma Plot 9.0 Software
(Systat Software, Inc., San Jose, CA, USA).
BrdU incorporation assay for cell
proliferation
The proliferative activities intrinsic to AsPC-1,
Capan-1 and HPAC cells were assessed with the BrdU incorporation
assay. Briefly, cells were seeded 5,000 cells/well) in 96-well
plates in quadruplicate in DMEM containing 10% FBS overnight.
Subsequently, the cells were grown in the same medium but without
FBS for an additional 24 h; this serum-depleted growth condition
was maintained throughout the assay. Cells were either treated with
IL-8 (10 or 25 ng/ml) alone for 24 h at 37°C or pre-treated with
various concentrations of reparixin or SCH527123 (40, 60 or 80
μM) at 37°C for 4 h and subsequently triggered to undergo
robust proliferation with the addition of IL-8 (25 ng/ml) to the
medium for 24 h at 37°C. Cells treated with DMSO were used as a
control. Following 24 h of growth induction, the cells were further
cultivated with 1X BrdU reagent at 37°C for 1 h and the number of
cells that incorporated BrdU (that is, proliferating cells actively
synthesizing DNA) were quantified according to the manufacturer's
protocol.
Colony formation assay
HPAC and AsPC-1 cells were seeded (2,000 cells/well)
in 6-well plates and incubated overnight. Cells were subsequently
incubated at 37°C with DMSO, or were treated with various doses of
reparixin or SCH527123 (20, 40 or 60 μM). The culture medium
with the respective treatments was changed every 3 days. After 1
week, the culture medium containing the drugs was removed and
changed to fresh cell culture medium without IL-8 inhibitors, and
the medium was subsequently changed once every week without IL-8
inhibitors for another two weeks. After 2 weeks, the cells were
washed twice with PBS, fixed with cold methanol for 30 min at 4°C
and stained with 1% crystal violet dye (dissolved in 25% methanol)
at room temperature for 1 h. The plates were washed with distilled
water and dried.
Wound-healing assay for cell
migration
HPAC cells were seeded (8.9×105
cells/well) in 6-well plates and incubated at 37°C overnight. When
the cells reached 100% confluence, the monolayer was scratched to
create a wound using a pipette tip. The monolayer was washed with
sterilized PBS, and cells were treated with DMSO, reparixin or
SCH527123 at the doses of 60, 80 or 100 μM, and incubated
for 23 h at 37°C; images were captured with a Nikon Eclipse TS100
microscope at 0 and 23 h. The width of the scratch line was
quantified by three independent observers and the measurements were
used as an indication of cell migration. The relative wound-healing
ability was calculated using the following formula: Percent wound
healing = [(width at 0 h − width at 23 h) / (width at 0 h)] × 100;
the average was calculated from 5 replicates. Under this setting,
the DMSO-treated control cells exhibited 100% wound-healing ability
after 23 h. The MTT assay was performed to determine if the effects
of reparixin and SCH527123 on cell migration may have been affected
by reduced viability. HPAC cells were seeded (20,000 cells/well) in
96-well microtiter plates and grown to 100% confluency at 37°C
overnight. Cells were treated with the same concentrations of
reparixin and SCH527123 as those in the wound-healing assay, and
were also incubated for 23 h at 37°C. The MTT assay was conducted
as aforementioned.
Western blot analysis
HPAC cells were grown to 50–60% confluence and
treated with DMSO or with reparixin or SCH527123 at 40, 60 and 80
μM for 24 h at 37°C. Subsequently, the cells were harvested
and lysed in ice-cold cell lysis buffer (10X; ca. no. 9803; Cell
Signaling Technology, Inc.) containing protease and phosphatase
inhibitors. Total protein was quantified using the Micro BCA
Protein assay kit (cat. no. 23252; Thermo Fisher Scientific, Inc.)
Protein lysates (60 μg) were separated by 10% SDS-PAGE, and
the resolved proteins were transferred to a polyvinylidene fluoride
membrane (cat. no. 10600023; GE Healthcare Life Sciences, Shanghai,
China). Membranes were incubated overnight at 4°C with primary
antibodies (1:1,000; all from Cell Signaling Technology, Inc.)
against phosphorylated (p)-ERK1/2 (T202/Y204; cat. no. 4695),
p-STAT3 (Y705; cat. no. 9145), pan-STAT3 (cat. no. 4904), p-AKT
(S473; cat. no. 4060), S6 (cat. no. 2217) and GAPDH (cat. no.
2118). The membranes were washed 3 times, 5 min each, with TBS +
0.1% Tween-20 (TBST), followed by incubation with horseradish
peroxidase-conjugated goat anti-rabbit immunoglobulin G secondary
antibody (1:10,000; cat. no. 7074; Cell Signaling Technology, Inc.)
for 1.5 h at room temperature. The membranes were washed with TBST
3 times, 5 min each protein bands were visualized using SuperSignal
West Femto Maximum Sensitivity Substrate (cat. no. 34096; Thermo
Fisher Scientific, Inc.).
Statistical analysis
Statistical analyses were conducted using GraphPad
Prism 5 software. Differences were analyzed with one-way analysis
of variance followed by Tukey's post hoc test for multiple
comparisons. Data are presented as the mean ± standard error of the
mean. P<0.05 was considered to indicate a statistically
significant difference.
Results
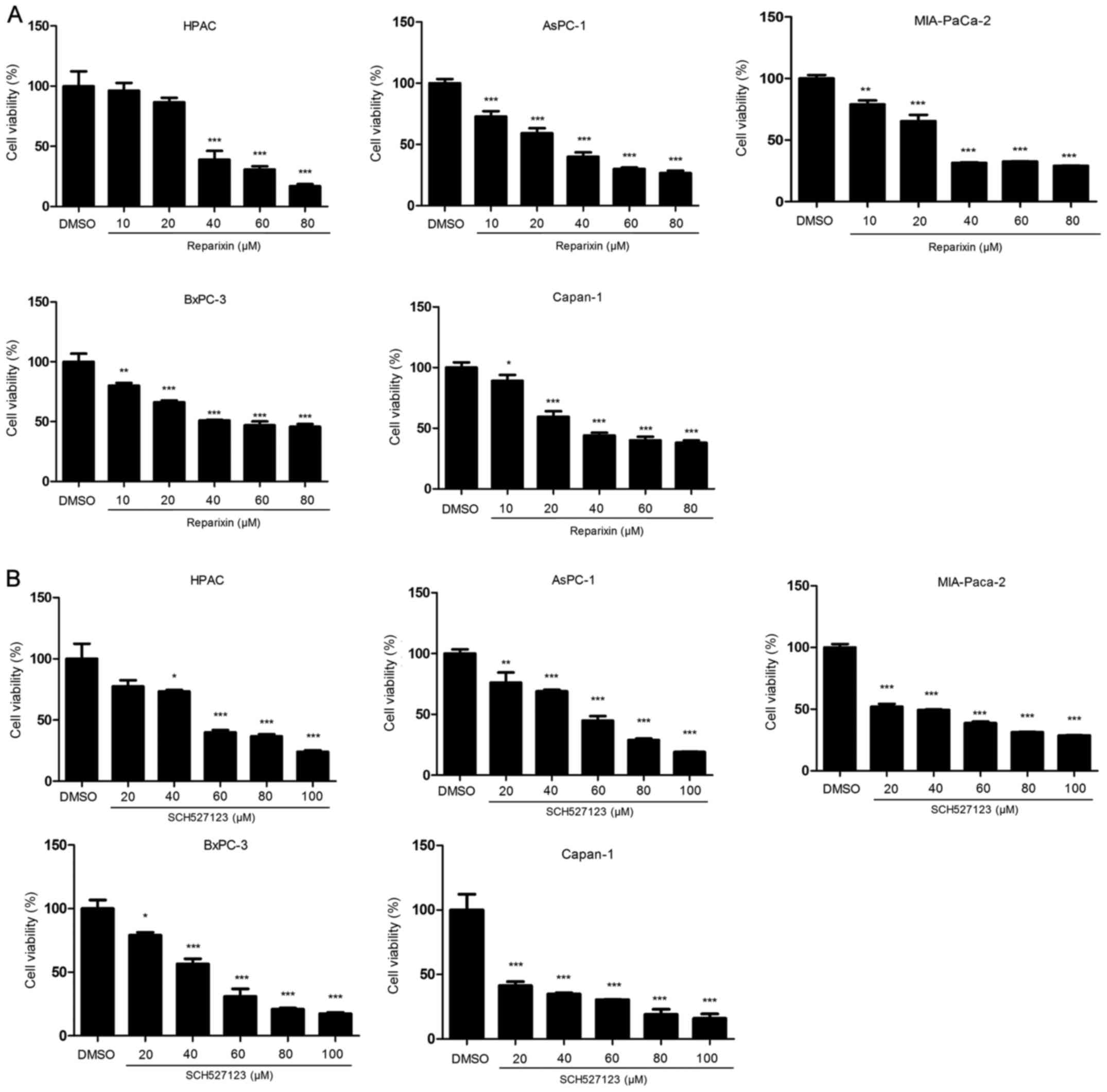
Reparixin and SCH527123 reduce cell
viability
Pancreatic cancer cell lines have been shown to
express IL-8 and IL-8 receptor (26–28).
Owing to the variation in cell densities used in the following
experiments, the higher the cell densities used, the more autocrine
IL-8 were secreted to medium; therefore, higher concentrations of
reparixin or SCH527123 were needed to efficiently suppress the
malignant features of pancreatic cancer cells. For cell viability,
the IC50 value was calculated following treatments with
five different concentrations of reparixin (10, 20, 40, 60 and 80
μM) or SCH527123 (20, 40, 60, 80 or 100 μM) to be
able to determine the optimum inhibitory effect. Inhibition of
IL-8/CXCR1/2 signaling with either reparixin or SCH527123 resulted
in a significant reduction in the viability of pancreatic cancer
cell lines HPAC, AsPC-1, MIA PaCa-2, BxPC-3 and Capan-1 (Fig. 1A and B, respectively). The
inhibitory effects were in a dose-dependent manner following 72-h
treatment, and the IC50 values for reparixin were
determined to be 37.66 μmol/l in HPAC, 27.45 μmol/l
in AsPC-1, 30.40 μmol/l in MIA PaCa-2, 52.94 μmol/l
in BxPC-3 and 34.48 μmol/l in Capan-1 cells. For cells
treated with SCH527123 the IC50 values were calculated
to be 53.49 μmol/l in HPAC, 48.54 μmol/in AsPC-1,
14.65 μmol/l in MIA PaCa-2, 42.14 μmol/l in BxPC-3
and 15.63 μmol/l in Capan-1 cells.
Reparixin and SCH527123 suppress
IL-8-stimulated cell proliferation
As the inhibition of CXCR1/2 signaling was
demonstrated to reduce PDAC cell viability (Fig. 1), and that this signaling cascade
was previously reported to regulate cell proliferation (16), the effects of reparixin and
SCH527123 on the proliferative activities associated with
pancreatic cancer cells was examined by the BrdU incorporation
assay. As demonstrated in Fig. 2A,
IL-8 treatment stimulated Capan-1, AsPC-1 and HPAC cell
proliferation in a dose-dependent manner; Subsequently, cells were
treated with DMSO or with either reparixin or SCH527123 at various
concentrations for 4 h, followed by treatment with IL-8 (25 ng/ml)
for 24 h. The inhibitory effects on the drug-treated cells were
assessed by BrdU incorporation assays; IL-8-induced cell
proliferation was reduced in cells treated with reparixin (Fig. 2B) or with SCH527123 (Fig. 2C) in a dose-dependent manner.
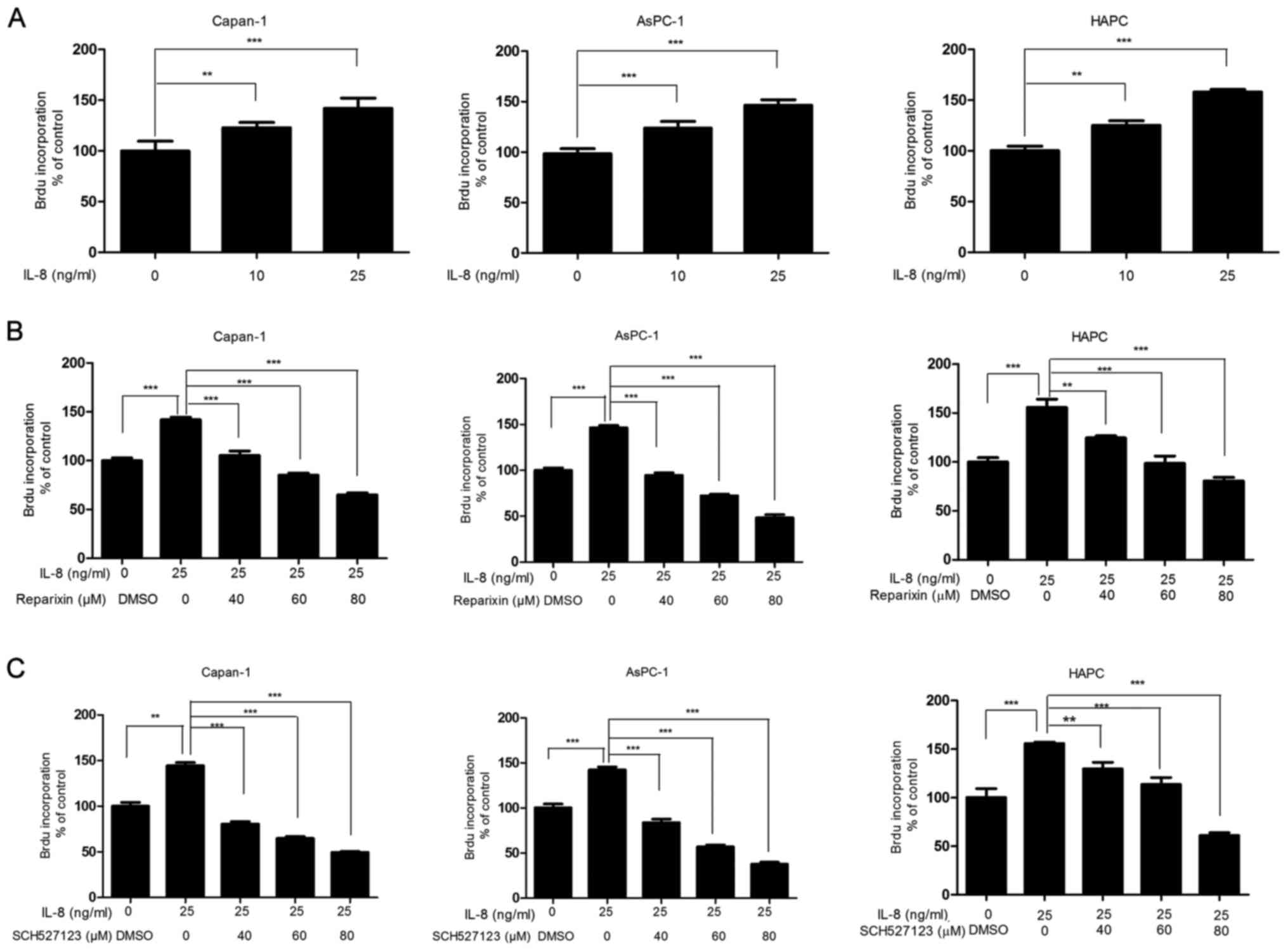 | Figure 2IL-8-induced human pancreatic cancer
cell proliferation is inhibited by reparixin or SCH527123. Capan-1,
AsPC-1 and HPAC were seeded at a density of 5,000 cells/well at
96-well plates. They were serum-depleted for 24 h and subsequently
treated with various concentrations of IL-8 for 24 h. BrdU was
added for 1 h and the absorbance, indicative of DNA proliferation,
was read at 450 nm. (A) Cell proliferation was induced by IL-8 in
dose-dependent manner. (B and C) To test the effects inhibitors on
cell proliferation, cells were pretreated with various
concentrations of either reparixin or SCH527123, or with DMSO for 4
h and subsequently triggered to undergo robust proliferation with
the addition of IL-8 for 24 h followed by BrdU incorporation assay.
*P<0.05, **P<0.01 and
***P<0.001 vs. DMSO. BrdU, bromodeoxyuridine; DMSO,
dimethyl sulfoxide; IL, interleukin. |
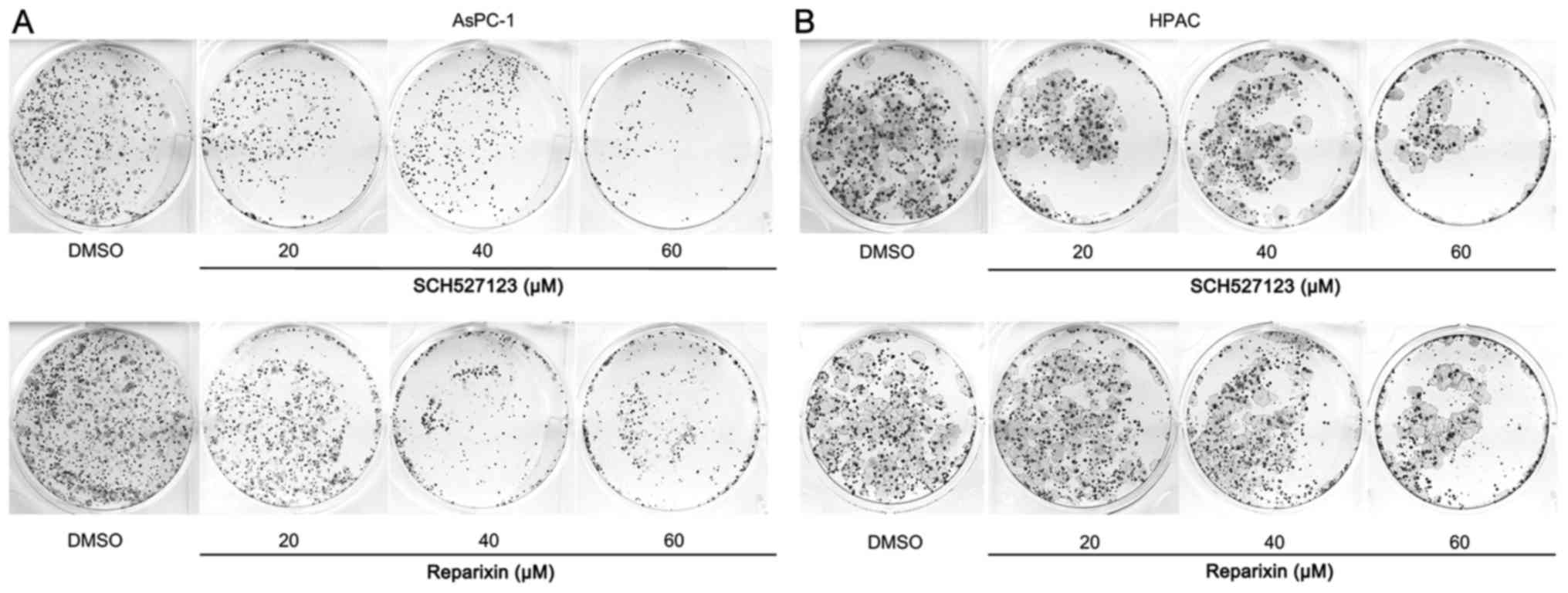
Reparixin and SCH527123 reduce the
ability of PDAC cell lines to grow and form colonies
The effects of reparixin and SCH527123 on the
intrinsic ability of pancreatic cancer cells to grow and form
colonies were examined. AsPC-1 and HPAC cells, seeded in six-well
plates, were treated with either DMSO or with either inhibitor,
reparixin or SCH527123. The colonies grew in the DMSO control
treated cells, ~3 week, and the plates were stained and images of
colony growth were captured (Fig.
3). Consistent with the results from viability and
proliferation assays aforementioned, the inhibition of CXCR1/2 by
reparixin or SCH527123 treatment reduced the colony-forming ability
of PDAC cell lines.
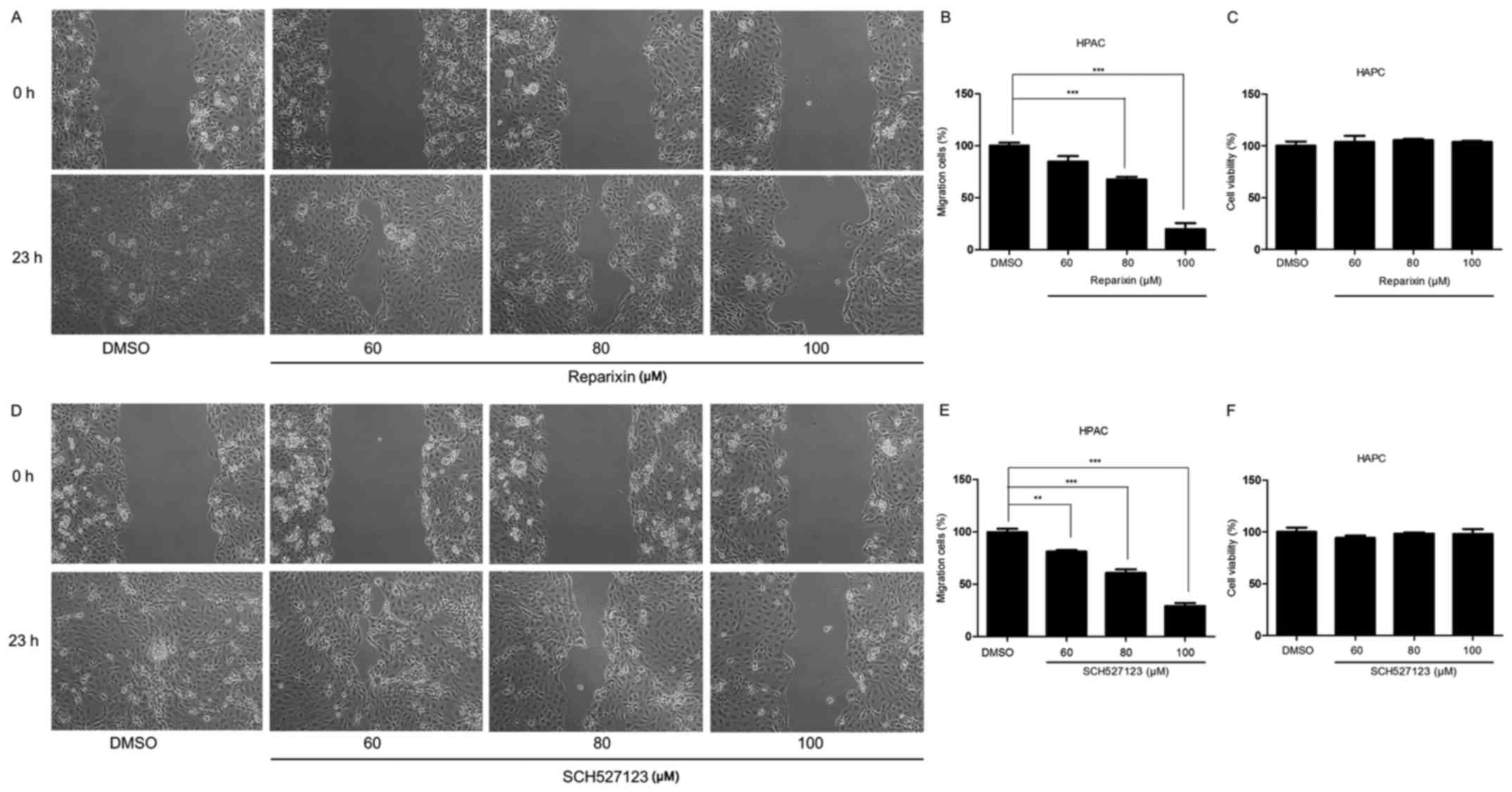
Reparixin and SCH527123 inhibit cell
migration
Previous studies have indicated that CXCR1/2
signaling was crucial for cell migration, which is important for
tumor invasion and metastasis (29,30).
Therefore, the present study examined the effects of CXCR1/2
signaling inhibition on the migratory ability of HPAC cells
(Fig. 4). Following treatment with
either reparixin or SCH527123, cell migratory ability was examined
by wound healing assays. The results demonstrated that the
migratory potential was reduced in cells treated with either
reparixin (Fig. 4A and B) or
SCH527123 (Fig. 4D and E) in a
dose-dependent manner, compared with the control cells treated with
DMSO. Subsequent MTT assay experiments indicated that the effects
of CXCR1/2 inhibition on migration were not a result of reduced
viability (Fig. 4C and F). These
data demonstrated that inhibition of CXCR1/2 by reparixin or
SCH527123 may block the migratory ability of pancreatic cell
lines.
Inhibition of CXCR1/2 signaling pathway
reduces activation of downstream effectors p-AKT, p-ERK, p-STAT3
and p-S6
Previous reports have indicated that IL-8 advances
tumor progression by activating downstream effectors, including
phosphatidylinositol 3-kinase (PI3K)/AKT (31), Ras/mitogen-activated protein kinase
(MAPK) (32–34), Janus kinase (JAK)/STAT3 (35,36)
and S6 (37) pathways. To
determine the underlying mechanism of how reparixin and SCH527123
are able to reduce the overall malignant features in pancreatic
cancer cells, the present study investigated whether these
inhibitors disrupted the activation of downstream effectors that
are regulated by IL-8/CXCR1/2.
Western blot analysis was used to examine the
changes in phosphorylation status of the targeted effectors, which
was indicative of the activation status of the effectors. The
results demonstrated that HPAC cells treated with either reparixin
or SCH527123 exhibited reduced phosphorylation levels of ERK and
AKT, with the greatest reduction observed in cells treated with 80
μM SCH527123 (Fig. 5A and
B, respectively). Similarly, the phosphorylation of S6, which
is known to regulate cell growth and proliferation through the
selective translation of particular classes of mRNA (38), was inhibited in a dose-dependent
manner by both inhibitors, with SCH527123 appearing to be more
potent compared with reparixin (Fig.
5).
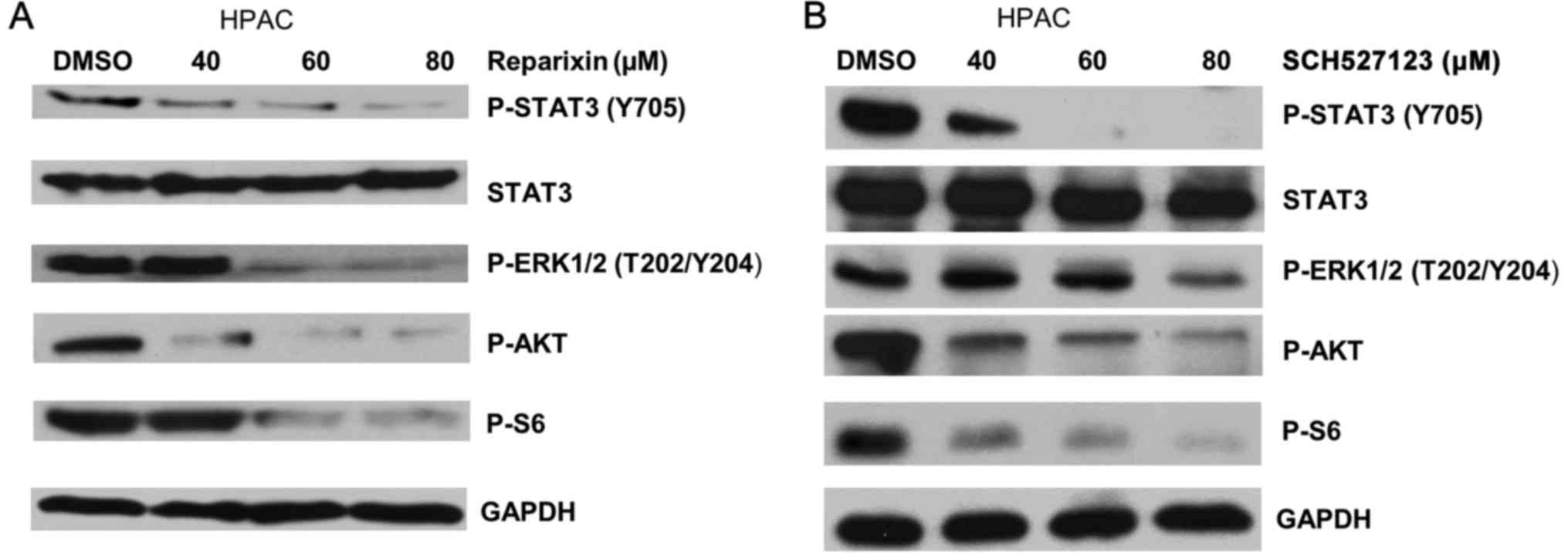 | Figure 5Reparixin and SCH527123 inhibit the
phosphorylation of STAT3, ERK, AKT and S6 in HPAC cells. (A and B)
HPAC pancreatic cancer cells were treated with either (A) reparixin
or (B) SCH527123, or with DMSO as a control. The cells were
harvested after 24 h and changes in the phosphorylation levels of
STAT3 (Y705), AKT (S473), ERK (T202/Y204) and ribosomal S6 were
examined by western blotting. AKT, RAC-α serine/threonine-protein
kinase; DMSO, dimethyl sulfoxide; ERK, extracellular
signal-regulated kinase; p, phosphorylated; S6, ribosomal protein
S6; STAT3, signal transducerand activator of transcription 3. |
Neither inhibitor treatment affected the overall
expression of total STAT3 proteins (Fig. 5). However, these inhibitors
remarkably impaired STAT3 phosphorylation at Y705 in a
dose-dependent manner (Fig. 5).
These results suggested for the first time, to the best of our
knowledge, that reparixin and SCH527123 impaired STAT3 activation
by inhibiting its phosphorylation rather than by suppressing
overall protein expression. The present study results demonstrated
that reparixin and SCH527123 function to suppress protein
phosphorylation, thus inhibiting the activation of AKT, ERK, S6 and
STAT3 in the HPAC cancer cell line, in a dose-dependent manner.
Discussion
The present study demonstrated that inhibition of
the CXCR1/2 pathway by reparixin and SCH527123 treatment reduced
pancreatic cancer neoplastic features, such as cell viability,
proliferation, colony formation and migratory potential in PDAC
cell lines. Molecular investigations revealed that the antagonists
also suppressed the phosphorylation that led to activation of
downstream targets STAT3, AKT, ERK and ribosomal S6.
Consistent with previous studies conducted on
melanoma and colon cancer (13,14),
the present study results demonstrated that IL-8 stimulation
accelerated PDAC cell proliferation. Treatments with the inhibitory
agents reparixin and SCH527123, which block the IL-8/CXCR1/2
signaling pathway, were able to reduce the aforementioned malignant
phenotypes. The tumor-promoting effects of IL-8 not only stimulated
growth, but also promoted cancer cell migration and invasion in a
colon cancer model (14). The
migratory ability that is intrinsic to pancreatic cancer cells, and
which preludes tumor metastasis, was revealed to be inhibited by
reparixin and SCH527123 in the present study. A previous study
reported that IL-8 exposure enhanced pancreatic cancer cell
invasion and that this effect was reversed by treatment with an
antagonizing CXCR1-specific antibody (39). Similarly, an in vivo study
using animal models further confirmed the cancer inhibitory effects
exerted by reparixin and SCH527123 treatment (15,20–23).
A pre-clinical study by Brandolini et al demonstrated that
reparixin was able to target breast cancer cells in xenograft
models and led to prominent tumor regression along with reduced
metastasis (15).
Notably, pre-clinical trials using reparixin
presented negligible adverse effects and no obvious cytotoxicity
was observed. For example, in a double-blinded, placebo-controlled
pilot study designed to evaluate the safety and efficacy of
reparixin for treating IRI and inflammation in patients undergoing
coronary artery bypass grafting (CABG), no apparent side-effects
were noted, which indicated that reparixin treatment in patients
with CABG appeared to be safe and well tolerated (19). Similarly, SCH527123 was revealed to
be safe and well tolerated in otherwise healthy individuals
suffering from severe asthma (25,40).
The safety and efficacy of reparixin to treat cancer
have also been evaluated. The outcomes from a phase Ib clinical
trial that examined the dose and safety of oral reparixin treatment
combined with paclitaxel in women with metastatic breast cancer
revealed that the oral administration of reparixin three times per
day appeared to be safe and tolerable (20). These data provided promising
support for the use of reparixin as an anticancer drug. Results
from the present study demonstrated that reparixin inhibited
pancreatic cancer cell proliferation, colony formation, migration
and cell viability. These results, in conjunction with the safety
and efficacy determined by previous studies, indicated the
potential benefit of administering reparixin as a novel therapeutic
agent for treating pancreatic cancer. Similarly, the inhibitor
SCH527123 was used in a pre-clinical xenograft model of colorectal
cancer and was reported to inhibit tumor growth (24). Co-treatment of SCH527123 with
oxaliplatin resulted in reductions in cell proliferation, tumor
growth and angiogenesis that were greater than treatment with
either agent alone (24). Taken
together, these results indicated that SCH527123 may be a novel
targeted therapeutic agent that may be used to treat pancreatic
cancer.
To better understand the mechanism of action of
these two inhibitor compounds, the expression levels of the main
effectors downstream of the IL-8/CXCR1/2 pathway were examined in
the present study. Previous studies reported that PI3K was a main
target of IL-8 signaling, and that activated PI3K pathway resulted
in increased phosphorylation of its substrate AKT (31). The phosphorylation of AKT initiated
by IL-8 signaling has been examined in a number of cancer cell
lines, which resulted in modulations of cell survival, cell
migration and angiogenesis (41,42).
The present study demonstrated that treatment with either reparixin
or SCH527123 notably reduced the phosphorylation levels of AKT.
These data suggested that reparixin or SCH527123 may be able to
inhibit pancreatic cancer cell viability, proliferation and
migration through the inhibition of AKT signaling, which is in
agreement with a previous study (21).
A number of previous studies using cancer cells have
also reported that IL-8 regulates cell survival, cell proliferation
and invasion through the MAPK signaling pathway and the subsequent
phosphorylation of the downstream effectors ERK1/2 (33,37,43),
which was previously reported to serve an important role in
enhancing oncogenic behavior and increasing the invasive potential
of melanoma cells (32,34). Results from the present study
demonstrated that HPAC cells treated with either reparixin or
SCH527123 exhibited a reduction in the level of ERK1/2
phosphorylation. These data suggested a role for the ERK pathway in
enhancing survival, proliferation and invasion of pancreatic cancer
cells.
A previous study has also revealed that functional
inactivation of STAT3 led to significant inhibition of pancreatic
cancer cell proliferation in vitro and led to reduced tumor
growth in vivo, and activated STAT3 contributed to the
malignant phenotype of human pancreatic cancer in part by promoting
cellular proliferation through accelerated G1/S-phase progression
(44). STAT3 activation can be
stimulated by IL-8 signaling through CXCR2, as demonstrated by a
previous study that used an IL-8 neutralizing antibody to
downregulate S727-phosphorylation of STAT3 in hepatoma cells
(36). Results from the present
study indicated that reparixin and SCH527123 may reduce cell
proliferation and colony formation by inhibiting STAT3
phosphorylation, rather than by suppressing its overall protein
expression level. These data demonstrated a potentially crucial
role of the STAT3 pathway in IL-8-CXCR1/2 signaling and in
stimulating pancreatic cancer cell growth.
S6 was previously reported to modulate cellular
metabolic events, including protein and lipid synthesis,
transcription, translation, cell metabolism and cell growth
(45). IL-8 was revealed to induce
the phosphorylation of ribosomal S6 kinase, which results in the
subsequent phosphorylation and activation of S6 (34). Results from the present study
demonstrated that reparixin and SCH527123 suppressed the expression
of p-S6 in a dose-dependent manner, by blocking the signaling
transduction conveyed from the IL-8/CXCR1/2 pathway.
In conclusion, the present study demonstrated that
the CXCR1/2 antagonists, reparixin and SCH527123, exhibited
antitumor activities in PDAC cell lines. The present study was the
first, to the best of our knowledge, to demonstrate that the
antitumor activity of reparixin and SCH527123 on cancer cell
growth, motility and migration may function through the
STAT3/AKT/ERK/S6 signaling pathways. Based on these results,
reparixin and SCH527123 may be promising therapeutic candidates for
treating human pancreatic cancer. Future in vivo
investigations of the effects of these drugs on tumor-bearing
laboratory animals should be conducted prior to clinical trials in
patients with PDAC.
Acknowledgments
We wish to thank Dr Rich Eckert at the Department of
Biochemistry and Molecular Biology at The University of Maryland
(Baltimore, MD, USA) for providing the microscope used for cell
migration analysis.
Funding
This study was supported by The University of
Maryland School of Medicine and the Comprehensive Cancer Center
start-up fund.
Availability of data and materials
All data generated and analyzed during this study
are included in this published article.
Authors' contributions
JL conceived and designed the experiments, and
contributed reagents, materials and analytical tools. SF and XC
performed the experiments. SF, HJL and JL analyzed the data and
wrote the paper.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
All authors declared that they have no competing
interests with regard to this work.
References
|
1
|
Vincent A, Herman J, Schulick R, Hruban RH
and Goggins M: Pancreatic cancer. Lancet. 378:607–620. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stathis A and Moore MJ: Advanced
pancreatic carcinoma: Current treatment and future challenges. Nat
Rev Clin Oncol. 7:163–172. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Edwards BK, Brown ML, Wingo PA, Howe HL,
Ward E, Ries LA, Schrag D, Jamison PM, Jemal A, Wu XC, et al:
Annual report to the nation on the status of cancer, 1975–2002,
featuring population-based trends in cancer treatment. J Natl
Cancer Inst. 97:1407–1427. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Riall TS, Nealon WH, Goodwin JS, Zhang D,
Kuo YF, Townsend CM Jr and Freeman JL: Pancreatic cancer in the
general population: Improvements in survival over the last decade.
J Gastrointest Surg. 10:1212–1223; discussion 1223–4. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moore MJ, Goldstein D, Hamm J, Figer A,
Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, et al;
National Cancer Institute of Canada Clinical Trials Group.
Erlotinib plus gemcitabine compared with gemcitabine alone in
patients with advanced pancreatic cancer: A phase III trial of the
National Cancer Institute of Canada Clinical Trials Group. J Clin
Oncol. 25:1960–1966. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Conroy T, Desseigne F, Ychou M, Bouché O,
Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de
la Fouchardière C, et al Groupe Tumeurs Digestives of Unicancer;
PRODIGE Intergroup: FOLFIRINOX versus gemcitabine for metastatic
pancreatic cancer. N Engl J Med. 364:1817–1825. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hertzer KM, Donald GW and Hines OJ: CXCR2:
A target for pancreatic cancer treatment? Expert Opin Ther Targets.
17:667–680. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pham NA, Schwock J, Iakovlev V, Pond G,
Hedley DW and Tsao MS: Immunohistochemical analysis of changes in
signaling pathway activation downstream of growth factor receptors
in pancreatic duct cell carcinogenesis. BMC Cancer. 8:432008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Holmes WE, Lee J, Kuang WJ, Rice GC and
Wood WI: Structure and functional expression of a human
interleukin-8 receptor. Science. 253:1278–1280. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Murphy PM and Tiffany HL: Cloning of
complementary DNA encoding a functional human interleukin-8
receptor. Science. 253:1280–1283. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Singh JK, Simões BM, Howell SJ, Farnie G
and Clarke RB: Recent advances reveal IL-8 signaling as a potential
key to targeting breast cancer stem cells. Breast Cancer Res.
15:2102013. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gabellini C, Trisciuoglio D, Desideri M,
Candiloro A, Ragazzoni Y, Orlandi A, Zupi G and Del Bufalo D:
Functional activity of CXCL8 receptors, CXCR1 and CXCR2, on human
malignant melanoma progression. Eur J Cancer. 45:2618–2627. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shen T, Yang Z, Cheng X, Xiao Y, Yu K, Cai
X, Xia C and Li Y: CXCL8 induces epithelial-mesenchymal transition
in colon cancer cells via the PI3K/Akt/NF-κB signaling pathway.
Oncol Rep. 37:2095–2100. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brandolini L, Cristiano L, Fidoamore A, De
Pizzol M, Di Giacomo E, Florio TM, Confalone G, Galante A, Cinque
B, Benedetti E, et al: Targeting CXCR1 on breast cancer stem cells:
Signaling pathways and clinical application modelling. Oncotarget.
6:43375–43394. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu Q, Li A, Tian Y, Wu JD, Liu Y, Li T,
Chen Y, Han X and Wu K: The CXCL8-CXCR1/2 pathways in cancer.
Cytokine Growth Factor Rev. 31:61–71. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chapman RW, Minnicozzi M, Celly CS,
Phillips JE, Kung TT, Hipkin RW, Fan X, Rindgen D, Deno G, Bond R,
et al: A novel, orally active CXCR1/2 receptor antagonist,
Sch527123, inhibits neutrophil recruitment, mucus production, and
goblet cell hyperplasia in animal models of pulmonary inflammation.
J Pharmacol Exp Ther. 322:486–493. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bertini R, Allegretti M, Bizzarri C,
Moriconi A, Locati M, Zampella G, Cervellera MN, Di Cioccio V,
Cesta MC, Galliera E, et al: Noncompetitive allosteric inhibitors
of the inflammatory chemokine receptors CXCR1 and CXCR2: Prevention
of reperfusion injury. Proc Natl Acad Sci USA. 101:11791–11796.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Opfermann P, Derhaschnig U, Felli A,
Wenisch J, Santer D, Zuckermann A, Dworschak M, Jilma B and
Steinlechner B: A pilot study on reparixin, a CXCR1/2 antagonist,
to assess safety and efficacy in attenuating ischaemia-reperfusion
injury and inflammation after on-pump coronary artery bypass graft
surgery. Clin Exp Immunol. 180:131–142. 2015. View Article : Google Scholar :
|
|
20
|
Schott AF, Goldstein LJ, Cristofanilli M,
Ruffini PA, McCanna S, Reuben JM, Perez RP, Kato G and Wicha M:
Phase ib pilot study to evaluate reparixin in combination with
weekly paclitaxel in patients with HER-2-Negative metastatic breast
cancer. Clin Cancer Res. 23:5358–5365. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ginestier C, Liu S, Diebel ME, Korkaya H,
Luo M, Brown M, Wicinski J, Cabaud O, Charafe-Jauffret E, Birnbaum
D, et al: CXCR1 blockade selectively targets human breast cancer
stem cells in vitro and in xenografts. J Clin Invest. 120:485–497.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Singh S, Sadanandam A, Nannuru KC, Varney
ML, Mayer-Ezell R, Bond R and Singh RK: Small-molecule antagonists
for CXCR2 and CXCR1 inhibit human melanoma growth by decreasing
tumor cell proliferation, survival, and angiogenesis. Clin Cancer
Res. 15:2380–2386. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Y, Liu J, Jiang Q, Deng J, Xu F, Chen
X, Cheng F, Zhang Y, Yao Y, Xia Z, et al: Human adipose-derived
mesenchymal stem cell-secreted CXCL1 and CXCL8 facilitate breast
tumor growth by promoting angiogenesis. Stem Cells. 35:2060–2070.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ning Y, Labonte MJ, Zhang W, Bohanes PO,
Gerger A, Yang D, Benhaim L, Paez D, Rosenberg DO, Nagulapalli
Venkata KC, et al: The CXCR2 antagonist, SCH-527123, shows
antitumor activity and sensitizes cells to oxaliplatin in
preclinical colon cancer models. Mol Cancer Ther. 11:1353–1364.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nair P, Gaga M, Zervas E, Alagha K,
Hargreave FE, O'Byrne PM, Stryszak P, Gann L, Sadeh J and Chanez P;
Study Investigators: Safety and efficacy of a CXCR2 antagonist in
patients with severe asthma and sputum neutrophils: A randomized,
placebo-controlled clinical trial. Clin Exp Allergy. 42:1097–1103.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kuwada Y, Sasaki T, Morinaka K, Kitadai Y,
Mukaida N and Chayama K: Potential involvement of IL-8 and its
receptors in the invasiveness of pancreatic cancer cells. Int J
Oncol. 22:765–771. 2003.PubMed/NCBI
|
|
27
|
Takamori H, Oades ZG, Hoch OC, Burger M
and Schraufstatter IU: Autocrine growth effect of IL-8 and GROalpha
on a human pancreatic cancer cell line, Capan-1. Pancreas.
21:52–56. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Miyamoto M, Shimizu Y, Okada K, Kashii Y,
Higuchi K and Watanabe A: Effect of interleukin-8 on production of
tumor-associated substances and autocrine growth of human liver and
pancreatic cancer cells. Cancer Immunol Immunother. 47:47–57. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li Z, Wang Y, Dong S, Ge C, Xiao Y, Li R,
Ma X, Xue Y, Zhang Q, Lv J, et al: Association of CXCR1 and 2
expressions with gastric cancer metastasis in ex vivo and tumor
cell invasion in vitro. Cytokine. 69:6–13. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jayatilaka H, Tyle P, Chen JJ, Kwak M, Ju
J, Kim HJ, Lee JSH, Wu PH, Gilkes DM, Fan R, et al: Synergistic
IL-6 and IL-8 paracrine signalling pathway infers a strategy to
inhibit tumour cell migration. Nat Commun. 8:155842017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Knall C, Worthen GS and Johnson GL:
Interleukin 8-stimulated phosphatidylinositol-3-kinase activity
regulates the migration of human neutrophils independent of
extracellular signal-regulated kinase and p38 mitogen-activated
protein kinases. Proc Natl Acad Sci USA. 94:3052–3057. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Smalley KS: A pivotal role for ERK in the
oncogenic behaviour of malignant melanoma? Int J Cancer.
104:527–532. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Venkatakrishnan G, Salgia R and Groopman
JE: Chemokine receptors CXCR-1/2 activate mitogen-activated protein
kinase via the epidermal growth factor receptor in ovarian cancer
cells. J Biol Chem. 275:6868–6875. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhuang L, Lee CS, Scolyer RA, McCarthy SW,
Palmer AA, Zhang XD, Thompson JF, Bron LP and Hersey P: Activation
of the extracellular signal regulated kinase (ERK) pathway in human
melanoma. J Clin Pathol. 58:1163–1169. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Burger M, Hartmann T, Burger JA and
Schraufstatter I: KSHV-GPCR and CXCR2 transforming capacity and
angiogenic responses are mediated through a JAK2-STAT3-dependent
pathway. Oncogene. 24:2067–2075. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhu B, Lin N, Zhang M, Zhu Y, Cheng H,
Chen S, Ling Y, Pan W and Xu R: Activated hepatic stellate cells
promote angiogenesis via interleukin-8 in hepatocellular carcinoma.
J Transl Med. 13:3652015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
MacManus CF, Pettigrew J, Seaton A, Wilson
C, Maxwell PJ, Berlingeri S, Purcell C, McGurk M, Johnston PG and
Waugh DJ: Interleukin-8 signaling promotes translational regulation
of cyclin D in androgen-independent prostate cancer cells. Mol
Cancer Res. 5:737–748. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nandagopal N and Roux PP: Regulation of
global and specific mRNA translation by the mTOR signaling pathway.
Transl Austin. 3:e9834022015.
|
|
39
|
Chen L, Fan J, Chen H, Meng Z, Chen Z,
Wang P and Liu L: The IL-8/CXCR1 axis is associated with cancer
stem cell-like properties and correlates with clinical prognosis in
human pancreatic cancer cases. Sci Rep. 4:59112014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Holz O, Khalilieh S, Ludwig-Sengpiel A,
Watz H, Stryszak P, Soni P, Tsai M, Sadeh J and Magnussen H:
SCH527123, a novel CXCR2 antagonist, inhibits ozone-induced
neutrophilia in healthy subjects. Eur Respir J. 35:564–570. 2010.
View Article : Google Scholar
|
|
41
|
Bi LK, Zhou N, Liu C, Lu FD, Lin TX, Xuan
XJ, Jiang C, Han JL, Huang H, Zhang CX, et al: Kidney cancer cells
secrete IL-8 to activate Akt and promote migration of mesenchymal
stem cells. Urol Oncol. 32:607–612. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li XJ, Peng LX, Shao JY, Lu WH, Zhang JX,
Chen S, Chen ZY, Xiang YQ, Bao YN, Zheng FJ, et al: As an
independent unfavorable prognostic factor, IL-8 promotes metastasis
of nasopharyngeal carcinoma through induction of
epithelial-mesenchymal transition and activation of AKT signaling.
Carcinogenesis. 33:1302–1309. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Luppi F, Longo AM, de Boer WI, Rabe KF and
Hiemstra PS: Interleukin-8 stimulates cell proliferation in
non-small cell lung cancer through epidermal growth factor receptor
transactivation. Lung Cancer. 56:25–33. 2007. View Article : Google Scholar
|
|
44
|
Scholz A, Heinze S, Detjen KM, Peters M,
Welzel M, Hauff P, Schirner M, Wiedenmann B and Rosewicz S:
Activated signal transducer and activator of transcription 3
(STAT3) supports the malignant phenotype of human pancreatic
cancer. Gastroenterology. 125:891–905. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Magnuson B, Ekim B and Fingar DC:
Regulation and function of ribosomal protein S6 kinase (S6K) within
mTOR signalling networks. Biochem J. 441:1–21. 2012. View Article : Google Scholar
|