Introduction
Prostate cancer (PCa) remains an important public
health concern and is the third leading cause of cancer-associated
mortality in males in the United States, with an estimated 1.11
million novel cases diagnosed per year worldwide and 16,1360 novel
cases diagnosed in the United States (1,2). PCa
has become the second most common type of carcinoma in males
worldwide and the most common malignancy in developed countries
(1,2). Established risk factors for this
disease include age, race, family history, genetics and obesity
(3). At present, radical
prostatectomy and radiotherapy are the gold standard treatments for
men with early-stage prostate cancer (4); however, >30% of patients treated
via these approaches will likely exhibit disease recurrence or
metastasis (5,6). Once the disease progresses, the first
line of therapy undertaken is androgen deprivation, which is not a
curative treatment for patients with advanced tumors (4). Hence, investigating signaling
pathways in detail and formulating novel targeted therapies is of
vital importance in enhancing the survival of patients with
advanced PCa.
The bromodomain and extra terminal (BET) family,
which includes bromodomain testis-specific, bromodomain-containing
protein (BRD)2, BRD3 and BRD4, encompasses epigenetic reader
proteins (7). Via their N-terminal
bromodomains (BD1 and BD2), the BET family of proteins exert
diverse physiological functions, including regulation of the cell
cycle, apoptosis and inflammation (8). BRD4, a well-reported member of the
BET family, is generally expressed as a nuclear protein that binds
to acetylated lysine residues on histones via its bromodomains
(9). BRD4 recruits chromatin
remodeling and transcription factors, as well as other associated
proteins, such as positive transcription elongation factor b, to
specific transcriptional sites, this leads to transcription
elongation via the modulation of RNA polymerase II (9,10).
BRD4 also interacts with RelA, a subunit of the nuclear factor-κB
transcriptional complex, inducing an inflammatory response
(10). BRD4 has been reported to
serve vital roles in cell proliferation and cardiac hypertrophy
(9,10). In addition, the aberrant expression
of BRD4 has been frequently reported in a variety of diseases, such
as cancer (11). Carcinogenesis
induced by the BRD4 protein was first reported in NUT midline
cancer (12). Studies have
subsequently revealed BRD4 to exert oncogenic functions in numerous
tumors and cancerous cells, including breast and lung cancers
(13,14). Several small-molecule inhibitors
have been thoroughly studied in recent years, including JQ1 and
IBET762, which possess a high specificity for the bromodomain of
the BET protein family (15). The
binding of these two molecules to the BET bromodomain leads to the
substitution of the protein from acetylated lysine residues on
chromatin (16,17). Of note, BET inhibitors have been
observed to exhibit significant antitumor effects in various
malignancies in vitro and in vivo (18,19).
BRD4 has been reported to be involved in androgen receptor
signaling and the progression of PCa (20,21);
however, other potential roles undertaken by this protein, as well
as the therapeutic efficacy of BET inhibitors in the treatment of
PCa, require further investigation.
In the present study, the roles of BRD4 in PCa were
determined and the potential efficacy of small molecules in the
binding of BET bromodomains were analyzed in vitro and in
vivo. The results of the present study revealed the expression
of BRD4 to be upregulated in PCa, which was positively associated
with metastatic spread and clinical stage. Functional assays
revealed that inhibition of BRD4 suppressed the proliferation,
migration and invasion, concomitantly with the induction of G0/G1
cell cycle arrest and apoptosis of PCa cells. In a mouse model, a
delay in tumor growth was also noted. The findings of the present
study revealed that the effects of BRD4 inhibition in PCa may be
mediated via the forkhead box protein O1 (FOXO1)-p21-Myc signaling
axis. In addition, the present study demonstrated a previously
unreported role of BRD4 in PCa and revealed a promising therapeutic
strategy for patients with PCa.
Materials and methods
Patient tissue specimens
A total of 46 pairs of fresh cancerous prostate
tissue and adjacent normal tissue specimens were obtained from
patients undergoing surgical treatment in the Department of
Urology, Renmin Hospital of Wuhan University (Wuhan, China) between
2016 and 2017. All the patients included in the present study were
male and aged between 59-82-years-old. Approval for the present
study was obtained from the Ethics Committee of the Renmin Hospital
of Wuhan University. Informed consent and relevant clinical
information were obtained from all patients prior to surgery.
Specimens were evaluated via clinicopathological examination and
all samples were classified according to the Tumor, Node,
Metastasis staging system (American Joint Committee on Cancer,
Chicago, IL, USA), preoperative prostate-specific antigen levels
and Gleason score (22). Blood
samples (5 ml) were collected from the arm of patients, then
centrifuged for 5 min (3,500 × g at room temperature) obtain the
serum. Finally, serum samples were detected with ADVIA Centaur XP
Immunoassay System (Siemens AG, Munich, Germany) to determine the
prostate-specific antigen levels. Clinicopathological data were
presented in Table I. Each tumor
sample or adjacent normal sample was divided into two sections; the
first part was snapfrozen in liquid nitrogen and stored at −80°C,
the other half was fixed in 4% paraformaldehyde for 24 h at room
temperature (Beyotime Institute of Biotechnology, Haimen, China).
Patients were also divided into high and low BRD4 expression groups
based on the determined mean cut-off expression value of 2.66.
 | Table IAssociation of relative BRD4
expression with the clinicopathological factors of patients with
prostate cancer. |
Table I
Association of relative BRD4
expression with the clinicopathological factors of patients with
prostate cancer.
| Factors | Patient
characteristics | BRD4 expression
| P-value |
|---|
| Low | High | Total |
|---|
| Age (years) | <70 | 11 | 10 | 21 | 0.264 |
| ≥70 | 9 | 16 | 25 | |
| Preoperative
PSA | <10 | 10 | 8 | 18 | 0.185 |
| ≥10 | 10 | 18 | 28 | |
| Gleason score | <7 | 7 | 15 | 22 | 0.127 |
| ≥7 | 13 | 11 | 24 | |
| Tumor stage | T1 | 12 | 8 | 20 | 0.047a |
| T2/T3 | 8 | 18 | 26 | |
| Metastasis | Absence | 16 | 11 | 27 | 0.010a |
| Presence | 4 | 15 | 19 | |
Cell lines and reagents
The human prostate epithelial cell line RWPE1 and
human prostate cancer cell lines, DU145, PC3 and LNCAP, employed in
the present study were purchased from the American Type Culture
Collection (Manassas, VA, USA). Human prostate cancer cells were
maintained in RPMI-1640 culture medium with 10% heat-inactivated
fetal bovine serum (FBS) (both from Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). RWPE1 cells were maintained in
keratinocyte-serum free medium (Gibco; Thermo Fisher Scientific,
Inc.) containing human recombinant epidermal growth factor (5
ng/ml) and bovine pituitary extract (50 µg/ml). All cells
were cultured in an incubator at 37°C and 5% CO2. JQ1
was purchased from Selleck Chemicals (50 mM, stock solution, cat.
no. S7110, Houston, TX, USA), dissolved in dimethyl sulfoxide and
stored at −80°C. DU145 and LNCAP cells were selected for subsequent
analysis as these cell lines exhibited significantly higher
expression levels of BRD4 than PC3 cells.
Short hairpin (sh)RNA construction and
lentiviral infection
BRD4-targeted shRNA was expressed in a
pGLV3/H1/green fluorescent protein (GFP) + Puro lentiviral vector
via BamHI and EcoRI; vectors were constructed by Shanghai
GenePharma Co., Ltd. (Shanghai, China). The shRNA sequence
targeting BRD4 was as follows: 5′-GCTCAAGACACTATGGAAACA-3′, the
shRNA sequence used as the negative control (sh-NC) was
5′-TTCTCCGAACGTGTCACGT-3′ (LV3-NC-GFP; Shanghai GenePharma Co.,
Ltd.). 293 cells (American Type Culture Collection) were cultured
in 10 cm plates at a density of 5×105 cells per well
with DMEM (Gibco; Thermo Fisher Scientific, Inc.) containing 10%
FBS in an incubator at 37°C and 5% CO2 for 48 h. Then
lentiviral shRNA-containing plasmids were transfected into 293
cells to generate lentiviruses; the lentiviral supernatant
containing the BRD4 shRNA was harvested and purified. DU145 and
LNCAP cells were then infected by application of viral supernatant
(multiplicity of infection 15 for DU145, multiplicity of infection
20 for LNCAP) in RPMI-1640 culture medium with 10% FBS. The
cationic polymer Polybrene (Shanghai GenePharma Co., Ltd.) was
added to facilitate transduction. After 24 h following transduction
the medium was exchanged and cells were cultured for an additional
48 h. Stable cell lines were selected via the application of 1
µg/ml puromycin (Shanghai GenePharma Co., Ltd.).
RNA interference
Small interfering (si)RNAs targeting FOXO1 and p21,
and a negative control (NC) were purchased from Wuhan Biossci Co.,
Ltd. (Wuhan, China). The target sequences were as follows: FOXO1
sense, 5′-CCAUGGACAACAACA GUAATT-3′ and antisense,
5′-UUACUGUUGUUGUCCAUG GTT-3′; p21 sense,
5′-CAGGCGGUUAUGAAAUUCATT-3′ and antisense,
5′-UGAAUUUCAUAACCGCCUGTT-3′; and NC sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and anti-sense,
5′-ACGUGACACGUUCGGAGAATT-3′. For knockdown experiments, DU145 and
LNCAP cells were cultured in 6-well plates at a density of
2×105 cells per well for 24 h, and siRNA (50 nM for
si-FOXO1 or si-NC, 30 nM for si-p21 or si-NC) transfections were
performed using Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.), according to manufacturer protocols.
After 6 h of incubation in an incubator at 37°C and 5%
CO2, the FBS free RMPI-1640 medium was replaced and
cells were cultured in RMPI-1640 medium containing 10% FBS for an
additional 42 h at 37°C. For co-transfection assay, DU145 and LNCAP
cells (a density of 2×105 cells per well) were cultured
in 6-well plates for 24 h at 37°C, subsequently transfected with
si-FOXO1 or si-p21or si-NC, then followed by transfection with
sh-BRD4 or sh-NC as aforementioned; analysis was conducted
following 24 h post-transfection and subsequent incubation for 48
h. Finally, reverse transcription-quantitative polymerase chain
reaction (RT-qPCR) and western blotting assays were used to
determine the efficiency of transfection.
BRD4 mRNA analysis in different cancer
types
The Cancer Genome Atlas (TCGA) and Starbase version
2.0 was used to investigate the expression of BRD4; data from the
Pan-Cancer Analysis Platform (http://starbase.sysu.edu.cn/panCancer.php) indicated
that BRD4 was upregulated in numerous types of cancer (23,24).
Cell proliferation assay
DU145 and LNCAP cell lines were plated onto a
96-well plate at a density of 5,000 cells/100 µl per well
and incubated overnight in an incubator at 37°C. Following JQ1
(1.92, 3.2, 9.6, 16, 48, 80, 240 and 400 nM, and 1.2 2, 6, 10, 30
and 50 µM) or DMSO treatment, shBRD4 or sh-NC transfection,
10 µl cell counting kit-8 (CCK-8) reagents (Dojindo
Molecular Technology, Inc., Kumamoto, Japan) were added to each
well according to the manufacturer's protocols, and cells were
incubated in the dark for 2 h at 37°C. Untreated cells served as a
control. A spectrophotometer was used to measure the absorbance of
the generated optical density (OD) value at 450 nm. The
half-maximal inhibitory concentration (IC50) values
(15.9 µM for DU145, 175 nM for LNCAP) were determined using
GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA, USA).
Colony formation assay
Cells were plated onto 6-well dishes. Following
attachment, cells were treated with JQ1 (15.9 µM for DU145,
175 nM for LNCAP) for 48 h or transfected with shBRD4 as
aforementioned. Cells were then harvested via 0.25% trypsinization
at 37°C for 45 sec and re-suspended in culture medium at 37°C.
Subsequently, 1,000 cells/per well were seeded in a 6-well plate
and cultured in a drug-free medium for 10-14 days. Then, cells were
washed with PBS, fixed with 100% methanol for 15 min, and stained
with 0.5% crystal violet solution at room temperature for 15 min
(Beyotime Institute of Biotechnology). A light microscope
(magnification, ×100; Olympus Corporation, Tokyo, Japan) was used
to count the number of positive colonies (>50 cells/colony).
Observations were performed in triplicate.
Flow cytometry analysis of the cell
cycle
Cells (2×105) were cultured in 6-well
dishes. Following attachment, cells were treated with JQ1 (10, 20,
40 µM for DU145, 100, 200, 400 nM for LNCAP) for 48 h or
transfected with shBRD4 as aforementioned. Cell suspensions were
harvested using 0.25% trypsin for 45 sec at 37°C and placed in
precooled 70% ethanol for fixation at −20°C overnight. Then, a
ribozyme (50 µg/ml) and propidium iodide (50 µg/ml,
PI; BD Biosciences, San Jose, CA, USA) were added to the cells,
which were incubated in the dark at room temperature for 30 min.
Samples were subsequently evaluated with a flow cytometer (BD
FACSCalibur; BD Biosciences, Franklin Lakes, NJ, USA) and data were
analyzed by ModFit LT 2.0 software (Verity Software House, Inc.,
Topsham, ME, USA).
Flow cytometry analysis of apoptosis
Cells (2×105) were plated in 6-well
dishes. Following attachment, cells were treated with JQ1 (10, 20,
40 µM for DU145, 100, 200, 400 nM for LNCAP) for 48 h or
transfected with shBRD4 as aforementioned. Cell suspensions were
harvested via 0.25% trypsinization for 45 sec at 37°C without EDTA,
washed with PBS, and resuspended in 250 ml binding buffer [Hangzhou
Multisciences (Lianke) Biotech Co., Ltd., Hangzhou, China]. Cell
suspensions were stained with 5 µl PI and 5 µl
Annexin V-fluorescein isothiocyanate solution (BD Biosciences) and
incubated at room temperature for 15 min in the dark. Finally, a
flow cytometer (BD FACSCalibur; BD Biosciences) was used to
quantify apoptotic cells. CellQuest 3.0 software (BD Biosciences)
was used to analyze the data. The rate of apoptosis was obtained
from three repeats, and determined with GraphPad Prism 6.
Wound healing assay
Cells (2×105) were seeded in 6-well
dishes and incubated in complete medium (RPMI-1640 culture medium
with 10% FBS) overnight. Cells were treated with JQ1 (15.9
µM for DU145, 175 nM for LNCAP) for 48 h or transfected with
shBRD4 as aforementioned. A 200-µl sterile pipette tip was
scraped across the wells and cell debris was removed via washing
with PBS. The supernatant was subsequently replaced with serum-free
medium (RPMI-1640 culture medium). Finally, the scratch wounds were
analyzed at various time points (0, 24 and 48 h) under a microscope
(magnification, ×200; Olympus Corporation).
Cell invasion assays
Cells (2×105) were seeded on a 6-well
plate, treated with JQ1 (15.9 µM for DU145, 175 nM for
LNCAP) for 48 h or transfected with shBRD4, and harvested via 0.25%
trypsinization for 45 sec at 37°C. Cells were then re-suspended in
serum-free RPMI-1640 culture medium. Subsequently, 1×105
cells/100 µl per well were inoculated on a Transwell chamber
pre-coated with Matrigel in the top chamber at 37°C. In the lower
chamber, 10% FBS-containing RPMI-1640 medium was used as an
attractant. Cells on the top chamber membrane were removed with a
cotton swab after 24 h of incubation at 37°C; invading cells in the
lower side of the chamber were fixed using 4% paraformaldehyde for
15 min and stained with 0.5% crystal violet solution for 15 min at
room temperature. Finally, cells were assessed under a light
microscope (magnification, ×200; Olympus Corporation).
Immunohistochemistry
Tissues from clinical specimens and in vivo
assays (described below) were fixed in 4% paraformaldehyde for 24 h
at room temperature, embedded in paraffin and then sliced into
4-µm sections. The sections were then deparaffinized with
xylene and rehydrated via a descending alcohol series (100, 95, 85
and 75%). The sections were incubated in Tris-EDTA Buffer (10 mM
Tris Base, 1 mM EDTA solution, 0.05% Tween-20, pH 9.0) for 15 min
at 95°C for antigen retrieval. While endogenous peroxidase activity
was neutralized with a methanol/H2O2
solution. Subsequently, sections were incubated with rabbit
monoclonal primary antibodies at 4°C overnight. Following
incubation with horseradish peroxidase (HRP)-conjugated goat and
anti-rabbit secondary antibodies at room temperature for 45 min
(cat. no. GB23303; 1:200; Wuhan Servicebio Technology Co., Ltd.,
Wuhan, China). Finally, all sections were stained with
3,3-diaminobenzidine at room temperature for 3 min (Wuhan
Servicebio Technology Co., Ltd.) and visualized via light
microscopy (magnifications, ×400 and 1,000; Olympus Corporation).
For quantitation, Image-Pro Plus software (ver 6.0; Media
Cybernetics, Inc., Rockville, MD, USA) was used to determine the
average integrated OD (IOD) obtained by analyzing three fields per
slide. The relative average IOD was obtained from the average IODs
of cancerous prostate tissue divided by the average IOD of adjacent
normal tissue specimens. For the analysis of animal tissue, the
average IOD was obtained from the average IODs of the JQ1 treatment
or sh-BRD4 groups divided by the average IOD of the control or
sh-NC groups, respectively. The primary antibodies employed in the
present study, including BRD4 (cat. no. ab128874; 1:200), Ki-67
(cat. no. ab16667; 1:100), p21 (cat. no. ab109520; 1:100) for
Immunohistochemistry were obtained from Abcam (Cambridge, UK), and
FOXO1 antibody (cat. no. 2880; 1:100) were obtained from Cell
Signaling Technology, Inc., Danvers, TX, USA).
RT-qPCR assay
Total RNA from prostate cell lines and tissue
specimens was isolated using TRIzol® reagent (Thermo
Fisher Scientific, Inc.). Spectrophotometry was used to measure the
concentration and purity of RNA, while the PrimeScript RT Reverse
transcriptase Reagent Kit (Takara Bio, Inc., Otsu, Japan) was
applied to synthesize complementary DNA according to the
manufacturer protocols. Briefly, RNA (2 µg) was incubated
for 15 min at 37°C and for 5 sec at 85°C. qPCR analysis was
conducted using the SYBR Premix Ex Taq kit (Takara Bio, Inc.) and
the StepOnePlus Real-Time PCR system (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The conditions of PCR were listed as
follows: 95°C for 10 sec, then following 40 cycles of 95°C for 5
sec, 58°C for 20 sec. Expression levels were normalized to that of
GAPDH; the relative expression of genes was determined using the
2−ΔΔCq method (25).
All assays were performed in triplicate. Primers were synthesized
by Sangon Biotech (Shanghai, China) and were presented in Table II.
| Table IIReverse transcription-quantitative
polymerase chain reaction primer sequences. |
Table II
Reverse transcription-quantitative
polymerase chain reaction primer sequences.
| Gene | Forward
(5′-3′) | Reverse
(5′-3′) |
|---|
| BRD4 |
TGAGTCGGAGGAAGAGGACAAGTG |
CGCAGTGTGGACGGCTTCAG |
| FOXO1 |
TGTCCTACGCCGACCTCATCAC |
GCACGCTCTTGACCATCCACTC |
| p21 |
GCTGAGCCGCGACTGTGATG |
CCTCCAGTGGTGTCTCGGTGAC |
| c-Myc |
CGAGGAGAATGTCAAGAGGCGAAC |
GCTTGGACGGACAGGATGTATGC |
| Bcl-2 |
GAACCGGCACCTGCACACC |
AGAGTCTTCAGAGACAGCCAGGAG |
| p53 |
ACCGGCGCACAGAGGAAGAG |
GCCTCATTCAGCTCTCGGAACATC |
| GAPDH |
CAACGTGTCAGTGGTGGACCTG |
GTGTCGCTGTTGAAGTCAGAGGAG |
Western blotting
Tissues and cells were lysed with
radioimmunoprecipitation assay buffer-containing PMSF (Beyotime
Institute of Biotechnology) to extract total proteins. A
Bicinchoninic Acid assay kit (Beyotime Institute of Biotechnology)
was used to determined protein concentration. Equal amounts of
proteins (20 µg) were separated via 10% polyacrylamide gel
electrophoresis (Beyotime Institute of Biotechnology), and proteins
were transferred onto polvinylidene difluoride (PVDF) membranes
(Wuhan Servicebio Technology Co., Ltd.). Blocking was performed in
Tris-buffered saline and 0.05% Tween-20 containing 5% nonfat milk
for 1.5 h at 37°C. The PVDF membranes were washed with TBST three
times, then incubated with rabbit polyclonal primary antibodies at
4°C overnight. Subsequently, the membrane was incubated with HRP
conjugated secondary antibodies (1:10,000) for 1 h at 37°C. Lastly,
an enhanced chemiluminescence kit [Hangzhou Multisciences (Lianke)
Biotech Co., Ltd., Hangzhou, China] was used to analyze the
membranes according to the instruction of manufacturer. Protein
bands were scanned with Molecular Imager® ChemiDoc™ XRS+
Imaging System (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The
optical densities a selection of bands were detected using ImageJ
software 1.45 (National Institutes of Health, Bethesda, MD, USA).
All assays were performed in triplicate. The primary antibodies of
BRD4 (ab128874; 1:500), p21 (cat. no. ab109520; 1:1,000), c-Myc
(cat. no. ab39688; 1:500), Bcl-2 (cat. no. ab182858; 1:1,000) and
GAPDH (cat. no. ab9485; 1:2500) were purchased from Abcam. FOXO1
(cat. no. 2880; 1:1,000), p53 (cat. no. 2527S; 1:1,000) and cyclin
D1 (cat. no. 2978S; 1:1,000) antibodies were purchased from Cell
Signaling Technology, Inc. Goat and anti-rabbit secondary
antibodies were obtained from Wuhan Boster Biological Technology,
Ltd. (Wuhan, China).
Tumor xenografting and in vivo treatment
assay
All experimental procedures were approved by the
Ethics Committee of Wuhan University and the animals received
humane care. All 4-5-week-old male BALB/c nude mice (Beijing Vital
River Laboratory Animal Technology, Co., Ltd., Beijing, China) were
housed in a pathogen-free environment at the Animal Center of
Renmin Hospital of Wuhan University. Briefly, a total of 40 mice
(15-18 g) were housed in a pathogen-free environment (22-27°C;
humidity, 40-70%, under a 12 h light/dark cycle with free access to
sterilized feed and water). DU145 cells were prepared by suspending
1×107 cells in 200 µl PBS, and subcutaneously
injected into the left upper extremity of mice. Once the tumor
volume was measurable (100 mm3), animals were randomized
into treated or negative control groups (10 mice/group). For the
drug treated groups, DMSO-containing JQ1 was diluted with
physiological saline to obtain a final solution of 5 mg/ml. Each
mouse was administered an intraperitoneal injection of JQ1, 50
mg/kg per day on day 9. Mice of the control group were administered
an equal amount of physiological saline containing 5% DMSO per day.
Mice were treated with JQ1 or vehicle for 19 days. In the BRD4
knockdown group, 1×107 stable shRNA BRD4-expressing
LNCAP or control cells (sh-NC) in 200 µl PBS were implanted
into the same anatomical sites of mice (10 mice/group). The tumor
size of each mouse were followed-up using Vernier calipers every 4
days and tumor volume was approximated as 0.5 × width2 ×
length (mm3). After 4 weeks, all animals were
sacrificed, the experiment was terminated, and tumors were excised,
images and weighed.
Statistical analysis
GraphPad Prism software (version 6) or SPSS 20.0
(IBM Corp., Armonk, NY, USA) were used for statistical analysis.
Data are presented as the mean ± standard deviation. All
experiments were performed in triplicate. A Student's t-test,
χ2 or one-way analysis of variance were used to evaluate
whether statistically significant differences were present. A
Bonferroni post-hoc test for multiple comparisons was also applied.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Overexpression of BRD4 in PCa cell lines
and tumor specimens
In the present study, the role served by BRD4 in
other types of cancer was analyzed. Data from the Pan-Cancer
Analysis Platform indicated that BRD4 was upregulated in numerous
types of cancer (Fig. 1A).
Additionally, 46 pairs of prostate cancer specimens and surrounding
normal tissue were collected and evaluated via RT-qPCR. The results
revealed that, compared with the surrounding normal prostate
tissue, the expression of BRD4 mRNA was significantly upregulated
in PCa samples (Fig. 1B). In
addition, BRD4 expression was positively associated with metastasis
(Fig. 1C). Data obtained from
immunohistochemical analysis revealed BRD4 to be primarily
expressed in the nucleus of PCa cells. Furthermore, tumor samples
were stained to a higher degree than in adjacent normal tissue as
presented by the IOD values (Fig.
1D), consistent with previous findings (20). The present study also observed
that, compared with RWPE1 cells, the mRNA expression levels of BRD4
were significantly increased in PCa cell lines (DU145, PC3 and
LNCAP); BRD4 protein expression levels were significantly
upregulated (Fig. 1E and F). To
further investigate the association between BRD4 expression and the
clinical characteristics of patients with PCa, the clinical data
obtained from 46 patients with PCa were analyzed. The results
revealed that the levels of BRD4 expression exhibited a significant
association with the tumor clinical stages and metastasis of PCa
(Table I).
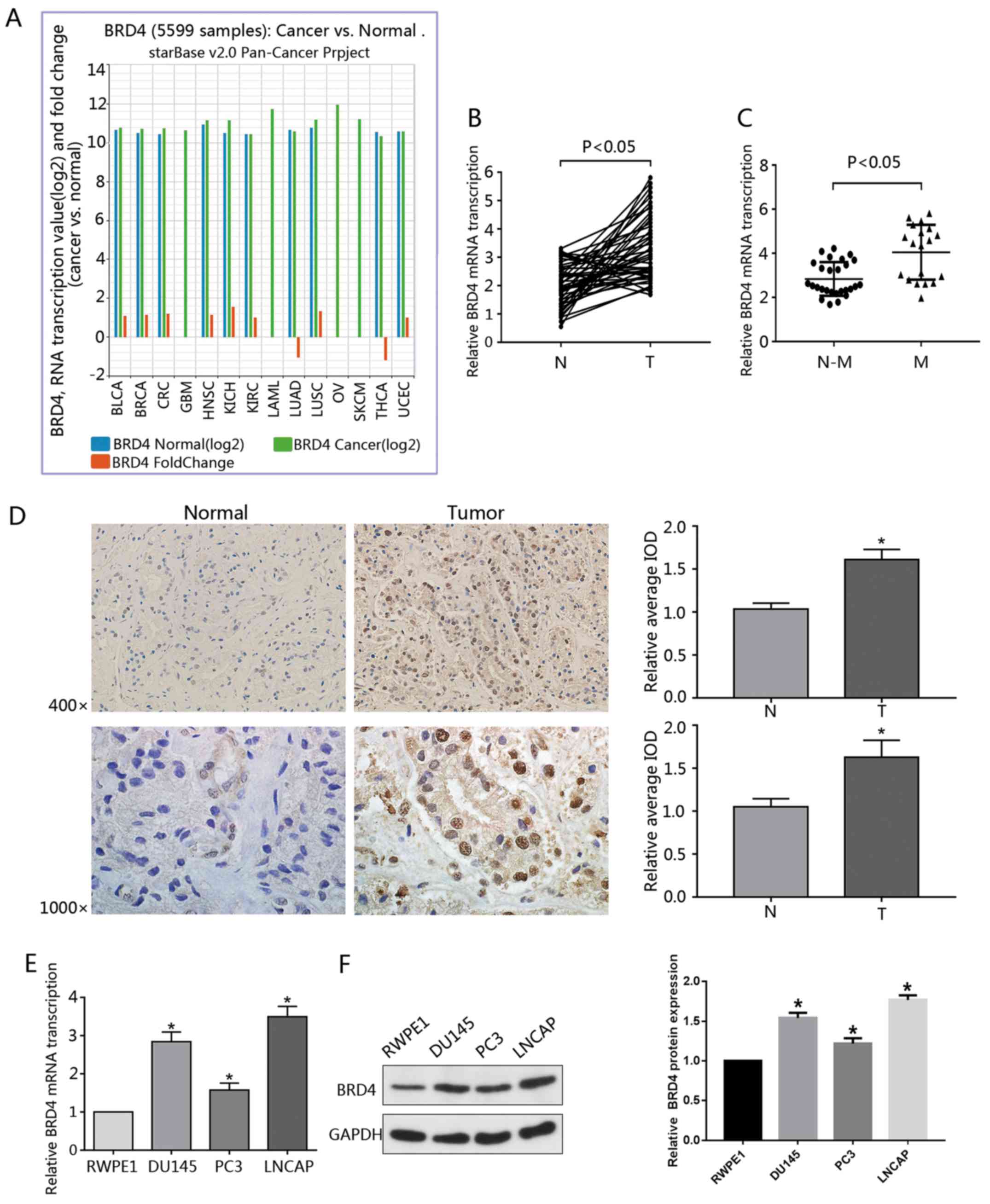 | Figure 1Overexpression of BRD4 in PCa cell
lines and tumor specimens. (A) Data regarding the expression of
BRD4 in 14 types of cancer was gathered from the Pan-Cancer
Analysis Platform in TCGA Data Portal (starBase version 2.0). (B)
RT-qPCR was used to analyze the transcription of BRD4 mRNA in human
cancerous and paired normal prostate tissues from 46 patients with
PCa. (C) Expression of BRD4 mRNA was significantly higher in
patients with M-PCa compared with N-M PCa specimens. (D)
Immunohistochemical analysis of BRD4 in PCa samples and surrounding
normal prostate tissue. Magnification, ×400 and ×1,000. The average
IOD was analyzed by Image-Pro Plus software. *P<0.05
vs. N prostate tissues. (E and F) mRNA and protein expression
levels of BRD4 in RWPE1 cells (human prostate epithelial cell
line), and DU145, PC3 and LNCAP cells (human prostate cancer cell
lines) were evaluated by RT-qPCR and western blotting.
*P<0.05 vs. RWPE1 cells. Data are presented as the
mean ± standard deviation. Experiments were performed in
triplicate. BRD4, bromodomain-containing protein 4; IOD, integrated
optical density; M, metastatic; N, normal; N-M, non-metastatic;
PCa, prostate cancer; RT-qPCR, reverse transcription-quantitative
polymerase chain reaction. |
BRD4 inhibition suppresses cellular
proliferation of PCa cell lines
To determine the effects of BRD4 on PCa cells,
BRD4-targeted shRNA was constructed and transduced into DU145 and
LNCAP cells. RT-qPCR and western blotting assays were used to
determine BRD4 expression in PCa cell lines. The results revealed
that BRD4 mRNA expression was significantly decreased in DU145 and
LNCAP cells compared with in the control, following transduction
with BRD4-targeted shRNA; the protein expression levels of BRD4
were notably downregulated in the BRD4 knockdown group compared
with in the control (Fig. 2A and
B). Cell proliferation was assessed via a CCK-8 assay. The
findings suggested that inhibition of BRD4 via shRNA significantly
attenuated cell proliferation of DU145 and LNCAP cells compared
with in the controls (Fig. 2C). In
addition, the present study reported that treatment with JQ1
decreased cell proliferation in dose-and time-dependent manners
(Fig. 2E). In addition, the
IC50 value of JQ1 in each cell line was determined
(Fig. 2D). The colony formation
capacity of DU145 and LNCAP cells treated with JQ1 or transfected
with shBRD4 was evaluated. The results revealed that BRD4
inhibition significantly suppressed colony formation compared with
in the negative control (Fig. 2F and
G). The findings of the present study suggested that inhibition
of BRD4 suppressed the proliferative ability of DU145 and LNCAP
cells.
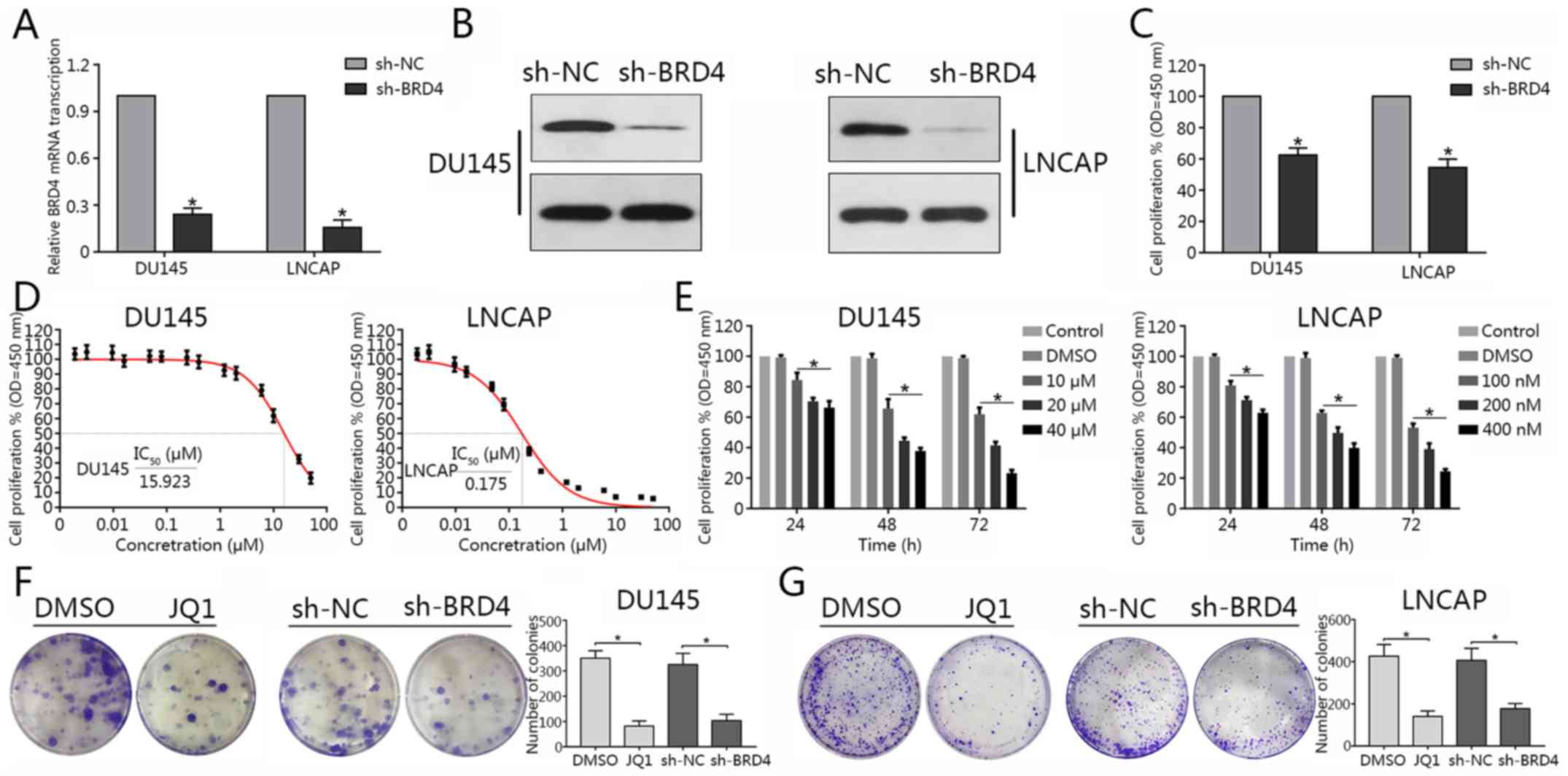 | Figure 2BRD4 inhibition suppresses cellular
proliferation of prostate cancer cell lines. (A and B) Reverse
transcription-quantitative polymerase chain reaction and western
blotting were conducted to determine transduction efficiency of
DU145 and LNCAP cells transduction with shBRD4. (C) Proliferative
ability of DU145 and LNCAP cells was assessed via a CCK-8 assay
following shBRD4 transduction. (D) After 72 h following JQ1
treatment (1.92, 3.2, 9.6, 16, 48, 80, 240 and 400 nM, and 1.2 2,
6, 10, 30 and 50 μM), the CCK-8 assay was performed to assess
cellular proliferation; and the corresponding IC50 values were
quantified. (E) Proliferation of DU145 and LNCAP cells following
treatment with different concentrations of JQ1 at 24, 48 and 72 h
was assessed via a CCK-8 assay. *P<0.05 vs. DMSO and
control group.. (F and G) DU145 and LNCAP cells were transduction
with shBRD4 or treated with JQ1 at their respective IC50
concentrations (15.9 µM for DU145, 175 nM for LNCAP) and
representative images of colony formation were obtained. The number
of colonies was subsequently calculated. Experiments were performed
in triplicate. *P<0.05. BRD4, bromodomain-containing
protein 4; CCK-8, cell counting kit-8; DMSO, dimethyl sulfoxide;
IC50, half-maximal inhibitory concentration; NC,
negative control; OD, optical density; sh, short hairpin RNA. |
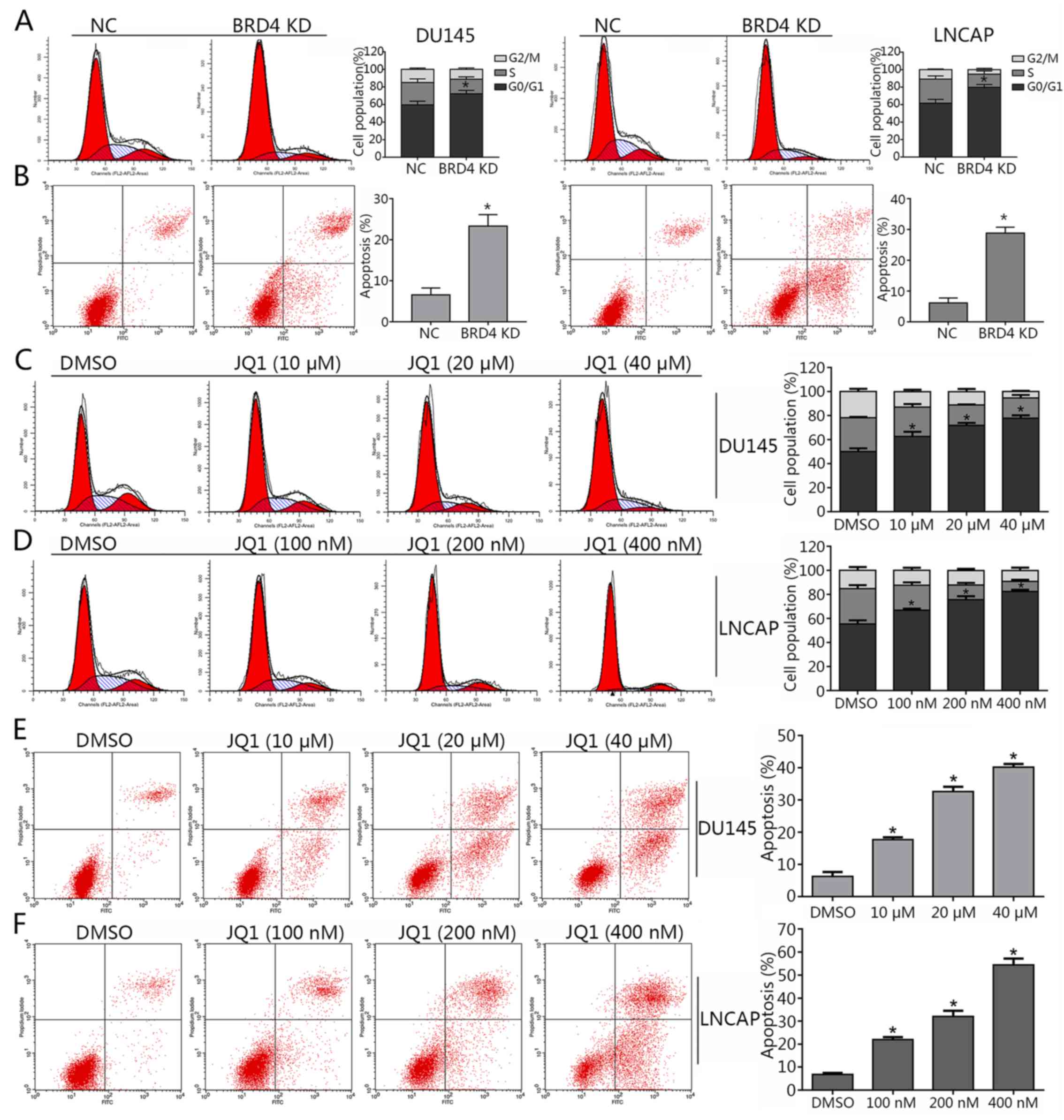
Inhibition of BRD4 induces cell cycle
arrest and apoptosis, and mitigates the invasion of PCa cell
lines
As BRD4 was suggested to exhibit pro-cancer effects
in PCa cells in (Figs. 1 and
2), the present study investigated
whether BRD4 inhibition may induce cell cycle arrest in DU145 and
LNCAP cells via JQ1 treatment or transduction with shRNA. The
results of cell cycle analysis revealed that the majority of
shBRD4-treated cells were in the G0/G1 phase; S and G2/M phase
cells were markedly less apparent compared with in the control
(Fig. 3A). In addition, the
apoptosis of cells in different groups was analyzed via flow
cytometry. The results demonstrated that BRD4 inhibition via shRNA
transduction significantly increased apoptosis compared with in the
control (Fig. 3B). Cells treated
with various concentrations of JQ1 exhibited a significant increase
in the number of cells in the G0/G1 phase compared with in the
control (Fig. 3C and D). The
apoptosis of JQ1-treated cells increased in a dose-dependent manner
compared with in the control (Fig. 3E
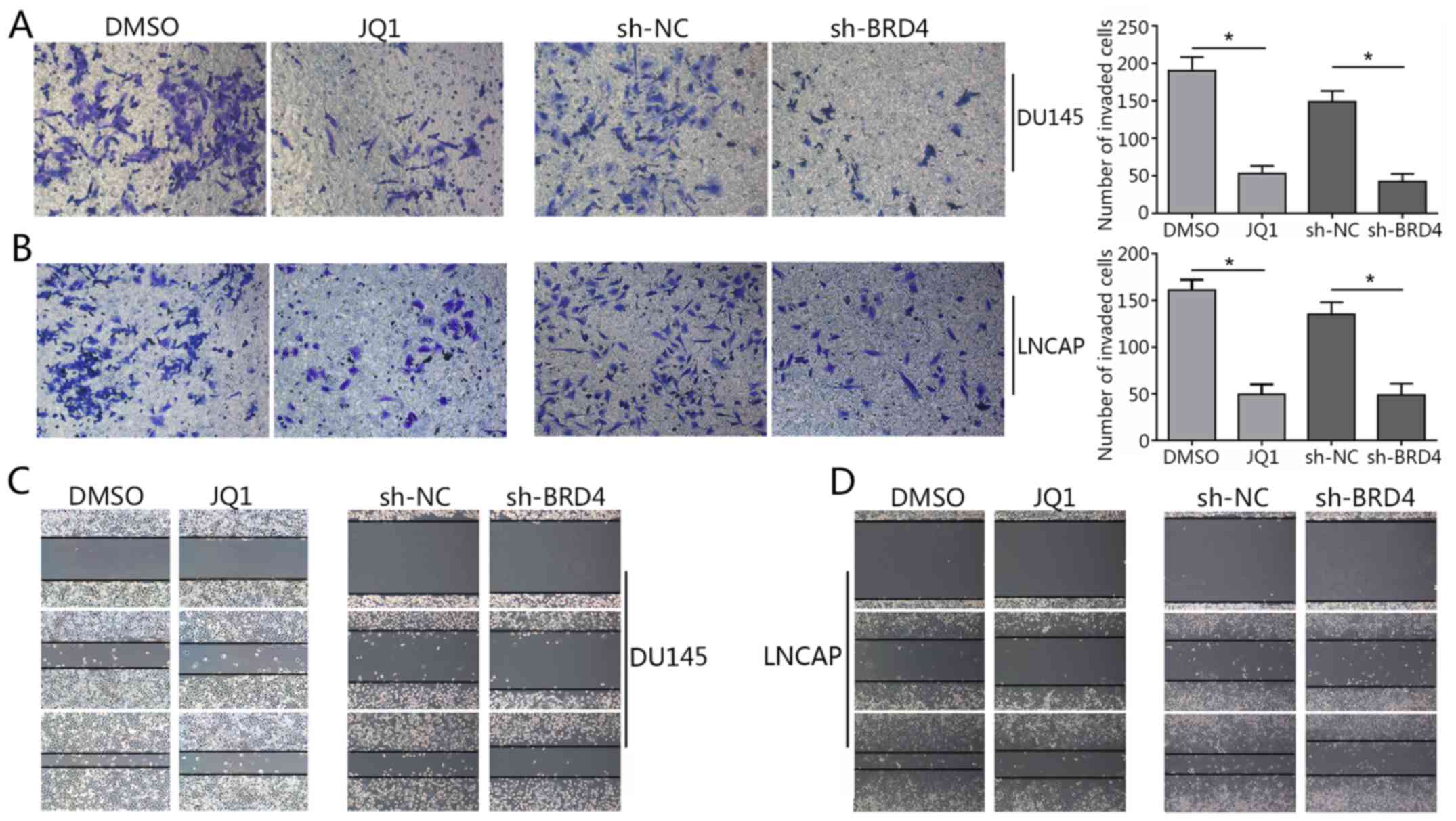
and F). The Transwell assay revealed a significant decrease in
invading cells treated with JQ1 or transfected with shBRD4 compared
with in the corresponding control (Fig. 4A and B). The wound healing assay
demonstrated that BRD4 inhibition mitigated cell migration in PCa
cells compared with in the corresponding controls (Fig. 4C and D). The findings of the
present study indicated that inhibition of BRD4 induced cell cycle
arrest and apoptosis, and mitigated the invasion of PCa cells.
Reduction of c-Myc expression by BRD4
inhibition involves p21
Recent studies have reported c-Myc to be a
downstream effector of BRD4 (19,26).
To determine whether c-Myc expression is affected by BRD4 in DU145
and LNCAP lines, these cells were transfected with shBRD4 or
treated with JQ1 in the present study. The results revealed that
BRD4 inhibition via JQ1 treatment or shBRD4 transduction
significantly decreased c-Myc mRNA expression levels compared with
in the control (Fig. 5A and B).
Additionally, as BRD4 inhibition was also reported to induce cell
cycle arrest and apoptosis (26,27).
Therefore, Bcl-2 was investigated in the present study. The results
revealed that the suppression of BRD4 significantly inhibited the
expression levels of Bcl-2 mRNA compared with in the control. Of
note, the present study observed that BRD4 inhibition via JQ1
treatment or shBRD4 transduction significantly promoted p21 mRNA
expression (Fig. 5).
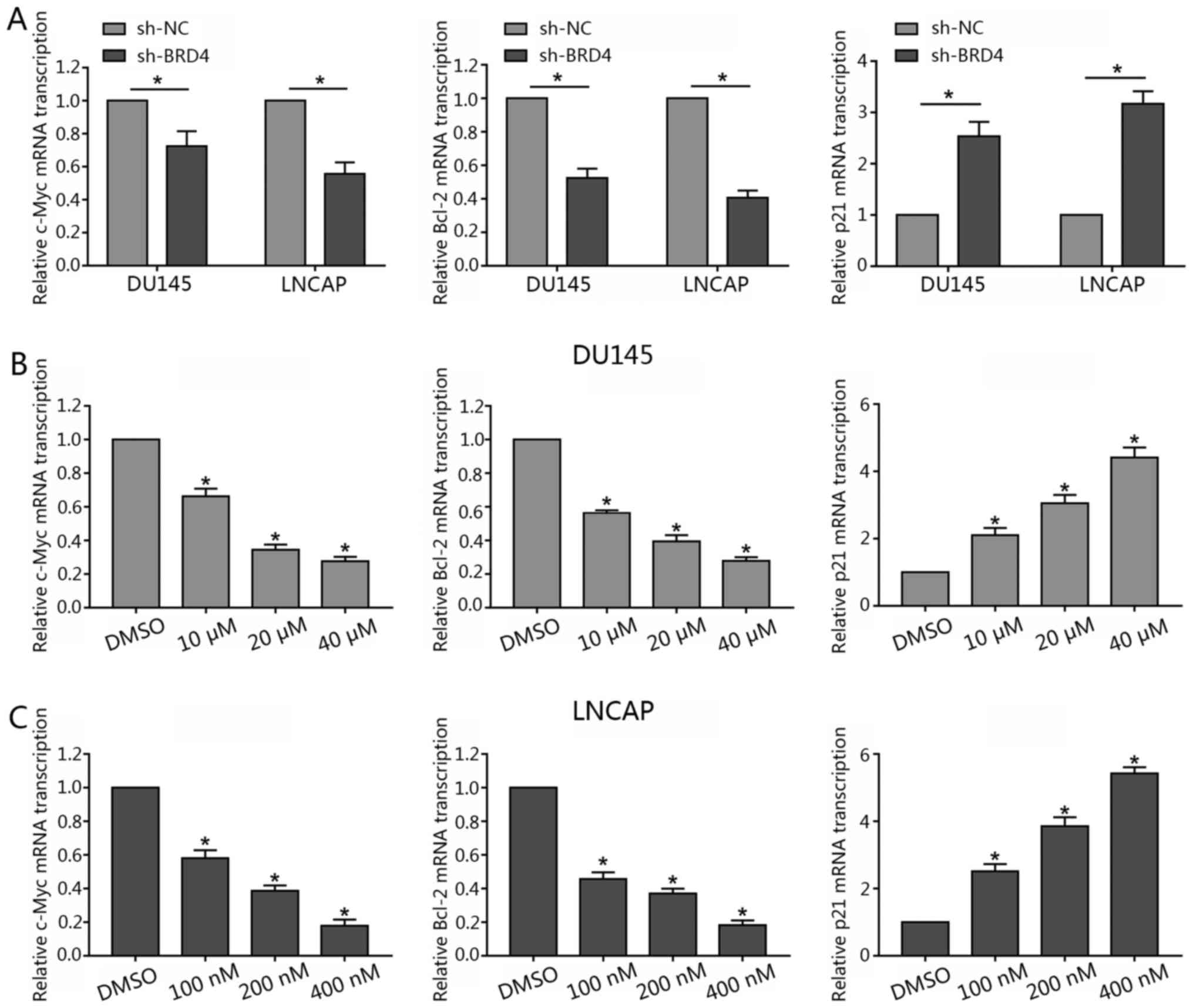 | Figure 5Inhibition of BRD4 decreases c-Myc
and Bcl-2 mRNA transcription, but increases p21 mRNA transcription
in prostate cancer lines. (A) DU145 and LNCAP cells were
transfected with either shBRD4 or NC shRNA, and the relative mRNA
transcription levels of c-Myc, Bcl-2 and p21 were analyzed by
RT-qPCR. (B and C) DU145 cells were treated either with JQ1 (10, 20
or 40 µM) or DMSO, and LNCAP were treated with either JQ1
(100, 200 or 400 nM) or DMSO for 48 h; the relative mRNA
transcription levels of c-Myc, Bcl-2 and p21 and were analyzed by
RT-qPCR. Data are presented as the mean ± standard deviation.
*P<0.05 vs. NC or DMSO group. BRD4,
bromodomain-containing protein 4; DMSO, dimethyl sulfoxide; NC,
negative control; RT-qPCR, reverse transcription-quantitative
polymerase chain reaction; sh, short hairpin RNA. |
Western blotting revealed that the expression levels
of p21 and cyclin D1 were notably increased in response to BRD4
inhibition; c-Myc and Bcl-2 protein expression levels were markedly
decreased compared with in the control (Fig. 6A). In addition, the expression
levels of p21 and cyclin D1 markedly increased in a dose-dependent
manner in response to JQ1 treatment; c-Myc and Bcl-2 protein
expression notably decreased in a dose-dependent manner (Fig. 6B).
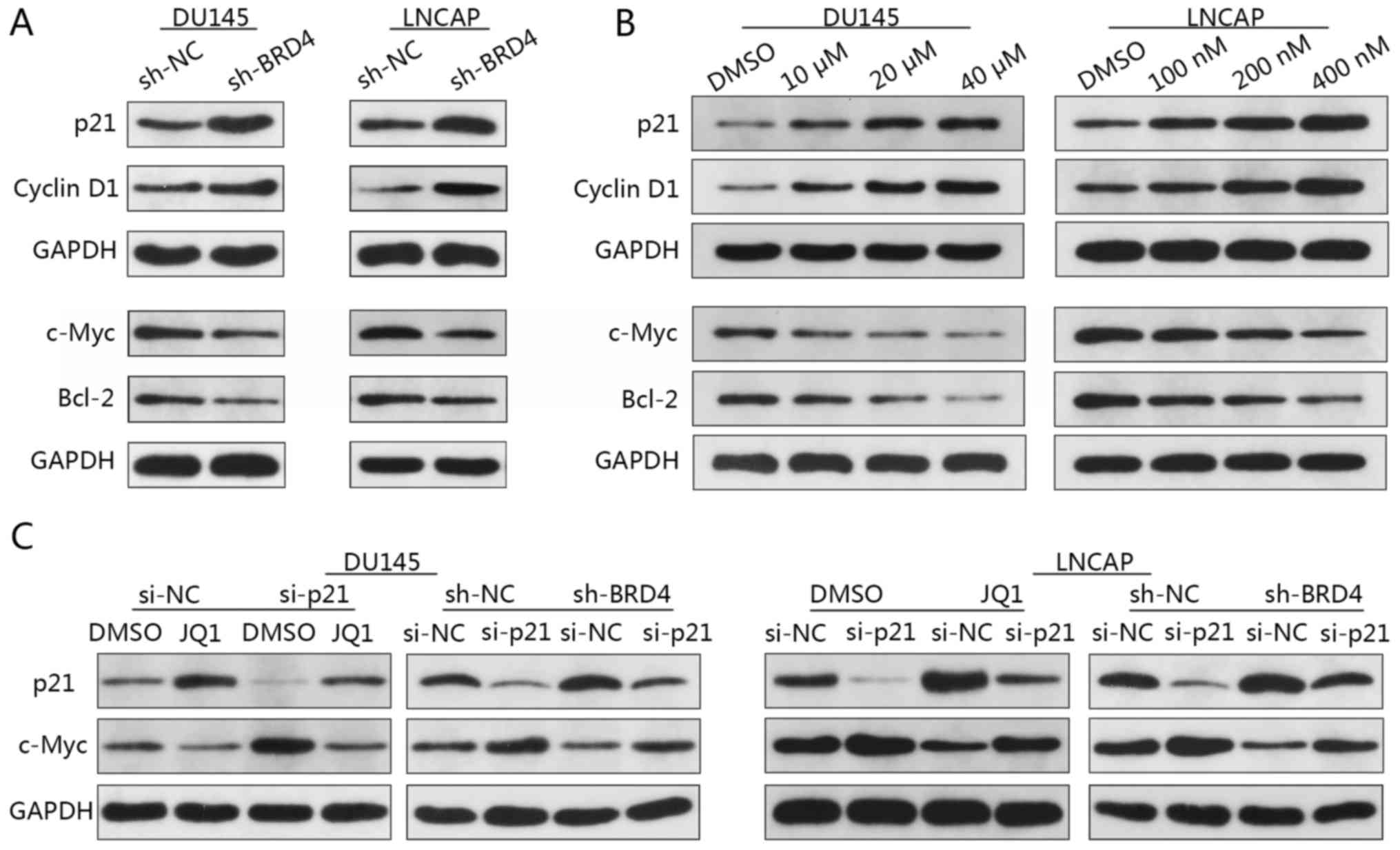 | Figure 6Reduction of c-Myc expression via
BRD4 inhibition involves p21. (A) DU145 and LNCAP cells were
transduced with shBRD4 or shRNA NC, and the relative expression
levels of c-Myc, Bcl-2, p21 and cyclin D1 were analyzed by western
blotting. (B) DU145 cells were treated either with JQ1 (10, 20 or
40 µM) or DMSO, and LNCAP cells were treated with JQ1 (100,
200 or 400 nM) or DMSO for 48 h. The relative expression levels of
c-Myc, Bcl-2, p21 and cyclin D1 were analyzed by western blotting.
(C) Relative p21 and c-Myc expression in DU145 and LNCAP cells.
DU145 cells were transfected with either p21 siRNA or NC siRNA, and
treated with JQ1 (15.9 µM) or DMSO; LNCAP cells transfected
with p21 siRNA or NC siRNA were treated with JQ1 (175 nM) or DMSO.
Bcl-2, B cell lymphoma-2; BRD4, bromodomain-containing protein 4;
DMSO, dimethyl sulfoxide; NC, negative control; sh, short hairpin
RNA; siRNA, small interfering RNA. |
Previous studies have reported that increased p21
and decreased c-Myc expression were concomitant upon BRD4
inhibition (28,29). To determine whether p21 and c-Myc
are associated in PCa cells, DU145 and LNCAP cells were firstly
transfected with p21-specific siRNA then followed by transfected
with sh-BRD4. Western blotting revealed increased c-Myc expression
when p21 was inhibited (Fig. 6C).
Additionally, a reduction in the expression of c-Myc via BRD4
inhibition was notably abrogated by the knockdown of p21 in DU145
and LNCAP cells compared with in the controls (Fig. 6C). These findings suggested c-Myc
to be a downstream target of p21 in PCa. To investigate whether
reductions in c-Myc expression are mediated via p21, DU145 and
LNCAP cells transfected with p21-specific siRNA then were treated
with JQ1. Western blotting demonstrated that treatment with JQ1
resulted in c-Myc downregulation, whereas p21 inhibition-induced
upregulation of c-Myc was abrogated by treatment with JQ1 (Fig. 6C). These findings suggested that
the reduction of c-Myc expression via BRD4 inhibition may involve
p21, and that p21 is an upstream regulator of c-Myc, which may be
directly affected by BRD4 inhibition.
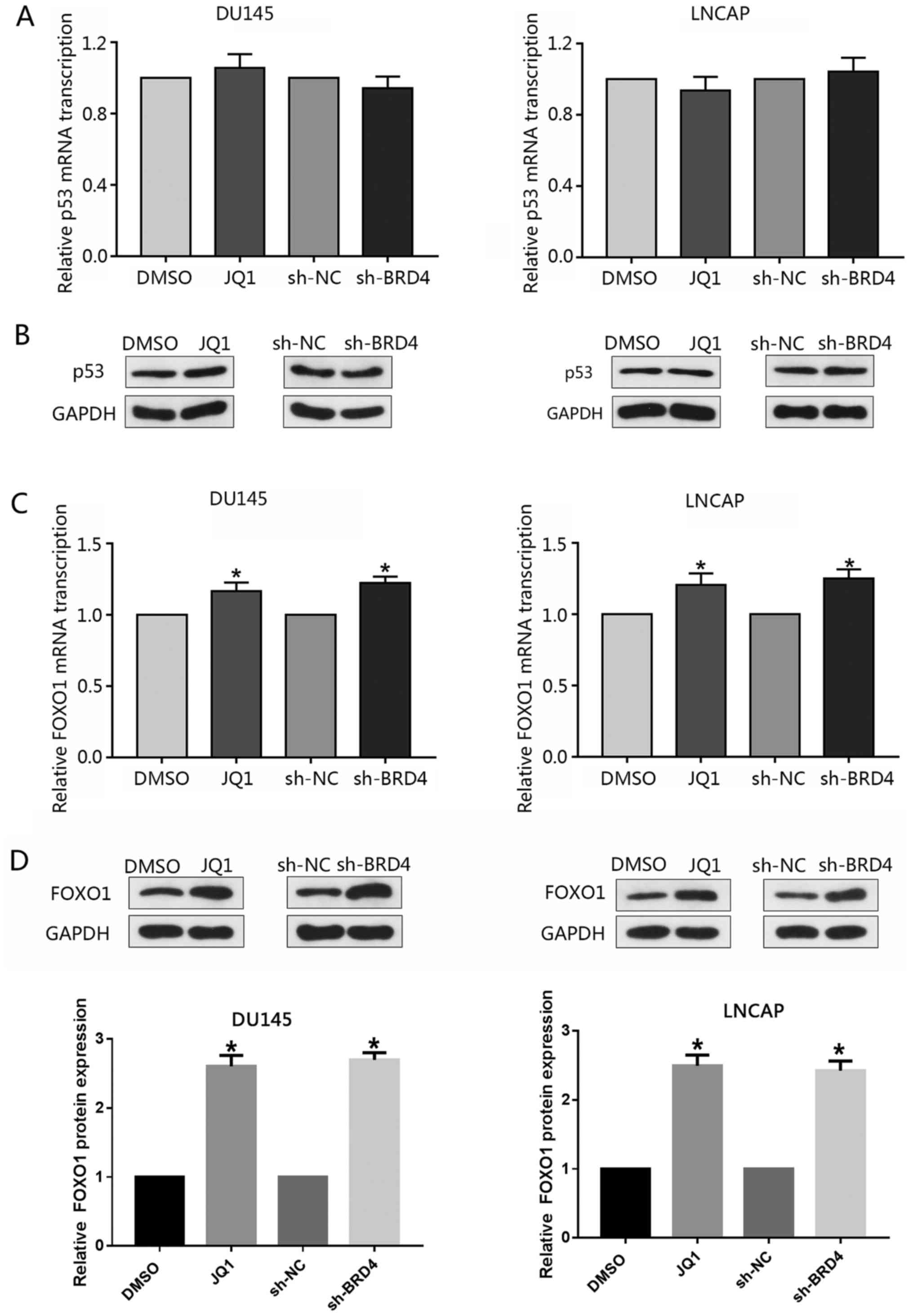
Induction of p21 via the inhibition of
BRD4 is mediated by FOXO1
The tumor-suppressor p53 is a well-reported upstream
regulator of p21 (30); however,
the present study investigated whether knockdown of BRD4 or
treatment with JQ1 may affect the expression p53. RT-qPCR or
western blotting did not reveal any notable alterations in the
expression levels of p53 following inhibition of BRD4 (Fig. 7A and B). Previous studies have
reported FOXO1 to inhibit PCa tumorigenesis and that FOXO1 binds to
the p21 promoter region resulting in cell cycle arrest (31,32).
Therefore, the present study investigated whether BRD4 inhibition
affected the expression of FOXO1 in PCa cells. The findings
demonstrated that inhibition of BRD4 only moderately increased
FOXO1 mRNA expression levels compared with in the control; the
expression levels of FOXO1 protein were signficantly increased in
response to BRD4 downregulation; similar findings were observed in
response to JQ1 treatment (Fig. 7C and
D). The present study proposed that the stability of the FOXO1
protein may be affected by post-transcriptional modifications, such
as post-BRD4 knock-down phosphorylation.
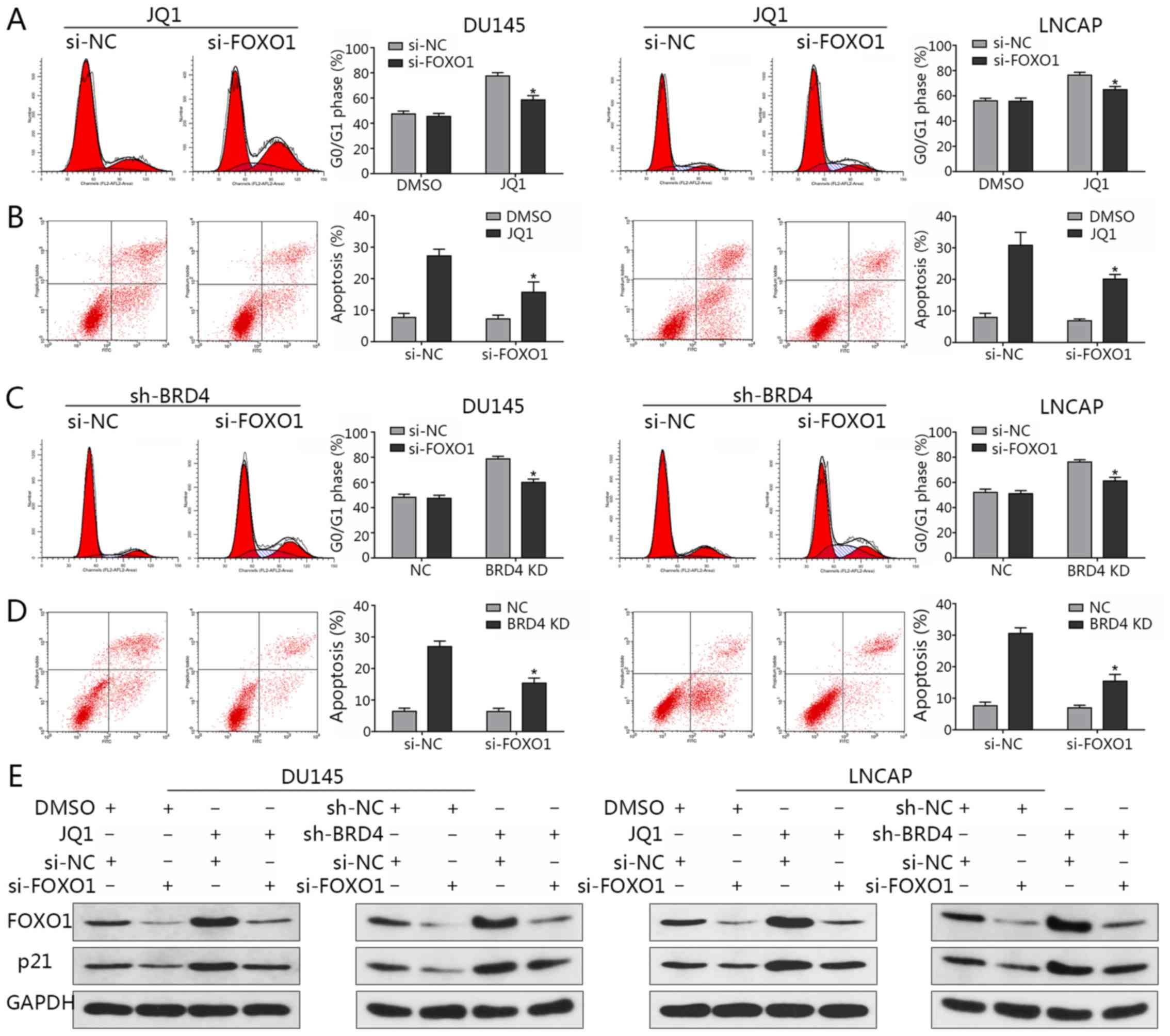
Following the knockdown of FOXO1 expression via
siRNA transfection, flow cytometry was applied to analyze the cell
cycle and evaluate the rates of apoptosis. The observations of the
present study revealed that the distribution of cells in the G0/G1
phase induced by JQ1 treatment were significantly decreased
compared with in the JQ1-treated control (Fig. 8A); the percentage of apoptosis was
significantly reduced when FOXO1 was downregulated compared with in
the JQ1-treated control (Fig. 8B).
Similar findings were observed in the presence of BRD4 inhibition
via shBRD4 transduction (Fig. 8C and
D). In addition, the results indicated that FOXO1
downregulation led to a reduction in p21 expression, while an
induction of p21 expression via BRD4 inhibition was abrogated by
the knockdown of FOXO1 (Fig. 8E).
This in turn suggested that FOXO1 may be a transcription factor
that induces p21 when BRD4 is suppressed; the induction of p21 by
JQ1 treatment was also abrogated by FOXO1 inhibition (Fig. 8E). The results indicated that the
induction of p21 by BRD4 inhibition may not be mediated by p53, but
by FOXO1.
Knockdown of BRD4 delays tumor growth in
PCa mouse models
DU145 cells were subcutaneously injected into male
nude mice. When tumor sizes were palpable, mice were randomized
into JQ1 treatment or NC groups. JQ1-treated mice exhibited a
significant reduction in tumor volumes compared with in the control
(Fig. 9A). To study effects of
BRD4 knockdown, stable LNCAP cells transfected with shBRD4 or
control were injected into mice. Nude mice injected with
shBRD4-transfected cells exhibited decreased tumor volumes and
weights compared to the control group; a significant decrease in
tumor weights was observed in the JQ1 treatment group and
shBRD4-transfected cells compared with in the control (Fig. 9B and C). In addition, metastatic
lymph nodes or distal invasion were not detected in any of the
groups at the termination of the experiment. Western blotting
demonstrated that FOXO1 and p21 expression levels were markedly
upregulated, while that of c-Myc were downregulated after BRD4
inhibition (Fig. 9D).
Immunohistochemical staining revealed the expression of Ki-67 in
the JQ1 treatment and shBRD4 groups to be significantly lower than
that of the control group, while FOXO1 and p21 expression were
significantly increased compared with the control group (Fig. 9E and F). These findings were
consistent with the results of in vitro analysis in the
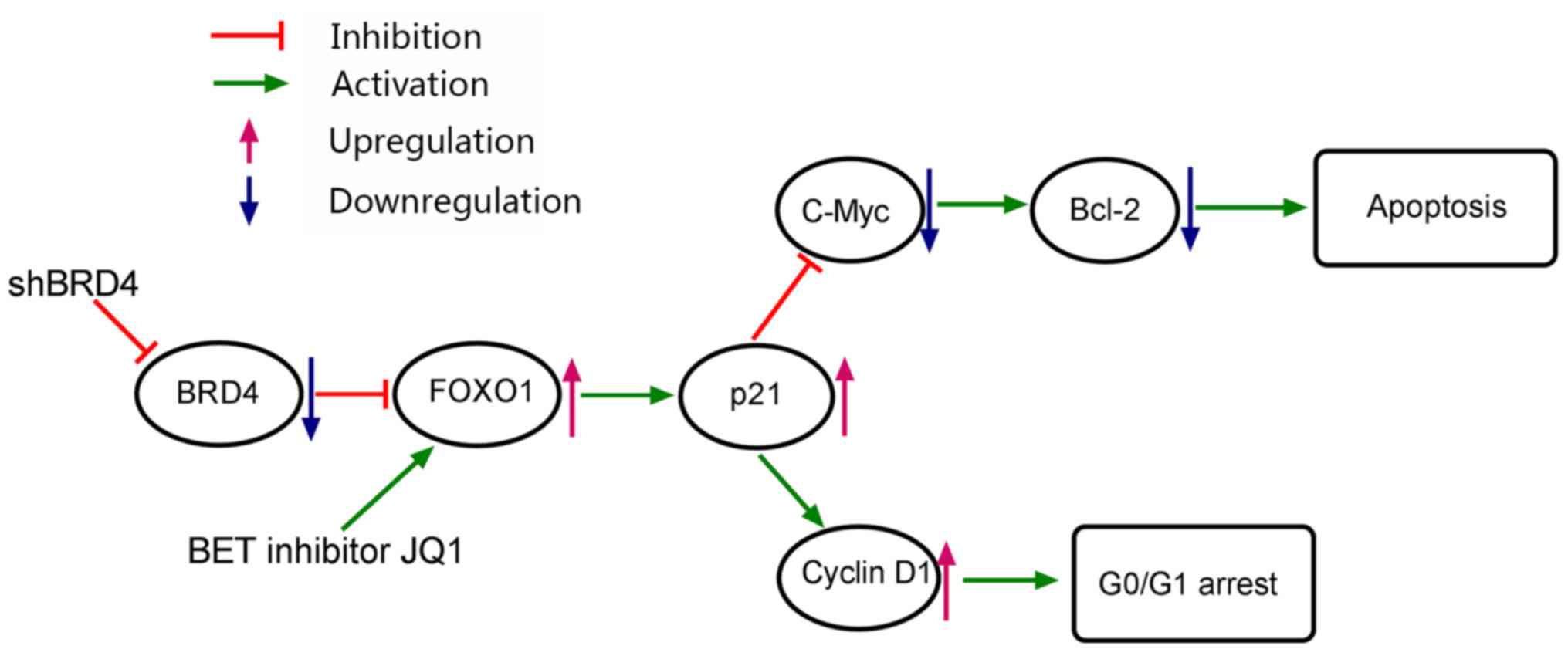
present study. Finally, a working model to demonstrate the possible
mechanism of cell cycle arrest and apoptosis in PCa cells as
induced by BRD4 inhibition was generated (Fig. 10).
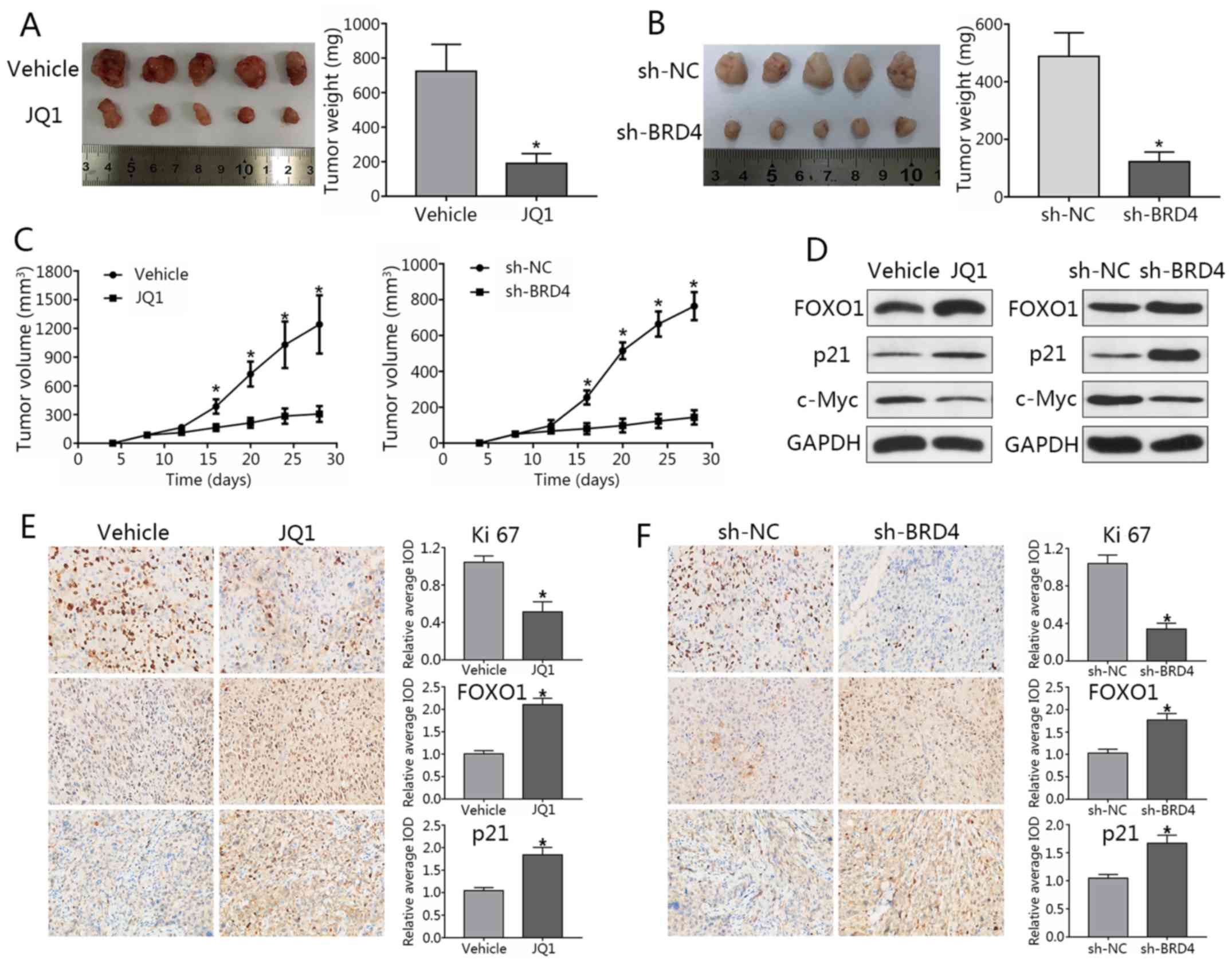 | Figure 9Knockdown of BRD4 delays tumor growth
in prostate cancer mouse models. (A) Image of tumors collected from
mice. Mice were treated with JQ1 or vehicle at day 9 post-seeding.
Four weeks later, mice were sacrificed, and tumors were excised.
Weights of tumors grown in mice were assessed and analyzed.
*P<0.05 vs. NC group. (B) Stable LNCAP cells
transduced with shBRD4 or negative control were implanted into
mice. (C) Mouse tumor volume curve as in response to JQ1 treatment
or shBRD4 transduction. *P<0.05 vs. NC group. (D)
Expression of FOXO1, p21 and c-Myc in xenograft tumors was assessed
by western blotting. (E and F) Immunohistochemical analyses of
Ki-67, FOXO1, and p21 in xenograft specimens. *P<0.05
vs. NC. The average IOD was analyzed by Image-Pro Plus software.
Magnification, ×400. Data are presented as the mean ± standard
deviation. BRD4, bromodomain-containing protein 4; FOXO1, forkhead
box protein O1; IOD, integrated optical density; NC, negative
control; si, small interfering RNA; sh, short hairpin RNA. |
Discussion
The prevention of PCa progression remains difficult
to achieve; the targeting of androgen receptor signaling remains
the treatment of choice in advanced stages of this disease
(33). Enzalutamide, the novel
nonsteroidal androgen receptor inhibitor, has been approved for the
treatment of patients with castrate-resistant PCa at present
(34,35). Unfortunately, the efficacy of
enzalutamide is also limited. Several studies have reported that
dysregulation of BRD4 markedly influences tumor growth and
progression (18,36); the biological functions of BRD4 in
PCa require further investigation for the development of potential
therapeutic strategies.
Aberrant expression of BRD4 was confirmed in
numerous types of cancers (11,36).
For example, the expression of BRD4 was observed to be upregulated
in kidney cancer and exerted a pro-oncogenic function in this
particular disease (11). In
squamous carcinoma of the skin, BRD4 was reported to be upregulated
compared with normal skin keratinocytes and fibroblasts, with
modeled overexpression of BRD4 promoting cell proliferation
(36). In the present study, the
expression and roles served by BRD4 in PCa were determined. In
accordance with previous findings, the present study revealed that
BRD4 expression was significantly increased in PCa samples compared
with in adjacent normal prostate tissue (20). In addition, high levels of BRD4
expression were positively associated with clinical stage and
metastasis in the present study. These findings indicated that BRD4
protein may be closely associated with the initiation of PCa and
exerts cancer-promoting functions in PCa. Inhibition of BRD4 may
therefore become a novel therapeutic strategy in the management of
this disease.
The present study reported that inhibition of BRD4,
via shRNA transduction or JQ1 treatment, decreased cell
proliferation, promoted cell cycle arrest and induced the apoptosis
of PCa cells; BRD4 inhibition also impaired tumor growth in mice.
Treatment with JQ1 or knockdown of BRD4 significantly inhibited
cell invasion and migration abilities of PCa cells. Therefore,
knockdown of BRD4 may be an additional therapeutic approach for
patients with PCa.
A previous study reported JQ1 to disrupt the
interactions between BRD4 and the N-terminal domain of androgen
receptors (21); however, the
molecular mechanisms underlying the anticancer effects of BRD4
inhibition remain unknown. BRD4 has been observed to specifically
regulate a series of genes, including c-Myc; c-Myc, in turn,
regulates numerous cellular functions, including cell growth,
survival and apoptosis (37).
Overexpression of c-Myc has also been detected in numerous types of
cancer, such as PCa (38).
Downregulation of c-Myc expression resulted in decreased cell
viability or apoptosis, via the reduction of Bcl-extra large and
Bcl-2 expression (39). The
present study reported that BRD4 inhibition suppressed PCa cell
proliferation by decreasing the expression of c-Myc, in
vitro and in vivo, and that reductions c-Myc expression
were accompanied with the downregulation of Bcl-2. The results of
the present study suggested that BRD4 inhibition may be associated
with the downregulation of Bcl-2.
Induction of p21 by BRD4 inhibition has been
demonstrated in other cancer cell types, including thyroid cancer
and glioblastoma cell lines (27,40),
with p21 serving as an important downstream gene affected by JQ1
treatment (27,28,40).
p21, a member the CIP/kip family of cyclin-dependent kinase
inhibitors, serves a major role not only as a regulator of the
cell-cycle, but also as a mediator of apoptosis and transcription
(41). In addition, p21 directly
interacts with and induces cyclin D1 associated-CDK4 and CDK6
expression, thereby inhibiting G1/S progression (42). In the present study, it was
reported that following BRD4 inhibition, p21 and cyclin D1
expression levels were significantly increased in PCa cells, which
indicated a mechanism by which JQ1 inhibits cell growth by
suppressing cell cycle progression.
Recent studies have reported c-Myc to be critical in
the suppression of p21 transcription (41,43).
To determine whether c-Myc expression is associated with p21 levels
following BRD4 inhibition, the expression of p21 was downregulated
in the present study, which suggested p21 to regulate the
expression of c-Myc. In addition, p21 was proposed to be the
upstream regulator of c-Myc and may be directly affected by BRD4
inhibition in the present study. A recent study indicated p21 as a
critical downstream effector of wildtype p53 (30); however, the present study reported
that the expression levels of p53 protein were notably altered in
response to BRD4 inhibition, which may be due to DU145 and LNCAP
cell lines harboring a mutant p53; however, further investigation
is required.
FOXO1, an important member of the FOX transcription
factor family, was observed to suppress tumor formation by
activating numerous target genes (31). Imatinib, a tyrosine kinase
inhibitor, upregulated the expression of FOXO1, and induced cell
cycle arrest and apoptosis in chronic myelogenous leukemia
(44). Similarly, the present
study reported that knockdown of BRD4 induced FOXO1 expression and
demonstrated that FOXO1 is required for the regulation of cell
apoptosis and cycle arrest. Additionally, FOXO1 was determined to
be the upstream regulator of p21 and was directly influenced by
BRD4 inhibition in the present study. BRD4 regulates gene
expression (18); however, the
mechanism underlying the inhibition of BRD4 upregulates FOXO1
remains unclear. In the present study, FOXO1 transcriptional levels
were only moderately increased by BRD4 inhibition, while that of
FOXO1 protein expression were notably increased. The present study
proposed that the stability of the FOXO1 protein may be affected by
post-transcriptional modifications, such as post-BRD4 knockdown
phosphorylation; however, further investigation is required to
elucidate these interactions. The findings of the present study
suggested that BRD4 serves a critical role in the initiation of PCa
and that the bromodomain inhibitor JQ1 should be further
investigated as a potential therapeutic agent in this disease. In
conclusion, the present study determined BRD4 to be significantly
increased in PCa tissue specimens and that BRD4 may possess an
important role in regulating the pathogenesis of PCa. In addition,
it was reported that knockdown of BRD4, via shRNA transduction or
JQ1 treatment, suppressed PCa proliferation in vitro and
in vivo. This effect may be attributable, at least in part,
to the induction of G0/G1 cell cycle arrest and apoptosis resulting
from the upregulation of FOXO1 and p21, and downregulation of c-Myc
and Bcl-2. To the best of our knowledge, the present study is the
first to report of the molecular mechanisms underlying the
pathogenesis of PCa and these findings may contribute to the
development of potential therapeutic strategies for the management
of this disease in the future.
Acknowledgments
Not applicable.
Funding
This study was supported by Application and Basic
Research Project Of Wuhan City (grant no. 2015060101010049), Wuhan
Morning Light Plan of Youth Science and Technology (grant no.
2017050304010281), Hubei Province Health and Family. Planning
Scientific Research Project (grant nos. WJ2017M025 and WJ2017Z005),
Natural Science Foundation of Hubei Province (grant nos. 2016CFB114
and 2017CFB181) and Research Project of Wuhan University (grant no.
2042017kf0097).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request
Authors' contributions
YT conceived and designed this study. YT, XL and LW
wrote and revised the manuscript. YT, LW, YD, ZC, XW, JG and MW
conducted the experiments and analyzed the data. YT, LW, HC and XW
collected the clinical samples. XL supervised the entire experiment
project. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Renmin Hospital of Wuhan University (Wuhan,
China). Informed consent was obtained from all participants.
Patient consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
3
|
Zhu X, Albertsen PC, Andriole GL, Roobol
MJ, Schroder FH and Vickers AJ: Risk-based prostate cancer
screening. Eur Urol. 61:652–661. 2012. View Article : Google Scholar
|
|
4
|
Attard G, Parker C, Eeles RA, Schroder F,
Tomlins SA, Tannock I, Drake CG and de Bono JS: Prostate cancer.
Lancet. 387:70–82. 2016. View Article : Google Scholar
|
|
5
|
Mullins JK, Feng Z, Trock BJ, Epstein JI,
Walsh PC and Loeb S: The impact of anatomical radical retropubic
prostatectomy on cancer control: The 30-year anniversary. J Urol.
188:2219–2224. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wilt TJ, Brawer MK, Jones KM, Barry MJ,
Aronson WJ, Fox S, Gingrich JR, Wei JT, Gilhooly P, Grob BM, et al:
Radical prostatectomy versus observation for localized prostate
cancer. N Engl J Med. 367:203–213. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jung M, Gelato KA, Fernandez-Montalvan A,
Siegel S and Haendler B: Targeting BET bromodomains for cancer
treatment. Epigenomics-Uk. 7:487–501. 2015. View Article : Google Scholar
|
|
8
|
Meloche J, Potus F, Vaillancourt M,
Bourgeois A, Johnson I, Deschamps L, Chabot S, Ruffenach G, Henry
S, Breuils-Bonnet S, et al: Bromodomain-Containing protein 4: The
epigenetic origin of pulmonary arterial hypertension. Circ Res.
117:525–535. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen R, Yik JH, Lew QJ and Chao SH: Brd4
and HEXIM1: Multiple roles in P-TEFb regulation and cancer. Biomed
Res Int. 2014:2328702014.PubMed/NCBI
|
|
10
|
Noguchi-Yachide T: BET bromodomain as a
target of epigenetic therapy. Chem Pharm Bull (Tokyo). 64:540–547.
2016. View Article : Google Scholar
|
|
11
|
Wu X, Liu D, Gao X, Xie F, Tao D, Xiao X,
Wang L, Jiang G and Zeng F: Inhibition of BRD4 suppresses cell
proliferation and induces apoptosis in renal cell carcinoma. Cell
Physiol Biochem. 41:1947–1956. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
French C: NUT midline carcinoma. Nat Rev
Cancer. 14:149–150. 2014. View Article : Google Scholar
|
|
13
|
Andrieu G, Tran AH, Strissel KJ and Denis
GV: BRD4 regulates breast cancer dissemination through
Jagged1/Notch1 signaling. Cancer Res. 76:6555–6567. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liao YF, Wu YB, Long X, Zhu SQ, Jin C, Xu
JJ and Ding JY: High level of BRD4 promotes non-small cell lung
cancer progression. Oncotarget. 7:9491–9500. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ferri E, Petosa C and McKenna CE:
Bromodomains: Structure, function and pharmacology of inhibition.
Biochem Pharmacol. 106:1–18. 2016. View Article : Google Scholar
|
|
16
|
Filippakopoulos P and Knapp S: Targeting
bromodomains: Epigenetic readers of lysine acetylation. Nat Rev
Drug Discov. 13:337–356. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sahai V, Redig AJ, Collier KA, Eckerdt FD
and Munshi HG: Targeting BET bromodomain proteins in solid tumors.
Oncotarget. 7:53997–54009. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li GQ, Guo WZ, Zhang Y, Seng JJ, Zhang HP,
Ma XX, Zhang G, Li J, Yan B, Tang HW, et al: Suppression of BRD4
inhibits human hepatocellular carcinoma by repressing MYC and
enhancing BIM expression. Oncotarget. 7:2462–2474. 2016.
|
|
19
|
Togel L, Nightingale R, Chueh AC,
Jayachandran A, Tran H, Phesse T, Wu R, Sieber OM, Arango D,
Dhillon AS, et al: Dual targeting of bromodomain and extraterminal
domain proteins, and WNT or MAPK signaling, inhibits c-MYC
expression and proliferation of colorectal cancer cells. Mol Cancer
Ther. 15:1217–1226. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Urbanucci A, Barfeld SJ, Kytola V, Itkonen
HM, Coleman IM, Vodak D, Sjoblom L, Sheng X, Tolonen T, Minner S,
et al: Androgen receptor deregulation drives bromodomain-mediated
chromatin alterations in prostate cancer. Cell Rep. 19:2045–2059.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Asangani IA, Dommeti VL, Wang X, Malik R,
Cieslik M, Yang R, Escara-Wilke J, Wilder-Romans K, Dhanireddy S,
Engelke C, et al: Therapeutic targeting of BET bromodomain proteins
in castration-resistant prostate cancer. Nature. 510:278–282. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mohler JL, Armstrong AJ, Bahnson RR,
D'Amico AV, Davis BJ, Eastham JA, Enke CA, Farrington TA, Higano
CS, Horwitz EM, et al: Prostate Cancer, Version 1.2016. J Natl
Compr Canc Netw. 14:19–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li JH, Liu S, Zhou H, Qu LH and Yang JH:
StarBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA
interaction networks from large-scale CLIP-Seq data. Nucleic Acids
Res. 42:D92–D97. 2014. View Article : Google Scholar
|
|
24
|
Yang JH, Li JH, Shao P, Zhou H, Chen YQ
and Qu LH: StarBase: A database for exploring microRNA-mRNA
interaction maps from Argonaute CLIP-Seq and Degradome-Seq data.
Nucleic Acids Res. 39:D202–D209. 2011. View Article : Google Scholar
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
26
|
Wu X, Liu D, Tao D, Xiang W, Xiao X, Wang
M, Wang M, Luo L, Li G, Zeng YF, et al: BRD4 regulates EZH2
transcription through upregulation of C-MYC and represents a novel
therapeutic target in bladder cancer. Mol Cancer Ther.
15:1029–1042. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cheng Z, Gong Y, Ma Y, Lu K, Lu X, Pierce
LA, Thompson RC, Muller S, Knapp S and Wang J: Inhibition of BET
bromodomain targets genetically diverse glioblastoma. Clin Cancer
Res. 19:1748–1759. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee DH, Qi J, Bradner JE, Said JW, Doan
NB, Forscher C, Yang H and Koeffler HP: Synergistic effect of JQ1
and rapamycin for treatment of human osteosarcoma. Int J Cancer.
136:2055–2064. 2015. View Article : Google Scholar
|
|
29
|
Kumar K, Raza SS, Knab LM, Chow CR, Kwok
B, Bentrem DJ, Popovic R, Ebine K, Licht JD and Munshi HG:
GLI2-dependent c-MYC upregulation mediates resistance of pancreatic
cancer cells to the BET bromodomain inhibitor JQ1. Sci Rep.
5:94892015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Georgakilas AG, Martin OA and Bonner WM:
P21: A two-faced genome guardian. Trends Mol Med. 23:310–319. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang Y, Blee AM, Wang D, An J, Pan Y, Yan
Y, Ma T, He Y, Dugdale J, Hou X, et al: Loss of FOXO1 cooperates
with TMPRSS2-ERG overexpression to promote prostate tumorigenesis
and cell invasion. Cancer Res. 77:6524–6537. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tinkum KL, White LS, Marpegan L, Herzog E,
Piwnica-Worms D and Piwnica-Worms H: Forkhead box O1 (FOXO1)
protein, but not p53, contributes to robust induction of p21
expression in fasted mice. J Biol Chem. 288:27999–28008. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Litwin MS and Tan HJ: The diagnosis and
treatment of prostate cancer: A review. JAMA. 317:2532–2542. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Basch E, Loblaw DA, Oliver TK, Carducci M,
Chen RC, Frame JN, Garrels K, Hotte S, Kattan MW, Raghavan D, et
al: Systemic therapy in men with metastatic castration-resistant
prostate cancer: American Society of Clinical Oncology and Cancer
Care Ontario clinical practice guideline. J Clin Oncol.
32:3436–3448. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ning YM, Brave M, Maher VE, Zhang L, Tang
S, Sridhara -R, Kim G, Ibrahim A and Pazdur R: U.S. Food and drug
administration approval summary: Enzalutamide for the treatment of
patients with chemotherapy-naive metastatic castration-resistant
prostate cancer. Oncologist. 20:960–966. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xiang T, Bai JY, She C, Yu DJ, Zhou XZ and
Zhao TL: Bromodomain protein BRD4 promotes cell proliferation in
skin squamous cell carcinoma. Cell Signal. 42:106–113. 2018.
View Article : Google Scholar
|
|
37
|
Pan J, Deng Q, Jiang C, Wang X, Niu T, Li
H, Chen T, Jin J, Pan W, Cai X, et al: USP37 directly
deubiquitinates and stabilizes c-Myc in lung cancer. Oncogene.
34:3957–3967. 2015. View Article : Google Scholar
|
|
38
|
Rebello RJ, Pearson RB, Hannan RD and
Furic L: Therapeutic approaches targeting MYC-Driven prostate
cancer. Genes (Basel). 8. pp. E712017, View Article : Google Scholar
|
|
39
|
Kelly PN, Grabow S, Delbridge AR, Strasser
A and Adams JM: Endogenous Bcl-xL is essential for Myc-driven
lymphoma-genesis in mice. Blood. 118:6380–6386. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gao X, Wu X, Zhang X, Hua W and Zhang Y,
Maimaiti Y, Gao Z and Zhang Y: Inhibition of BRD4 suppresses tumor
growth and enhances iodine uptake in thyroid cancer. Biochem
Biophys Res Commun. 469:679–685. 2016. View Article : Google Scholar
|
|
41
|
Karimian A, Ahmadi Y and Yousefi B:
Multiple functions of p21 in cell cycle, apoptosis and
transcriptional regulation after DNA damage. DNA Repair (Amst).
42:63–71. 2016. View Article : Google Scholar
|
|
42
|
Hydbring P, Malumbres M and Sicinski P:
Non-canonical functions of cell cycle cyclins and cyclin-dependent
kinases. Nat Rev Mol Cell Biol. 17:280–292. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Delbridge AR, Grabow S, Bouillet P, Adams
JM and Strasser A: Functional antagonism between pro-apoptotic BIM
and anti-apoptotic BCL-XL in MYC-induced lymphomagenesis. Oncogene.
34:1872–1876. 2015. View Article : Google Scholar
|
|
44
|
Pellicano F, Scott MT, Helgason GV,
Hopcroft LE, Allan EK, Aspinall-O'Dea M, Copland M, Pierce A,
Huntly BJ, Whetton AD, et al: The antiproliferative activity of
kinase inhibitors in chronic myeloid leukemia cells is mediated by
FOXO transcription factors. Stem Cells. 32:2324–2337. 2014.
View Article : Google Scholar : PubMed/NCBI
|