Introduction
Gastric cancer (GC) is currently the fourth most
common type of cancer, with the second highest mortality rate
worldwide (1,2). It is estimated that ~1,000,000 new GC
cases are diagnosed and ~738,000 patients succumb to GC annually
worldwide (3). Despite the
significant advances in the diagnosis, prevention and treatment of
GC, its prognosis remains poor, with an overall 5-year survival
rate of ~20% (2).
Systemic chemotherapy is a relatively effective
therapeutic approach to patients with advanced and relapsed GC,
which account for 80-90% of all GC patients (4). Innate or acquired chemoresistance
represents a major challenge for the treatment of GC (5,6).
Cisplatin (CDDP), a known antitumor drug, has been widely used for
the treatment of multiple malignancies, such as ovarian and lung
cancer (7). Furthermore, CDDP in
combination with other drugs (e.g., cetuximab, capecitabine,
trastuzumab and sunitinib) has been applied to clinical trials of
GC (8). Autophagy, a
‘self-digestion’ process, degrades and catabolizes
unnecessary/excessive proteins and aged/damaged intracellular
organelles to maintain/restore metabolic homeostasis (9). Prior studies also demonstrated that
autophagy plays a potential oncogenic or tumor-suppressive role in
the development of GC (10).
Furthermore, autophagy was found to be closely associated with drug
resistance in cancer therapy (11,12).
Additionally, CDDP treatment has been shown to improve autophagic
activity in certain tumors (e.g., cervical, breast, liver and
anaplastic thyroid cancer) (13,14).
Long non-coding RNAs (lncRNAs), a group of
transcripts longer than 200 nucleotides (nt) without protein-coding
potential, have been identified as positive or negative regulators
of autophagy and chemoresistance in cancer (15,16).
LncRNA metastasis-associated lung adenocarcinoma transcript 1
(MALAT1), also referred to as nuclear-enriched abundant transcript
2 (NEAT2), has been reported to be an oncogene in several cancers,
including lung cancer and GC (17,18).
Moreover, earlier studies revealed that MALAT1 may enhance
chemoresistance of cancer cells by promoting autophagy (19,20).
The aim of the present study was to elucidate the
role and underlying molecular mechanisms of MALAT1 in the
resistance of GC cells to CDDP.
Materials and methods
Cell culture
The human GC cell line AGS was obtained from
American Type Culture Collection (ATCC, Manassas, VA, USA). The
human GC cell line HGC-27 and the 293T cell line were purchased
from the cell bank of the Chinese Academy of Sciences (Shanghai,
China). AGS cells were maintained in F-12K medium (ATCC)
supplemented with 10% fetal bovine serum (FBS; Invitrogen; Thermo
Fisher Scientific, Inc., Carlsbad, CA, USA). HGC-27 cells were
cultured in RPMI-1640 medium (Invitrogen; Thermo Fisher Scientific,
Inc.) containing 1.5 g/l NaHCO3 (Sigma-Aldrich; Merck
KGaA, St. Louis, MO, USA), 2.5 g/l glucose (Sigma-Aldrich; Merck
KGaA), 0.11 g/l sodium pyruvate (Sigma-Aldrich; Merck KGaA) and 20%
FBS (Invitrogen; Thermo Fisher Scientific, Inc.). 293T cells were
cultured in Dulbecco's modified Eagle's medium (Invitrogen; Thermo
Fisher Scientific, Inc.) containing 10% FBS (Invitrogen; Thermo
Fisher Scientific, Inc.). HGC-27 and AGS cells were continuously
exposed to gradually increasing concentrations of CDDP (0.5-10
µg/ml) for >6 months to establish a CDDP-resistant HGC-27
cell line (HGC-27/CDDP) and a CDDP-resistant AGS (AGS/CDDP) cell
line.
Reagents and cell transfection
miR-30b mimic and its scramble control (miR-NC),
miR-30b inhibitor and its negative control (anti-miR-30b), small
interfering RNA (siRNA) targeting MALAT1 (si-MALAT1) and its
negative control (si-NC), were synthesized by GenePharma (Shanghai,
China). Full-length sequences of MALAT1 or autophagy-related gene 5
(ATG5) were constructed into a pcDNA3.1 vector (Invitrogen; Thermo
Fisher Scientific, Inc.) to generate pcDNA-MALAT1 (MALAT1) or
pcDNA-ATG5 (ATG5) overexpression plasmid, respectively. All
plasmids or oligonucleotides were transfected into GC cells by
Lipofectamine 2000 reagent (Invitrogen; Thermo Fisher Scientific
Inc.) according to the instructions of the manufacturer. CDDP was
purchased from MedChem Express Co., Ltd. (Monmouth Junction, NJ,
USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) assay
Total RNA (mRNAs and microRNAs) was isolated from GC
or CDDP-resistant GC cells using TRIzol reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) and quantified by NanoDrop ND 1000
(NanoDrop Technologies, Wilmington, DE, USA). The analysis of
miR-30b expression was performed using TaqMan® MicroRNA
Real-Time PCR Assay reagents and primers (Applied Biosystems;
Thermo Fisher Scientific, Inc., Foster City, CA, USA) with RNU6B
(primers also purchased from Applied Biosystems; Thermo Fisher
Scientific, Inc.) as an endogenous control. For MALAT1 expression
analysis, RNAs were reversely transcribed into first-strand cDNA
using the High Capacity cDNA Reverse Transcription kit (Applied
Biosystems; Thermo Fisher Scientific, Inc.) and random primers,
followed by qPCR detection by SYBR® Premix Ex Taq™
reagent (Takara, Otsu, Japan) and specific quantitative primers
(for MALAT1 or GAPDH). GAPDH was employed to normalize the
expression of MALAT1. The primers for MALAT1 or GAPDH were as
follows: MALAT1, 5'-CTTAAGCGCAGCGCCATTTT-3' (forward) and
5'-CCTCCAAACCCCAAGACCAA-3' (reverse) ; GAPDH, 5 ' -
CAACTCCCTCAAGATTGTCAGCAA-3' (forward) and
5'-GGCATGGACTGTGGTCATGA-3' (reverse).
Cell Counting Kit-8 (CCK-8) assay
For the determination of half maximal inhibitory
concentration (IC50), cells (104 cells/well)
were seeded into 96-well plates and then treated with varying
concentrations of CDDP (0-25 µg/ml) for 48 h. For the
detection of drug sensitivity, untransfected or transfected cells
were treated with CDDP (5 µg/ml) for 48 h. Next, cell
viability was determined by the CCK-8 assay kit (Dojindo Molecular
Technologies, Rockville, MD, USA) following the manufacturer's
instructions. Briefly, 10 µl of CCK-8 solution was added
into each well of the 96-well plates. After 3 h of incubation, cell
absorbance was examined at 450 nm.
Western blot assay
Total proteins were extracted using RIPA buffer
(Beyotime Institute of Biotechnology, Shanghai, China) and
quantified using Pierce BCA Protein Assay kit (Thermo Fisher
Scientific, Rockford, IL, USA). Subsequently, equal amounts of
protein (50 µg) were separated by 12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and electrophoretically
transferred onto nitrocellulose membranes (Pall, New York, NY,
USA). After blocking with 5% skimmed milk for 1 h at room
temperature, the membranes were probed overnight at 4°C with rabbit
monoclonal antibody against ATG5 (ab108327, 1:2,000 dilution;
Abcam, Cambridge, UK), mouse monoclonal antibody against p62
(ab56416, 1:1,000 dilution; Abcam), rabbit polyclonal antibody
against LC3B (ab51520, 1:3,000 dilution; Abcam, containing LC3-I
and LC3-II), or rabbit polyclonal antibody against β-actin (ab8227,
1:2,000 dilution; Abcam). Next, the membranes were incubated with
horseradish peroxidase-conjugated goat anti-rabbit (ab6721, 1:5,000
dilution; Abcam) or goat anti-mouse (ab6789, 1:5,000 dilution;
Abcam) secondary antibody for 1 h at room temperature. Finally,
protein signals were detected by Pierce™ ECL Western Blotting
Substrate (Thermo Fisher Scientific, Inc.) and analyzed by Quantity
One software (Bio-Rad Laboratories, Hercules, CA, USA).
Luciferase assay
Partial sequences of MALAT1 or ATG5 3'-untranslated
region containing predicted miR-30b-binding sites were constructed
into a psiCHECK-2 luciferase vector (Promega Corporation, Madison,
WI, USA) to generate MALAT1 wild-type (WT) or ATG5 WT luciferase
reporter, respectively. Also, MALAT1 mutant (MUT) or ATG5 MUT
luciferase reporter containing mutant miR-30b-binding sites were
also produced using GeneArt™ Site-Directed Mutagenesis System
(Invitrogen; Thermo Fisher Scientific, Inc.). Then, the constructed
luciferase reporters were transfected into 293T cells in
combination with mimics or plasmids. At 48 h after transfection,
luciferase activities were determined using a dual luciferase
reporter assay kit (Promega Corporation).
RNA immunoprecipitation (RIP) assay
RIP assay was performed using Magna RIP™ RNA-Binding
Protein Immunoprecipitation kit (EMD Millipore, Bedford, MA, USA)
and primary antibody against IgG (EMD Millipore) or Argonaute 2
(Ago2; EMD Millipore) according to the instructions of the
manufacturer.
GFP-LC3 puncta experiment
GC cells and CDDP-resistant GC cells were
transfected with pSelect-GFP-hLC3 plasmid (InvivoGen, San Diego,
CA, USA) and other plasmids or oligomers (mimics, inhibitors or
siRNAs), followed by treatment with CDDP (5 µg/ml) for 48 h.
The cells were then fixed using 4% paraformaldehyde for 20 min at
4°C and stained using DAPI solution (1 µg/ml; Beyotime
Institute of Biotechnology) for 15 min. Finally, the cells were
visualized using a confocal laser scanning microscope (TCS-TIV;
Leica, Nussloch, Germany). The percentage of GFP-LC3-positive cells
containing >20 puncta was counted in 50 randomly selected
fields.
Statistical analysis
Data were obtained from at least 3 independent
experiments with the results expressed as mean ± standard
deviation. Student's t-test was used to compare the difference
between two groups, and one-way analysis of variance followed by
Tukey's post-host test was employed to evaluate the differences
among multiple (>2) groups. In all cases, P<0.05 indicated
that the difference was statistically significant.
Results
MALAT1 is highly expressed in
CDDP-resistant GC cells
First, CDDP-resistant GC cell lines (AGS/CDDP and
HGC-27/CDDP) were established to explore whether MALAT1 was
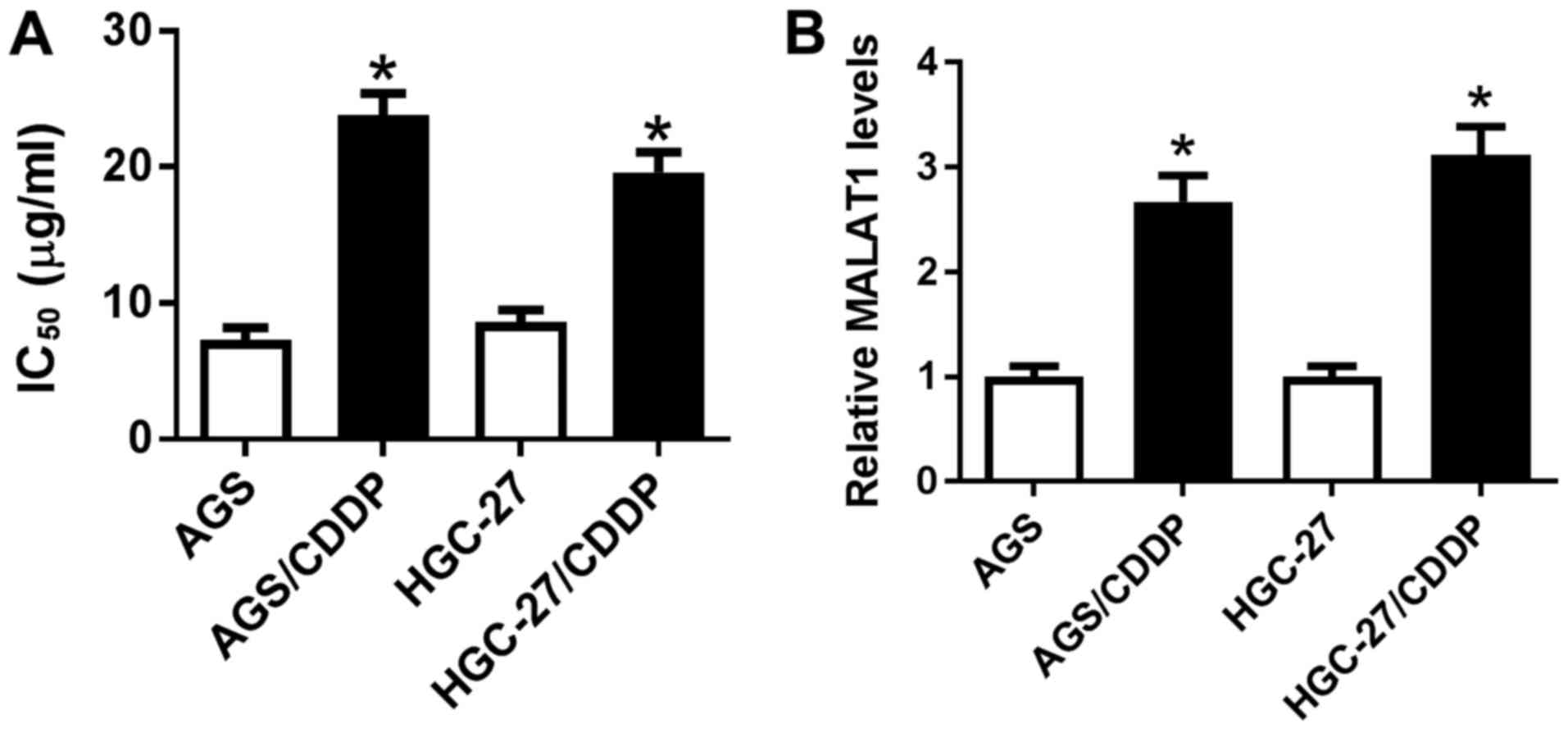
associated with CDDP resistance in GC. As shown in Fig. 1A, the IC50 of CDDP was
markedly increased in AGS/CDDP and HGC-27/CDDP cells when compared
with that in the corresponding parental cell lines (AGS and HGC-27,
respectively), indicating that CDDP-resistant GC cell lines were
successfully established. Then, it was further demonstrated that
MALAT1 expression was notably upregulated in AGS/CDDP and
HGC-27/CDDP cell lines relative to the respective parental cell
lines (Fig. 1B), indicating that
MALAT1 may be correlated with CDDP resistance in GC.
MALAT1 potentiates CDDP resistance by
improving autophagic activity in CDDP-resistant GC cell lines
RT-qPCR analysis further demonstrated that the level
of MALAT1 was notably increased in AGS/CDDP cells transfected with
MALAT1 overexpression plasmid, but was strikingly reduced in
HGC-27/CDDP cells transfected with si-MALAT1 (Fig. 2A). Therefore, MALAT1 overexpression
plasmid and si-MALAT1 were used for the following gain-of-function
and loss-of-function assays, respectively. Moreover, MALAT1
overexpression induced an obvious increase in AGS/CDDP cell
viability (Fig. 2B). Conversely,
the viability of MALAT1-silenced HGC-27/CDDP cells was markedly
reduced (Fig. 2B). The CCK-8 assay
further demonstrated that AGS/CDDP and HGC-27/CDDP cells were more
resistant to CDDP compared with their respective parental cells
(Fig. 2C). Moreover, ectopic
expression of MALAT1 markedly enhanced the resistance of AGS/CDDP
cells to CDDP (Fig. 2C).
Conversely, MALAT1 knockdown weakened CDDP resistance of
HGC-27/CDDP cells (Fig. 2C).
Previous studies indicated that MALAT1 improved drug resistance of
cancer cells by promoting autophagy (19,20).
Hence, the effect of MALAT1 on autophagy was further investigated.
The results revealed that cell autophagic activity was markedly
increased in CDDP-resistant GC cell lines, as evidenced by an
increased percentage of GFP-LC3-positive cells (Fig. 2D), the LC3-II/LC3-I protein ratio
was increased (Fig. 2E) and p62
protein expression was reduced (Fig.
2E)in AGS/CDDP and HGC-27/CDDP cells relative to their
respective parental cells. Moreover, MALAT1 overexpression
facilitated cell autophagy in AGS/CDDP cells, while MALAT1
knockdown suppressed autophagy in HGC-27/CDDP cells (Fig. 2D and E). In summary, these results
demonstrated that MALAT1 potentiated CDDP resistance by promoting
autophagy in CDDP-resistant GC cells.
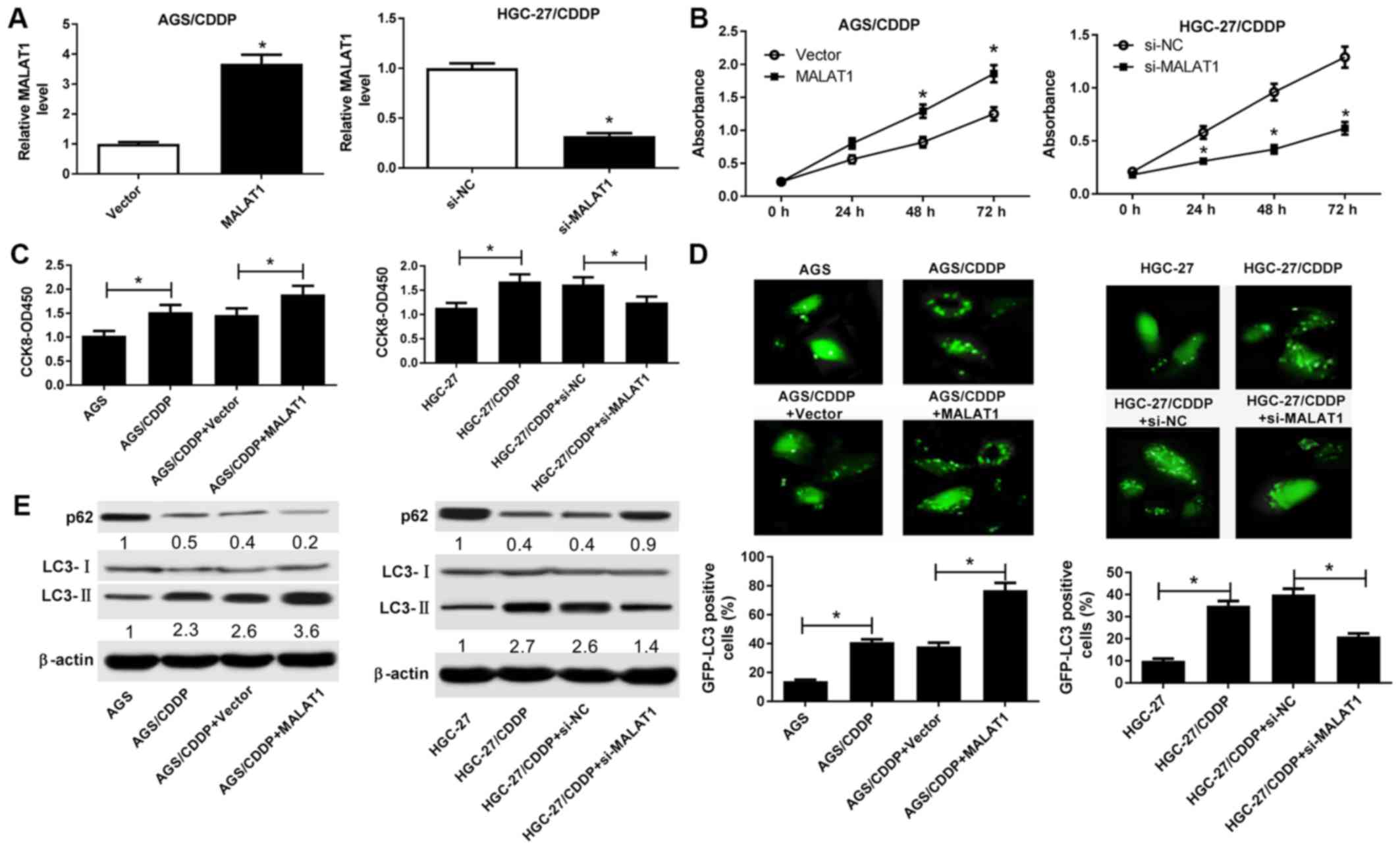 | Figure 2(A-E) MALAT1 potentiated CDDP
resistance by promoting autophagy in CDDP-resistant GC cell lines.
(A and B) AGS/CDDP cells were transfected with pcDNA3.1 vector or
MALAT1 overexpression plasmid, and HGC-27/CDDP cells were
transfected with si-NC or si-MALAT1. At 48 h a post-transfection,
the MALAT1 level was determined by RT-qPCR analysis and cell
viability was measured by the CCK-8 assay. (C and E) AGS/CDDP cells
were transfected with pcDNA3.1 vector or MALAT1 overexpression
plasmid, and HGC-27/CDDP cells were transfected with si-NC or
si-MALAT1. At 24 h post-transfection, transfected or untransfected
cells were treated with CDDP (5 µg/ml) for another 48 h.
Then, (C) cell viability was determined by the CCK-8 assay and (E)
the protein expression of p62, LC3-I and LC3-II was measured by
western blotting. (D) The cells in the AGS, AGS/CDDP, AGS/CDDP +
vector and AGS/CDDP + MALAT1 groups were transfected with
pSelect-GFP-hLC3 plasmid, pSelect-GFP-hLC3 plasmid,
pSelect-GFP-hLC3 plasmid + pcDNA3.1 vector and pSelect-GFP-hLC3
plasmid + MALAT1, respectively. At 24 h post-transfection, the
cells were treated with CDDP (5 µg/ml) for another 48 h,
followed by detection of the percentage of GFP-LC3-positive cells.
*P<0.05. MALAT1, metastasis-associated lung
adenocarcinoma transcript 1; CDDP, cisplatin; GC, gastric cancer;
RT-qPCR, reverse transcription-quantitative polymerase chain
reaction; CCK-8, Cell Counting Kit-8; GFP, green fluorescent
protein. |
MALAT1 inhibits miR-30b expression by
direct interaction
Accumulating evidence supports the hypothesis that
lncRNAs may act as competing endogenous RNAs (ceRNAs) to regulate
the expression of miRNAs and miRNA target genes in GC (21). Hence, bioinformatics analysis was
performed by miRcode online website to identify miRNAs that may
interact with MALAT1. Among candidate miRNAs, miR-30b was selected
due to its critical role in GC progression and autophagy (22-24)
(Fig. 3A). Subsequent luciferase
assay demonstrated that miR-30b overexpression markedly reduced
luciferase activity of the wild-type MALAT1 reporter, but did not
affect luciferase activity of the mutant MALAT1 reporter,
suggesting that MALAT1 may interact with miR-30b by putative
binding sites (Fig. 3B). To
further investigate the spatial interaction between MALAT1 and
miR-30b, RIP assay was performed in AGS/CDDP cell lysates using an
antibody against Ago2, which is an essential component of the
RNA-induced silencing complex (RISC). The results revealed that
miR-30b and MALAT1 were substantially enriched in Ago2
immunoprecipitation complexes (Fig.
3C), indicating that MALAT1 was able to spatially interact with
miR-30b. RT-qPCR assay further demonstrated that miR-30b expression
was notably downregulated in MALAT1-overexpressing AGS/CDDP cells,
but was markedly upregulated in MALAT1-silenced AGS/CDDP cells
(Fig. 3D). In summary, these
results indicate that MALAT1 inhibits miR-30b expression by direct
interaction.
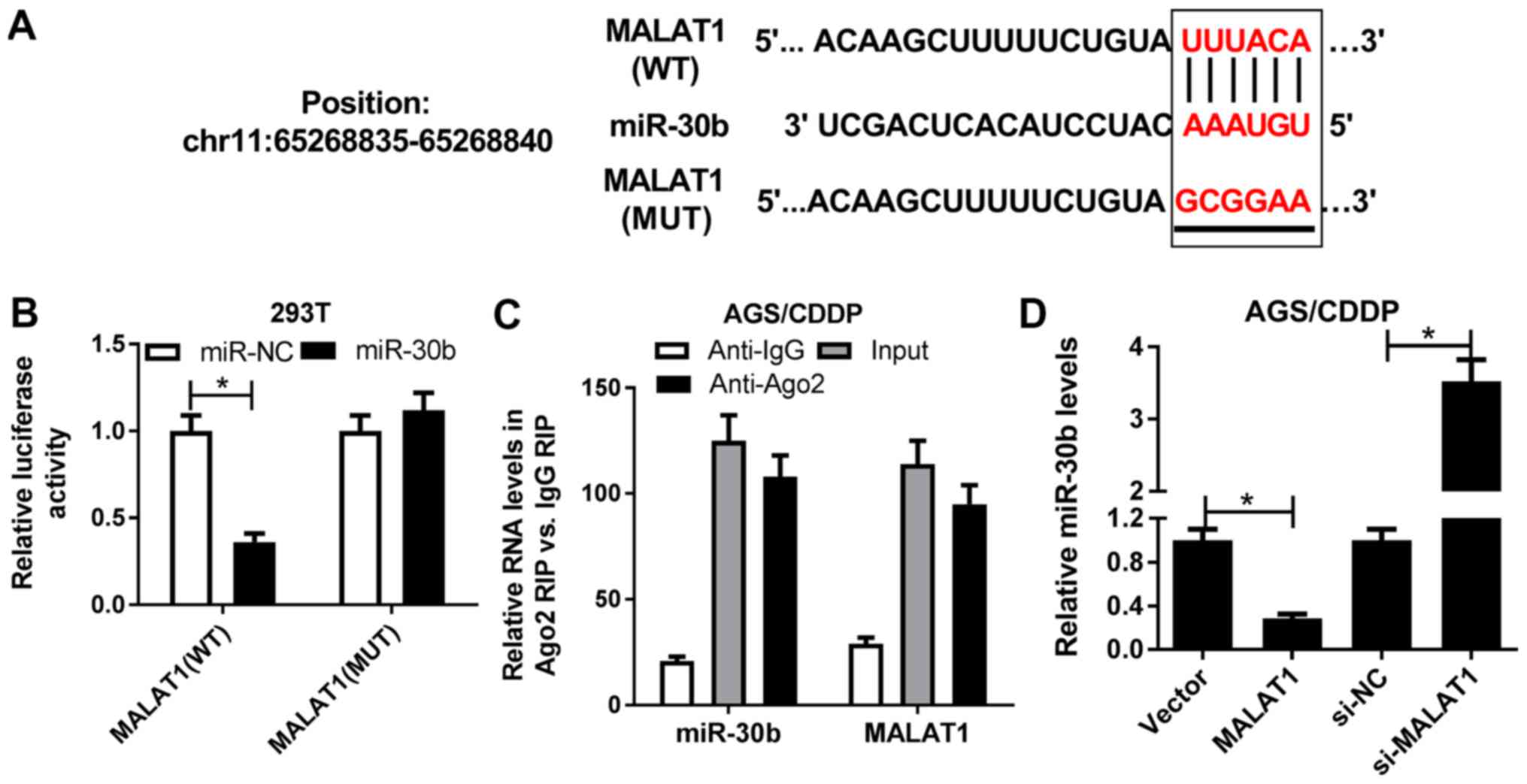 | Figure 3MALAT1 inhibited miR-30b expression
by direct interaction. (A) Predicted interaction sites between
MALAT1 and miR-30b by miRcode online website, and MUT sites in
MALAT1 reporter. (B) 293T cells were co-transfected with MALAT1 WT
or MALAT1 MUT reporter and miR-NC or miR-30b. At 48 h
post-transfection, luciferase activity was detected by luciferase
assay. (C) RIP assay was performed in AGS/CDDP cell lysates using
the antibody against Ago2 or IgG. Then, the expression of miR-30b
and MALAT1 in Ago2 or IgG immunoprecipitation complexes was
measured via RT-qPCR assay. (D) AGS/CDDP cells were transfected
with pcDNA3.1 vector, MALAT1, si-NC or si-MALAT1, followed by the
examination of miR-30b level using RT-qPCR assay at 48 h
post-transfection. *P<0.05. MUT, mutant; WT,
wild-type; MALAT1, metastasis-associated lung adenocarcinoma
transcript 1; RIP, RNA immunoprecipitation; Ago2, Argonaute 2;
RT-qPCR, reverse transcription-quantitative polymerase chain
reaction. |
miR-30b abolishes MALAT1-induced CDDP
resistance by inhibiting autophagy in CDDP-resistant GC cell
lines
Next, restoration experiments were performed to
determine whether MALAT1 enhanced CDDP resistance by regulating
miR-30b expression in CDDP-resistant GC cells. First, it was
demonstrated that the introduction of miR-30b mimic markedly
ablated the inhibitory effect of MALAT1 on miR-30b expression in
AGS/CDDP cells. Conversely, the transfection of miR-30b inhibitor
notably abolished si-MALAT1-induced miR-30b upregulation in
HGC-27/CDDP cells (Fig. 4A).
Functional analyses revealed that miR-30b overexpression
effectively reduced MALAT1-mediated CDDP resistance in AGS/CDDP
cells (Fig. 4B). By contrast, the
resistance of HGC-27/CDDP cells to CDDP was notably enhanced in
si-MALAT1-transfected HGC-27/CDDP cells following miR-30b
downregulation (Fig. 4B).
Therefore, miR-30b depletion abrogated the suppressive effect of
MALAT1 knockdown on CDDP resistance in HGC-27/CDDP cells. Rescue
assays further demonstrated that the restoration of miR-30b
attenuated MALAT1-induced autophagy in AGS/CDDP cells, as evidenced
by a reduced GFP-LC3-positive cell percentage (Fig. 4C), decreased the LC3-II/LC3-I
protein ratio and increased p62 protein expression (Fig. 4D) in MALAT1-enforced AGS/CDDP cells
after the introduction of miR-30b mimic. By contrast, the loss of
miR-30b relieved si-MALAT1-mediated autophagy inhibition in
HGC-27/CDDP cells (Fig. 4C and D).
In summary, these data demonstrated that miR-30b weakened
MALAT1-induced CDDP resistance by inhibiting autophagy in
CDDP-resistant GC cells.
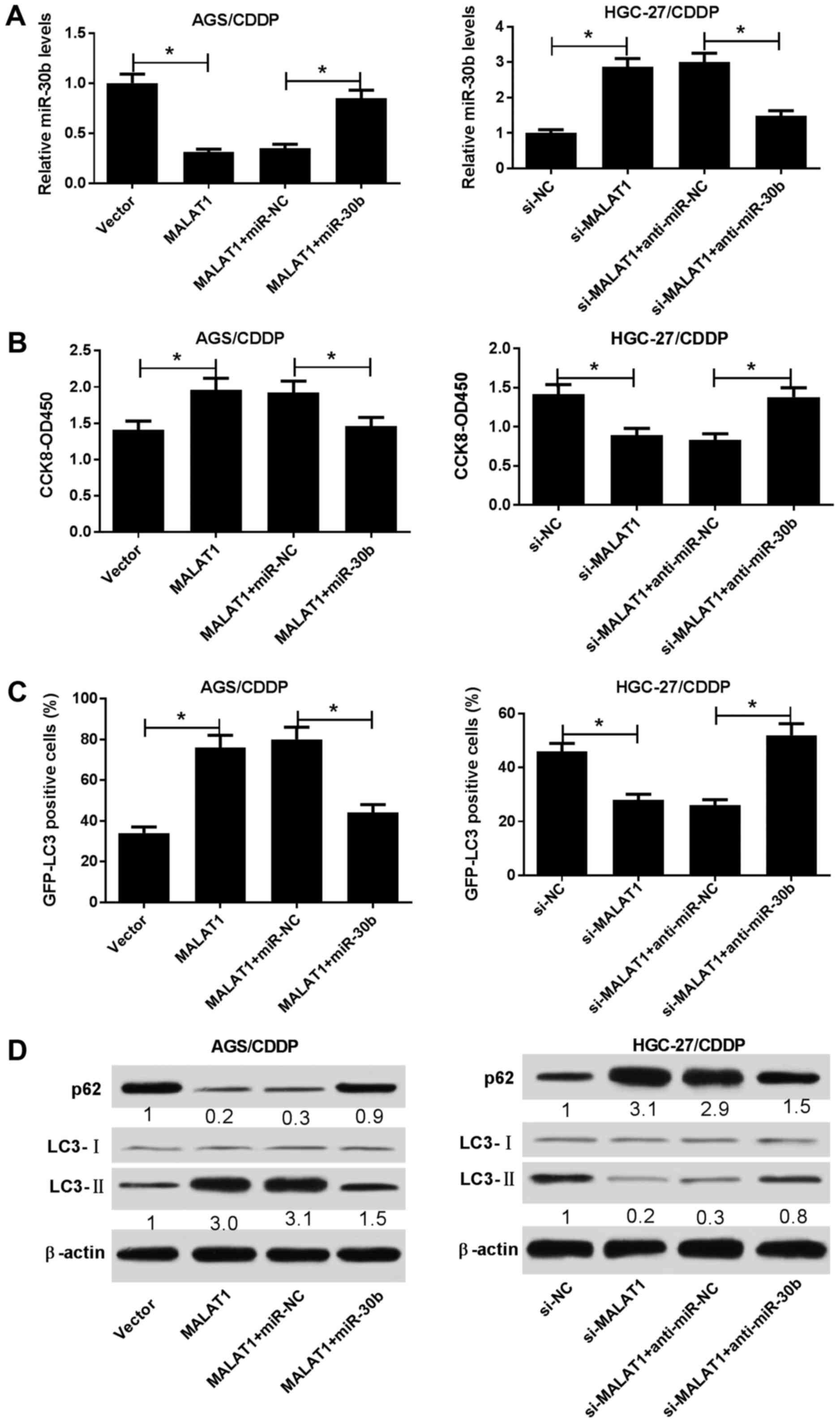 | Figure 4Restoration of miR-30b abolished
MALAT1-induced CDDP resistance by inhibiting autophagy in
CDDP-resistant GC cell lines. (A-D) AGS/CDDP cells were transfected
with pcDNA3.1 vector, MALAT1, MALAT1 + miR-NC, or MALAT1 + miR-30b.
HGC-27/CDDP cells were transfected with si-NC, si-MALAT1, si-MALAT1
+ anti-miR-NC, or si-MALAT1 + anti-miR-30b. The cells in panel C
were co-transfected with pSelect-GFP-hLC3 plasmid. At 24 h
post-transfection, the cells were treated with CDDP (5
µg/ml) for 48 h, followed by the determination of (A)
miR-30b level, (B) cell viability, (C) percentage of
GFP-LC3-positive cells and (D) protein expression of p62, LC3-I and
LC3-II. *P<0.05. ATG5, autophagy-related gene 5;
CDDP, cisplatin; GC, gastric cancer; GFP, green fluorescent
protein. |
ATG5 is a target of miR-30b
The TargetScan online website was then employed to
search for targets of miR-30b. Among candidate targets, ATG5 was
selected on account of its key roles in autophagy and
chemoresistance (11,25) (Fig.
5A). A luciferase assay revealed that the introduction of
miR-30b mimic resulted in a marked reduction of luciferase activity
of the wild-type ATG5 reporter (Fig.
5B). However, miR-30b expression exerted no effect on
luciferase activity of the mutant ATG5 reporter (Fig. 5B). These data indicated that
miR-30b may interact with ATG5 by predicted binding sites.
Moreover, miR-30b upregulation strikingly inhibited ATG5 expression
in AGS/CDDP cells (Fig. 5C).
Conversely, miR-30b depletion induced a marked increase of the ATG5
level in AGS/CDDP cells (Fig. 5C).
Generally, these results confirmed that ATG5 is a target of
miR-30b.
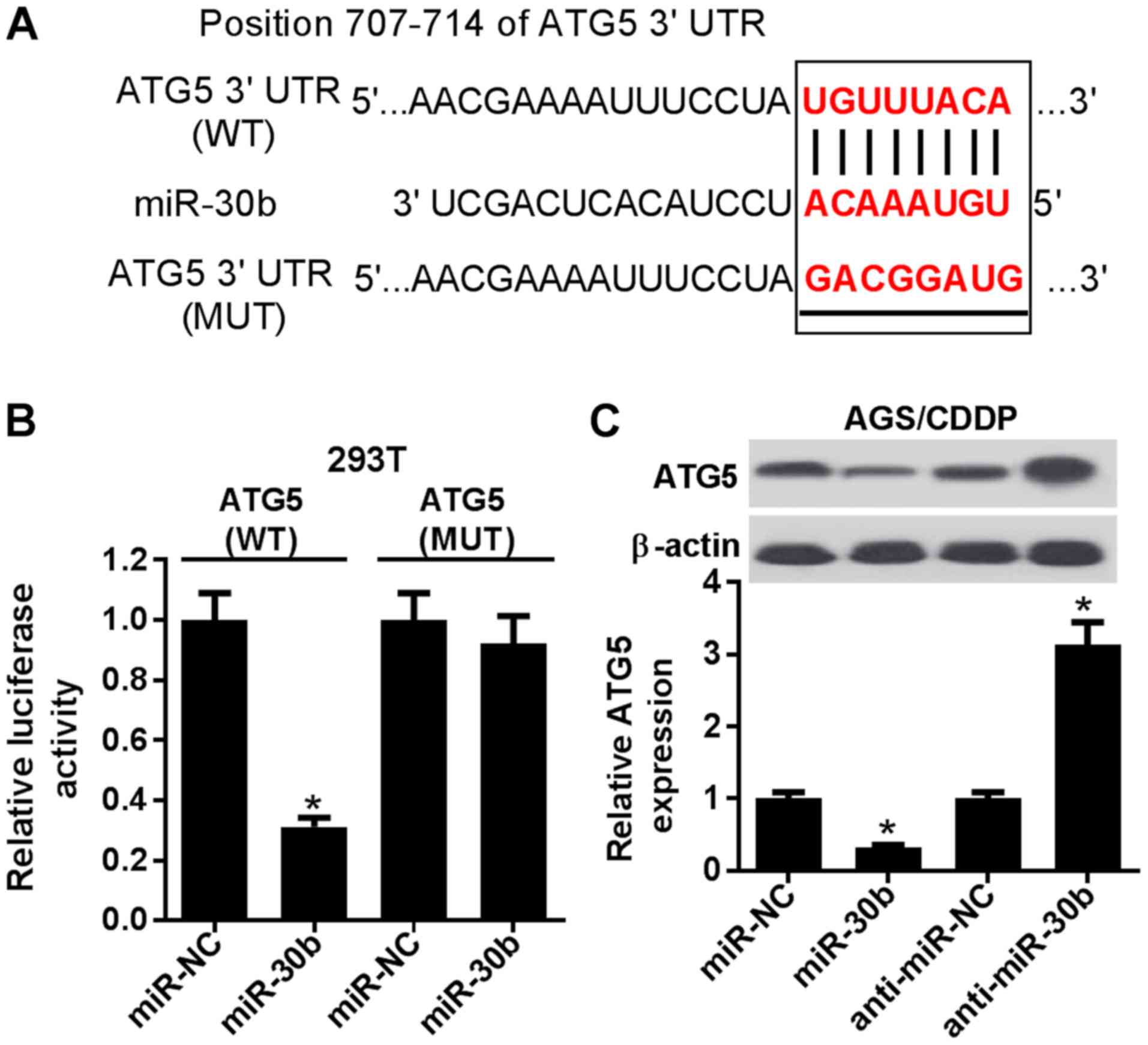 | Figure 5ATG5 was a target of miR-30b. (A)
Predicted binding sites between miR-30b and the ATG5
3'-untranslated region by TargetScan online website, and MUT sites
in ATG5 reporter. (B) 293T cells were co-transfected with ATG5 WT
or ATG5 MUT reporter and miR-NC or miR-30b mimic, followed by the
measurement of luciferase activity at 48 h post-transfection. (C)
AGS/CDDP cells were tranfected with miR-NC, miR-30b, anti-miR-NC,
or anti-miR-30b. At 48 h post-transfection, the ATG5 protein level
was tested using western blot assay. *P<0.05. ATG5,
autophagy-related gene 5; CDDP, cisplatin; MUT, mutant; WT,
wild-type. |
miR-30b-mediated inhibitory effects on
CDDP resistance and autophagy are abrogated by ATG5 in
CDDP-resistant GC cells
It was next demonstrated that ATG5 overexpression
reversed the miR-30b-mediated inhibitory effect on ATG5 expression,
and ATG5 silencing weakened anti-miR-30b-induced ATG5 upregulation
(Fig. 6A). Functional analyses
revealed that miR-30b upregulation resulted in reduced CDDP
resistance (Fig. 6B), reduced
GFP-LC3-positive cell percentage (Fig.
6C), decreased the LC3-II/LC3-I protein ratio and increased p62
protein expression (Fig. 6D) in
AGS/CDDP cells. Therefore, miR-30b overexpression alleviated CDDP
resistance and inhibited autophagy in AGS/CDDP cells, while these
effects were abolished by increased ATG5 expression (Fig. 6B-D). By contrast, ATG5 knockdown
rescued anti-miR-30b-induced CDDP resistance and autophagy in
HGC-27/CDDP cells (Fig. 6B-D). In
summary, these results revealed that miR-30b reduced CDDP
resistance and autophagic activity of AGS/CDDP and HGC-27/CDDP
cells via targeting ATG5.
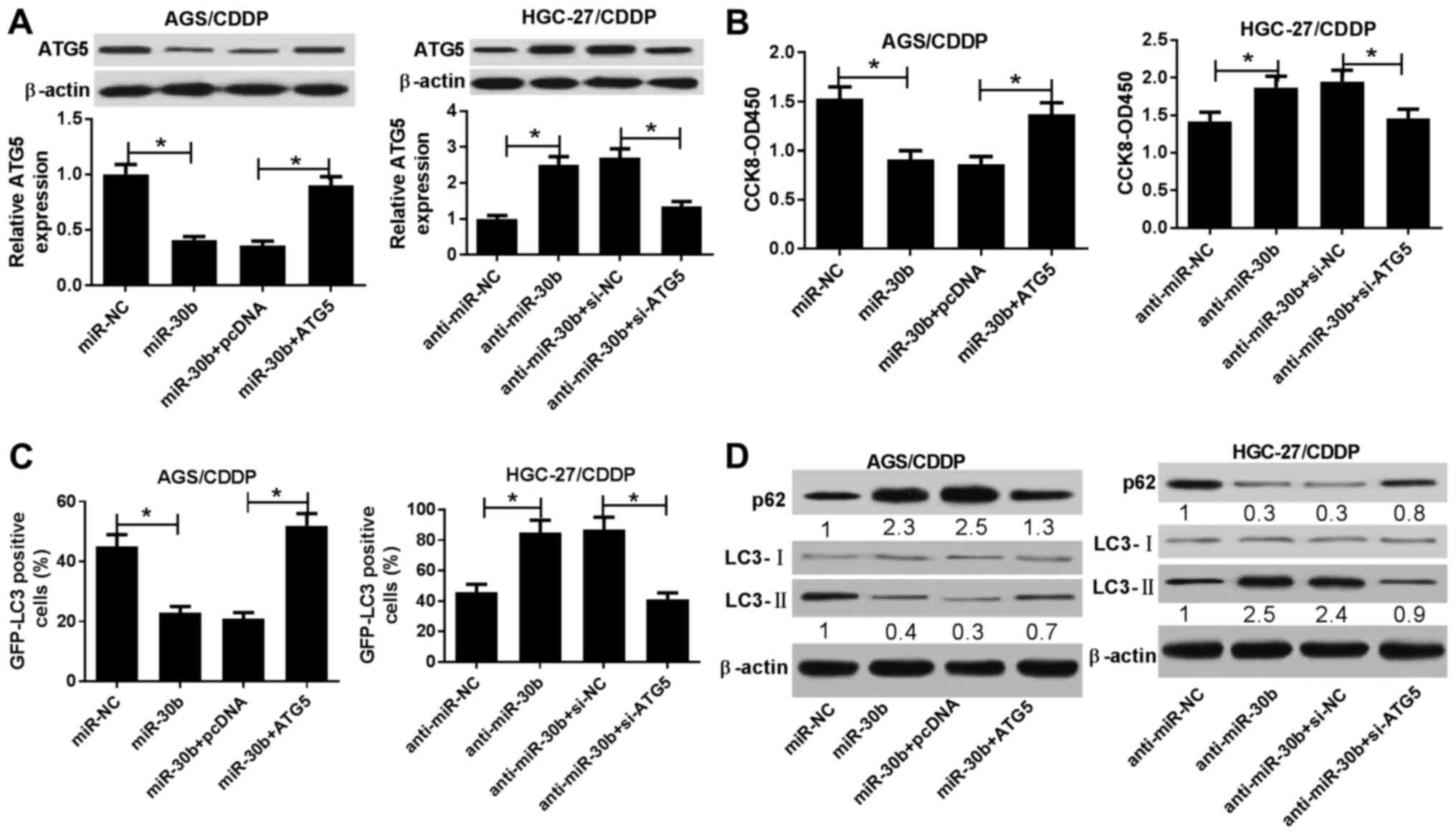 | Figure 6miR-30b reduced CDDP resistance and
autophagic activity of AGS/CDDP and HGC-27/CDDP cells via targeting
ATG5. (A-D) AGS/CDDP cells were transfected with miR-NC, miR-30b,
miR-30b + pcDNA3.1, or miR-30b + ATG5. HGC-27/CDDP cells were
transfected with anti-miR-NC, anti-miR-30b, anti-miR-30b + si-NC,
or anti-miR-30b + si-ATG5. Also, cells in panel C were
co-transfected with pSelect-GFP-hLC3 plasmid. At 24 h
post-transfection, cells were treated with CDDP (5 µg/ml)
for 48 h, followed by the determination of (A) ATG5 level, (B) cell
viability, (C) percentage of GFP-LC3-positive cells and(D) protein
expression of p62, LC3-I and LC3-II. *P<0.05. CDDP,
cisplatin; ATG5, autophagy-related gene 5; GFP, green fluorescent
protein. |
MALAT1 promotes ATG5 expression by acting
as a ceRNA of miR-30b in CDDP-resistant GC cell lines
We further investigated whether MALAT1 could act as
a ceRNA of miR-30b to increase ATG5 expression in AGS/CDDP and
HGC-27/CDDP cells. As shown in Fig.
7A, miR-30b overexpression attenuated luciferase activity of
the wild-type ATG5 reporter, while this effect was rescued by
increased MALAT1 expression in 293T cells. Western blot assay
further revealed that ectopic expression of MALAT1 induced the
upregulation of the ATG5 protein expression, while this effect was
weakened by increased miR-30b expression in AGS/CDDP cells
(Fig. 7B). By contrast, ATG5
expression was decreased in MALAT1-silenced HGC-27/CDDP cells and
the downregulation of miR-30b alleviated the inhibitory effect of
MALAT1 knockdown on ATG5 expression in HGC-27/CDDP cells (Fig. 7C). In conclusion, these data
demonstrated that MALAT1 sequestered miR-30b from ATG5, resulting
in the upregulation of ATG5 expression in CDDP-resistant GC
cells.
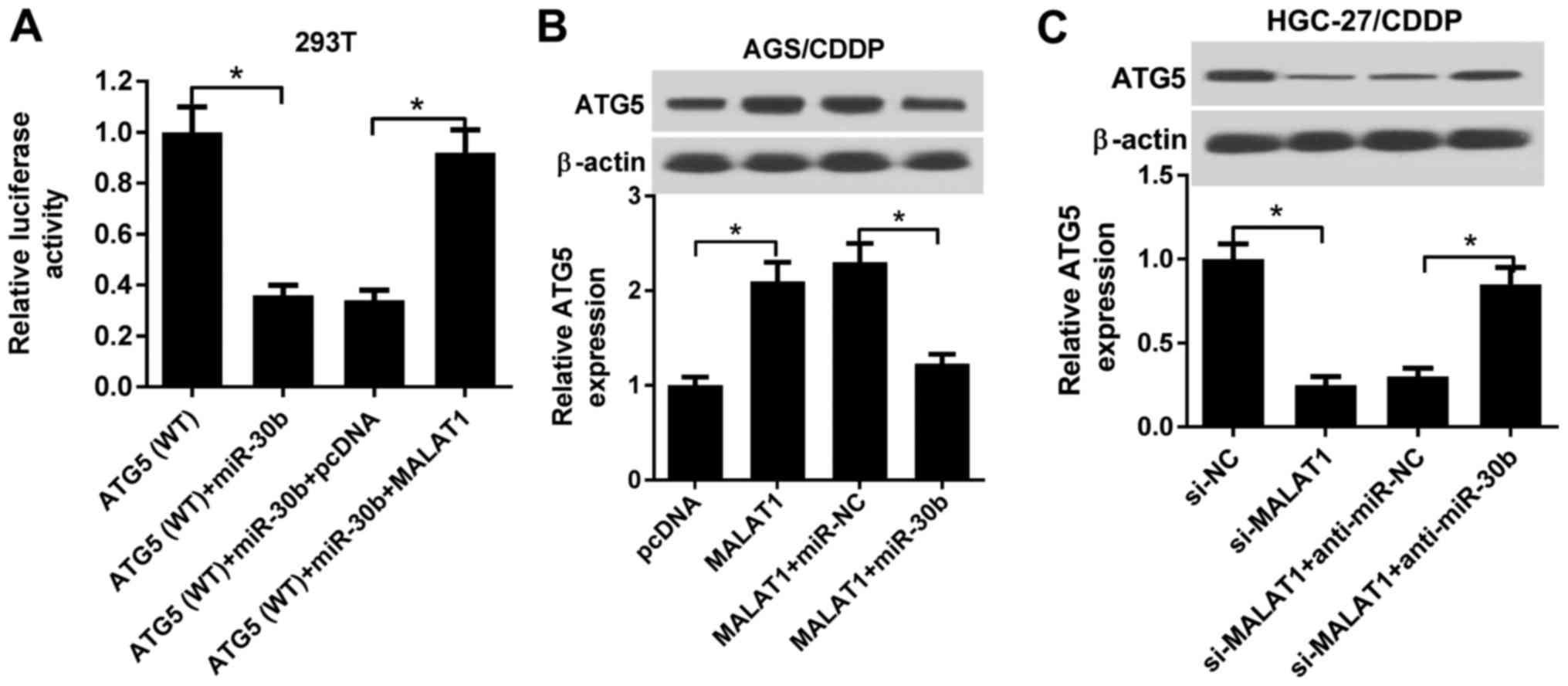 | Figure 7MALAT1 promoted ATG5 expression by
acting as a ceRNA of miR-30b in CDDP-resistant GC cell lines. (A)
293T cells were co-transfected with ATG5 WT reporter, ATG5 WT
reporter + miR-30b, ATG5 WT reporter + miR-30b + pcDNA3.1 vector,
or ATG5 WT reporter +miR-30b + MALAT1, followed by the measurement
of luciferase activity at 48 h post-transfection. (B) AGS/CDDP
cells were transfected with pcDNA3.1 vector, MALAT1, MALAT1 +
miR-NC, or MALAT1 + miR-30b. Then, ATG5 protein level was detected
at 48 h post-transfection. (C) HGC-27/CDDP cells were transfected
with si-NC, si-MALAT1, si-MALAT1 + anti-miR-NC, or si-MALAT1 +
anti-miR-30b, followed by measurement of ATG5 protein level at 48 h
post-transfection. *P<0.05. MALAT1,
metastasis-associated lung adenocarcinoma transcript 1; ATG5,
autophagy-related gene 5; ceRNA, competingendogenous RNA; CDDP,
cisplatin; WT, wild-type. |
Discussion
Current therapeutic strategies (e.g., surgical
resection and adjuvant chemotherapy) are generally not curative for
the majority of GC patients, particularly those diagnosed with
metastatic and advanced GC, due to various reasons, including
innate and acquired chemoresistance (6). Consequently, it is imperative to
elucidate the molecular mechanisms underlying GC chemoresistance in
order to design more effective interventions. Autophagy,
characterized by the conversion of LC3-I to LC3-II and the
formation of autolysosomes (the complex of autophagosomes and
lysosomes), plays a dual role (oncogenic or antitumor) in cancer
initiation and progression (26,27).
Moreover, autophagy exerts a dual effect on the therapeutic
efficacy of chemotherapy drugs: It may enhance the therapeutic
efficacy of drugs, or it may reduce therapeutic efficacy and
enhance drug resistance (11,12).
Accumulating evidence indicates that miRNAs and
lncRNAs are closely correlated with chemoresistance and autophagy
in cancer (15,28,29).
LncRNA MALAT1 was found to be aberrantly highly expressed in GC,
whereas MALAT1 knockdown inhibited GC progression (30-32).
Moreover, MALAT1 has been reported to be implicated in
chemoresistance and autophagy in certain types of cancer (19,20).
For example, Yuan et al demonstrated that the depletion of
MALAT1 reduced chemoresistance of multidrug-resistant
hepatocellular cancer cells to 5-fluorouracil (5-FU), adriamycin or
mitomycin C by inhibiting autophagy (19). Additionally, YiRen et al
observed that MALAT1 knockdown weakened the chemoresistance of
vincristine (VCR)-resistant GC cells to 5-FU, VCR and CDDP by
suppressing autophagy via regulating the miR-23b-3p/ATG12 axis
(20).
In the present study, it was demonstrated that
MALAT1 expression was notably upregulated in CDDP-resistant GC
(AGS/CDDP and HGC-27/CDDP) cells. YiRen et al also reported
that MALAT1 was highly expressed in VCR-resistant SGC7901
(SGC7901/VCR) cells compared with parental cells (20). Moreover, cell autophagic activity
was improved in CDDP-resistant GC cells compared with parental
cells. Increased autophagic activity was also observed in
SGC7901/VCR cells compared with that in parental SGC7901 cells
(20). Functional analyses
revealed that MALAT1 overexpression potentiated CDDP resistance of
AGS/CDDP cells by promoting autophagy. Conversely, MALAT1 knockdown
reduced the resistance of HGC-27/CDDP cells to CDDP by inhibiting
autophagy.
miR-30b is a member of the miR-30 family, which also
includes miR-30a, miR-30c, miR-30d and miR-30e (33). It was previously demonstrated that
other members of the miR-30 family were implicated in mediating
CDDP resistance by regulating autophagy in certain types of cancer.
For example, miR-30a overexpression weakened CDDP-induced
autophagy, but boosted CDDP-induced apoptosis in cervical cancer
(HeLa) cells (13). Therefore,
miR-30a promoted sensitivity of HeLa cells to CDDP by inhibiting
autophagy (13). Zhang et
al also demonstrated that miR-30d increased the sensitivity of
anaplastic thyroid cancer cells to CDDP by reducing autophagic
activity (14). miR-30b has been
reported to be downregulated in GC and was shown to inhibit the
development of GC by targeting plasminogen activator inhibitor-1
(22). Moreover, miR-30b
overexpression inhibited high phosphorus (Pi)-induced autophagy by
reducing the expression of autophagy-related genes, such as ATG5,
in vascular smooth muscle cells (24). Additionally, miR-30b suppressed
autophagy to abate hepatic ischemia-reperfusion injury by reducing
ATG12-ATG5 conjugates (23). The
abovementioned findings indicated that miR-30b may regulate CDDP
resistance and autophagy. In the present study, it was further
demonstrated that MALAT1 inhibited miR-30b expression by direct
interaction in AGS/CDDP cells. Furthermore, restoration assays
revealed that miR-30b upregulation abrogated MALAT1-induced CDDP
resistance by inhibiting autophagy in CDDP-resistant GC cells.
Accumulating evidence shows that miRNAs may exert
their effects by regulating target gene expression (34). Hence, ATG5 as a target of miR-30b
was validated by bioinformatics analysis and luciferase assay, and
the results were in line with those of previous studies (35,36).
ATG5, a key regulator of autophagy (25), has been generally considered as a
protective factor of tumor cells against chemotherapy (11). ATG5 expression was markedly
upregulated in CDDP-resistant GC cells (SGC7901/CDDP) and the
depletion of ATG5 decreased chemoresistance of SGC7901/CDDP cells
to CDDP. The findings of the present study revealed that miR-30b
weakened autophagy-related CDDP resistance by targeting ATG5 in
CDDP-resistant GC cells.
It was further confirmed that MALAT1 may act as a
ceRNA of miR-30b to sequester miR-30b from ATG5, resulting in the
upregulation of ATG5 expression in CDDP-resistant GC cells.
However, YiRen et al reported that MALAT1 knockdown exerted
no effect on ATG5 mRNA level in SGC7901/VCR cells (20), whereas Li et al demonstrated
that MALAT1 knockdown resulted in the increase of ATG5 expression
in diffuse large B-cell lymphoma cells (37). It may be hypothesized that this
difference among reported results may be due to the different
intracellular environments.
Collectively, the findings of the present study
revealed that MALAT1 potentiated CDDP resistance by inducing
autophagy via regulating the miR-30b/ATG5 axis in CDDP-resistant GC
cells, improving our understanding of the role and molecular
function of MALAT1 in drug resistance, and indicating potential
therapeutic strategies to prevent or reverse drug resistance.
Funding
No funding was received.
Availability of data and materials
All the data sets generated and analyzed in this
study are available from the corresponding author on reasonable
request.
Authors' contributions
This study was conceived and designed by ZX and JS.
The experiments were carried out by JN and ZX. The manuscript was
prepared by ZX, JS and JN. All the authors have read and approved
the final version of this manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests to disclose.
Acknowledgments
Not applicable.
References
|
1
|
Jemal A, Center MM, DeSantis C and Ward
EM: Global patterns of cancer incidence and mortality rates and
trends. Cancer Epidemiol Biomarkers Prev. 19:1893–1907. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Karimi P, Islami F, Anandasabapathy S,
Freedman ND and Kamangar F: Gastric cancer: Descriptive
epidemiology, risk factors, screening, and prevention. Cancer
Epidemiol Biomarkers Prev. 23:700–713. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar
|
|
4
|
Wagner AD, Unverzagt S, Grothe W, Kleber
G, Grothey A, Haerting J and Fleig WE: Chemotherapy for advanced
gastric cancer. Cochrane Database Syst Rev. 3:CD0040642010.
|
|
5
|
Charalampakis N, Economopoulou P,
Kotsantis I, Tolia M, Schizas D, Liakakos T, Elimova E, Ajani JA
and Psyrri A: Medical management of gastric cancer: A 2017 update.
Cancer Med. 7:123–133. 2018. View Article : Google Scholar :
|
|
6
|
Zhang D and Fan D: Multidrug resistance in
gastric cancer: Recent research advances and ongoing therapeutic
challenges. Expert Rev Anticancer Ther. 7:1369–1378. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cohen SM and Lippard SJ: Cisplatin: From
DNA damage to cancer chemotherapy. Prog Nucleic Acid Res Mol Biol.
67:93–130. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xu W, Yang Z and Lu N: Molecular targeted
therapy for the treatment of gastric cancer. J Exp Clin Cancer Res.
35:12016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mizushima N and Klionsky DJ: Protein
turnover via autophagy: Implications for metabolism. Annu Rev Nutr.
27:19–40. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qian HR and Yang Y: Functional role of
autophagy in gastric cancer. Oncotarget. 7:17641–17651. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sui X, Chen R, Wang Z, Huang Z, Kong N,
Zhang M, Han W, Lou F, Yang J, Zhang Q, et al: Autophagy and
chemotherapy resistance: A promising therapeutic target for cancer
treatment. Cell Death Dis. 4:e8382013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kumar A, Singh UK and Chaudhary A:
Targeting autophagy to overcome drug resistance in cancer therapy.
Future Med Chem. 7:1535–1542. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zou Z, Wu L, Ding H, Wang Y, Zhang Y, Chen
X, Chen X, Zhang CY, Zhang Q and Zen K: MicroRNA-30a sensitizes
tumor cells to cisplatinum via suppressing beclin 1-mediated
autophagy. J Biol Chem. 287:4148–4156. 2012. View Article : Google Scholar
|
|
14
|
Zhang Y, Yang WQ, Zhu H, Qian YY, Zhou L,
Ren YJ, Ren XC, Zhang L, Liu XP, Liu CG, et al: Regulation of
autophagy by miR-30d impacts sensitivity of anaplastic thyroid
carcinoma to cisplatin. Biochem Pharmacol. 87:562–570. 2014.
View Article : Google Scholar :
|
|
15
|
Sun T: Long noncoding RNAs act as
regulators of autophagy in cancer. Pharmacol Res. 129:151–155.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Majidinia M and Yousefi B: Long non-coding
RNAs in cancer drug resistance development. DNA Repair (Amst).
45:25–33. 2016. View Article : Google Scholar
|
|
17
|
Gutschner T, Hämmerle M and Diederichs S:
MALAT1-a paradigm for long noncoding RNA function in cancer. J Mol
Med (Berl). 91:791–801. 2013. View Article : Google Scholar
|
|
18
|
Li Y, Wu Z, Yuan J, Sun L, Lin L, Huang N,
Bin J, Liao Y and Liao W: Long non-coding RNA MALAT1 promotes
gastric cancer tumorigenicity and metastasis by regulating
vasculogenic mimicry and angiogenesis. Cancer Lett. 395:31–44.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yuan P, Cao W, Zang Q, Li G, Guo X and Fan
J: The HIF-2α-MALAT1-miR-216b axis regulates multi-drug resistance
of hepatocellular carcinoma cells via modulating autophagy. Biochem
Biophys Res Commun. 478:1067–1073. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
YiRen H, YingCong Y, Sunwu Y, Keqin L,
Xiaochun T, Senrui C, Ende C, XiZhou L and Yanfan C: Long noncoding
RNA MALAT1 regulates autophagy associated chemoresistance via
miR-23b-3p sequestration in gastric cancer. Mol Cancer. 16:1742017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hu Y, Tian H, Xu J and Fang JY: Roles of
competing endogenous RNAs in gastric cancer. Brief Funct Genomics.
15:266–273. 2016. View Article : Google Scholar
|
|
22
|
Zhu ED, Li N, Li BS, Li W, Zhang WJ, Mao
XH, Guo G, Zou QM and Xiao B: miR-30b, down-regulated in gastric
cancer, promotes apoptosis and suppresses tumor growth by targeting
plasminogen activator inhibitor-1. PLoS One. 9:e1060492014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li SP, He JD, Wang Z, Yu Y, Fu SY, Zhang
HM, Zhang JJ and Shen ZY: miR-30b inhibits autophagy to alleviate
hepatic ischemia-reperfusion injury via decreasing the Atg12-Atg5
conjugate. World J Gastroenterol. 22:4501–4514. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang J, Sun YT, Xu TH, Sun W, Tian BY,
Sheng ZT, Sun L, Liu LL, Ma JF, Wang LN, et al: MicroRNA-30b
Regulates High Phosphorus Level-Induced Autophagy in Vascular
Smooth Muscle Cells by Targeting BECN1. Cell Physiol Biochem.
42:530–536. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Klionsky DJ: Autophagy: from phenomenology
to molecular understanding in less than a decade. Nat Rev Mol Cell
Biol. 8:931–937. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tanida I, Ueno T and Kominami E: LC3 and
Autophagy. Methods Mol Biol. 445:77–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
White E, Mehnert JM and Chan CS:
Autophagy, Metabolism, and Cancer. Clin Cancer Res. 21:5037–5046.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pan B, Yi J and Song H: MicroRNA-mediated
autophagic signaling networks and cancer chemoresistance. Cancer
Biother Radiopharm. 28:573–578. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ayers D and Vandesompele J: Influence of
microRNAs and Long Non-Coding RNAs in Cancer Chemoresistance. Genes
(Basel). 8:952017. View Article : Google Scholar
|
|
30
|
Wang J, Su L, Chen X, Li P, Cai Q, Yu B,
Liu B, Wu W and Zhu Z: MALAT1 promotes cell proliferation in
gastric cancer by recruiting SF2/ASF. Biomed Pharmacother.
68:557–564. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qi Y, Ooi HS, Wu J, Chen J, Zhang X, Tan
S, Yu Q, Li YY, Kang Y, Li H, et al: MALAT1 long ncRNA promotes
gastric cancer metastasis by suppressing PCDH10. Oncotarget.
7:12693–12703. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xia H, Chen Q, Chen Y, Ge X, Leng W, Tang
Q, Ren M, Chen L, Yuan D, Zhang Y, et al: The lncRNA MALAT1 is a
novel biomarker for gastric cancer metastasis. Oncotarget.
7:56209–56218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ouzounova M, Vuong T, Ancey PB, Ferrand M,
Durand G, Le-Calvez Kelm F, Croce C, Matar C, Herceg Z and
Hernandez-Vargas H: MicroRNA miR-30 family regulates non-attachment
growth of breast cancer cells. BMC Genomics. 14:1392013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu Z, Wei X, Zhang A, Li C, Bai J and
Dong J: Long non-coding RNA HNF1A-AS1 functioned as an oncogene and
autophagy promoter in hepatocellular carcinoma through sponging
hsa-miR-30b-5p. Biochem Biophys Res Commun. 473:1268–1275. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen Z, Jin T and Lu Y: AntimiR-30b
Inhibits TNF-α Mediated Apoptosis and Attenuated Cartilage
Degradation through Enhancing Autophagy. Cell Physiol Biochem.
40:883–894. 2016. View Article : Google Scholar
|
|
37
|
Li LJ, Chai Y, Guo XJ, Chu SL and Zhang
LS: The effects of the long non-coding RNA MALAT-1 regulated
autophagy-related signaling pathway on chemotherapy resistance in
diffuse large B-cell lymphoma. Biomed Pharmacother. 89:939–948.
2017. View Article : Google Scholar : PubMed/NCBI
|