Introduction
Pancreatic ductal adenocarcinoma (PDAC) is an
aggressive tumor with a very poor prognosis (1). The 5-year survival rate of patients
with PDAC is approximately 6% with a mean survival of 6 months
(2). The curative potential is
optimal when surgical resection is involved; however, only 15-20%
of patients are eligible for surgery at the time of diagnosis, as
the majority of patients with PDAC present with either locally
advanced disease (which is rarely resectable) or metastatic disease
(3).
Gemcitabine (GEM) is a difluorinated analog of the
naturally-occurring nucleoside, deoxycytidine (4). GEM has been widely used in the
treatment of diverse carcinomas, including non-small cell lung
cancer, bladder cancer and breast cancer, and has become the
standard chemotherapy for PDAC. Although recently some modern
chemotherapeutics, such as FOLFIRINOX (5) and nab-paclitaxel (PTX) plus GEM
(6) have been used as first-line
chemotherapy, GEM remains a key drug for use in the treatment of
PDAC, particularly for second-line treatment. However, acquired
resistance of PDAC to GEM has become a major issue. When resistance
to GEM develops, clinicians usually have to switch treatment to a
third-line drug, and there are no effective therapies available for
patients that fail third-line therapy.
Metformin (MET) is the first-line treatment for type
2 diabetes mellitus and is the most widely prescribed drug
worldwide. Recently, several epidemiological studies have indicated
the potential anti-tumor effects of metformin against various types
of cancer, including PDAC (7-10).
Furthermore, basic research has revealed the mechanisms underlying
the anticancer effects of MET, which mainly involve the induction
of AMP-activated protein kinase (AMPK) (11). Tumor growth inhibition by MET is
mediated primarily by the inhibition of mammalian target of
rapamycin (mTOR) signaling (12).
The mTOR signaling cascade serves as a master regulator of
metabolism, cell growth and proliferation. According to a previous
study, 15-20% of patients with PDAC have very high levels of active
phosphorylated mTORS2448, and patients with such tumors
have a significantly reduced survival (13). Therefore, targeted anti-mTOR
therapies may offer a clinical benefit for patients with PDAC.
A previous study also reported that mTOR-dependent
signals stimulated hypoxia-inducible factor 1α (HIF-1α)
accumulation and HIF-1-mediated transcription in cells exposed to
hypoxic conditions (14). Vascular
endothelial growth factor (VEGF) is primarily induced during
hypoxia and is regulated by HIF-1 (15). The majority of PDACs are
characterized by a hypoxic tumor microenvironment (16). The oxygen pressure in a solid tumor
is generally lower than in the surrounding interstitial tissue, and
tumors exhibiting extensive hypoxia have been shown to be more
aggressive than corresponding tumors that are better oxygenized
(17,18). Based on these data, in this study,
we evaluated the activities of HIF-1α, and its downstream target,
VEGF, following treatment with MET.
Recently, we reported a method for cloning
GEM-resistant cell lines and established multiple GEM-resistant
clones derived from the human pancreatic cancer cell line, BxPC-3
(19). In this study, we used a
moderately GEM-resistant cell line, BxG30. This study was designed
to evaluate the anti-tumor effects of MET against GEM-resistant
PDAC in a mouse xenograft model. Our results demonstrated that the
anti-tumor effect of MET was mediated by the suppression of the
function of mTOR. Furthermore, we provide evidence that MET
inhibits HIF-1α and VEGF activation in hypoxic cells. These
findings are of significant clinical interest and reveal the
potential use of MET as an anti-tumor agent in the treatment of
PDAC.
Materials and methods
Chemotherapeutic drugs
MET was provided free of charge by Sumitomo
Dainippon Pharma Co., Ltd. (Osaka, Japan). GEM was purchased from
Eli Lilly Japan (Hyogo, Japan).
Cells and cell culture
The human pancreatic cancer cell line, BxPC-3, was
purchased from the American Type Culture Collection (ATCC,
Manassas, VA, USA) and cultured in RPMI-1640 medium (Wako Pure
Chemical Industries Ltd., Osaka, Japan) supplemented with 10% (v/v)
heat-inactivated fetal bovine serum (FBS; Invitrogen/Thermo Fisher
Scientific, Waltham, MA, USA) in a humidified 5% CO2 incubator at
37°C. BxG30, a moderately GEM-resistant cell line, was established
as previously described (18).
Briefly, the BxPC-3 cells were allowed to acclimatize by stepwise
exposure to increasing concentrations of GEM beginning at 1.5 ng/ml
and increasing by 1-10 ng/ml increments at every passage for 6
months. A BxPC-3-derived cell line cultured in the presence of a
final concentration of 30 ng/ml GEM was named BxG30.
Xenograft models
Animals were maintained according to institutional
regulations in facilities approved by the Animal Care Committee of
Kitasato University (Tokyo, Japan) and in accordance with Japanese
government guidelines for animal experiments. The protocol of the
animal experiments was reviewed and approved by The Laboratory
Animal care and use Committees of Kitasato University on April 1st,
2013 (approval no. 13023). Animal experiments were performed in
accordance with the ethical guidelines of the Kitasato Institute.
Isoflurane was used as an inhaled anesthetic. Humanitarian
endpoints, such as performing euthanasia treatment at an
appropriate time, when unbearable pain was involved, were
considered. The Institutional Animal Care and Use Committee
Guidebook (20) was referred to as
regards the humanitarian endpoints. In this study, no mice had to
be euthanized due to poor conditions before the end of the
experiment.
During a preliminary toxicity test of MET
monotherapy, certain adverse events, such as appetite loss and body
weight loss, occurred at doses of 800 mg/kg and increased
significantly from the dose of 1,500 mg/kg. Finally, we decided on
a 600 mg/kg dose of MET, as this induced a consistent anti-tumor
effect without any adverse events. For the purposes of this study,
96 male BALB/c Slc-nu/nu mice (6 weeks old) were obtained from CLEA
Japan Inc. (Tokyo, Japan). The body weights of the mice ranged from
15 to 19 g on arrival. The mice were maintained in a
specific-pathogen-free condition (temperature, 23±2°C; humidity,
55±10%) and were provided with autoclaved food and water. After
being allowed to acclimatize for 1 week, 2×106 BxPC-3
and BxG30 cells in 0.1 ml of 1% phosphate-buffered saline (PBS)
were injected into both the flanks of the mice. The mice were
randomly divided into 4 groups as follows: i) The control group [no
treatment, final tumor number (n)=10]; ii) the GEM-treated group
(80 mg/kg, n=11); iii) the MET-treated group (600 mg/kg, n=11); and
iv) the combination treatment group (GEM + MET, n=12). The BxG30
cells were injected into the other mice, and the mice were divided
into 4 groups according to the treatments (control group, n=14; GEM
group, n=13; MET group, n=10; GEM + MET group, n=14). Treatment was
initiated 2 weeks following implantation. MET was diluted in PBS to
600 mg/kg and administered orally every day for 4 weeks. GEM was
dissolved in PBS and injected intraperitoneally at a dose of 80
mg/kg every week (on days 15, 22, 29 and 36). Estimated tumor
volumes and body weights were measured each week. The estimated
tumor volume was calculated using the Battelle Columbus
Laboratories Protocol according to the following formula (21): TV = (A×B2)/2, where TV =
tumor volume, A = major axis and B = minor axis.
Following sacrifice on day 42, the tumors were
dissected and weighed. The estimated tumor volumes of the control
group (C) and treatment groups (T) were calculated as described
above. The relative tumor volumes (TRW and CRW) were calculated at
defined time points by evaluating the ratio of (T) and (C) to the
estimated tumor volumes at the beginning of treatment, and the
treatment to the control ratio (T/C%) was calculated as TRW divided
by CRW. The minimal T/C% over the treatment period was assessed and
used to calculate the therapeutic effect. A value of T/C% <50%
was required for the treatment to be considered effective.
Although some tumor ulceration was observed, all
mice exhibiting ulceration, bleeding or necrosis also exhibited
good general conditions without any signs of infection, anemia or
abnormal behavior. Thus, no treatment was administered for the
ulcerations as it was not deemed necessary.
Cytotoxicity assays
The BxG30 cells were seeded at a density of
2.2×103 cells per well in 96-well plates containing
culture medium supplemented with 10% FBS. After 24 h, the cultures
were washed and incubated in medium alone (control) or medium
containing MET or GEM at various concentrations (final
concentrations of MET: 0, 1.65, 2.475, 3.3 and 4.125 µg/ml;
final concentrations of GEM: 0, 2, 3, 4, 5 and 10 ng/ml) in
quadruplicate. The number of viable cells was counted after 72 h
using a Cell Counting Kit-8 (Nacalai Tesque, Inc., Kyoto, Japan)
according to the manufacturer’s instructions. The assay reagent was
a tetrazolium compound (WST-8) that is reduced by live cells to
produce a colored formazan product whose absorbance can be measured
at 450 nm. The quantity of the formazan product is directly
proportional to the number of live cells in the culture. The
inhibition index (I.I., %) was calculated using the following
formula: I.I. (%) = (b−c)/(b−a) ×100, where a = the optical density
(OD) of the cells, b = the OD of the cells + WST-8 reagent, and c =
the OD of the cells + WST-8 reagent + anti-tumor agent) and
IC50 values were calculated. A classical isobologram was
used to evaluate the synergistic effects of GEM and MET. All
experiments were repeated at least 3 times.
Determination of protein concentrations
in culture supernatants
The BxG30 cells were pre-cultured at a density of
1.5×105 cells per well in RPMI-1640 medium containing
10% FBS for 24 h. After removing the culture medium and washing
with PBS, the cells were treated with MET and GEM (final
concentrations of MET: 0, 0.825, 1.65 and 3.3 µg/ml; final
concentrations of GEM: 0, 1, 2, 4 and 10 ng/ml). The cells were
cultured for 24 h and lysed in lysis buffer [Cell Signaling
Technology (CST) Japan, Tokyo, Japan] containing Protease Inhibitor
Cocktail Set III (Wako Pure Chemical Industries, Osaka, Japan) and
Phosphatase Inhibitor Cocktail Solution I (Wako Pure Chemical
Industries). The lysates were centrifuged at 23,000 × g for 20 min
at 4°C and the supernatants were collected. The samples were stored
at -80°C and used for western blot analysis.
Western blotting of pS6 and HIF-1α
For protein extraction, cells were plated into
6-well cell culture plates (Corning, Inc. Corning, NY, USA) at a
density of 1.5×105 cells in serum-containing medium and
then incubated for 24 h at 37°C. The cells were rinsed twice with
PBS and scraped into Cell Lysis Buffer (CST) and Phosphatase
Inhibitor Cocktail Solution I (Wako, Osaka, Japan) was added.
Following incubation for 20 min on ice, cell lysates were cleared
by 20 min of centrifugation at 15,000 × g at 4°C. Protein
concentration was determined using the BCA Protein Assay Reagent
(Pierce, Rockford, IL, USA), and conditioned to 15 µg per
lane. Samples were boiled for 3 min at 100°C and 4X LDS Sample
Buffer (Life Technologies Japan Ltd., Tokyo, Japan), and 5%
2-mercaptoethanol (Nacalai Tesque Inc.). Equal amounts of total
protein were separated on a 12.5% SDS polyacrylamide gel at a
constant current of 20 mA. Separated proteins were transferred to
PVDF membranes (Life Technologies Japan Ltd.) at 30V by using
iBlot® Gel Transfer Device (Life Technologies Japan
Ltd.). The membrane was blocked with 5% dry-milk with 0.1% Tris
Buffered Saline-Tween-20 (TBS-T) for 60 min. The membrane was then
incubated overnight at 4°C and with primary antibody, and then
probed with secondary antibodies for 1 h at room temperature.
Chemiluminescence was developed using the ECL Select Western
Blotting Detection System and the band intensities were then
detected using Image Quant LAS 500 (both from GE Healthcare Japan,
Tokyo, Japan) and analyzed using NIH Image J software. The BxG30
cells were treated with several concentrations of MET and GEM
(final concentrations of MET: 0, 0.825, 1.65 and 3.3 µg/ml;
final concentrations of GEM: 0, 1, 2, 4 and 10 ng/ml).
The following antibodies were used for western
blotting: pS6 rabbit antibody, S6 mouse antibody, HIF-1α rabbit
antibody (all from CST; #2317S, #3716S) and
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) rabbit polyclonal
IgG (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA;
#sc-25778) as primary antibodies, and horseradish peroxidase
(HRP)-conjugated anti-rabbit IgG antibody (CST; #7074P2) or
HRP-conjugated anti-mouse IgG antibody (GE Healthcare Japan, Tokyo,
Japan; NA931-100UL) as secondary antibodies. Each sample was
analyzed 3 times independently.
Exposure to hypoxia
On the day of the experiment, the culture medium was
replaced with fresh RPMI-1640 medium containing 10% FBS. Culture
dishes were placed in a modular incubator chamber
(Billups-Rothenberg Inc., San Diego, CA, USA), a humidified
airtight chamber with air flow valves. To simulate hypoxic
conditions, the cells were incubated with 1% O2, 5%
CO2 and 94% N2 for 48 to 78 h.
ELISA for the detection of VEGF
ELISA was performed to quantify secreted VEGF from
BxG30 cells using a Novex ELISA kit (Life Technologies Japan Ltd.)
according to the manufacturer’s instructions. The BxG30 cells were
incubated under hypoxic conditions as described above for 72 h with
various concentrations of MET (final concentrations: 0.825, 1.65
and 3.3 µg/ml) and GEM (final concentrations: 1, 2 and 4
ng/ml). At the end of the ELISA procedure, stop solution was added
and the absorbance of each well was measured at 450 nm using a
Multiskan FC instrument (Thermo Fisher Scientific). Each sample was
analyzed 3 times independently.
Statistical analysis
Dunnett’s test was used to compare the results from
the treatment groups with the control group. One-way factorial
ANOVA followed by the Dunnett’s test as a post hoc test was used to
analyze multiple comparisons between the control group and each of
the treatment groups. Differences were considered statistically
significant at P<0.05.
Results
Anti-tumor activity of MET in the BxPC-3
xenograft model
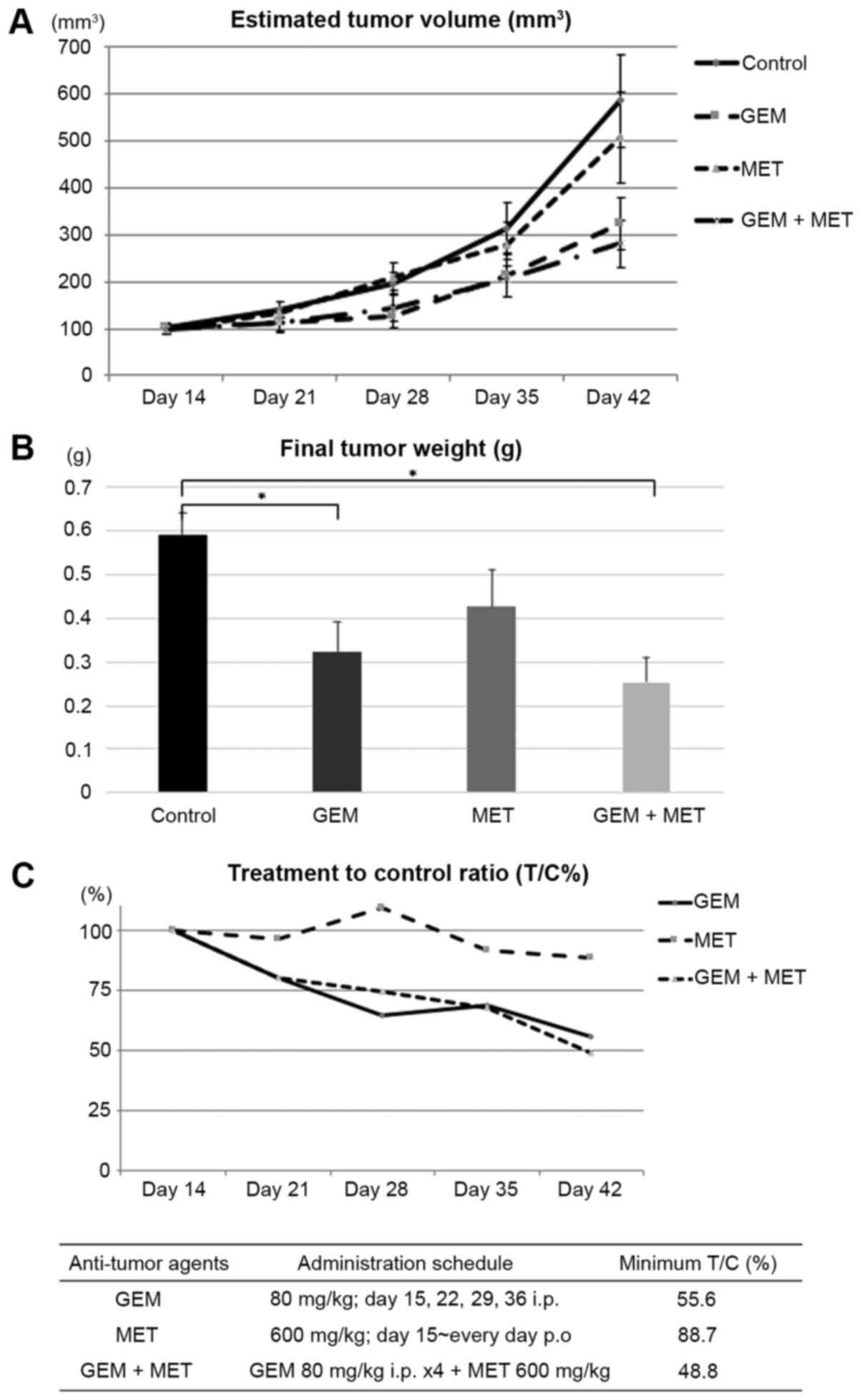
The estimated tumor volumes and body weights of the
mice were measured each week following implantation of the BxPC-3
xenografts. The final tumor weights were 0.59±0.05 g (mean ± SEM)
in the control group (n=10), 0.32±0.07 g in the GEM-treated mice
(n=11), 0.42±0.08 g in the MET-treated mice (n=11) and 0.23±0.06 g
in the mice treated with GEM and MET (n=12) (Fig. 1A and B). Both the GEM-treated
groups exhibited significantly reduced tumor weights compared with
the control group. However, there were no differences in tumor
weight between the mice treated with MET alone and the control
animals. The minimal T/C% on day 42 was 55.6% in the GEM-treated
mice, 88.7% in the MET-treated mice and 48.8% in the mice treated
with GEM and MET (Fig. 1C).
Anti-tumor activity of MET in the BxG30
xenograft model
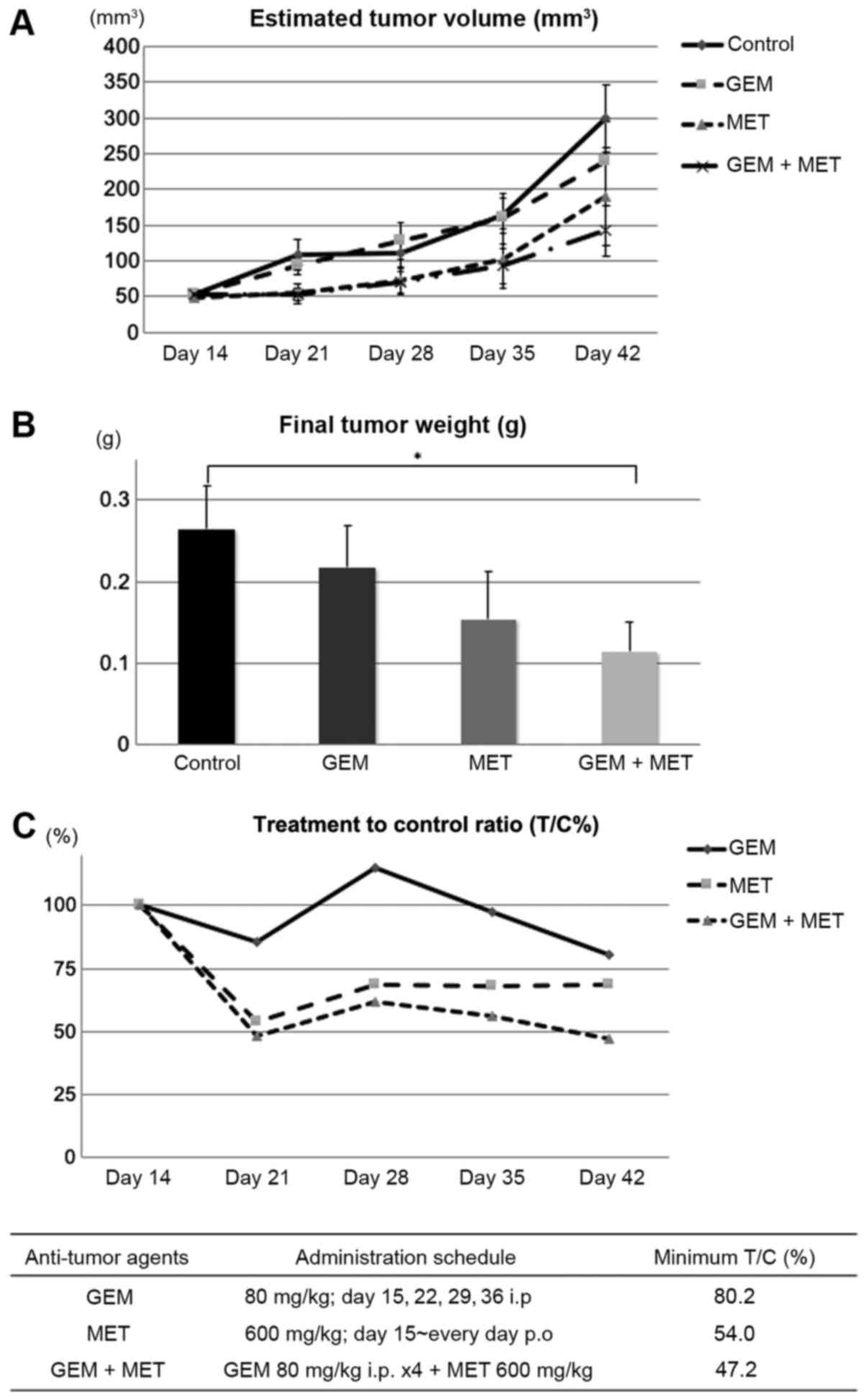
The anti-tumor activity of MET against BxG30, a
GEM-resistant PDAC cell line, was evaluated using a BxG30 xenograft
model. The final tumor weights were 0.26±0.05 g in the control mice
(n=14), 0.21±0.05 g in the GEM-treated mice (n=13), 0.15±0.06 g in
the MET-treated mice (n=10) and 0.11±0.03 g in the mice treated
with GEM and MET (n=14). Compared with the control animals, the
final tumor volumes were significantly decreased only in mice
treated with both GEM and MET (Fig. 2A
and B). The T/C% was 80.2% in the GEM-treated mice, 54.0% in
the MET-treated mice and 47.2% in the mice treated with both GEM
and MET. The anti-tumor effect of GEM against BxG30 cells was
clearly limited. The MET-treated animals exhibited a satisfactory
inhibition of tumor growth, although the minimal T/C% was <50%
on day 42 only in the group of mice treated with both GEM and MET
(Fig. 2C). These data indicated
that combination therapy with GEM and MET exerted marked anti-tumor
activity against GEM-resistant PDAC.
It was decided that the specimens were inadequate
for use in further experiments, as some specimens harvested from
both the BxPC-3 and BxG30 xenograft model were necrotic in some
places with ulceration of the skin and/or bleeding from tumors.
Cytotoxicity assay
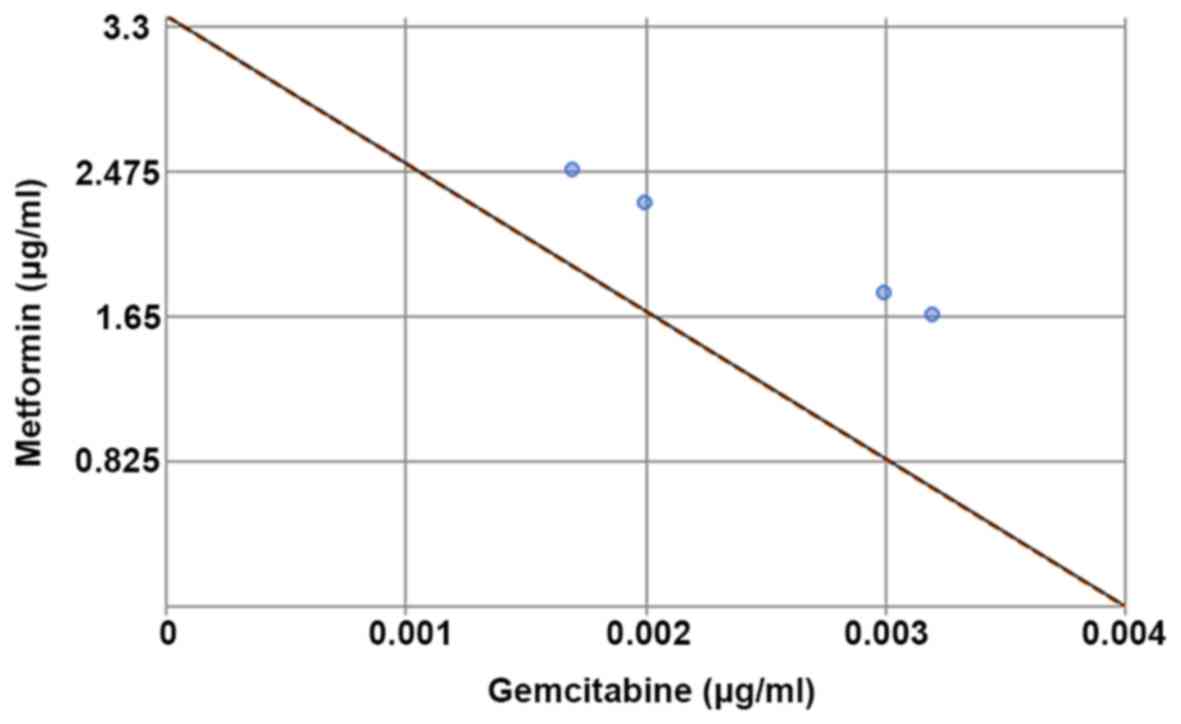
We initially evaluated the cytotoxic effects of MET
and GEM in BxG30 cells using an in vitro WST cytotoxicity
assay. Both agents inhibited cell growth in a concentration- and
time-dependent manner (data not shown). The IC50 values
(means ± SEM) of MET and GEM were 3.36±0.13 µg/ml and
3.75±0.22 ng/ml, respectively. The IC50 values of MET
and GEM were connected by a dotted line in a classical isobologram
to evaluate potential synergistic effects (Fig. 3), but none were observed.
Western blot analysis of pS6 and
HIF-1α
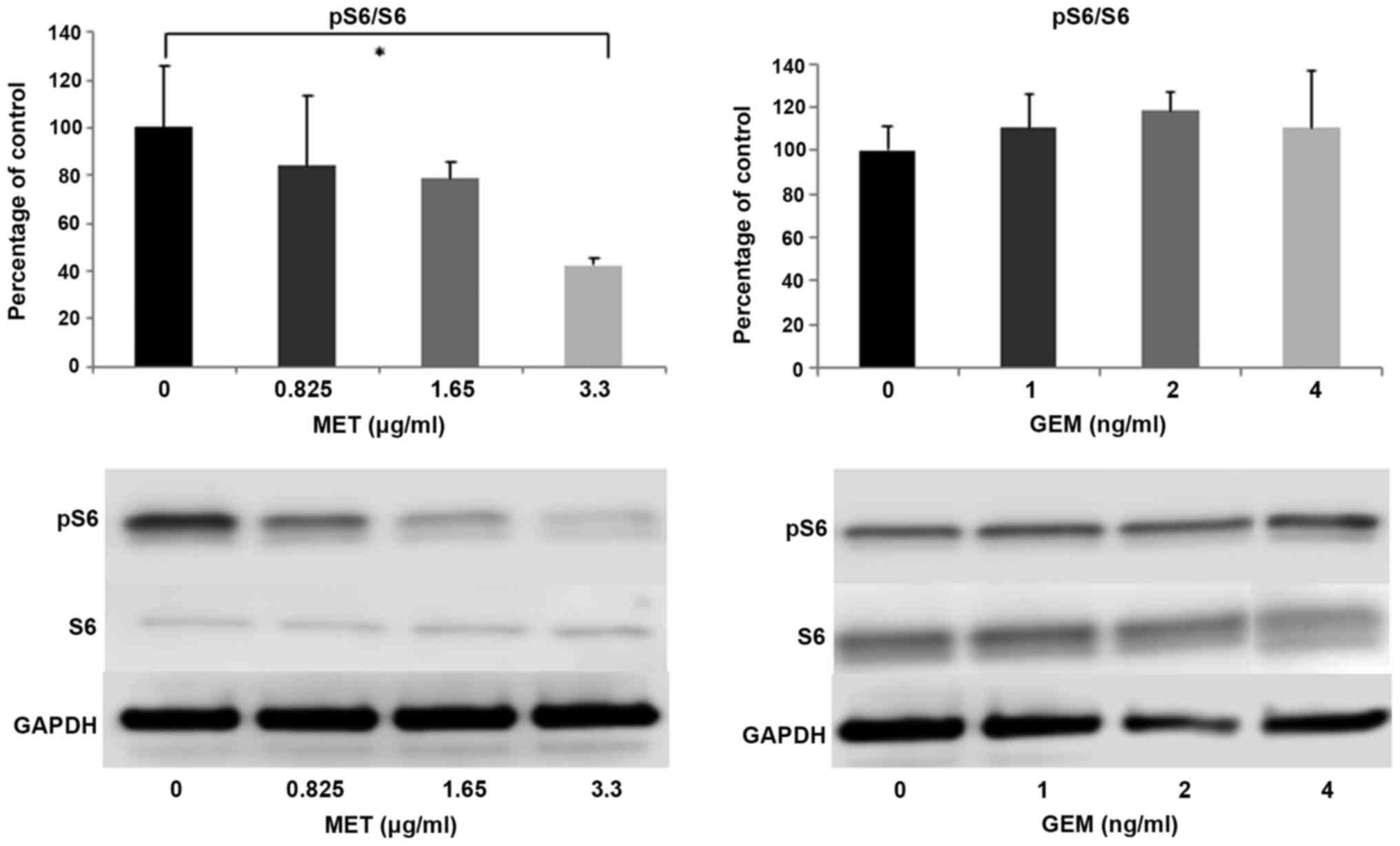
The phosphorylation of ribosomal protein S6 (pS6),
one of the most important downstream effectors of the mTOR
signaling pathway, was assessed by western blot analysis. The
relative expression of pS6 and total S6 (pS6/S6) was markedly
decreased in a dose-dependent manner in the cells treated with MET.
S6 phosphorylation was inhibited by incubation with MET, but not
with GEM (Fig. 4A and B). The
pS6/S6 ratios of each MET concentration were 84.08±29.66% (0.825
µg/ml), 78.48±6.74% (1.65 µg/ml) and 42.39±2.74% (3.3
µg/ml) relative to the control, respectively. Treatment with
3.3 µg/ml of MET significantly inhibited the activation of
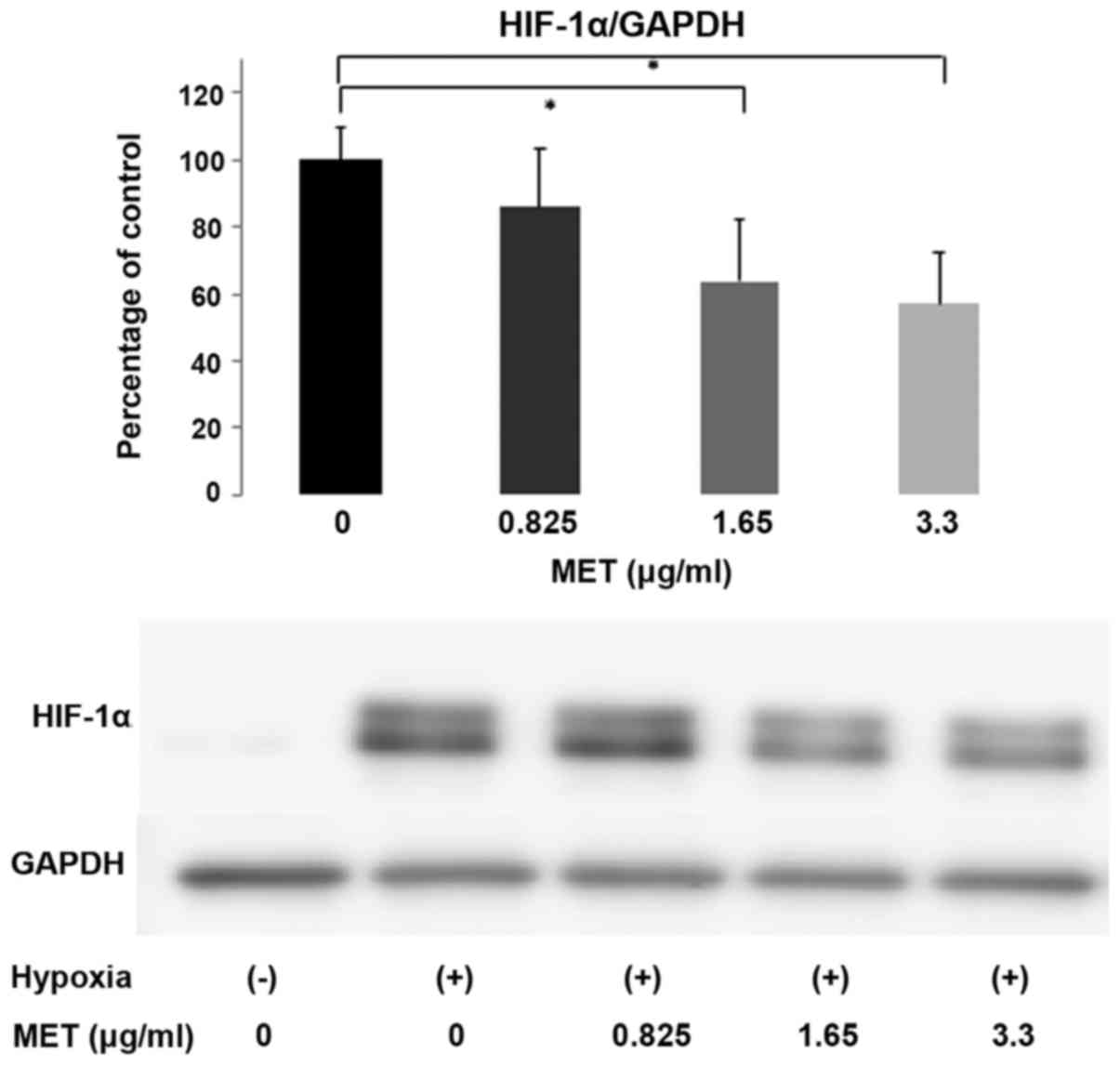
S6 compared with the control cells (P<0.05). HIF-1α is a
well-known downstream target of mTOR and its expression level was
also evaluated by western blot analysis. Hypoxic conditions clearly
induced the overexpression of HIF-1α. When the BxG30 cells were
treated with various concentrations of MET, the expression of
HIF-1α was suppressed in a dose-dependent manner. Treatment with
>1.65 µg/ml of MET significantly suppressed HIF-1α
expression, even under hypoxic conditions (Fig. 5).
Detection of VEGF production by
ELISA
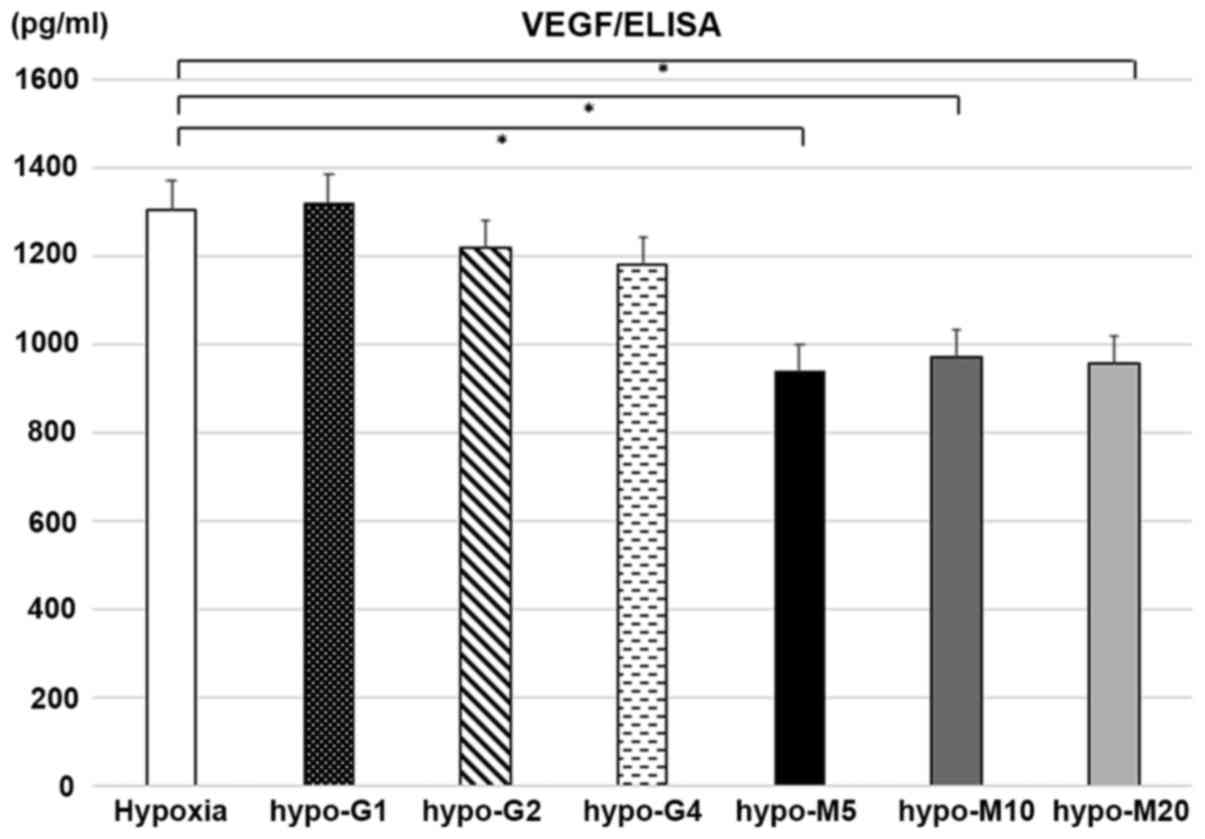
The production of VEGF by the BxG30 cells was
evaluated by ELISA under hypoxic conditions. The results revealed
that VEGF production was suppressed by MET treatment in comparison
with the untreated cells (P<0.01). However, no significant
difference was observed in the secretion of VEGF between the
GEM-treated and untreated cells (Fig.
6).
Discussion
The results of this study revealed that MET exerted
anti-tumor activity against PDAC. Combination therapy with GEM and
MET exerted potent anti-tumor activity not only against wild-type
PDAC xenografts, but also against GEM-resistant PDAC xenografts.
The data of this study also demonstrated that MET suppressed the
expression of HIF-1α and VEGF in tumor cells through the inhibition
of the mTOR signaling pathway. These results suggested that the
anti-tumor activity of MET was mediated through the inhibition of
angiogenesis, which is a very different mechanism of action from
that of GEM.
Pancreatic cancer is the third-leading cause of
cancer-related mortality in the United States, with a 5-year
survival rate of approximately 7-8% (22). Although novel modern
chemotherapeutics have been developed, such as FOLFIRINOX and
nab-PTX + GEM, the prognosis of patients with PDAC remain dismal.
Contemporary drug therapy for PDAC centers on combining multiple
cytotoxic agents with overlapping dose-limiting toxicities, and
thus there is an urgent need for the development of novel drugs
with anti-tumor mechanisms different from those of the cytotoxic
drugs currently in use.
Up to 85% of patients with PDAC have diabetes or
hyperglycemia, which frequently manifest as early as 2 to 3 years
prior to PDAC diagnosis (23).
Additionally, pancreatic surgery is becoming more common in the
developed world, with an increasing number of patients developing
diabetes subsequent to either partial or total pancreatectomy.
Secondary diabetes following pancreatectomy has been reported in
5-10% of patients in western countries (24,25).
MET, an anti-hyperglycemic drug, is the first-line treatment for
type II diabetes and is a widely prescribed anti-diabetic drug;
hence, one might expect that MET is used in a large number of
patients with PDAC. Numerous epidemiological studies have indicated
that the administration of MET in patients with type II diabetes is
associated with a reduced cancer incidence and cancer-related
mortality (7,8,26).
Therefore, MET has been regarded as a potential therapeutic agent,
particularly for diabetic patients with PDAC. GEM is still one of
the key agents used for the chemotherapy of PDAC; however, acquired
resistance to GEM has become a major concern. Thus, in this study,
we assessed the anti-tumor effects of MET against PDAC, which we
expected should involve a mechanism of action different from that
of GEM. This assumption was based on observations from intractable
PDAC cases in patients with diabetes and from experiments using a
GEM-resistant PDAC cell line.
We previously reported the cloning of a
GEM-resistant cell line derived from the wild-type PDAC cell line,
BxPC-3 (19). GEM-resistant clones
overexpressed ribonucleotide reductase subunit M1 (RRM1), an enzyme
involved in metabolism of GEM. When the expression of RRM1 was
high, resistance to GEM was high.
In mice implanted with wild-type BxPC-3 xenografts,
MET treatment alone exerted limited inhibitory effects on tumor
growth compared with GEM treatment alone. GEM treatment exerted
more favorable anti-tumor effects on wild-type BxPC-3 xenografts;
however, only combination therapy with MET and GEM resulted in
effective anti-tumor activity as measured using the T/C%, the
minimal treated-control ratio, which revealed that inhibition was
<50% on day 42. According to these data, combination therapy
with MET and GEM was regarded as the promising treatment.
We also evaluated the anti-tumor effect of MET in
mice implanted with GEM-resistant xenografts. The results indicated
that the anti-tumor effects of GEM on BxG30 xenografts were
limited, while MET treatment markedly inhibited tumor growth.
Combination therapy with MET and GEM resulted in <50% of minimal
T/C% on day 42, confirming that combination therapy exerted potent
anti-tumor effects even against GEM-resistant tumors.
We subsequently evaluated the cytotoxic effects of
MET and GEM on BxG30 cells using an in vitro WST assay. The
results of the classical isobologram revealed that combination
treatment with MET and GEM did not exert a synergistic effect
against BxG30 cells. This discrepancy between the results of the
in vivo and in vitro experiments suggested that the
anti-tumor effect of MET may not result from cytotoxicity, but
through indirect effects on the tumor microenvironment in
vivo.
It is well known that MET activates AMPKα, resulting
in the inhibition of the mTOR signaling pathway and its downstream
effectors (27-30). Indeed, MET has been shown to
inhibit the constitutive and induced activation of mTOR in several
pancreatic cancer cell lines (31,32).
mTOR is a highly evolutionarily conserved protein kinase that plays
a key role in the integration of signals from growth factors,
nutrients and the energy status of cells (33). mTOR signaling plays a pivotal role
in the proliferation and survival of PDAC cells and is activated in
pancreatic cancer tissues (34-36).
One of the downstream effectors of mTOR is ribosomal S6 kinase
(S6K1), which enhances and phosphorylates pS6, leading to the
translation of mRNA (37). A
previous study indicated that the S6K1 pathway is the major
mTOR-dependent downstream mediator of mTOR-regulated G1-phase
progression (38). This pathway
also mediates the mTOR-dependent control of cell growth and cell
size (39). Consequently, mTOR has
emerged as an attractive therapeutic target in PDAC.
In this study, the results of western blot analysis
revealed a marked decrease in the pS6/S6 ratio in MET-treated
cells, indicating the significant suppression of mTOR activity by
MET. By contrast, we observed no suppressive effect of GEM on mTOR
activity. This alternative anti-tumor effect of MET, which differs
from other cytotoxic anti-cancer drugs, may have resulted in the
discrepant anti-tumor effects of combination treatment observed in
the in vivo and in vitro experiments.
Hypoxic conditions have been detected in several
human malignancies, including PDAC (17). Tumor hypoxia occurs when the
consumption of oxygen exceeds its delivery by the vascular system.
In experimental studies, hypoxia predicts aggressive growth and
spontaneous metastasis formation in a PDAC xenograft (40). Hypoxic conditions contribute to the
induction of HIF-1, a key regulator of the cellular response to
oxygen deprivation (41). HIF-1 is
a heterodimeric transcription factor containing an
inducibly-expressed HIF-1α subunit and a constitutively-expressed
HIF-1β subunit. HIF-1α is well known as one of the most important
downstream effectors of the mTOR signaling pathway, and plays a
major role in tumor progression. Thus, the expression of HIF-1α was
evaluated by western blot analysis. The results revealed the
significant inhibition of HIF-1α expression by MET, but not by GEM,
under hypoxic conditions.
The inhibition of HIF-1α activity leads to the
suppression of VEGF expression. In this study, ELISA experiments
revealed the suppression of VEGF production by MET treatment
compared with the untreated cells. By contrast, GEM treatment did
not suppress VEGF production in BxG30 cells. PDAC is often
considered as a hypoxic cancer. Hence, the majority of pancreatic
cancer cells induce a high expression of HIF-1α via the activation
of the mTOR signaling cascade. Additionally, the overexpression of
HIF-1α leads to the induction of VEGF expression. The findings of
this study suggest that MET may inhibit tumor progression through
the suppression of the mTOR/HIF-1α/VEGF signaling cascade; this
mechanism is in marked contrast with GEM, which does not achieve
anti-tumor activity by suppressing this signaling cascade.
To summarize the above-mentioned results, the
findings of this study demonstrated that combination therapy
exerted excellent anti-tumor effects even for GEM-resistant PDAC in
the animal model. Our findings also demonstrated that MET
downregulated mTOR activity, through the evaluation of the
phosphorylation of ribosomal protein S6. Furthermore, we
demonstrated that HIF-1α expression was inhibited by MET treatment,
leading to the suppression of VEGF production under hypoxic
conditions. Moreover, the cytotoxic effect of MET was not proven by
cytotoxic assay, suggesting that the anti-tumor effects of MET are
not produced through cytotoxicity, but rather through the
environment surrounding the tumor, particularly the inhibition of
angiogenesis via the inhibition of VEGF activity. Similarly, a
previous study reported that mTOR may play an important role as an
upstream activator of HIF-1 function in cancer cells and may carry
out its anti-tumor activity through mTOR/HIF1α/VEGF cascade
(40).
In conclusion, the data of the present study
demonstrated that MET restricts PDAC tumor growth by suppressing
mTOR/HIF-1 signaling, which is a different anti-tumor mechanism
than that of GEM. These results are of clinical interest and reveal
the potential use of MET in the treatment of PDAC.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article or are available from the
corresponding author on reasonable request.
Authors’ contributions
KS, OT, YS and YK participated in the conception and
design of the study. KS and OT conducted the experiments. KS and OT
performed data analysis. KS wrote or contributed to the writing of
the manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The protocol of the animal experiments was reviewed
and approved by The Laboratory Animal Care and Use Committees of
Kitasato University on April 1st, 2013 (approval no. 13023). Animal
experiments were performed in accordance with the ethical
guidelines of the Kitasato Institute.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
MET
|
metformin
|
|
GEM
|
gemcitabine
|
|
PDAC
|
pancreatic ductal adenocarcinoma
|
|
mTOR
|
mammalian target of rapamycin
|
|
HIF-1α
|
hypoxia-inducible factor 1α
|
|
VEGF
|
vascular endothelial growth factor
|
Acknowledgments
The authors are grateful to the members of the
Department of Surgery of Kitasato Institute Hospital (Tokyo, Japan)
for their helpful suggestions and assistance. The authors would
also like to thank the students of the Department of Research and
Education Center for Clinical Pharmacy, Kitasato University School
of Pharmacy, for assisting with basic experiments. In addition, the
authors would like to thank Ms. Renee Mosi from Edanz Group
(www.edanzediting.com/ac) for editing a
draft of this manuscript.
References
|
1
|
Yadav D and Lowenfels AB: The epidemiology
of pancreatitis and pancreatic cancer. Gastroenterology.
144:1252–1261. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
DeSantis CE, Lin CC, Mariotto AB, Siegel
RL, Stein KD, Kramer JL, Alteri R, Robbins AS and Jemal A: Cancer
treatment and survivorship statistics, 2014. CA Cancer J Clin.
64:252–271. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Heinemann V: Present and future treatment
of pancreatic cancer. Semin Oncol. 29(Suppl 9): 23–31. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hertel LW, Boder GB, Kroin JS, Rinzel SM,
Poore GA, Todd GC and Grindey GB: Evaluation of the antitumor
activity of gemcitabine (2′,2′-difluoro-2′-deoxycytidine). Cancer
Res. 50:4417–4422. 1990.PubMed/NCBI
|
|
5
|
Conroy T, Desseigne F, Ychou M, Bouché O,
Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de
la Fouchardière C, et al Groupe Tumeurs Digestives of Unicancer;
PRODIGE Intergroup: FOLFIRINOX versus gemcitabine for metastatic
pancreatic cancer. N Engl J Med. 364:1817–1825. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Von Hoff DD, Ervin T, Arena FP, Chiorean
EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et
al: Increased survival in pancreatic cancer with nab-paclitaxel
plus gemcitabine. N Engl J Med. 369:1691–1703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Evans JM, Donnelly LA, Emslie-Smith AM,
Alessi DR and Morris AD: Metformin and reduced risk of cancer in
diabetic patients. BMJ. 330:1304–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Libby G, Donnelly LA, Donnan PT, Alessi
DR, Morris AD and Evans JM: New users of metformin are at low risk
of incident cancer: A cohort study among people with type 2
diabetes. Diabetes Care. 32:1620–1625. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Decensi A, Puntoni M, Goodwin P, Cazzaniga
M, Gennari A, Bonanni B and Gandini S: Metformin and cancer risk in
diabetic patients: A systematic review and meta-analysis. Cancer
Prev Res (Phila). 3:1451–1461. 2010. View Article : Google Scholar
|
|
10
|
Singh S, Singh PP, Singh AG, Murad MH,
McWilliams RR and Chari ST: Anti-diabetic medications and risk of
pancreatic cancer in patients with diabetes mellitus: A systematic
review and meta-analysis. Am J Gastroenterol. 108:510–519; quiz
520. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jalving M, Gietema JA, Lefrandt JD, de
Jong S, Reyners AK, Gans RO and de Vries EG: Metformin: Taking away
the candy for cancer? Eur J Cancer. 46:2369–2380. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zakikhani M, Dowling R, Fantus IG,
Sonenberg N and Pollak M: Metformin is an AMP kinase-dependent
growth inhibitor for breast cancer cells. Cancer Res.
66:10269–10273. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Morran DC, Wu J, Jamieson NB, Mrowinska A,
Kalna G, Karim SA, Au AY, Scarlett CJ, Chang DK, Pajak MZ, et al
Australian Pancreatic Cancer Genome Initiative (APGI): Targeting
mTOR dependency in pancreatic cancer. Gut. 63:1481–1489. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhong H, Chiles K, Feldser D, Laughner E,
Hanrahan C, Georgescu MM, Simons JW and Semenza GL: Modulation of
hypoxia-inducible factor 1alpha expression by the epidermal growth
factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human
prostate cancer cells: Implications for tumor angiogenesis and
therapeutics. Cancer Res. 60:1541–1545. 2000.PubMed/NCBI
|
|
15
|
Forsythe JA, Jiang BH, Iyer NV, Agani F,
Leung SW, Koos RD and Semenza GL: Activation of vascular
endothelial growth factor gene transcription by hypoxia-inducible
factor 1. Mol Cell Biol. 16:4604–4613. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Büchler P, Reber HA, Lavey RS, Tomlinson
J, Büchler MW, Friess H and Hines OJ: Tumor hypoxia correlates with
metastatic tumor growth of pancreatic cancer in an orthotopic
murine model. J Surg Res. 120:295–303. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vaupel P: The role of hypoxia-induced
factors in tumor progression. Oncologist. 9(Suppl 5): 10–17. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Semenza GL: Defining the role of
hypoxia-inducible factor 1 in cancer biology and therapeutics.
Oncogene. 29:625–634. 2010. View Article : Google Scholar :
|
|
19
|
Yoneyama H, Takizawa-Hashimoto A, Takeuchi
O, Watanabe Y, Atsuda K, Asanuma F, Yamada Y and Suzuki Y: Acquired
resistance to gemcitabine and cross-resistance in human pancreatic
cancer clones. Anticancer Drugs. 26:90–100. 2015. View Article : Google Scholar
|
|
20
|
Office of Laboratory Animal Welfare
(OLAW): Institutional Animal Care and Use Committee Guidebook. OLAW
National Institutes of Health; Bethesda, MD: 2002
|
|
21
|
Ovejera AA, Houchens DP and Barker AD:
Chemotherapy of human tumor xenografts in genetically athymic mice.
Ann Clin Lab Sci. 8:50–56. 1978.PubMed/NCBI
|
|
22
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sah RP, Nagpal SJ, Mukhopadhyay D and
Chari ST: New insights into pancreatic cancer-induced
paraneoplastic diabetes. Nat Rev Gastroenterol Hepatol. 10:423–433.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cui Y and Andersen DK: Pancreatogenic
diabetes: Special considerations for management. Pancreatology.
11:279–294. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ewald N, Kaufmann C, Raspe A, Kloer HU,
Bretzel RG and Hardt PD: Prevalence of diabetes mellitus secondary
to pancreatic diseases (type 3c). Diabetes Metab Res Rev.
28:338–342. 2012. View Article : Google Scholar
|
|
26
|
Zhang ZJ, Zheng ZJ, Kan H, Song Y, Cui W,
Zhao G and Kip KE: Reduced risk of colorectal cancer with metformin
therapy in patients with type 2 diabetes: A meta-analysis. Diabetes
Care. 34:2323–2328. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cerezo M, Tichet M, Abbe P, Ohanna M,
Lehraiki A, Rouaud F, Allegra M, Giacchero D, Bahadoran P,
Bertolotto C, et al: Metformin blocks melanoma invasion and
metastasis development in AMPK/p53-dependent manner. Mol Cancer
Ther. 12:1605–1615. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim HG, Hien TT, Han EH, Hwang YP, Choi
JH, Kang KW, Kwon KI, Kim BH, Kim SK, Song GY, et al: Metformin
inhibits P-glycoprotein expression via the NF-κB pathway and CRE
transcriptional activity through AMPK activation. Br J Pharmacol.
162:1096–1108. 2011. View Article : Google Scholar :
|
|
29
|
Rozengurt E, Sinnett-Smith J and Kisfalvi
K: Crosstalk between insulin/insulin-like growth factor-1 receptors
and G protein-coupled receptor signaling systems: A novel target
for the antidiabetic drug metformin in pancreatic cancer. Clin
Cancer Res. 16:2505–2511. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sinnett-Smith J, Kisfalvi K, Kui R and
Rozengurt E: Metformin inhibition of mTORC1 activation, DNA
synthesis and proliferation in pancreatic cancer cells: Dependence
on glucose concentration and role of AMPK. Biochem Biophys Res
Commun. 430:352–357. 2013. View Article : Google Scholar :
|
|
31
|
Karnevi E, Said K, Andersson R and
Rosendahl AH: Metformin-mediated growth inhibition involves
suppression of the IGF-I receptor signalling pathway in human
pancreatic cancer cells. BMC Cancer. 13:2352013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Soares HP, Ni Y, Kisfalvi K, Sinnett-Smith
J and Rozengurt E: Different patterns of Akt and ERK feedback
activation in response to rapamycin, active-site mTOR inhibitors
and metformin in pancreatic cancer cells. PLoS One. 8:e572892013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Asano T, Yao Y, Zhu J, Li D, Abbruzzese JL
and Reddy SA: The rapamycin analog CCI-779 is a potent inhibitor of
pancreatic cancer cell proliferation. Biochem Biophys Res Commun.
331:295–302. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pham NA, Schwock J, Iakovlev V, Pond G,
Hedley DW and Tsao MS: Immunohistochemical analysis of changes in
signaling pathway activation downstream of growth factor receptors
in pancreatic duct cell carcinogenesis. BMC Cancer. 8:432008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ito D, Fujimoto K, Mori T, Kami K, Koizumi
M, Toyoda E, Kawaguchi Y and Doi R: In vivo antitumor effect of the
mTOR inhibitor CCI-779 and gemcitabine in xenograft models of human
pancreatic cancer. Int J Cancer. 118:2337–2343. 2006. View Article : Google Scholar
|
|
37
|
Gingras AC, Raught B and Sonenberg N:
Regulation of translation initiation by FRAP/mTOR. Genes Dev.
15:807–826. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fingar DC, Richardson CJ, Tee AR, Cheatham
L, Tsou C and Blenis J: mTOR controls cell cycle progression
through its cell growth effectors S6K1 and 4E-BP1/eukaryotic
translation initiation factor 4E. Mol Cell Biol. 24:200–216. 2004.
View Article : Google Scholar :
|
|
39
|
Fingar DC, Salama S, Tsou C, Harlow E and
Blenis J: Mammalian cell size is controlled by mTOR and its
downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 16:1472–1487.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chang Q, Jurisica I, Do T and Hedley DW:
Hypoxia predicts aggressive growth and spontaneous metastasis
formation from orthotopically grown primary xenografts of human
pancreatic cancer. Cancer Res. 71:3110–3120. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Semenza GL, Agani F, Feldser D, Iyer N,
Kotch L, Laughner E and Yu A: Hypoxia, HIF-1, and the
pathophysiology of common human diseases. Adv Exp Med Biol.
475:123–130. 2000. View Article : Google Scholar : PubMed/NCBI
|