Introduction
Autophagy serves roles in a variety of physiological
and pathophysiological processes, including starvation response,
intracellular protein and organelle clearance, development,
anti-aging, elimination of microorganisms, cell death, tumor
suppression and antigen presentation (1). Dysfunction of autophagy is associated
with cancer, neurodegeneration, microbial infection and aging
(2). Autophagy has long been
characterized as a non-selective degradative pathway activated by
starvation. However, it has become evident and important that
autophagy enforces intracellular quality-control (QC) by selective
disposal of damaged proteins and organelles for maintaining normal
cellular homeostasis, and this nutrient-independent selective
autophagy occurs at basal, constitutive levels (3,4). It
is now generally accepted that selective autophagy receptors,
including mammalian p62/SQSTM1, determine exquisite target
specificity (5,6). p62 has LC3- and ubiquitin-binding
domains, allowing it to mediate selective recognition of
polyubiquitinated protein aggregates. The mechanism of selective
autophagosome formation consists of a series of molecular events
beginning with the modification of the cargo by ubiquitination,
followed by its recognition by the polymeric autophagy receptors,
including p62, contributing toward its further modification and the
recruitment of specific autophagy-related (ATG) proteins, leading
to the fusion of the completed autophagosome with a lysosome, and
degradation of the cargo and receptor (6). Autophagy is a complex process that
requires a tight coordination among distinct molecular systems
(7,8). In addition to the core
autophagy-related proteins, the secretory and endocytic pathways,
and the cyto-skeletal network also serve important roles during
autophagy, including facilitating autophagosome transport and
enabling clearance of autophagic substrates (9).
Heterogeneous nuclear ribonucleoprotein K (hnRNPK)
is a DNA/RNA binding protein shuttling between the cell nucleus and
cytoplasm. Its functions are mainly associated with the regulation
of gene expression by controlling the RNA metabolism, including the
synthesis, splicing, nuclear export, translation and decay of
various mRNAs (10–13). Recent studies have also
demonstrated that hnRNPK may regulate multiple cell signaling
pathways by physical or/and functional interactions with various
components of these pathways, including PKC, Src, ERK and GSK-3β
kinases (14–21). Notably, abnormal expression of
hnRNPK has been observed in various types of cancer, including
non-small cell lung cancer (22),
colorectal cancer (23), prostate
cancer (24), squamous cell
carcinoma (25) and nasopharyngeal
carcinoma (26). An increasing
volume of evidence has suggested that hnRNPK serves critical roles
in numerous cellular processes, including cell proliferation,
differentiation, apoptosis and motility (27,28).
Notable, Zhang et al (29)
recently demonstrated that hnRNPK contributed toward drug
resistance in acute myeloid leukemia through the regulation of
autophagy (29). However, the
mechanism by which hnRNPK modulates autophagy remains to be
elucidated.
Our previous study demonstrated that
hnRNPK-knockdown could significantly decrease the acetylation of
α-tubulin on Lys40 in RAW264.7 cells, which may influence the
microtubule stability and thereby regulate the formation and
dynamic patterning of podosomes (30). Recent reports have highlighted the
contribution of acetylation toward autophagy control and revealed
that acetylated microtubules are essential for the fusion of
autophagosomes with lysosomes (31,32).
α-tubulin is specifically acetylated on Lys40 (K40), which is
mainly catalyzed by a conserved α-tubulin acetyltransferase (α-TAT)
(33). The X-ray crystal structure
of human α-TAT and the chemical basis for α-tubulin K40
acetylation, together with the catalytic mechanism of α-TAT
elucidated through a detailed enzymatic analysis has indicated that
α-TAT selectively acetylates α-tubulin K40 (34,35).
Hubbert et al (36)
reported that HDAC6 functions as a tubulin deacetylase. This
aforementioned study indicated that overexpression of HDAC6 leads
toward a global deacetylation of α-tubulin, whereas a decrease in
the expression levels of HDAC6 increased α-tubulin acetylation
in vivo. Furthermore, another previous study demonstrated
that recombinant HDAC6 protein potently deacetylates α-tubulin in
assembled microtubules in vitro. These convergent results
prompted the aim of the present study to investigate whether hnRNPK
regulates autophagy by modulation of α-tubulin acetylation and to
investigate the related regulatory mechanisms.
The present study used the 293 cell line as a
cellular model to study the role of hnRNPK in basal or induced
autophagy. It was revealed that hnRNPK modulated selective
quality-control autophagy but was dispensable for non-selective,
starvation-or rapamycin-induced autophagy. hnRNPK overexpression
partially reversed chloroquine (CQ)-inhibited autophagy, but had no
effect on the 3-methyladenine (3-MA)-mediated inhibition of
autophagy, suggesting its role at the late stage of autophagy
instead of the early stage. Using a stable hnRNPK single allele
knockout (KO) (hnRNPK+/−) 293 cell line established
using the CASPR-Cas 9 system, it was demonstrated that hnRNPK
deficiency led to an increase in HDAC6 mRNA and protein levels,
together with a decrease in α-tubulin K40 acetylation. A
mCherry-GFP-LC3 reporter assay demonstrated that
autophagosome-lysosome fusion in hnRNPK+/− cells was
significantly enhanced in these cells compared with the wild-type
cells, which could be effectively suppressed by treatment with
tubastatin A (Tub A), a selective inhibitor of HDAC6. In addition,
a prominent co-localization of ubiquitin-positive aggregates with
LC3-positive autophagosomes was observed in hnRNPK+/−
cells, but not in the wild-type group or hnRNPK+/− cells
treated with Tub A. Taken together, these results suggested that
hnRNPK may serve a pivotal role in the basal autophagy by
transcriptionally regulating the expression levels of HDAC6 to
influence autophagosome-lysosome fusion.
Materials and methods
Cell lines and reagents
The 293 cell line (Cell Resource Center, Institute
of Life Science Chinese Academy of Sciences, Shanghai, China) was
cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (FBS; GE Healthcare, Chicago, IL, USA) at 37°C
in a humidified atmosphere containing 5% CO2. 293 cells
at a confluence of 80–90% were transfected using Lipofectamine 3000
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). Rapamycin,
3-methyladenine, MG132, Tubulin-Tracker Red and SDS lysis buffer
were purchased from Beyotime Institute of Biotechnology (Haimen,
China). Tubastatin A HCl was purchased from Sigma-Aldrich; Merck
KGaA (Darmstadt, Germany). Chloroquine was purchased from Selleck
Chemicals (Houston, TX, USA). The mCherry-GFP-LC3B plasmid and
pCMV-flag-hnRNPK plasmids were constructed in our laboratory.
Primary antibodies against the following proteins were used: hnRNPK
(cat. no. sc-28380; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA), LC-3B (cat. no. 2775S; CST Biological Reagents Co., Ltd.,
Shanghai, China), HDAC6 (cat. no. 12834-1-AP; ProteinTech Group,
Inc., Chicago, IL, USA), p62/SQSTM1 (cat. no. 66184-1-AP;
ProteinTech Group, Inc.), α-TAT1 (cat. no. ab58742; Abcam,
Cambridge, MA, USA), α-tubulin (cat. no. 11224-1-AP; ProteinTech
Group, Inc.), β-tubulin (cat. no. 10094-1-AP; ProteinTech Group,
Inc.), acetylated α-tubulin (cat. no. T7451; Sigma-Aldrich; Merck
KGaA), ubiquitin (cat. no. ab134953; Abcam, Inc.) and GAPDH (cat.
no. ZS-25778, OriGene Technologies, Inc., Beijing, China). The
antibodies were diluted in Tris-buffered saline with Tween-20
buffer (dilution, 1:2,000; cat. no. 9997; CST Biological Reagents
Co., Ltd.) containing 5% dry skimmed milk.
CRISPR/Cas9-mediated hnRNPK KO in 293
cells
The CRISPR/Cas9 plasmid was purchased from Genloci
Biotechnologies, Inc. (Jiangsu, China). The sequences of gRNA were
as follows: Strand A, 5′-caccGATGATGTTTGAT GACCGTCG-3′ and strand
B, 5′-aaacCGACGGTCATCAAAC ATCATC-3′. The sequences of non-targeting
gRNA (negative control) were as follows: Strand A,
5′-caccGGGTCTTC-GAGAAGACCT-3′ and strand B, 5′-aaacAGGTCTTCTCGA
AGACCC-3′. These gRNAs were synthesized by Shanghai GenePharma Co.,
Ltd. (Shanghai, China). According to the manufacturer’s protocol,
transfection was performed with CRISPR/Cas9 expressing plasmid
carrying hnRNPK gRNA in a 6-well plate with a seeding density of
3×106 cells/well. A mixture of 2.5 µg/well DNA
and Lipofectamine 3000 was added, followed by incubation for 48 h.
A total of 2 µg/ml puromycin was used for the selection. The
established 293 hnRNPK-knockdown cell line was cultured in DMEM
with 10% FBS and 2 µg/ml puromycin. The medium was changed
twice per week. Isolation of cell colony populations from the
selected cells was performed by serial dilutions and cells with a
single allele KO were harvested.
hnRNPK siRNA transfection
293 cells were transfected in suspension with 100 nM
hnRNPK siRNA or non-targeting siRNA. Briefly, 2.0×105
cells in a 200 µl suspension were mixed with 200 µl
opti-MEM plus and 5 µl Lipofectamine 2000, and incubated at
room temperature for 10 min, prior to being seeded into a 6-well
plate with 200 µl complete DMEM supplemented with 10% FBS.
For an effective hnRNPK-knockdown, a mixture of two siRNAs with a
final concentration of 100 nM was used, including the following
sequences: hnRNPK siRNA-1 forward, 5′-UAUUAAGGCUCUCCGUA CATT-3′ and
reverse, 5′-UGUACGGAGAGCCUUAAU ATT-3′; hnRNPK-siRNA-2 forward,
5′-CCUUAUGAUCCC AACUUUUTT-3′ and reverse, 5′-AAAAGUUGGGAUCAU
AAGGTT-3′; negative control siRNA (NC): forward, 5′-UUC
UCCGAACGUGUCACGUTT-3′ and reverse, 5′-ACGUGAC ACGUUCGGAGAATT-3′.
The treatment was performed 24 h after transfection and the siRNAs
were synthetized by Shanghai GenePharma Co., Ltd.
Detection of protein expression by
western blotting
Cells were harvested and lysed with a lysis buffer
[20 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X-100, sodium
pyro-phosphate, β-glycerophosphate, EDTA,
Na3VO4, leupeptin and 1% protease inhibitor
cocktail (Roche Applied Science, Penzberg, Germany)]. Protein
concentrations were measured using a bicinchoninic acid protein
assay (Pierce; Thermo Fisher Scientific, Inc.). A total of 50
µg protein were separated by 10 or 12% SDS-PAGE and
transferred to polyvinylidene difluoride membranes (EMD Millipore,
Billerica, MA, USA). Following blocking in 5% dry skimmed milk for
60 min at room temperature, membranes were incubated with the
primary antibodies (dilution, 1:2,000) overnight at 4°C. These
antibodies were as follows: hnRNPK, LC-3B, HDAC6, p62, α-TAT1,
α-tubulin, β-tubulin, acetylated α-tubulin and GAPDH. Next,
membranes were washed with TBST buffer and incubated with
horseradish peroxidase-conjugated secondary antibodies (dilution,
1:5,000; cat. no. SA00001-1 or SA00001-2; ProteinTech Group, Inc.)
for 60 min at room temperature. All antibodies used in the present
study were diluted in 5% dry skimmed milk in TBST buffer. An
enhanced chemiluminescence kit (Beyotime Institute of
Biotechnology) was used to detect the expression of proteins.
GAPDH, α-tubulin or β-tubulin quantification was used to correct
for variations in total protein loading. The western blotting bands
were quantified using ImageJ software (version number, 1.42;
National Institutes of Health, Bethesda, MD, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cultured cells using
TRIzol reagent (Thermo Fisher Scientific, Inc.) and DNA was removed
using the recombinant DNaseI. cDNA was prepared from 1 µg
total RNA, using reverse transcriptase and an iScript™ cDNA
synthesis kit (cat. no. 1708891; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) according to the manufacturer’s protocol. All
PCR reactions were performed using SsoFast™ Eva Green Supermix
(cat. no. 172-5202; Bio-Rad Laboratories, Inc.), with the following
39 thermal cycles: 95°C for 15 sec; 60°C for 15 sec; and 72°C for
20 sec. The following primers were used for PCR: HDAC6 forward,
5′-GGCTTCAG TTTCCTGTGCTC-3′ and reverse, 5′-CTTCCTCCTCGCTC
TCCTCT-3′; and GAPDH (control) forward, 5′-AACGGATTT GGTCGTATTG-3′
and reverse, 5′-GGAAGATGGTGATGGG ATT-3′. The relative concentration
of HDAC6 mRNA was calculated using the 2−ΔΔCq method
(37-39).
Confocal immunofluorescence
Cells cultured on glass cover-slips were washed with
PBS, fixed with 4% paraformaldehyde in PBS for 30 min at room
temperature, washed with 2 mg/ml glycine in PBS, permeabilized with
0.2% Triton X-100 for 10 min and blocked with 10% goat serum in PBS
for 60 min at room temperature. The cells were subsequently
incubated with primary antibodies diluted at 1:100 in 2% goat serum
PBS overnight at 4°C. Following extensive washes with a washing
buffer (0.05% Tween-20, 1% BSA, PBS), cells were incubated with
fluorescent secondary antibodies (dilution, 1:200) for an
additional 60 min in the dark at room temperature and washed again
with a washing buffer. These fluorescent secondary antibodies were
as follows: Alexa Fluor 594 goat anti-rabbit IgG (cat. no. ZF-0516;
ZSGB-BIO Co., Ltd., Beijing, China), Alexa Fluor 594 goat
anti-mouse IgG (cat. no. ZF-0513; ZSGB-BIO Co., Ltd.), Alexa Flour
488 goat anti-rabbit IgG (cat. no. ZF-0511; ZSGB-BIO Co., Ltd.) and
Alexa Fluor 488 goat anti-mouse IgG (cat. no. ZF-0512; ZSGB-BIO
Co., Ltd.). The nuclei were counterstained with 4,
6-diamidino-2-phenylindole (Sigma-Aldrich; Merck KGaA) for 10 min
at room temperature and washed with PBS. The imaging experiments
were performed using laser scanning confocal microscopes (LSM700;
Zeiss GmbH, Jena, Germany) equipped with a Zeiss Plan-Neofluar 40×
NA 0.75 or 63× NA 1.25 Oil Dic objective. The magnification was
×400 or ×630.
Statistical analysis
All statistical analyses were performed using
GraphPad Prism 6 software (GraphPad Software, Inc., La Jolla, CA,
USA). The significant differences between two groups were analyzed
by the two-tailed unpaired Student’s t-tests (40,41).
Significant differences between multiple groups were analyzed by
two-way analysis of variance and Holm-Sidak multiple comparisons
tests. Data are expressed as the mean ± standard deviation, n≥3,
unless otherwise stated. P<0.05 was considered to indicate a
statistically significant difference.
Results
hnRNPK negatively regulates basal
autophagy in 293 cells
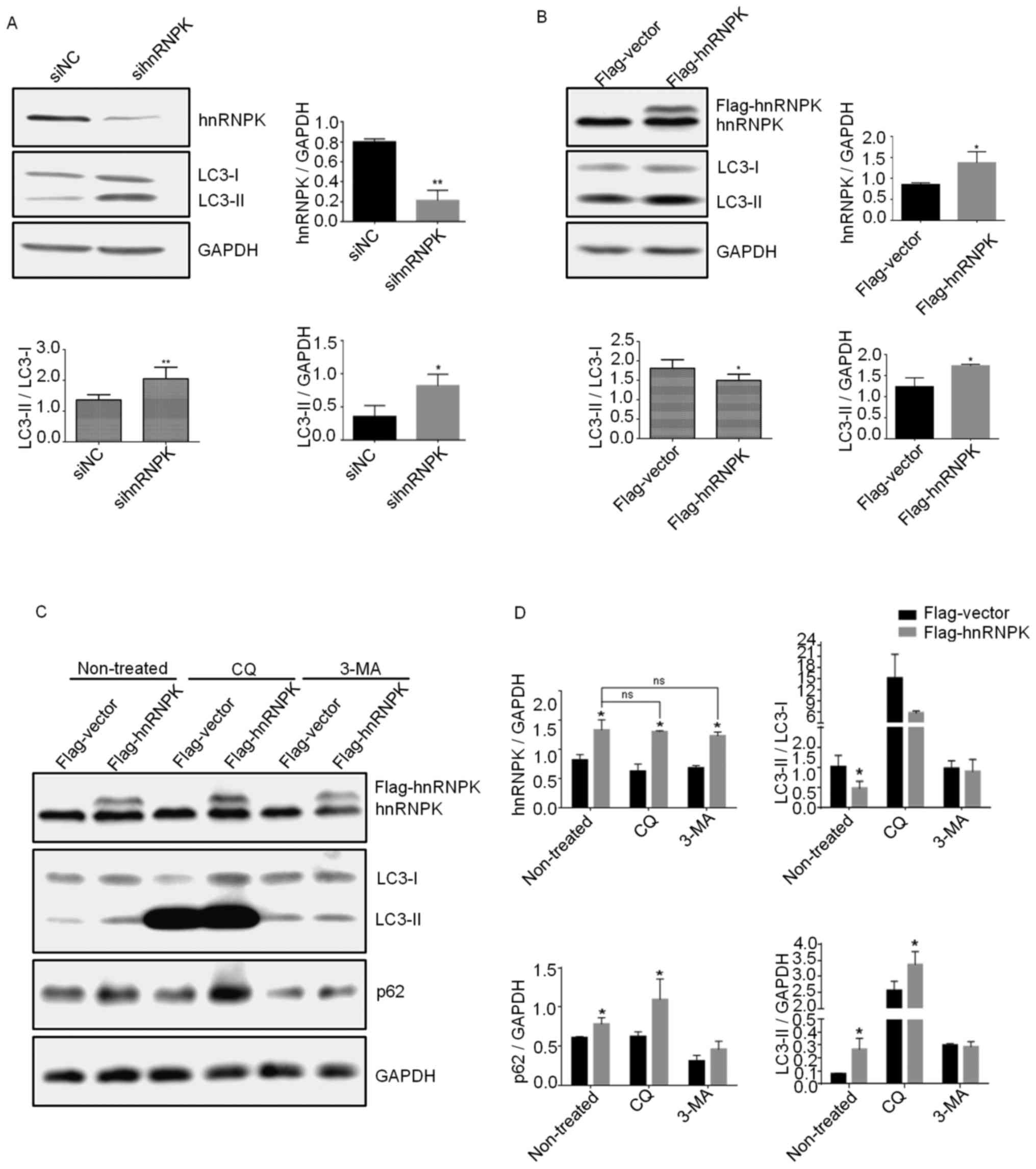
To investigate the role of hnRNPK in autophagy,
hnRNPK was either knocked down with a specific siRNA or
overexpressed by transfection with a Flag-hnRNPK plasmid in 293
cells. A total of 48 h after transfection, autophagy flux
activation was assessed by the ratio of LC3B-II/LCB3-I using
western blotting with a specific anti-LC3B antibody. As shown in
Fig. 1A and B, hnRNPK-knockdown
significantly increased the LC3B-I to LCB3-II conversion, whereas
its overexpression inhibited this process, suggesting a negative
role of hnRNPK in the basal autophagy of 293 cells. Furthermore, it
was found that LC3-II, the lipidated form of LC3 and an
autophagosome marker, was increased by hnRNPK-knockdown and
-overexpression. Taken together, the aforementioned experimental
results suggested that hnRNPK may inhibit autophagy flux in the
late stage of the process at the level of lysosomal degradation. In
order to further confirm this result and determine the precise
function of hnRNPK in the autophagic flux, experiments were
performed by combining hnRNPK-overexpression with two autophagy
inhibitors, including 3-MA, which inhibits the formation of
autophagic vacuoles at the initial stage of the autophagic process,
and CQ, an inhibitor of autophagy at the late stage, which blocks
autophagosome fusion and degradation. 293 cells were transfected
with pCMV-flag-hnRNPK or pCMV-flag empty vector for 24 h and then
treated with 25 µM CQ or 1 mM 3-MA. The results indicated
that hnRNPK-overexpression could reverse the inhibitory effect of
CQ on autophagy, whereas 3-MA-inhibited autophagy remained
unaltered (Fig. 1C and D). A
double band could be seen on the western blot when cells were
transfected with pCMV-flag-hnRNPK and incubated with the hnRNPK
antibody. The protein band of lower molecular weight represented
the endogenous hnRNPK, while the upper band represented the
exogenous flag-hnRNPK, with the quantification of
hnRNPK-overexpression shown in Fig.
1B. In addition, the increased levels of p62 further confirmed
the inhibition of selective autophagy via hnRNPK-overexpression.
This result suggested that hnRNPK may serve a role in autophagy at
the stage involving autophagosome fusion.
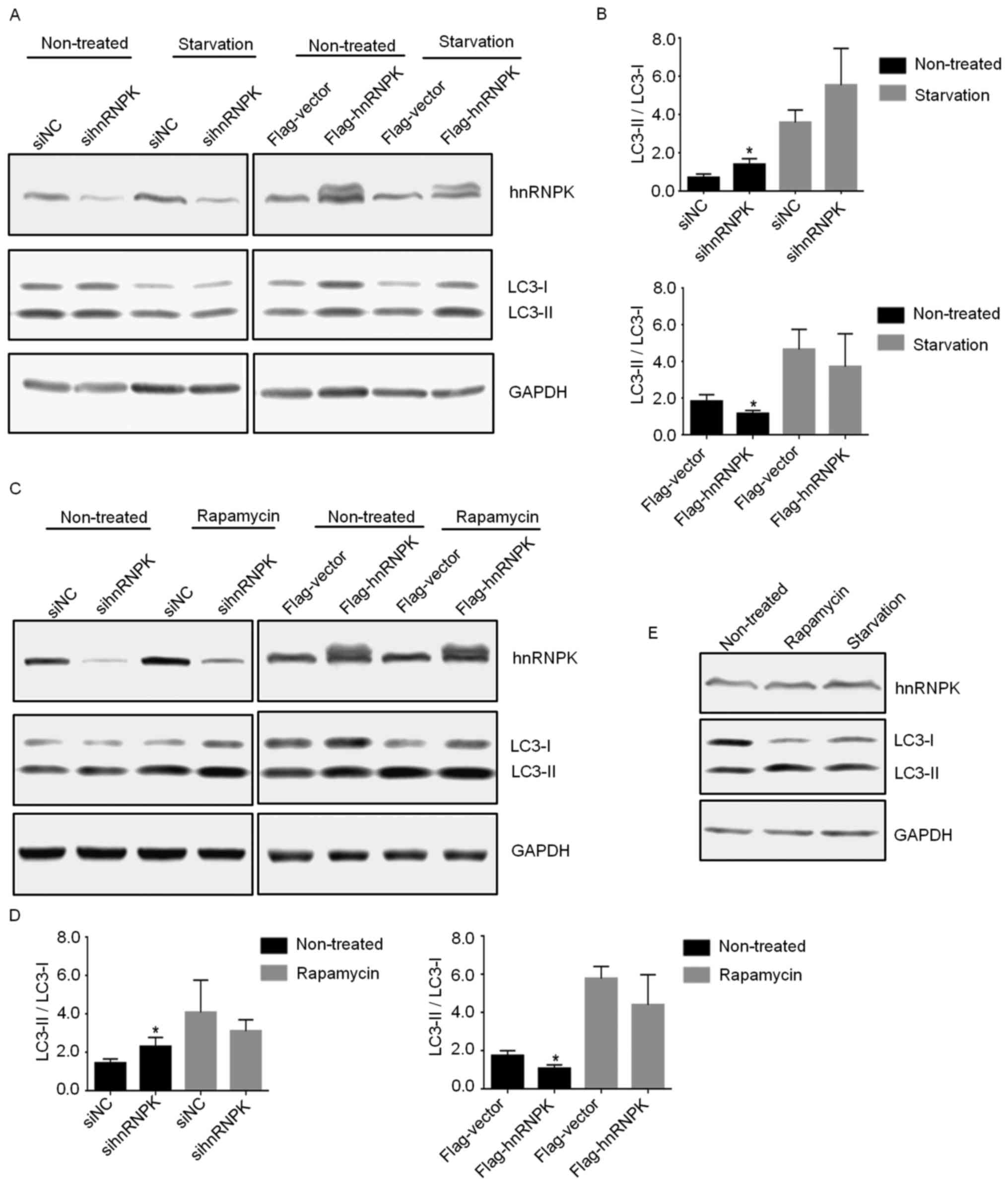
Subsequently, the role of hnRNPK in serum
starvation-or rapamycin- induced autophagy was investigated. Serum
starvation and rapamycin are the two most commonly used conditions
for the induction of non-selective autophagy in which cytosolic
contents and organelles are degraded to supply cells with essential
macromolecules, and to provide the energy required for survival.
293 cells were either transfected with specific hnRNPK-targeting
siRNA or pCMV-flag-hnRNPK plasmid for 24 h, and then treated with
100 nM rapamycin for 24 h or subjected to serum starvation for 6 h.
Cells were subsequently collected and subjected to western blotting
for the verification of hnRNPK-knockdown or -overexpression and
determination of the expression levels of LC3-I and LC3-II.
Notably, compared with their respective negative controls, as
indicated, neither knockdown nor overexpression of hnRNPK in 293
cells exerted notable effects on the serum starvation- or
rapamycin-induced autophagy, as demonstrated in Fig. 2A–D. In addition, the present study
verified that while rapamycin and serum starvation induced
significant autophagy in the treated 293 cells, these treatments
had no notable effect on the endogenous expression of hnRNPK
(Fig. 2E), further excluding the
possible involvement of hnRNPK in these two non-selective autophagy
processes. Taken together, these results suggested that hnRNPK is
likely to be involved in nutrient-independent basal autophagy by
intervening at the later stage, but is dispensable in serum
starvation- or rapamycin-induced autophagy.
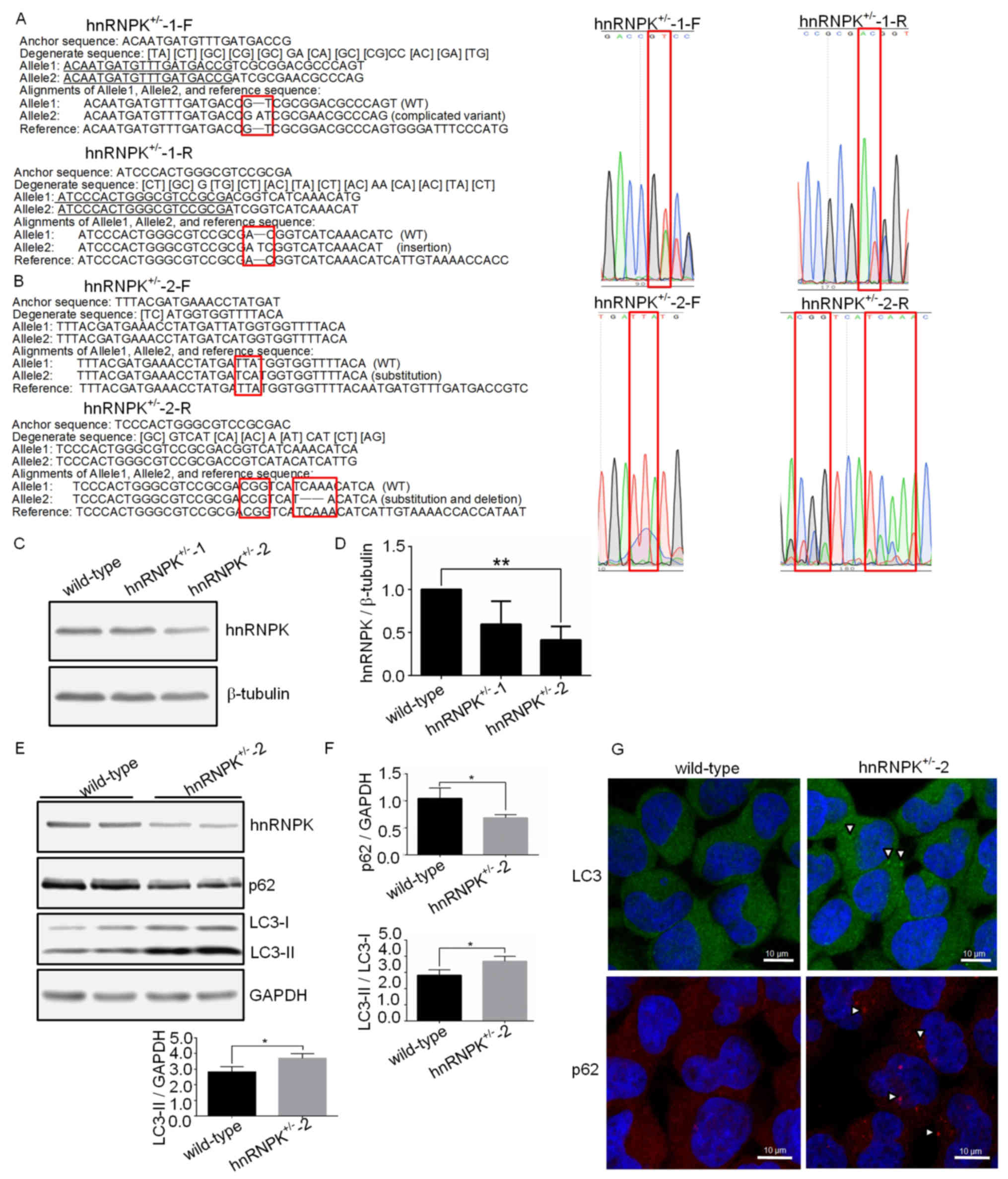
Establishment of a stable hnRNPK KO 293
cell line
In order to further confirm the function of hnRNPK
in basal autophagy and provide a stable cell model for the
associated mechanistic studies, a hnRNPK KO 293 cell line was
generated using the prokaryotic type II CRISPR-Cas9 system
(42,43). Two clones have been isolated
through selection with puromycin. The first clone
(hnRNPK+/−-1) displayed a 2 base-pair insertion
(Fig. 3A), whereas the other clone
(hnRNPK+/−-2) contained a deletion of 3 base pairs and a
2 base-pair replacement (Fig. 3B).
The two clones are single allele KO clones. The fact that only
single allele KO clones could be recovered may be explained by the
essential role of hnRNPK in the survival of the cell, in which case
the complete depletion of hnRNPK may have been lethal. In support
of this hypothesis, it has been recently reported that
double-allele KO of hnRNPK was lethal for the mouse embryo
(27). Due to the single allele KO
in the two clones used in the present study, a decrease in the
hnRNPK expression levels rather than complete depletion was
expected. This was verified by the western blotting shown in
Fig. 3C and D, where an improved
knockdown efficiency of hnRNPK+/−-2 has been observed
compared with the hnRNPK+/−-1 clone. Sequencing analysis
revealed that, in allele 2 of the hnRNPK+/−-2 cells, 2
amino acid changes occurred at Y236 and D245 positions, along with
the deletion of the F243 amino acid residue. Y236 is a binding site
between hnRNPK and tyrosine kinases, including Src, Lyn, Fyn and
Lck, mostly involved in gene expression and signal transduction
(17). F243 and D245 were located
in the K-protein-interactive region (KI) responsible for a number
of the known K protein interactions (11,24).
These mutations did not result in a frameshift; however, they may
lead to the conformational alteration and instability of the hnRNPK
protein. hnRNPK+/−-2 was therefore selected for the
subsequent experiments. The autophagy status in the
hnRNPK+/−-2 cells was firstly examined by western
blotting. As shown in Fig. 3E and
F, significant augmentation of the LC3-II /LC3-I ratio and
LC3-II protein level, along with a decrease in the p62 level could
be detected in the hnRNPK+/−-2 cells, compared with the
wild-type cells. Furthermore, more prominent LC3-positive dots and
p62 punctae were observed in hnRNPK+/−-2 cells by
immunofluorescence (Fig. 3G).
These results further confirmed the important role of hnRNPK in the
basal autophagy of 293 cells.
hnRNPK inhibits the expression of HDAC6
to maintain α-tubulin K40 acetylation in basal autophagy
The present study subsequently aimed to investigate
the mechanism of action of hnRNPK in autophagy under normal
nutrient conditions. Our previous study had demonstrated that
hnRNPK regulated the α-tubulin K40 acetylation in multi-nucleated
mature osteoclasts (30). As
expected, it was revealed that the α-tubulin K40 acetylation level
was significantly decreased in the hnRNPK+/−-2 cells
compared with the wild-type 293 cells, while the total α-tubulin
protein level remained unchanged (Fig.
4A and B). α-tubulin K40 acetylation is mainly regulated by two
enzymes with opposite functions, the acetyltransferase α-TAT and
the deacetylase HDAC6 (44,45).
Based on the function of hnRNPK in the regulation of gene
expression, the present study further examined if hnRNPK may
influence α-tubulin K40 acetylation through regulating the
expression of these two enzymes. Western blot assays were performed
with protein extracts from the wild-type 293 and
hnRNPK+/−-2 cells using specific anti-α-TAT or
anti-HDAC6 antibodies. It was revealed that, while hnRNPK
deficiency had no effect on the α-TAT protein expression level, the
HDAC6 protein level was notably increased in the
hnRNPK+/−-2 cells, compared with the wild-type 293 cells
in basal autophagy. This result was subsequently confirmed by
RT-qPCR, which indicated a 2.5-fold increase in the HDAC6 mRNA
level in the hnRNPK+/−-2 cells compared with the
wild-type 293 cells, suggesting that hnRNPK regulated the activity
of HDAC6 by modulating its mRNA synthesis or metabolism, thereby
controlling the acetylation of α-tubulin K40 (Fig. 4C). In order to determine whether
autophagy regulation by the hnRNPK-HDAC6 axis is an event specific
to basal autophagy, the wild-type 293 cells and
hnRNPK+/−-2 cells were treated with rapamycin or
subjected to serum starvation, and subsequently collected and
subjected to qPCR and western blot analyses for the assessment of
the HDAC6 expression levels. As shown in Fig. 4D–I, neither mRNA nor protein levels
of HDAC6 in the wild-type or hnRNPK+/−-2 293 cells
exhibited marked changes in the serum starvation- or
rapamycin-induced autophagy. In accordance with these results, the
hnRNPK single allele KO had no significant effect on the α-tubulin
K40 acetylation and α-TAT expression level in the serum starvation-
or rapamycin-induced autophagy. All these results further supported
the hypothesis that the hnRNPK-HDAC6 axis contributes toward basal
autophagy but not toward serum-starvation or rapamycin-induced
autophagy.
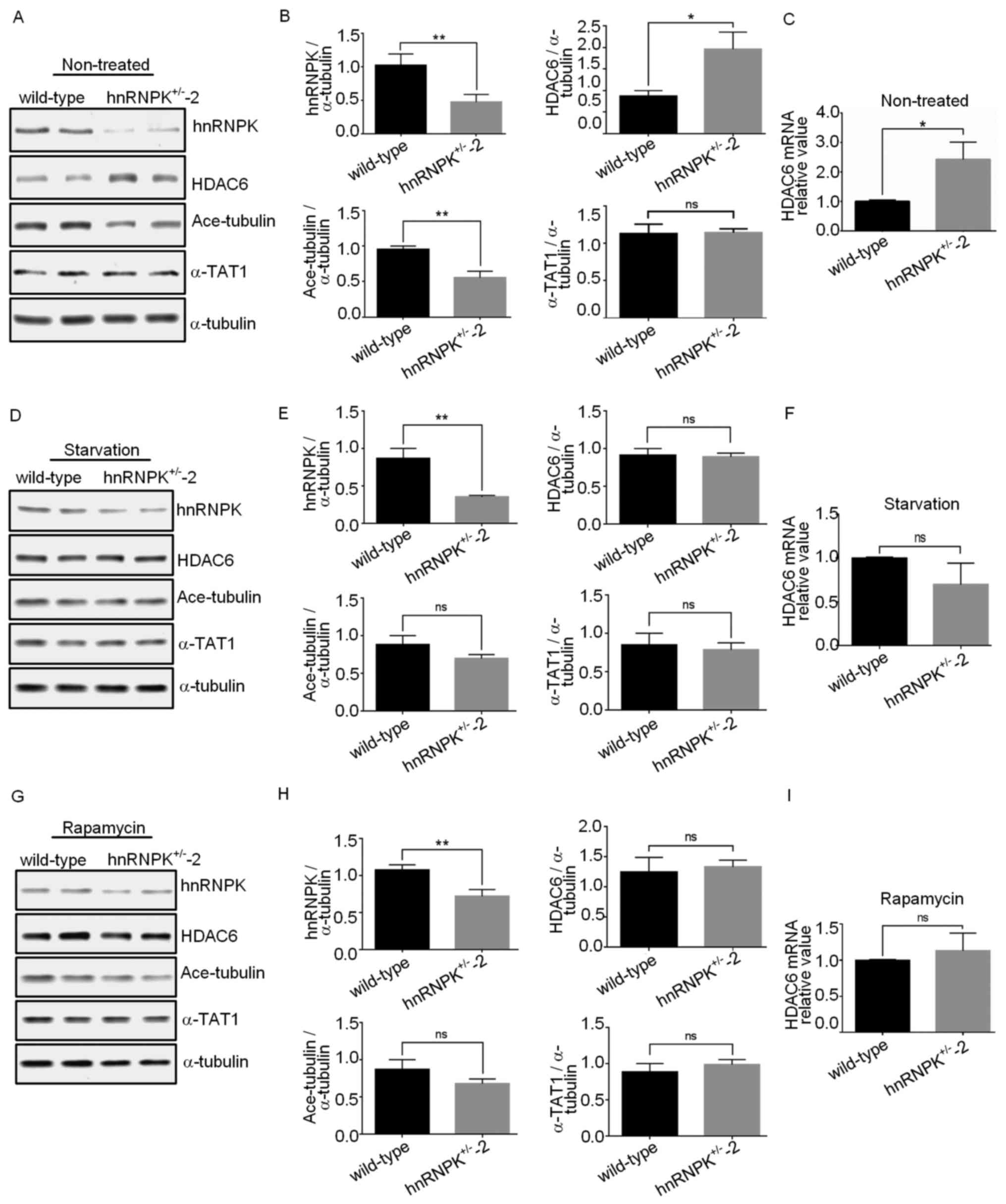 | Figure 4hnRNPK inhibits HDAC6 expression to
maintain α-tubulin K40 acetylation in basal autophagy. (A) Western
blot analyses were performed with proteins extracted from the
wild-type 293 and hnRNPK+/−-2 cells under normal
nutrient conditions using the indicated antibodies. (B) The signal
intensity from the immunoblot as described in (A) was quantified
and α-tubulin was used for the normalization of the expression
value. Data are presented as the mean ± standard deviation, n≥3.
*P<0.05, **P<0.01; ns, no significance.
The error bars represent the standard deviation. (C) Total mRNA was
extracted from the wild-type or hnRNPK+/−-2 cells, and
HDAC6 mRNA levels were quantified by reverse
transcription-quantitative polymerase chain reaction. The results
are presented as the fold-changes following normalization using
GAPDH and compared with the control group. Data are presented as
the mean ± standard deviation, n≥3, *P<0.05. The
error bars represent the standard deviation. 293 cells and
hnRNPK+/−-2 cells were (D) subjected to serum starvation
for 6 h or (G) treated with rapamycin for 24 h, and protein
expression levels were analyzed by western blotting using the
indicated antibodies. The signal intensities from the immunoblots
as described in (D) and (G) were quantified, and α-tubulin was used
for the normalization of the value. The results are presented as
the mean ± standard deviation of three independent experiments (E
and H). (H) Total mRNA was extracted from the wild-type or
hnRNPK+/−-2 cells described in (D and G), and HDAC6 mRNA
levels were quantified by reverse transcription-quantitative
polymerase chain reaction. The results are presented as the
fold-changes following normalization using GAPDH and compared with
the control group (F and I). Data are presented as the mean ±
standard deviation, n≥3, *P<0.05. The error bars
represent the standard deviation. |
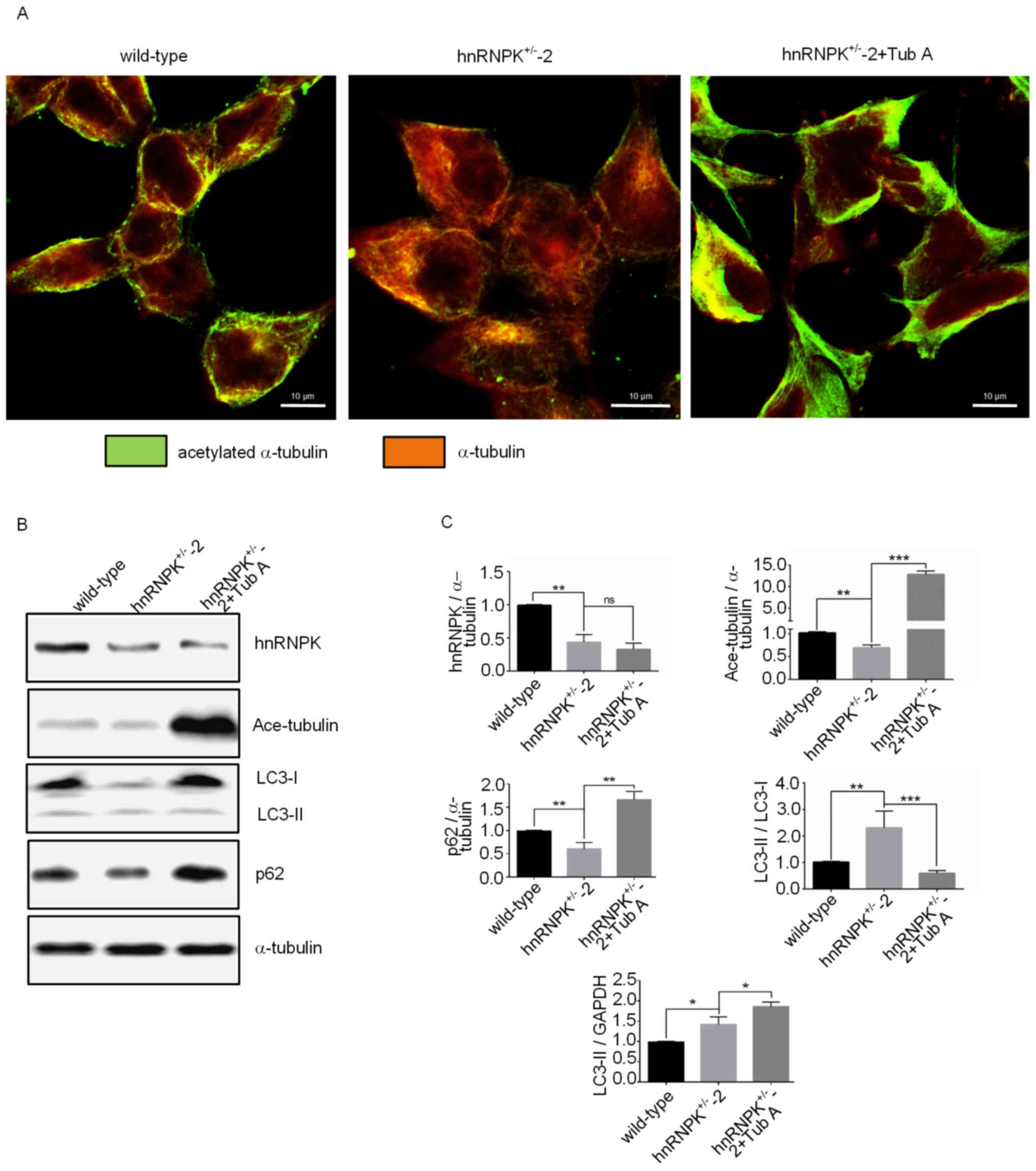
Immunofluorescence detection was used to confirm the
decrease in α-tubulin acetylation in cells. Furthermore, it was
demonstrated that this alteration stemmed from the hyperactivity of
HDAC6 since the treatment of cells with Tub A, a selective
inhibitor of HDAC6, could re-stimulate the α-tubulin K40
acetylation level (Fig. 5A). As
expected, variations in the α-tubulin acetylation of the parental
293 cells, hnRNPK+/−-2 cells and Tub A treated
hnRNPK+/−-2 cells was associated with the degree of
autophagy of the cells, as indicated by the LC3-II/LC3-I ratio, and
the expression levels of LC3-II and p62 determined by western
blotting (Fig. 5B and C).
hnRNPK-HDAC6 axis regulates
autophagosome-lysosome fusion associated with QC autophagy
Autophagosomes must fuse with lysosomes to degrade
the aggregates, and HDAC6 is required for an efficient fusion of
autophagosomes and lysosomes in mouse embryonic fibroblasts (MEFs)
(3). In order to determine whether
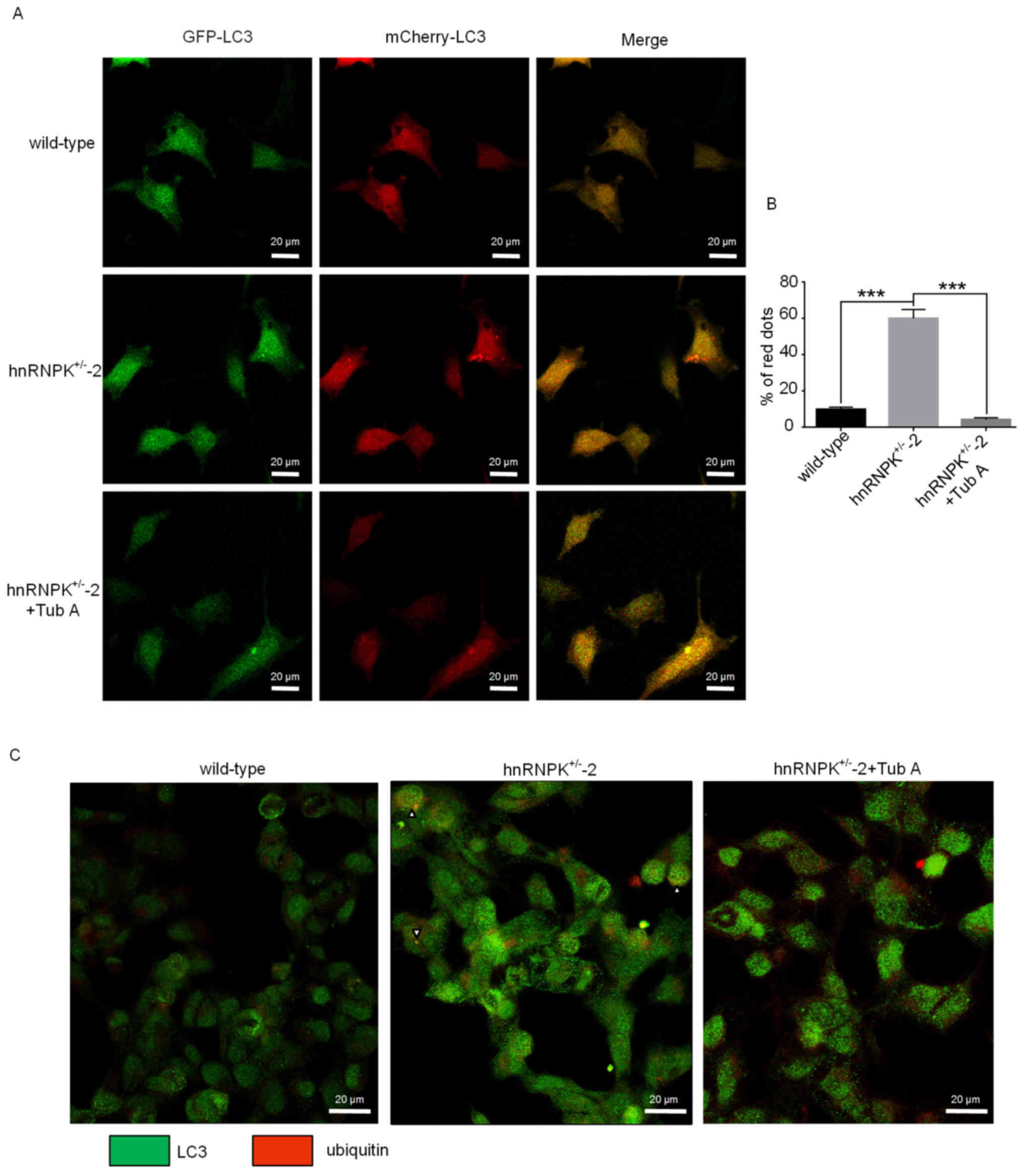
hnRNPK may serve a role in such a process, the mCherry-GFP-LC3
reporter assay was used to evaluate the autophagosome-lysosome
fusion efficiency in the wild-type 293 cells and
hnRNPK+/−-2 cells treated or untreated with Tub A. As
shown in Fig. 6A, the number of
yellow fluorescence-labelled vesicles was decreased, while an
increased number of red fluorescence-labeled vesicles appeared in
the hnRNPK+/−-2 293 cells compared with the wild-type
control cells. This result indicated an increased efficiency of
lysosome-autophagosome fusion (Fig.
6A, upper and middle panels). Accordingly,
autophagosome-lysosome fusion was effectively suppressed when the
hnRNPK+/−-2 cells were treated with Tub A, as indicated
by the increased appearance of yellow fluorescence-labelled
vesicles compared with cells untreated with Tub A (Fig. 6A, middle and lower panels).
Accumulation of red vesicles in the hnRNPK+/−-2 cells
strongly indicated that HDAC6 is required for efficient
autophagosome-lysosome fusion under normal nutrient conditions
(Fig. 6B), which was in agreement
with a previous study (3). These
results demonstrated that the effect of hnRNPK on
autophagosome-lysosome fusion is HDAC6-dependent.
It has been hypothesized that QC autophagy removes
toxic protein aggregates and that cargo selection is partly
achieved by targeted ubiquitination for degradation (46–48).
To further demonstrate the functional role of hnRNPK in protein
aggregate clearance of QC autophagy, an immunofluorescence assay
was used to label the LC3-positive autophagosomes and the
ubiquitinated positive protein aggregates in the 3 aforementioned
types of cells under the treatment of MG132 to block the proteasome
degradation. As shown in Fig. 6C,
compared with the control cells, prominent LC3-positive autophagic
vesicles (green dots) were abundantly present and found
co-localized with the ubiquitin-positive protein aggregates (red
dots) in the hnRNPK+/−-2 293 cells, indicating a highly
efficient selective autophagy-dependent protein aggregate
degradation (Fig. 6C, middle
panels). When the same cells were treated with Tub A, a comparable
intensity of the LC3-positive autophagosome staining could be
observed; however, this staining was not co-localized with the
ubiquitin-positive aggregates, likely due to the deficiency in
docking and tethering ubiquitinated protein aggregates into
autophagosomes. Taken together, the results of the present study
suggested that hnRNPK could regulate the aggregate clearance in the
QC autophagy through a HDAC6-dependent mechanism.
Discussion
Autophagy not only prevents the accumulation of
damaged or unnecessary components, but also facilitates the
recycling of these components to sustain homoeostasis. The concepts
of ‘induced autophagy’ and ‘basal autophagy’ have been introduced
in several previous studies. While the former produces amino acids
in response to extracellular or intracellular stress and signals,
including starvation, growth factor deprivation and pathogen
infection, the latter of which, also referred to as QC autophagy,
is important for baseline turnover of cytosolic components
(9,49).
The process of autophagy consists of several
sequential steps, including induction, autophagosome formation,
degradation and amino acid/peptide generation. The microtubule (MT)
cytoskeleton has been reported to be important for all these steps
(31,50-53).
The MT cytoskeleton consists of highly dynamic tubular polymers
assembled from protofilaments of α/β-tubulin dimers, and is
essential for intracellular transport of organelles and
macromolecules (33). Previous
studies also reported that an intact microtubule cytoskeleton was
required for autophagy and that microtubule disruption interfered
with autophagy by impairing autolysosome formation (46,54).
Kochl et al (31)
demonstrated that, in primary rat hepatocytes, fusion of
autophagosomes with endosomes is inhibited by nocodazole and
vinblastine, which prevent the polymerization of microtubules. In
particular, protein aggregates are formed in the cytoplasm and
accumulate at the microtubule organizing center (MTOC) where they
are subsequently engulfed into autophagosomes to subsequently fuse
with lysosomes for protein degradation (46,55).
K40 acetylated α-tubulin is most abundant in stable microtubules,
but is absent from dynamic cellular structures; including neuronal
growth cones and the leading edges of fibroblasts (36). α-tubulin acetylation is mainly
regulated by acetyltransferase (α-TAT) and histone deacetylases
(HDACs). Previous studies have demonstrated that acetylated
microtubules and HDAC6 are required for autophagy (33-36).
Consistently, the deacetylase HDAC6 has not only been associated
with microtubule-dependent transport of protein aggregates and
lysosomes to the MTOC (46), but
also with the recruitment and assembly of the cortactin-dependent
F-actin cytoskeleton to stimulate the fusion of autophagosomes and
lysosomes, suggesting an important role of α-tubulin acetylation in
these processes. The aforementioned results, together with those
from the present study demonstrating that hnRNPK could regulate the
dynamic stability of microtubules through upregulating the
acetylation of K40 α-tubulin in the maturing osteoclasts, support a
hypothesis that hnRNPK may serve a critical role in the
microtubule-dependent process of autophagy. The results of the
present study have not only unraveled the regulatory function of
hnRNPK in selective QC autophagy, but also elucidated the
associated mechanism by which this protein modulates the expression
level of HDAC6 and consequently influences the microtubule dynamics
through the regulation of K40 α-tubulin acetylation. HDAC6, which
deacetylates α-tubulin in vivo and in vitro, was
often considered a key regulator of the stability of the dynamic
microtubules (56). An increasing
number of studies have confirmed the regulatory role of HDAC6 in QC
autophagy, but not in starvation-induced autophagy, through
recognizing poly-ubiquitinated proteins and controlling the fusion
process of autophagosomes and lyso-somes (3,47,57,58).
HDAC6 is a multidomain protein including a BUZ-finger domain, which
binds to polyubiquitin chains, and two deacetylase domains
interrupted by a motif that binds to cytoplasmic dynein (36), which enable efficient recruitment
and delivery of aggregates to autophagosomes via the microtubule
cytoskeleton (46,48). It has been shown to be involved in
aggresome formation and the fusion of autophagosomes with lysosomes
(31). Lee et al (3) demonstrated that HDAC6 is a central
component of basal autophagy that targets protein aggregates and
damaged mitochondria, and controls autophagosome maturation, which
is essential for ubiquitin-selective QC autophagy (3). In the present study, the effect of
hnRNPK-knockdown on QC autophagy could be partially reverted by Tub
A, an inhibitor of HDAC6 deacetylation activity. This highlights
the role of HDAC6 in the acetylation of α-tubulin in addition to
its binding to polyubiquitin chains, further confirming the dual
action of HDAC6 in the degradation of the ubiquitinated protein
aggregates during QC autophagy.
hnRNPK, shuttling between the nucleus and cytoplasm,
serves as a docking platform that integrates signal transduction
pathways and regulates the expression of specific genes in the cell
through the regulation of RNA metabolism and expression. It has
been demonstrated to control the expression of tumor suppressor
p53, which may have positive and negative regulatory roles in
autophagy induction (59-62). However, the present study
demonstrated that hnRNPK negatively regulated autophagy under
normal nutrient conditions, and seemed to be dispensable for
non-selective starvation- and rapamycin-induced autophagy, implying
that the regulation of p53 activity during the autophagy processes
is associated with a complex regulatory mechanism in which other
factors may also participate. Despite its long-term argumentation
toward the origin, phenotype, karyotype and tumorigenicity, the 293
cell line is one of the most commonly used cell models in cell
biology studies due to its reliable growth and propensity for
transfection. Therefore, the 293 cell line was used in the present
study as a general cell model, without any particular purpose
associated with the origin of the cell, for the study of cell
autophagy that serves important physiological roles in a variety of
cells (63–65).
To the best of our knowledge, our previous study was
the first to describe and discuss the regulation of the α-tubulin
K40 acetylation by hnRNPK and its functional impact in
multi-nucleated mature osteoclasts (30). The present study attempted to
confirm a similar function of hnRNPK in the context of autophagy
regulation in 293 cells, and to further investigate the underlying
mechanism through the regulation of HDAC6 expression. The results
of the present study demonstrated that hnRNPK negatively regulates
the expression levels of HDAC6, but the precise underlying
mechanism of this remains to be elucidated. Belonging to the hnRNP
family, hnRNPK has been most extensively studied as a DNA/RNA
binding protein regulating mRNA metabolisms, including the
synthesis, maturation, trafficking, stability and translational
modulation (10–13). The present study analyzed the mRNA
expression levels of HDAC6 in the hnRNPK+/− and
wild-type 293 cells and demonstrated that the mRNA and protein
expression of HDAC6 was increased in the hnRNPK+/−-2
cells compared with the wild-type cells, suggesting that hnRNPK may
regulate the synthesis or stability of HDAC6 mRNA rather than the
translation. Further studies would aid in elucidating the precise
mechanism of hnRNPK-mediated regulation of HDAC6 expression.
Taken together, the results of the present study
indicated that hnRNPK serves a key role in the ubiquitin-selective
QC autophagy by regulating the expression of HDAC6, which is
required for successful autophagic clearance of protein aggregates
and efficient fusion of autophagosomes with lysosomes, which
contributes toward the elucidation of the components of the
autophagic machinery.
Funding
The present study was supported by grants from the
Guangdong Natural Science Foundation, Guangdong, China (grant nos.
S2013030013315 and 2016A030313083) and the Science and Technology
Program of Guangzhou, Guangdong, China (grant no.
201607010175).
Availability of data and materials
All datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors’ contributions
LZ, LX and LL conceived and designed the
experiments. LZ, MJ and ZT performed the experiments. LZ, LX, GX
and LL analyzed the data. LX and LL wrote the main manuscript text,
and LZ and LX prepared the figures. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank members of the Key
Laboratory of Functional Protein Research of Guangdong Higher
Education Institutes for providing discussion and support
throughout the present study.
References
|
1
|
Mizushima N: Autophagy: Process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee JY, Koga H, Kawaguchi Y, Tang W, Wong
E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, et al: HDAC6
controls autophagosome maturation essential for ubiquitin-selective
quality-control autophagy. EMBO J. 29:969–980. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sun K, Deng W, Zhang S, Cai N, Jiao S,
Song J and Wei L: Paradoxical roles of autophagy in different
stages of tumorigenesis: Protector for normal or cancer cells. Cell
Biosci. 3:352013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rogov V, Dötsch V, Johansen T and Kirkin
V: Interactions between autophagy receptors and ubiquitin-like
proteins form the molecular basis for selective autophagy. Mol
Cell. 53:167–178. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Behrends C, Sowa ME, Gygi SP and Harper
JW: Network organization of the human autophagy system. Nature.
466:68–76. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Galluzzi L, Pietrocola F, Levine B and
Kroemer G: Metabolic control of autophagy. Cell. 159:1263–1276.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Michelotti EF, Tomonaga T, Krutzsch H and
Levens D: Cellular nucleic acid binding protein regulates the CT
element of the human c-myc protooncogene. J Biol Chem.
270:9494–9499. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Michelotti EF, Michelotti GA, Aronsohn AI
and Levens D: Heterogeneous nuclear ribonucleoprotein K is a
transcription factor. Mol Cell Biol. 16:2350–2360. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Miau LH, Chang CJ, Shen BJ, Tsai WH and
Lee SC: Identification of heterogeneous nuclear ribonucleoprotein K
(hnRNP K) as a repressor of C/EBPbeta-mediated gene activation. J
Biol Chem. 273:10784–10791. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen LC, Chung IC, Hsueh C, Tsang NM, Chi
LM, Liang Y, Chen CC, Wang LJ and Chang YS: The antiapoptotic
protein, FLIP, is regulated by heterogeneous nuclear
ribonucleoprotein K and correlates with poor overall survival of
nasopharyngeal carcinoma patients. Cell Death Differ. 17:1463–1473.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bomsztyk K, Denisenko O and Ostrowski J:
hnRNP K: One protein multiple processes. BioEssays. 26:629–638.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schullery DS, Ostrowski J, Denisenko ON,
Stempka L, Shnyreva M, Suzuki H, Gschwendt M and Bomsztyk K:
Regulated interaction of protein kinase Cdelta with the
heterogeneous nuclear ribonucleoprotein K protein. J Biol Chem.
274:15101–15109. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Habelhah H, Shah K, Huang L,
Ostareck-Lederer A, Burlingame AL, Shokat KM, Hentze MW and Ronai
Z: ERK phosphorylation drives cytoplasmic accumulation of hnRNP-K
and inhibition of mRNA translation. Nat Cell Biol. 3:325–330. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ostareck-Lederer A, Ostareck DH, Cans C,
Neubauer G, Bomsztyk K, Superti-Furga G and Hentze MW:
c-Src-mediated phosphorylation of hnRNP K drives translational
activation of specifically silenced mRNAs. Mol Cell Biol.
22:4535–4543. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Miyazaki T, Sanjay A, Neff L, Tanaka S,
Horne WC and Baron R: Src kinase activity is essential for
osteoclast function. J Biol Chem. 279:17660–17666. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wolf D, Witte V, Clark P, Blume K,
Lichtenheld MG and Baur AS: HIV Nef enhances Tat-mediated viral
transcription through a hnRNP-K-nucleated signaling complex. Cell
Host Microbe. 4:398–408. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chang JW, Koike T and Iwashima M: hnRNP-K
is a nuclear target of TCR-activated ERK and required for T-cell
late activation. Int Immunol. 21:1351–1361. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hutchins EJ and Szaro BG: c-Jun N-terminal
kinase phosphorylation of heterogeneous nuclear ribonucleoprotein K
regulates vertebrate axon outgrowth via a posttranscriptional
mechanism. J Neurosci. 33:14666–14680. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gao X, Feng J, He Y, Xu F, Fan X, Huang W,
Xiong H, Liu Q, Liu W, Liu X, et al: hnRNPK inhibits GSK3β Ser 9
phosphorylation, thereby stabilizing c-FLIP and contributes to
TRAIL resistance in H1299 lung adenocarcinoma cells. Sci Rep.
6:229992016. View Article : Google Scholar
|
|
23
|
Carpenter B, McKay M, Dundas SR, Lawrie
LC, Telfer C and Murray GI: Heterogeneous nuclear ribonucleoprotein
K is over expressed, aberrantly localised and is associated with
poor prognosis in colorectal cancer. Br J Cancer. 95:921–927. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Barboro P, Repaci E, Rubagotti A, Salvi S,
Boccardo S, Spina B, Truini M, Introini C, Puppo P, Ferrari N, et
al: Heterogeneous nuclear ribonucleoprotein K: Altered pattern of
expression associated with diagnosis and prognosis of prostate
cancer. Br J Cancer. 100:1608–1616. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Matta A, Tripathi SC, DeSouza LV, Grigull
J, Kaur J, Chauhan SS, Srivastava A, Thakar A, Shukla NK, Duggal R,
et al: Heterogeneous ribonucleoprotein K is a marker of oral
leukoplakia and correlates with poor prognosis of squamous cell
carcinoma. Int J Cancer. 125:1398–1406. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen LC, Hsueh C, Tsang NM, Liang Y, Chang
KP, Hao SP, Yu JS and Chang YS: Heterogeneous ribonucleoprotein K
and thymidine phosphorylase are independent prognostic and
therapeutic markers for nasopharyngeal carcinoma. Clin Cancer Res.
14:3807–3813. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gallardo M, Lee HJ, Zhang X, Bueso-Ramos
C, Pageon LR, McArthur M, Multani A, Nazha A, Manshouri T,
Parker-Thornburg J, et al: hnRNP K is a haploinsufficient tumor
suppressor that regulates proliferation and differentiation
programs in hematologic nalignancies. Cancer Cell. 28:486–499.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Barboro P, Ferrari N and Balbi C: Emerging
roles of heterogeneous nuclear ribonucleoprotein K (hnRNP K) in
cancer progression. Cancer Lett. 352:152–159. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang J, Liu X, Lin Y, Li Y, Pan J, Zong
S, Li Y and Zhou Y: HnRNP K contributes to drug resistance in acute
myeloid leukemia through the regulation of autophagy. Exp Hematol.
44:850–856. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fan X, Xiong H, Wei J, Gao X, Feng Y, Liu
X, Zhang G, He QY, Xu J and Liu L: Cytoplasmic hnRNPK interacts
with GSK3β and is essential for the osteoclast differentiation. Sci
Rep. 5:177322015. View Article : Google Scholar
|
|
31
|
Köchl R, Hu XW, Chan EY and Tooze SA:
Microtubules facilitate autophagosome formation and fusion of
autophagosomes with endosomes. Traffic. 7:129–145. 2006. View Article : Google Scholar
|
|
32
|
Xie R, Nguyen S, McKeehan WL and Liu L:
Acetylated microtubules are required for fusion of autophagosomes
with lysosomes. BMC Cell Biol. 11:892010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Al-Bassam J and Corbett KD: α-Tubulin
acetylation from the inside out. Proc Natl Acad Sci USA.
109:19515–19516. 2012. View Article : Google Scholar
|
|
34
|
Friedmann DR, Aguilar A, Fan J, Nachury MV
and Marmorstein R: Structure of the α-tubulin acetyltransferase,
αTAT1, and implications for tubulin-specific acetylation. Proc Natl
Acad Sci USA. 109:19655–19660. 2012. View Article : Google Scholar
|
|
35
|
Taschner M, Vetter M and Lorentzen E:
Atomic resolution structure of human α-tubulin acetyltransferase
bound to acetyl-CoA. Proc Natl Acad Sci USA. 109:19649–19654. 2012.
View Article : Google Scholar
|
|
36
|
Hubbert C, Guardiola A, Shao R, et al:
HDAC6 is a microtubule-associated deacetylase. Nature. 417:455–458.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kim SH, Shanware NP, Bowler MJ and
Tibbetts RS: Amyotrophic lateral sclerosis-associated proteins
TDP-43 and FUS/TLS function in a common biochemical complex to
co-regulate HDAC6 mRNA. J Biol Chem. 285:34097–34105. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhu Y, Qin Z, Gao J, Yang M, Qin Y, Shen T
and Liu S: Vitamin D therapy in experimental allergic
encephalomyelitis could be limited by opposing effects of
sphingosine 1-phosphate and gelsolin dysregulation. Mol Neurobiol.
50:733–743. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pan Z, Ding Q, Guo Q, Guo Y, Wu L, Wu L,
Tang M, Yu H and Zhou F: MORC2, a novel oncogene, is upregulated in
liver cancer and contributes to proliferation, metastasis and
chemoresistance. Int J Oncol. 53:59–72. 2018.PubMed/NCBI
|
|
40
|
Wyant GA, Abu-Remaileh M, Wolfson RL, Chen
WW, Freinkman E, Danai LV, Vander Heiden MG and Sabatini DM: mTORC1
activator SLC38A9 is required to efflux essential amino acids from
lysosomes and use protein as a nutrient. Cell. 171:642–654.e12.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Qian X, Li X, Cai Q, Zhang C, Yu Q, Jiang
Y, Lee JH, Hawke D, Wang Y, Xia Y, et al: Phosphoglycerate kinase 1
phosphorylates Beclin1 to induce autophagy. Mol Cell.
65:917–931.e6. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Barrangou R, Birmingham A, Wiemann S,
Beijersbergen RL, Hornung V and Smith A: Advances in CRISPR-Cas9
genome engineering: Lessons learned from RNA interference. Nucleic
Acids Res. 43:3407–3419. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hsu PD, Lander ES and Zhang F: Development
and applications of CRISPR-Cas9 for genome engineering. Cell.
157:1262–1278. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Matsuyama A, Shimazu T, Sumida Y, Saito A,
Yoshimatsu Y, Seigneurin-Berny D, Osada H, Komatsu Y, Nishino N,
Khochbin S, et al: In vivo destabilization of dynamic micro-tubules
by HDAC6-mediated deacetylation. EMBO J. 21:6820–6831. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kalebic N, Sorrentino S, Perlas E, Bolasco
G, Martinez C and Heppenstall PA: αTAT1 is the major α-tubulin
acetyltransferase in mice. Nat Commun. 4:19622013. View Article : Google Scholar
|
|
46
|
Iwata A, Riley BE, Johnston JA and Kopito
RR: HDAC6 and microtubules are required for autophagic degradation
of aggregated huntingtin. J Biol Chem. 280:40282–40292. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ouyang H, Ali YO, Ravichandran M, Dong A,
Qiu W, MacKenzie F, Dhe-Paganon S, Arrowsmith CH and Zhai RG:
Protein aggregates are recruited to aggresome by histone
deacetylase 6 via unanchored ubiquitin C termini. J Biol Chem.
287:2317–2327. 2012. View Article : Google Scholar :
|
|
48
|
Hook SS, Orian A, Cowley SM and Eisenman
RN: Histone deacetylase 6 binds polyubiquitin through its zinc
finger (PAZ domain) and copurifies with deubiquitinating enzymes.
Proc Natl Acad Sci USA. 99:13425–13430. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mizushima N: The pleiotropic role of
autophagy: From protein metabolism to bactericide. Cell Death
Differ. 12(Suppl 2): 1535–1541. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Monastyrska I, Rieter E, Klionsky DJ and
Reggiori F: Multiple roles of the cytoskeleton in autophagy. Biol
Rev Camb Philos Soc. 84:431–448. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kimura S, Noda T and Yoshimori T:
Dynein-dependent movement of autophagosomes mediates efficient
encounters with lysosomes. Cell Struct Funct. 33:109–122. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jahreiss L, Menzies FM and Rubinsztein DC:
The itinerary of autophagosomes: From peripheral formation to
kiss-and-run fusion with lysosomes. Traffic. 9:574–587. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Reunanen H, Marttinen M and Hirsimäki P:
Effects of griseofulvin and nocodazole on the accumulation of
autophagic vacuoles in Ehrlich ascites tumor cells. Exp Mol Pathol.
48:97–102. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mackeh R, Perdiz D, Lorin S, Codogno P and
Poüs C: Autophagy and microtubules - new story, old players. J Cell
Sci. (126): 1071–1080. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lamark T and Johansen T: Aggrephagy:
Selective disposal of protein aggregates by macroautophagy. Int J
Cell Biol. 2012.736905:2012.
|
|
56
|
Hubbert C, Guardiola A, Shao R, Kawaguchi
Y, Ito A, Nixon A, Yoshida M, Wang XF and Yao TP: HDAC6 is a
microtubule-associated deacetylase. Nature. 417:455–458. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lin TW, Chen MT, Lin LT, Huang PI, Lo WL,
Yang YP, Lu KH, Chen YW, Chiou SH and Wu CW: TDP-43/HDAC6 axis
promoted tumor progression and regulated nutrient
deprivation-induced autophagy in glioblastoma. Oncotarget.
8:56612–56625. 2017.PubMed/NCBI
|
|
58
|
Pandey UB, Nie Z, Batlevi Y, McCray BA,
Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA,
Schuldiner O, et al: HDAC6 rescues neurodegeneration and provides
an essential link between autophagy and the UPS. Nature.
447:859–863. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Galindo-Moreno M, Giráldez S, Sáez C,
Japón MA, Tortolero M and Romero F: Both p62/SQSTM1 DAC6-dependent
autophagy and the aggresome pathway mediate CDK1 degradation in
human breast cancer. Sci Rep. 7:100782017. View Article : Google Scholar
|
|
60
|
Blasius M and Bartek J: ATM targets hnRNPK
to control p53. Cell Cycle. 12:1162–1163. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zong WX and Moll U: p53 in autophagy
control. Cell Cycle. 7:2947–2948. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Balaburski GM, Hontz RD and Murphy ME: p53
and ARF: Unexpected players in autophagy. Trends Cell Biol.
20:363–369. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Yang Z and Klionsky DJ: Mammalian
autophagy: Core molecular machinery and signaling regulation. Curr
Opin Cell Biol. 22:124–131. 2010. View Article : Google Scholar :
|
|
64
|
Li D, Zhao K, Yang X, Xiao X and Tang S:
TCS2 increases olaquindox-induced apoptosis by upregulation of ROS
production and downregulation of autophagy in HEK293 cells.
Molecules. 22:5952017. View Article : Google Scholar
|
|
65
|
Stepanenko AA and Dmitrenko VV: HEK293 in
cell biology and cancer research: Phenotype, karyotype,
tumorigenicity, and stress-induced genome-phenotype evolution.
Gene. 569:182–190. 2015. View Article : Google Scholar : PubMed/NCBI
|