Introduction
Lung cancer is the leading cause of
cancer-associated mortality worldwide (1,2),
with an associated 5-year survival rate of <16% (3). Advancements in treatment have been
made recently (4); however, the
molecular mechanisms underlying the pathogenesis of lung cancer
remain unknown. Therefore, there is an urgent requirement to
identify the molecular mechanisms underlying lung cancer in order
to develop novel treatment strategies and improve patient
survival.
Plakoglobin is a member of the armadillo family of
proteins, and is a structural and functional homolog of β-catenin
(5-7). Notably, previous studies have
reported that a decreased level of plakoglobin is associated with
shorter disease-free survival (DFS) and worse overall survival (OS)
in non-small cell lung cancer (NSCLC) (8-10).
On the contrary, He et al (11) demonstrated that high cytoplasmic
plakoglobin expression in tumor tissues is associated with poor DFS
and OS in patients with resected lung adenocarcinoma. Unlike
β-catenin, which has an oncogenic function, plakoglobin generally
acts as a tumor/metastasis suppressor (12,13).
Plakoglobin is weakly expressed in numerous NSCLC cell lines and
overexpression of plakoglobin in these cell lines has been
demonstrated to inhibit the proliferation of lung cancer cells
(14). Overexpressing plakoglobin
in SCC-9 squamous carcinoma cells was observed to induce a
mesenchymal to epidermoid phenotypic transition (15). Decreased expression of plakoglobin
is regulated by promoter hypermethylation in renal cell carcinoma
(16), prostate cancer (17), embryonal rhabdomyosarcoma (18) and mammary carcinoma (19); however, the factors affecting low
plakoglobin expression are largely unknown, and whether plakoglobin
is involved in other signaling pathways in lung cancer remains
unclear.
Aberrant expression of histone deacetylases (HDACs)
is a hallmark of numerous types of cancer, and is involved in
mediating the proliferation, migration, invasion and genotoxic
chemotherapy resistance of cancer cells (20). HDACs are potential anticancer drug
targets that may be used to treat patients with lung cancer
(21,22). HDACs act as transcriptional
repressors by interacting with corepressor complexes and regulating
the acetylation state of histones, resulting in condensed chromatin
(23). Plakoglobin regulates HDAC4
expression by binding to its promoter via interactions with the
T-cell factor/lymphoid enhancer-binding factor family of
transcription factors (24).
Plakoglobin expression is reduced in tumor tissues compared with in
normal tissues, and HDACs have been demonstrated to inhibit
plakoglobin expression in cancers (25,26);
however, which HDACs directly inhibit plakoglobin expression in
lung cancer require further investigation.
In the present study, the role of HDACs in
regulating plakoglobin expression in lung cancer was investigated.
HDAC7 was demonstrated to directly bind to the promoter of
plakoglobin to deacetylate H3K9 and H4K5 and inhibit plakoglobin
expression in lung cancer. This novel HDAC7/plakoglobin axis may
serve as a promising target for treating patients with lung
cancer.
Materials and methods
Cells and reagents
A total of three human lung cancer cell lines (H460,
H1299 and A549) were purchased from the American Type Culture
Collection (ATCC, Manassas, VA, USA) and were cultured in
Dulbecco's modified Eagle's medium (DMEM; cat. no. 11965-092)
supplemented with 10% fetal bovine serum (FBS) (cat. no. 10270-106)
(both from Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
and maintained at 37°C in an incubator containing 5%
CO2. Dimethyl sulfoxide, Trichostatin A (TSA) and
nicotinamide (NAM) were purchased from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany), and 7.5 mM NAM or 0.5 nM TSA was applied to
A549, H1299 or H460 cells for 24 h at 37°C in an incubator
containing 5% CO2.
Plasmids
The full-length cDNA of human HDAC7 and plakoglobin
were cloned into the pSin-puro vector (Focus Bioscience Co., Ltd.,
Nanchang, China), and the restriction sites were EcoRI and
NheI. The full-length cDNA of human HDAC (1-11)
was constructed by inserting a Flag tag into the HDAC N-terminus in
a pcDNA3.1(+) vector (cat. no. V790-20, Invitrogen; Thermo Fisher
Scientific, Inc.) and the plasmids were transfected into H460 cells
using Lipofectamine 2000 (no. 11668019; Thermo Fisher Scientific,
Inc.). All recombinant plasmids were verified by DNA sequencing
(Ruiboxingke Biotech Co., Ltd., Beijing, China).
Antibodies
Human anti-plakoglobin (cat. no. 75550), anti-GAPDH
(cat. no. 5174), anti-HDAC7 (cat. no. 33418), anti-Flag (cat. no.
14793) and the Acetyl-Histone Antibody Sampler kits containing
anti-H3K9, -H3K18, -H3K27 and -histone 3 (cat. nos. 9927), and
anti-H4K5, H4K12, histone 4 (cat. no. 8346) were obtained from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Anti-β-actin (cat.
no. 66009-1-1g) was procured from ProteinTech Group, Inc. (Chicago,
IL, USA).
Stable lines
H1299 cells stably expressing scrambled (Scr) or
HDAC7 short hairpin RNA (shRNA) were established using the
Sigma-Aldrich shRNA system (Merck KGaA), according to the
manufacturer's protocols. Scr shRNA (with no known targets in the
human genome) with the following sequence:
5′-GGGCGAGGAGCTGTTCACCG-3′, and the oligonucleotides for human
HDAC7 shRNA#1 and #2 were 5′-GATCCGGGTGCAAGTAAATA-3′ and
5′-CCACTATTCCTGGCTCTGCAA-3′, respectively. A total of 3 µg
pSin-puro delivering HDAC7 or plakoglobin, 3 µg
pSin-puro-empty vector, 3 µg shRNA-NC, 3 µg
shRNA-HDAC7#1 or #2 was co-transfected with 3 µg pMD2.G and
3 µg psPAX2 into 293 cells (ATCC) for 48 h. using
Lipofectamine 2000. The recombinant viruses were subsequently
collected and applied to H1299, A549 or H460 cells cultured with 8
µg/ml Polybrene for 24 h. The stable lines were selected
with 1 µg/ml puromycin for 2 weeks.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
RT-qPCR was performed as described (27). Briefly, total RNA obtained from
cell lines was isolated using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols. First-strand cDNA was synthesized at 42°C
for 60 min using the Revert Aid™ First Strand cDNA Synthesis kit
(cat. no. 6110A; Takara Bio, Inc., Otsu, Japan). Subsequently, the
qPCR reaction was performed in a CFX96 Real-Time PCR Detection
system (Bio-Rad Laboratories, Inc., Hercules, CA, USA) using
SYBR®-Green mix (Tiangen Biotech Co., Ltd., Beijing,
China). Thermal cycling of the qPCR reaction was initiated with a
denaturation step at 95°C for 15 min, and consisted of 40 cycles
(denaturation at 95°C 15 sec, annealing at 60°C for 30 sec and
elongation at 72°C for 30 sec). The amplified products were
examined using the 2−ΔΔCq method (28), and each sample was calibrated to
the expression levels of the housekeeping gene GAPDH. The primers
used for amplifying HDAC7, plakoglobin and GAPDH were as follows:
HDAC7, forward, 5′-GGCGGCCCTAGAAAGAACAG-3′ and reverse,
5′-CTTGGGCTTATAGCGCAGCTT-3′; plakoglobin, forward,
5′-TCTCCAACCTGACATGCAACA-3′ and reverse,
5′-CATAGTTGAGACGCACAGAGTTC-3′; and GAPDH, forward,
5′-ACAGTCAGCCGCATCTTCTT-3′ and reverse,
5′-GACAAGCTTCCCGTTCTCAG-3′.
Cell proliferation assay
In vitro cell proliferation was assessed
using the Cell Counting Kit-8 (CCK-8) assay. For cell
proliferation, cells were seeded in 96-well plates at a density of
1,000 cells/well and incubated for 1, 2, 3, 4 or 5 days; 10
µl of CCK-8 reagent (Beyotime Institute of Biotechnology,
Haimen, China) was then added to each well, followed by incubation
for 1.5 h at 37°C. The absorbance value (optical density) of each
well was measured at 450 nm using an iMark microplate reader
(Bio-Rad Laboratories, Inc.).
Chromatin immunoprecipitation (ChIP)
assay
The ChIP assay was performed using the ChIP kit
(cat. no. 53008, Active Motif, Carlsbad, CA, USA) as described
previously (29). Briefly, to fix
the cells, Complete Cell Fixative Solution (included in kit) was
added to the existing culture medium for the cells at 80%
confluence at room temperature, and the fixation reaction was
stopped by adding Stop Solution (included in kit) to the existing
culture medium. The cells were collected by centrifugation at 1,000
× g for 5 min at 4°C. Subsequently, the nuclear pellet was
resuspended in ChIP Buffer (included in kit). The cell lysate was
subjected to shearing using a sonication instrument (Ningbo Scientz
Biotechnology Co., Ltd., Ningbo, China) to a fragment length of
200-500 bp. Total genomic DNA (input) was quantified and 20
µg of chromatin from each sample was immunoprecipitated
overnight at 4°C with 5 µg anti-HDAC7 (cat. no. 33418; Cell
Signaling Technology, Inc.), anti-acetyl-H3K9 (cat. no. 39917) or
anti-acetyl-H4K5 (cat. no. 39699) (both from Active Motif), or
normal IgG as a negative control. Then, nucleosome complexes were
isolated with the protein G agarose beads for 3 h at 4°C. Bound
DNA-protein complexes were eluted and cross-links were reversed
after a series of washes using the washing reagent contain in the
ChIP kit. Purified DNA was resuspended in TE buffer. Subsequently,
the PCR reaction was performed using PrimeSTAR® Max DNA
Polymerase (cat. no. R045A; Takara Bio, Inc.). Thermal cycling of
the qPCR reaction was initiated with a denaturation step at 94°C
for 2 min, and consisted of 35 cycles (denaturation at 98°C 10 sec,
annealing at 60°C for 15 sec and elongation at 72°C for 30 sec).
The primers for the plakoglobin were as follows: Plakoglobin-ChIP,
forward, 5′-GGACAGTCAGGCGAGATAGC-3′ and reverse,
5′-CGAACAAAAGGCGAAGAGAC-3′
Transwell assays
For the Transwell migration assay,
4.0×104 (A549) or 1.5×105 (H1299) cells in
200 µl serum-free DMEM were added to cell culture inserts
with an 8-µm microporous filter, without an extracellular
matrix coating (BD Biosciences, Franklin Lakes, NJ, USA). DMEM
medium containing 10% FBS (Gibco; Thermo Fisher Scientific, Inc.;
cat. no. 10270-106) was then added to the bottom chamber. After 24
h of incubation at 37°C in an incubator containing 5%
CO2, the cells on the lower surface of the filter were
fixed, stained and examined under a light microscope. The number of
migrated cells in three random optical fields (magnification, ×10)
from triplicate filters was averaged. For the Transwell invasion
assay, 8.0×104 (A549) or 3.0×105 (H1299)
cells resuspended in 200 µl serum-free DMEM were added to
cell culture inserts, which contained 8-µm microporous
filters and were coated with Matrigel (BD Biosciences; cat. no.
354480). DMEM containing 10% FBS was then added to the bottom
chamber. After 24 h of incubation at 37°C in an incubator
containing 5% CO2, the cells on the lower surface of the
filter were fixed with 10% formalin for 15 min at room temperature,
stained with 0.1% crystal violet for 60 min at room temperature and
examined under a light microscope. The number of migrated cells in
three random optical fields (magnification, ×100) from triplicate
filters was averaged.
Western blotting
Western blotting procedures were performed as
described previously (3),
including the experimental conditions of the gels. Briefly,
cultured cells from all cell lines were lysed in ice-cold
radioimmunoprecipitation assay lysis buffer (cat. no. P0013C;
Beyotime Institute of Biotechnology) at 4°C for 30 min. Following
centrifugation (12,000 × g) at 4°C for 20 min, the lysates were
obtained and protein concentration was determined with the BCA
method. Equal amounts of protein (30 µg/lane) were separated
by 10% SDS-PAGE. The conditions were as follows: Voltage of 100 V
for 2.5 h at room temperature. Proteins were then transferred to
polyvinylidene fluoride membranes with an electrical current of 250
mA at 4°C for 2 h. To block the non-specific binding sites, the
membranes were incubated with 5% non-fat milk (in Tris-buffered
saline with 0.1% Tween-20) at room temperature for 60 min, and then
membranes were then incubated with the following primary
antibodies: Human anti-plakoglobin (cat. no. 75550, anti-GAPDH
(cat. no. 5174), anti-HDAC7 (cat. no. 33418), anti-Flag (cat. no.
14793) and the Acetyl-Histone Antibody Sampler kit (cat. nos. 9927
and 8346) (all from Cell signaling Technology, Inc.), anti-β-actin
(cat. no. 66009-1-1g; ProteinTech Group, Inc.) at a dilution of
1:1,000 overnight at 4°C. Subsequently, the membranes were
incubated with anti-rabbit IgG Secondary antibody
peroxidase-conjugated (cat. no. W401B; Promega Corporation,
Madison, WI, USA) and anti-Mouse IgG Secondary Antibody Peroxidase
Conjugated (cat. no. W402B; Promega Corporation) with the dilution
1:10,000 at room temperature for 2 h. Specific protein bands were
visualized using an enhanced chemiluminescence detection system
(cat. no. P0018F; Beyotime Institute of Biotechnology) and exposed
to radiographic film (Carestream Health, Rochester, NY, USA; cat.
no. 6535876).
Animal experiments
All animal studies were performed in accordance with
protocols approved by the Research Animal Resource Center of Sun
Yat-Sen University (Guangzhou, China). The mice were maintained in
specific pathogen free conditions at a temperature of 20-25°C, a
50-70% humidity, under a light/dark cycle of 12 h, with free access
to water and food. A total of 18 male athymic nude mice at 4 weeks
of age were obtained from Shanghai Institutes for Biological
Sciences, Chinese Academy of Sciences (Shanghai, China). For
subcutaneous injection, 2×106 H1299 cells with stably
expressing Scr or shHDAC7-1 were mixed with 0.2 ml PBS (pH 7.4) and
30% (v/v) Matrigel matrix (BD Biosciences). Suspensions were
injected subcutaneously into the flanks of 4-week-old male athymic
nude mice, which were monitored over 5 weeks. For tail vein
injection, 3×106 H1299 cells with stably expressing Scr
or shHDAC7-1 were resuspended in 300 µl PBS (Biological
Industries, Beit Haemek, Israel) and injected into the lateral tail
vein of mice that did not receive a subcutaneous injection of cells
(3×106 cells/animal). Mice were sacrificed 7 weeks
following injection, and nodules were counted and compared between
the H1299-scr and H1299-shHDAC7-1 groups.
Immunohistochemistry (IHC) and
histological evaluation
Samples were fixed in 10% formalin for 10 h at room
temperature and embedded in paraffin. Sections (3-µm thick)
were prepared and mounted onto positively-charged glass slides.
Sections for HDAC7 staining were incubated in 10 mM citrate buffer
(pH 6.0) and boiled in a microwave oven for 15 min. After
incubation with Protein Block Serum-Free (Dako; Agilent
Technologies, Inc., Santa Clara, CA, USA), sections were incubated
overnight with anti-HDAC7 (1:250) at 4°C in a humidified container.
Following washing with PBS three times, the tissue slides were
treated with a non-biotin horseradish peroxidase detection system
according to the manufacturer's protocols (Dako; Agilent
Technologies, Inc.). IHC staining was evaluated by two independent
pathologists who were experts in diagnosing lung cancer. HDAC7
detection was quantified as described (3). Briefly, the HDAC7 signal was detected
in the cytoplasm and the nucleus, and the intensity in tissues was
categorized into four categories: 0, absent; 1, weak; 2, moderate;
or 3, strong. The percentage of stained cells was categorized as:
0, no staining; 1, 1-10%; 2, 11-50%; 3, 51-80%; and 4, 81-100%. The
staining score for each tissue was calculated by multiplying the
intensity value by the percentage value, and the average of the
scores from the two pathologists was used as the final score.
Clinical dataset analysis
The association between HDAC7, and the clinical
characteristics or survival of lung cancer patients were analyzed
using the online KMplot database (30). The median value of expression was
selected as the cutoff in the online database; Cox's proportional
hazards model was used to estimate the hazard ratio.
Study approval
A total of 35 pairs of specimens were collected from
patients with lung cancer (median age, 45 years; age range, 33-78
years; male/female ratio, 3:4) with resection between March 2014
and September 2016, and inclusion criteria were as follow: Initial
diagnosis of lung cancer. Exclusion criteria included: Previous
radiotherapy, chemotherapy or surgery, and previous malignancy or
other concomitant malignant disease. Normal lung tissues were
collected at >5 cm from the edge of the tumor. The use of human
lung cancer tissues was reviewed and approved by the Ethics
Committee of the Sun Yat-Sen University Cancer Center. Informed
consent was obtained from patients.
The correlation between plakoglobin and
HDAC7 expression
The raw data for the correlation between plakoglobin
and HDAC7 expression were obtained from the MethHC 1.0.3. software
(http://methhc.mbc.nctu.edu.tw/php/index.php).
Statistical analysis
All statistical analyses were performed using SPSS
for Windows, version 16.0 (SPSS, Inc., Chicago, IL, USA). All
values from the in vitro assays were expressed as the mean ±
standard deviation or standard error of the mean of at least three
independent experiments or replicates. A paired Student's t-test
was used for the comparison of two groups, multiple group
comparisons were performed by using one-way analysis of variance
followed by a Tukey-Kramer post-hoc test. The correlation between
plakoglobin and HDAC7 expression was analyzed by a Pearson's
correlation analysis. The Mann-Whitney test was used to analyze the
difference expression of HADC7 in lung cancer tissues and paired
normal tissues. P<0.05 was considered to indicate a
statistically significant difference.
Results
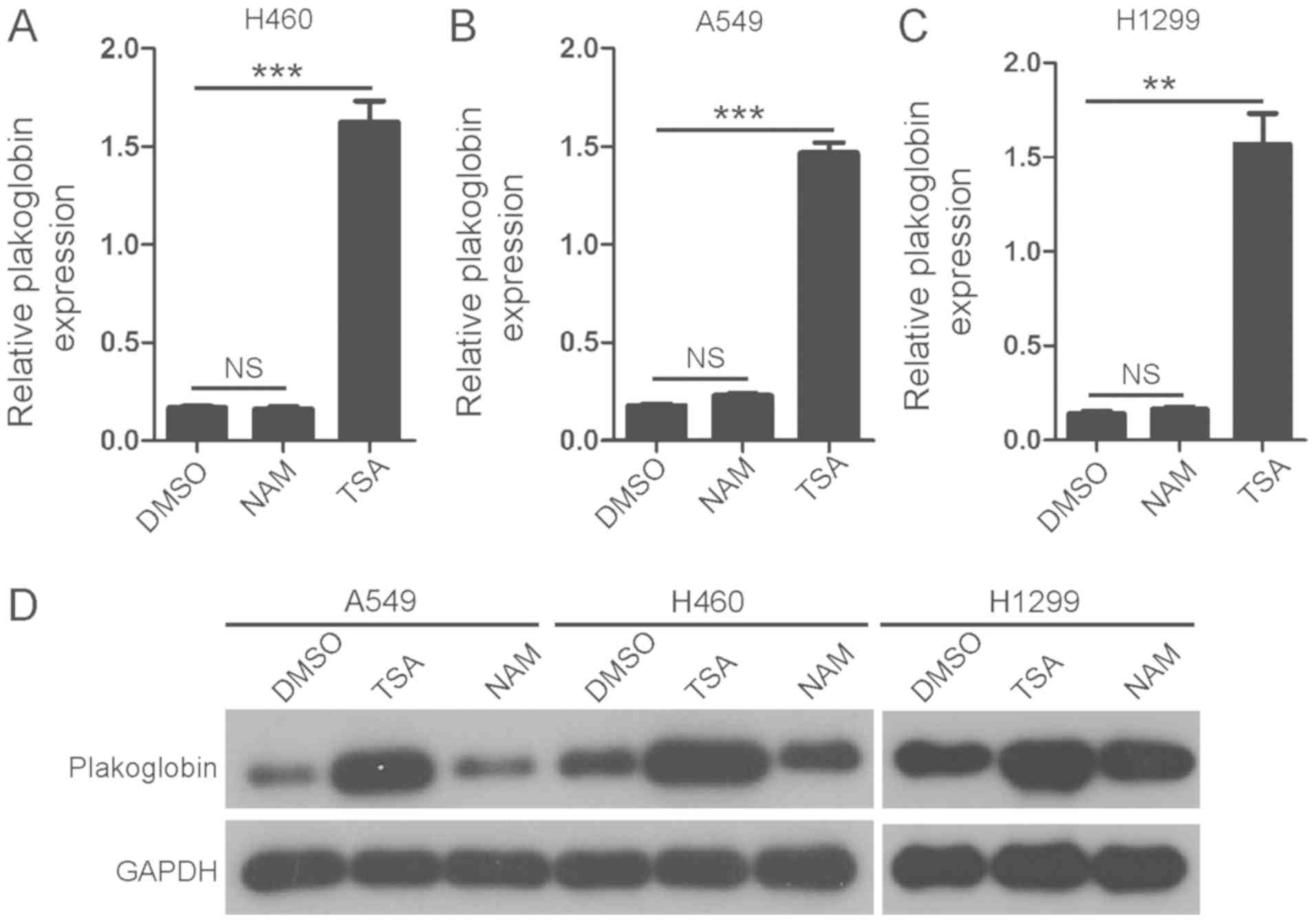
Treating lung cancer cells with TSA
increases the mRNA and protein expression of plakoglobin
H460, A549 and H1299 cells were treated with HDAC
inhibitors TSA and NAM for 24 h; these reagents are inhibitors of
class I and II HDACs and class III HDACs (31,32),
respectively. It was identified that the addition of TSA, but not
NAM, significantly increased the mRNA expression levels of
plakoglobin compared with the control (Fig. 1A-C); notable increases in the
protein expression levels of plakoglobin were observed following
TSA treatment. The results indicated that plakoglobin may be
regulated by class I and II HDACs in lung cancer cells.
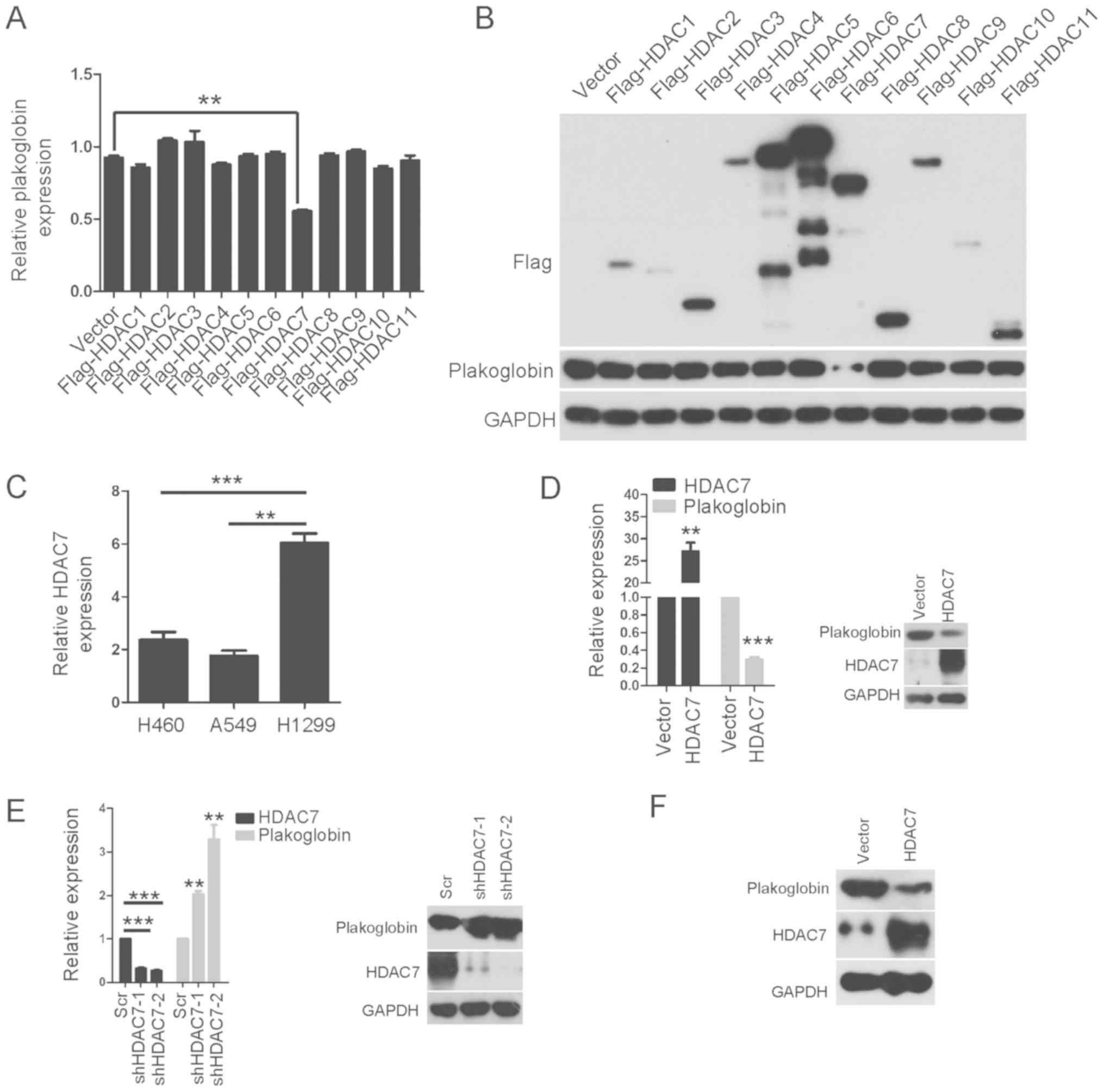
HDAC7 is a novel negative regulator of
plakoglobin in lung cancer
In order to determine which class I and II HDAC was
responsible for the suppression of plakoglobin expression, class I
and II HDACs were ectopically expressed in lung cancer cells. As
presented in Fig. 2A and B, only
ectopic HDAC7 significantly suppressed the mRNA levels of
plakoglobin in H460 cells; the protein expression levels of HDA7
were notably downregulated. Furthermore, the expression levels of
HDAC in three cancer cell lines was determined (Fig. 2C). It was evaluated whether
overexpression of HDAC7 could decrease plakoglobin expression in
other lung cancer cells according to the endogenous levels of HDAC7
in the H460, H1299 and A549 cell lines. RT-qPCR indicated that
stable expression of HDAC7 significantly suppressed the mRNA
expression levels of plakoglobin in A549 cells compared with the
control; the protein expression levels of plakoglobin following
HDAC7 overexpression were notably reduced (Fig. 2D). Thus, plakoglobin expression was
downregulated by overexpression of HDAC7. The opposite effect was
observed with HDAC7 knockdown in H1299 cells. As indicated in
Fig. 2E, the two shRNA sequences
effectively decreased the expression of endogenous HDAC7 levels
compared with the control, whereas the mRNA and protein expression
levels of plakoglobin increased. Furthermore, the protein
expression levels of plakoglobin decreased following stable
overexpression of HDAC7 in H1299 cells (Fig. 2F).
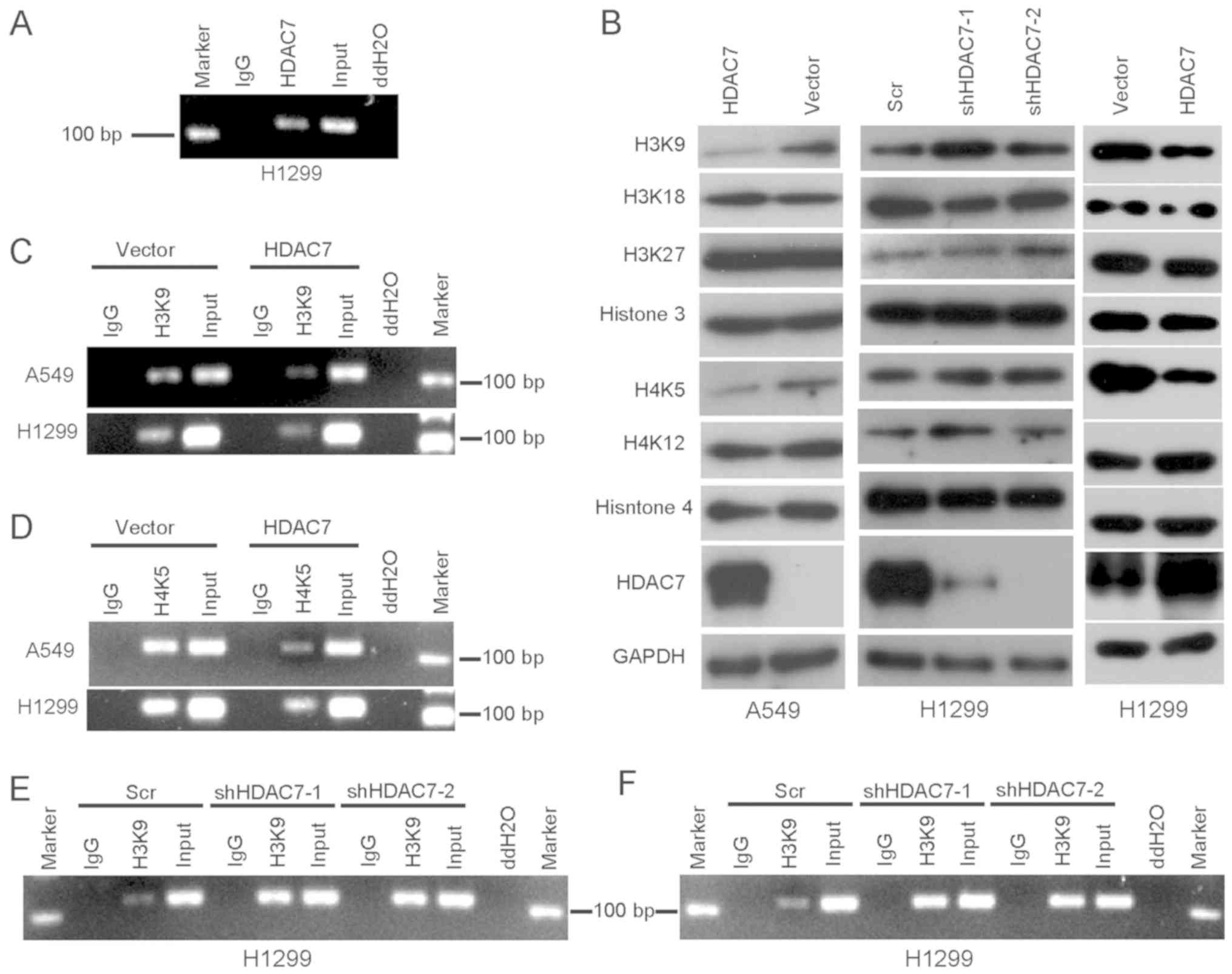
HDAC7 directly binds to the promoter
region of plakoglobin and deacetylates histones H3 and H4 in lung
cancer
To investigate the mechanisms by which HDAC7
participates in silencing plakoglobin expression, a ChIP assay was
performed to analyze whether HDAC7 directly binds to the
plakoglobin promoter. The results indicated that HDAC7 binds to the
plakoglobin promoter in H1299 cells (Fig. 3A). Acetylation of histones 3 and 4
was assessed by western blot analysis using antibodies targeting
lysine residues that are known to be acetylated. It was identified
that the levels of acetylation at H3K9 and H4K5 markedly increased
following knockdown of HDAC7 with shDAC7 #2 in H1299 cells compared
with the control (Fig. 3B);
however, overexpression of HDAC7 decreased the levels of the H3K9
and H4K5 acetylation in A549 and H1299 cells (Fig. 3B). To investigate whether H3K9 and
H4K5 acetylation was decreased in the promoter of plakoglobin, a
ChIP assay was performed to analyze H3K9 and H4K5 acetylation
within the plakoglobin promoter regions. The results indicated that
the degree of histone acetylation at this promoter was decreased in
HDAC7-overexpressing A549 and H1299 cells (Fig. 3C and D). By contrast, the level of
histone acetylation at this promoter increased in HDAC7-knocked
down H1299 cells (Fig. 3E and F).
Acetylation of H3K9 and H4K5 contributes to chromatin
decondensation (33), and the
enrichment of acetylated H3K9 and H4K5 near a transcription
initiation site is associated with the transcriptional activity of
the gene (33,34). Thus, HDAC7 may directly inhibit the
transcription of plakoglobin via binding to the promoter region of
plakoglobin and deacetylating histones H3K9 and H4K5 in lung
cancer.
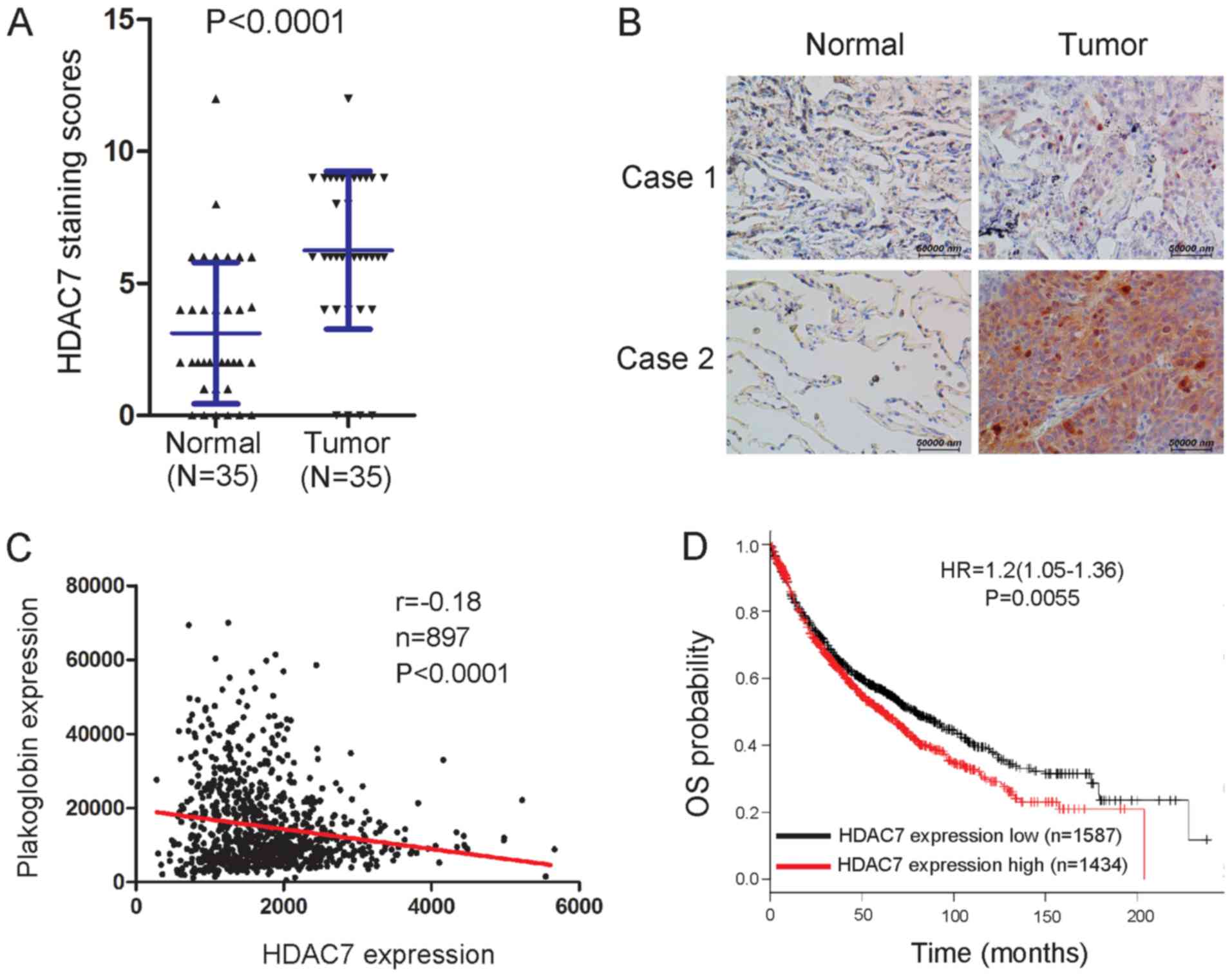
HDAC7 expression is increased in lung
cancer samples and higher HDAC7 levels predict a poor outcome
As plakoglobin is downregulated in lung cancer
tissues (9), the expression of
HDAC7 in lung cancer tissues was evaluated using IHC. The results
indicated that HDAC7 expression significantly increased in lung
cancer tissues compared with paired normal tissues (Fig. 4A); representative images were
presented (Fig. 4B). Using an
online database (35), an inverse
correlation was identified between HDAC7 and plakoglobin in tissues
(Fig. 4C). In addition, whether
the mRNA expression levels of HDAC7 is clinically relevant in lung
cancer was determined in the present study. Based on the HDAC7 mRNA
levels, lung cancer samples were subdivided into two groups, and
the associated OS was analyzed. Individuals with high HDAC7 levels
were observed to exhibit shorter OS compared with those with low
levels (Fig. 4D) using a large
public clinical microarray database of lung tumors from 2,170
patients (30). Therefore, HDAC7
expression levels may be a clinical predictor in lung cancer.
HDAC7 promotes lung cancer growth and
metastasis
Subsequently, it was investigated whether HDAC7
regulates the biological behaviors of lung cancer cells. Stable
cell lines with either ectopic expression of HDAC7 in A549 and
H1299 cells, or silenced HDAC7 in H1299 cells, were constructed
(Fig. 2C-E). Cell proliferation
assays indicated that the ectopic expression of HDAC7 significantly
promoted cell proliferation of A549 cells compared with the control
(Fig. 5A). The migration and
invasion of lung cancer cells was analyzed. The ectopic expression
of HDAC7 significantly enhanced cell migration and invasion
abilities of A549 cells compared with the control (Fig. 5B and C). The proliferative ability
was notably increased following HDAC7 overexpression in H1299 cells
(Fig. 5D); however, significant
increases in migration and invasion were observed compared with the
control (Fig. 5E and F). On the
contrary, knockdown of HDAC7 markedly reduced cell proliferation
(Fig. 5G), and significantly
inhibited H1299 cell migration and invasion compared with the
control (Fig. 5H and I). In
summary, these findings indicate that HDAC7 promotes lung cancer
cell proliferation, migration and invasion.
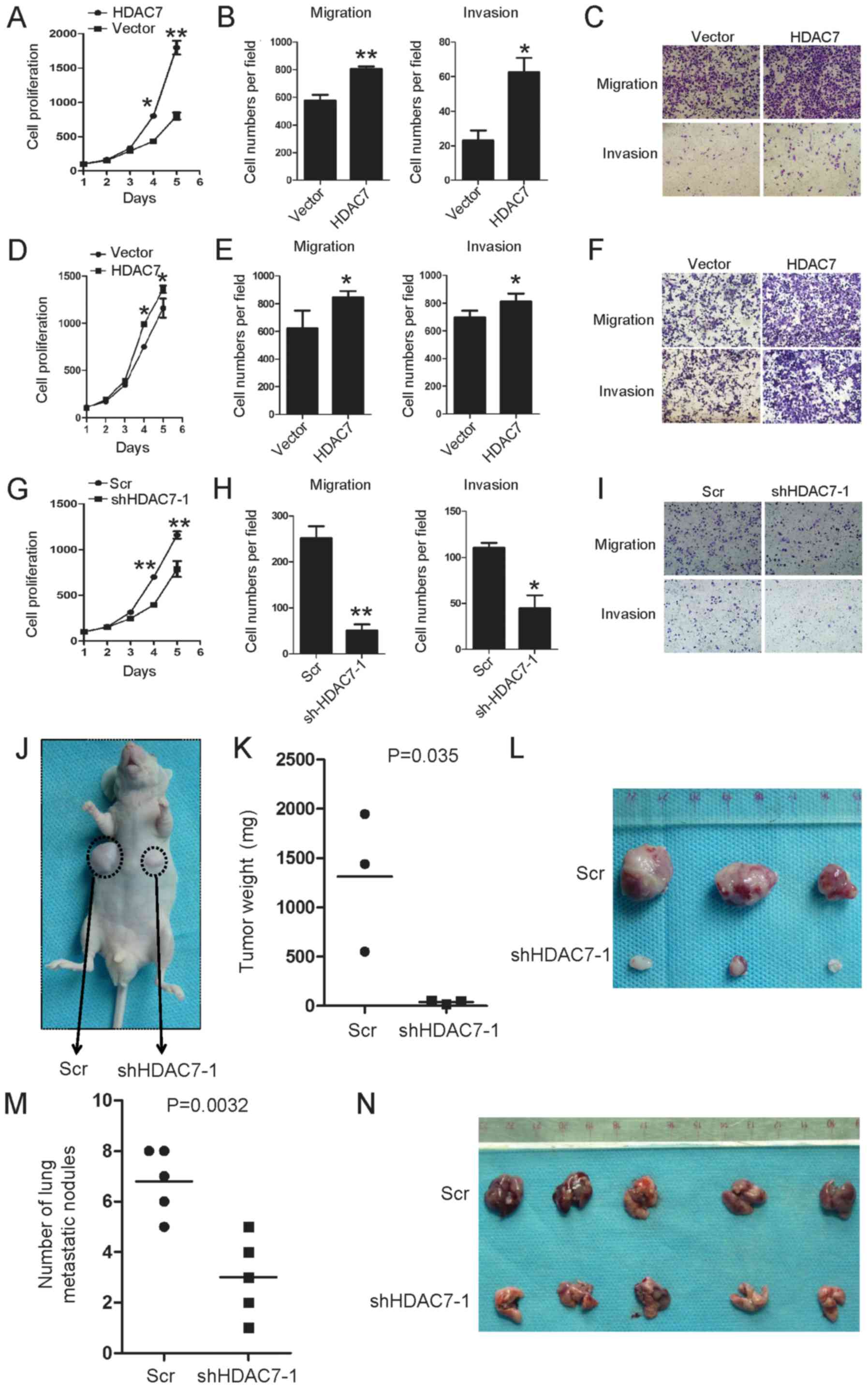 | Figure 5HDAC7 promotes tumor growth and
metastasis. (A and D) Cell proliferation of A549 and H1299 cells
transfected with HDAC7-overexpressing or empty vector in
vitro was measured at different time-points, as indicated by a
CCK-8 assay. Data are presented as the mean ± standard error.
*P<0.05, **P<0.01 vs. Vector. The
migration and invasion abilities of (B and C) A549 and (E and F)
H1299 cells transfected with HDAC7-overexpressing or empty vector
were measured at 24 h by Transwell assays. Data are presented as
the mean ± standard deviation. *P<0.05,
**P<0.01 vs. Vector . The number of cells passing via
the membrane in each cell was statistically analyzed in triplicate
and repeated three times with similar results. Magnification, ×100.
(C) and (F) are representative images. (G) Cell proliferation of
H1299 cells expressing sh-HDAC7-1 or Scr in vitro was
measured at different time-points, as indicated by a CCK-8 assay.
Data are presented as the mean ± standard error.
**P<0.01 vs. Scr. (H and I) Cell migration and
invasive abilities of H1299 cells expressing sh-HDAC7-1 and Scr
were measured at 24 h by Transwell assays. Data are presented as
the mean ± standard deviation. *P<0.05,
**P<0.01 vs. Scr. The number of cells passing via the
membrane in each cell was statistically analyzed in triplicate and
repeated three times with similar results. (I) Is a representative
image. (J-L) Stable H1299 with HDAC7 knockdown cells were injected
into nude mice, which were randomly divided into two groups. Mice
were sacrificed at 5 weeks following inoculation. Tumors were
excised from the mice and weighed. (M and N) Stable H1299 cells
with HDAC7 knockdown were injected via the tail vein, and the
number of visible lung metastases was counted 7 weeks later
(P=0.0032). HDAC, histone deacetylase; CCK-8, Cell Counting Kit-8;
Scr, scramble; sh, short hairpin RNA. |
As HDAC7 was observed to promote lung cancer cell
proliferation, migration and invasion in vitro, whether
HDAC7 promotes lung cancer cell tumorigenesis and metastasis was
investigated in vivo. To confirm the promoting effects of
HDAC7, an in vivo tumorigenesis study was performed by
inoculating shHDAC7 H1299 cells into nude mice. Mice in the
shHDAC7#1-H1299 and Scr groups were sacrificed 5 weeks after
inoculation, with average tumor weights of 0.04 and 1.313 g,
respectively (P<0.01; Fig.
5J-L). These results suggested a significant inhibitory effect
of decreased HDAC7 on in vivo tumorigenesis.
To further evaluate the role of HDAC7 in lung cancer
cell migration and invasion in vivo, lung cancer cells with
or without HDAC7 knockdown were injected into nude mice via the
tail vein. Cells with endogenous HDAC7 formed colonies in the lungs
within 7 weeks of injection, whereas the HDAC7 knockdown cells
resulted in significantly reduced pulmonary metastatic colonization
ability (Fig. 5M and N). These
results suggest that HDAC7 promotes lung tumor growth and
metastasis in vivo.
Suppression of plakoglobin by HDAC7
promotes cell proliferation, migration and invasion in lung
cancer
As the aforementioned experiments demonstrated that
plakoglobin was a direct target of HDAC7, whether loss of
plakoglobin contributes to the oncogenic role of HDAC7 in lung
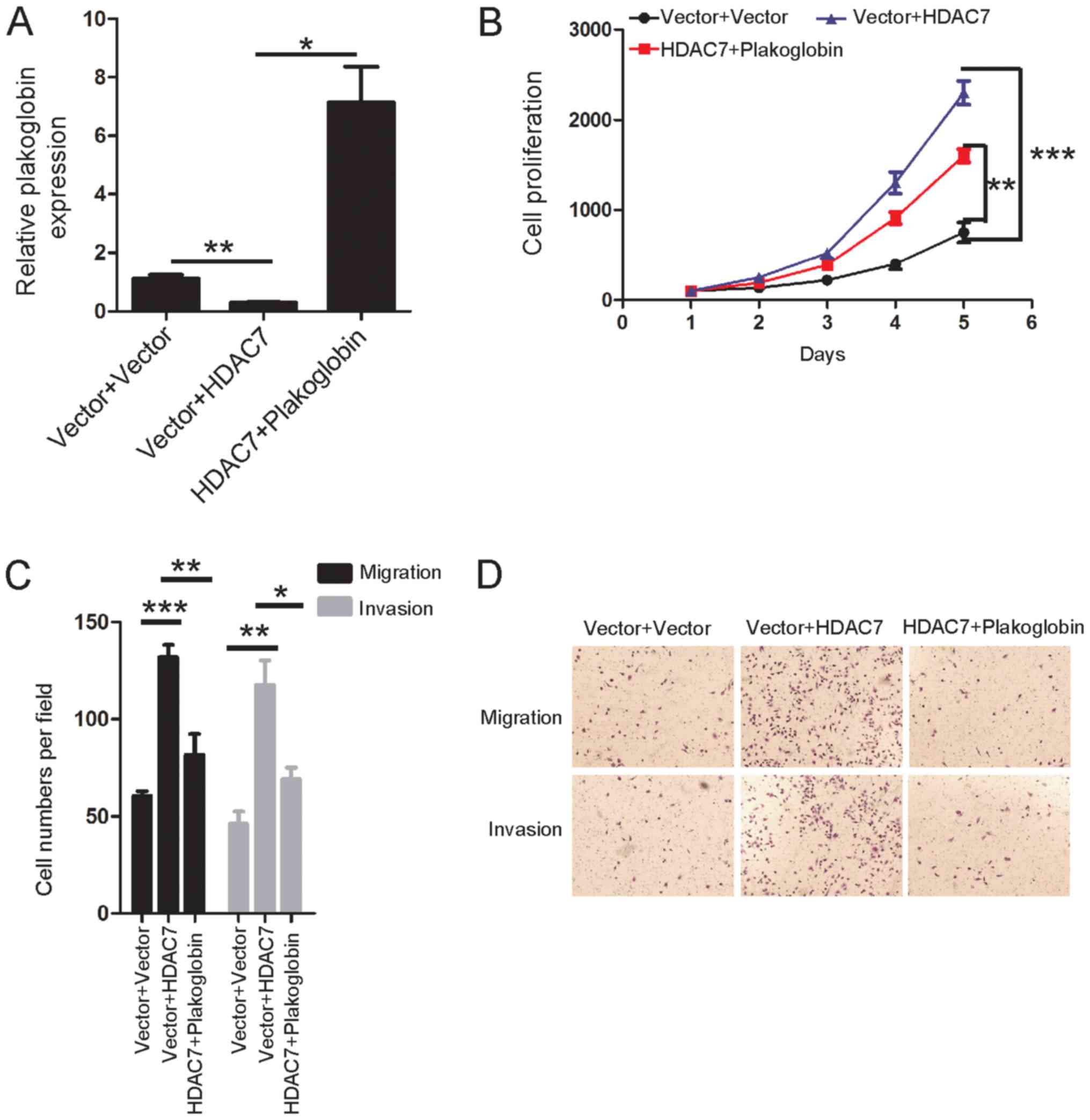
cancer was determined. Plakoglobin was stably induced into
HDAC7-overexpressing lung cancer cells to observe whether
restoration of plakoglobin expression could rescue the promoted
cell proliferation, migration and invasion of HDAC7-overexpressing
cells in vitro (Fig. 6A).
Notably, overexpression of plakoglobin significantly reduced the
effects of HDAC7 overexpression on cell proliferation, migration
and invasion compared with the control (Fig. 6B-D). These results indicated that
suppression of plakoglobin by HDAC7 promoted cell proliferation,
migration and invasion in lung cancer.
Discussion
Plakoglobin is a structural and functional homolog
of β-catenin (5); these proteins
serve two major roles in the cell, including the mediation of
cell-cell adhesion and cell signaling (5). As adhesive proteins, plakoglobin and
β-catenin can tether cadherin proteins to the cytoskeleton by
interacting with the cytoplasmic domain of cadherins (5). Unlike β-catenin, plakoglobin has been
demonstrated to be absent or weakly expressed in NSCLC cells and
tumor tissues (5).
Hypermethylation of the plakoglobin promoter mediates repression of
its transcription in lung cancer (18); however, the mechanisms that
contribute to the inactivation of plakoglobin expression in lung
cancer cells have not been fully investigated. The present study
aimed to determine which HDACs directly regulate plakoglobin
expression in lung cancer cells. The results indicated that HDAC7
could serve as a novel negative regulator of plakoglobin expression
in lung cancer.
HDACs (1, 2, 5, 7 and 10) are overexpressed in lung
cancer and promote lung cancer progression (36-40).
HDAC7 promotes the proliferation of cancer cells, including HeLa,
HCT116 and MCF-7 cells, which is likely to occur via stimulating
c-Myc and inhibiting p21 and p27 expression (41-44);
however, HDAC7 depletion had no effect on the proliferation of
glioblastoma U87 cells in vitro (45). Therefore, its role in cell
proliferation may vary in different cancer cell types and act via
different mechanisms. Lei et al (41) reported that HDAC7 has positive
effects on lung tumorigenesis in mice and cell proliferation/soft
agar colony formation of human lung cancer cell lines by inhibiting
signal transducer and activator 3 activation via its deacetylation.
The present study also indicated that HDAC7 promoted lung cancer
cell proliferation, migration and invasion in vitro, and
promoted H1299 cell tumor growth and metastasis in vivo.
Mechanistically, it was demonstrated that HDAC7 can directly
suppress plakoglobin expression in lung cancer by promoting the
deacetylation of H3 and H4 histones at the promoter region; a
negative correlation between HDAC7 and plakoglobin in lung cancer
tissues was reported in the present study. These findings indicate
a novel mechanism by which plakoglobin expression is lost in lung
cancer cells. The results also suggested that HDAC7 promotes lung
cancer cell proliferation, migration and invasion, at least partly
via inhibiting plakoglobin expression, as demonstrated by rescue
experiments. In addition, plakoglobin was identified as a target
gene of HDAC in human fibrosarcoma HT1080 cells (26), but the subtype of HDAC was unclear.
Therefore, further investigation was conducted in the present
study. The novel axis of HDAC7/plakoglobin may provide a basis for
the development of novel therapeutic strategies for human lung
cancer in the future.
To the best of our knowledge, this study is the
first to demonstrate plakoglobin is a target of HDAC7. It was also
demonstrated that plakoglobin inhibits HDAC7-mediated lung tumor
growth and metastasis, and this axis may be applied in the
development of novel therapeutic strategies for treating patients
with lung cancer.
Funding
This study was supported by the National Science
Foundation of China (grant no. 81660449 to YS; grant nos. 81460430
and 81760545 to SWL; grant no. 81372244 to YL) the Jiangxi
Provincial Natural Science Foundation of China (grant no.
20161ACB21001 to YS; grant no. 20171BCD40026 to YS), the Jiangxi
Provincial Health and Family Planning Commission Foundation (grant
nos. 20164005 and 2015A077 to YS) and the Guangxi Natural Science
Foundation of China (grant no. 2016GXNS FDA380037 to SWL).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YS, YL and SWL made substantial contributions to the
conception and design of the present the study. YS LS, YW and WY
developed the methodology, and acquired, analyzed and interpreted
the data. YL and SWL wrote and reviewed the manuscript. SWL
supervised the study. All authors read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Research
Animal Resource Center and the Ethical Committee of Sun Yat-Sen
University. Informed consent was obtained from patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Best MG, Sol N, In't Veld SG, Vancura A,
Muller M, Niemeijer AN, Fejes AV, Tjon Kon Fat LA, Huis In't Veld
AE, Leurs C, et al: Swarm intelligence-enhanced detection of
non-small-cell lung cancer using tumor-educated platelets. Cancer
Cell. 32:238–252.e239. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Serresi M, Gargiulo G, Proost N, Siteur B,
Cesaroni M, Koppens M, Xie H, Sutherland KD, Hulsman D, Citterio E,
et al: Polycomb repressive complex 2 is a barrier to KRAS-driven
inflammation and epithelial-mesenchymal transition in
non-small-cell lung cancer. Cancer Cell. 29:17–31. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lv XB, Liu L, Cheng C, Yu B, Xiong L, Hu
K, Tang J, Zeng L and Sang Y: SUN2 exerts tumor suppressor
functions by suppressing the Warburg effect in lung cancer. Sci
Rep. 5:179402015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Politi K, Ayeni D and Lynch T: The next
wave of EGFR tyrosine kinase inhibitors enter the clinic. Cancer
Cell. 27:751–753. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Aktary Z and Pasdar M: Plakoglobin: Role
in tumorigenesis and metastasis. Int J Cell Biol. 2012:1895212012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sechler M, Borowicz S, Van Scoyk M,
Avasarala S, Zerayesus S, Edwards MG, Kumar Karuppusamy Rathinam M,
Zhao X, Wu PY, Tang K, et al: Novel role for γ-catenin in the
regulation of cancer cell migration via the induction of hepatocyte
growth factor activator inhibitor type-1 (HAI-1). J Biol Chem.
290:15610–15620. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McCrea PD, Turck CW and Gumbiner B: A
homolog of the armadillo protein in Drosophila (plakoglobin)
associated with E-cadherin. Science. 254:1359–1361. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Passlick B, Pantel K, Stosiek P, Hosch S,
Thetter O and Izbicki JR: Expression of plakoglobin in bronchial
carcinomas: Incidence and significance for disease outcome.
Langenbecks Arch Chir Suppl Kongressbd. 113:810–813. 1996.In
German.
|
|
9
|
Pantel K, Passlick B, Vogt J, Stosiek P,
Angstwurm M, Seen-Hibler R, Häussinger K, Thetter O, Izbicki JR and
Riethmüller G: Reduced expression of plakoglobin indicates an
unfavorable prognosis in subsets of patients with non-small-cell
lung cancer. J Clin Oncol. 16:1407–1413. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pirinen RT, Hirvikoski P, Johansson RT,
Hollmén S and Kosma VM: Reduced expression of alpha-catenin,
beta-catenin, and gamma-catenin is associated with high cell
proliferative activity and poor differentiation in non-small cell
lung cancer. J Clin Pathol. 54:391–395. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
He X, Zhou T, Yang G, Fang W, Li Z, Zhan
J, Zhao Y, Cheng Z, Huang Y, Zhao H, et al: The expression of
plakoglobin is a potential prognostic biomarker for patients with
surgically resected lung adenocarcinoma. Oncotarget. 7:15274–15287.
2016.PubMed/NCBI
|
|
12
|
Alaee M, Nool K and Pasdar M: Plakoglobin
restores tumor suppressor activity of p53R175H mutant by
sequestering the oncogenic potential of β-catenin. Cancer Sci.
109:1876–1888. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Winn RA and Heasley LE: Gamma-catenin
expression is reduced or absent in a subset of human non-small cell
lung cancers, and its re-expression inhibits cell growth. Chest.
125(Suppl 5): 122S–123S. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Winn RA, Bremnes RM, Bemis L, Franklin WA,
Miller YE, Cool C and Heasley LE: gamma-Catenin expression is
reduced or absent in a subset of human lung cancers and
re-expression inhibits transformed cell growth. Oncogene.
21:7497–7506. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Parker HR, Li Z, Sheinin H, Lauzon G and
Pasdar M: Plakoglobin induces desmosome formation and epidermoid
phenotype in N-cadherin-expressing squamous carcinoma cells
deficient in plakoglobin and E-cadherin. Cell Motil Cytoskeleton.
40:87–100. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Breault JE, Shiina H, Igawa M,
Ribeiro-Filho LA, Deguchi M, Enokida H, Urakami S, Terashima M,
Nakagawa M, Kane CJ, et al: Methylation of the gamma-catenin gene
is associated with poor prognosis of renal cell carcinoma. Clin
Cancer Res. 11:557–564. 2005.PubMed/NCBI
|
|
17
|
Shiina H, Breault JE, Basset WW, Enokida
H, Urakami S, Li LC, Okino ST, Deguchi M, Kaneuchi M, Terashima M,
et al: Functional loss of the gamma-catenin gene through epigenetic
and genetic pathways in human prostate cancer. Cancer Res.
65:2130–2138. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gastaldi T, Bonvini P, Sartori F, Marrone
A, Iolascon A and Rosolen A: Plakoglobin is differentially
expressed in alveolar and embryonal rhabdomyosarcoma and is
regulated by DNA methylation and histone acetylation.
Carcinogenesis. 27:1758–1767. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shafiei F, Rahnama F, Pawella L, Mitchell
MD, Gluckman PD and Lobie PE: DNMT3A and DNMT3B mediate autocrine
hGH repression of plakoglobin gene transcription and consequent
phenotypic conversion of mammary carcinoma cells. Oncogene.
27:2602–2612. 2008. View Article : Google Scholar
|
|
20
|
Wang L, Syn NL, Subhash VV, Any Y, Thuya
WL, Cheow ES, Kong L, Yu F, Peethala PC, Wong AL, et al: Pan-HDAC
inhibition by panobinostat mediates chemosensitization to
carboplatin in non-small cell lung cancer via attenuation of EGFR
signaling. Cancer Lett. 417:152–160. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li F, Wang T, Wang Z, Chen X and Liu R:
Histone deacetylase inhibitor quisinostat activates caspase
signaling and upregulates p53 acetylation to inhibit the
proliferation of HepG2 cells. Mol Med Rep. 16:6094–6101. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu W, Lu W, Chen G, Cheng F, Su H, Chen Y,
Liu M and Pang X: Inhibition of histone deacetylases sensitizes EGF
receptor-TK inhibitor-resistant non-small-cell lung cancer cells to
erlotinib in vitro and in vivo. Br J Pharmacol. 174:3608–3622.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Farooqi AA, Naqvi SK, Perk AA, Yanar O,
Tabassum S, Ahmad MS, Mansoor Q, Ashry MS, Ismail M, Naoum GE, et
al: Natural agents-mediated targeting of histone deacetylases. Arch
Immunol Ther Exp (Warsz). 66:31–44. 2018. View Article : Google Scholar
|
|
24
|
Yim JH, Baek JH, Lee CW, Kim MJ, Yun HS,
Hong EH, Lee SJ, Park JK, Um HD and Hwang SG: Identification of
HDAC4 as a target of γ-catenin that regulates the oncogenic
K-Ras-mediated malignant phenotype of Rat2 cells. Biochem Biophys
Res Commun. 436:436–442. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bailey CK, Mittal MK, Misra S and
Chaudhuri G: High motility of triple-negative breast cancer cells
is due to repression of plakoglobin gene by metastasis modulator
protein SLUG. J Biol Chem. 287:19472–19486. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shim JS, Kim DH and Kwon HJ: Plakoglobin
is a new target gene of histone deacetylase in human fibrosarcoma
HT1080 cells. Oncogene. 23:1704–1711. 2004. View Article : Google Scholar
|
|
27
|
Sang Y, Zang R, Sun L, Kaddie C, Li SW,
Xiong L, Peng Y, Zeng L and Huang G: MORF4L1 suppresses cell
proliferation, migration and invasion by increasing p21 and
E-cadhering expression in nasopharyngeal carcinoma. Oncol Lett.
17:294–302. 2019.PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
29
|
Sang Y, Chen MY, Luo D, Zhang RH, Wang L,
Li M, Luo R, Qian CN, Shao JY, Zeng YX, et al: TEL2 suppresses
metastasis by down-regulating SERPINE1 in nasopharyngeal carcinoma.
Oncotarget. 6:29240–29253. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Győrffy B, Surowiak P, Budczies J and
Lánczky A: Online survival analysis software to assess the
prognostic value of biomarkers using transcriptomic data in
non-small-cell lung cancer. PLoS One. 8:e822412013. View Article : Google Scholar
|
|
31
|
Ding S, Khoury-Hanold W, Iwasaki A and
Robek MD: Epigenetic reprogramming of the type III interferon
response potentiates antiviral activity and suppresses tumor
growth. PLoS Biol. 12:e10017582014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vanhaecke T, Papeleu P, Elaut G and
Rogiers V: Trichostatin A-like hydroxamate histone deacetylase
inhibitors as therapeutic agents: Toxicological point of view. Curr
Med Chem. 11:1629–1643. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee BM and Mahadevan LC: Stability of
histone modifications across mammalian genomes: Implications for
'epigenetic' marking. J Cell Biochem. 108:22–34. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hayashi-Takanaka Y, Maehara K, Harada A,
Umehara T, Yokoyama S, Obuse C, Ohkawa Y, Nozaki N and Kimura H:
Distribution of histone H4 modifications as revealed by a panel of
specific monoclonal antibodies. Chromosome Res. 23:753–766. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huang WY, Hsu SD, Huang HY, Sun YM, Chou
CH, Weng SL and Huang HD: MethHC: A database of DNA methylation and
gene expression in human cancer. Nucleic Acids Res. 43:D856–D861.
2015. View Article : Google Scholar :
|
|
36
|
Ouaïssi M, Sielezneff I, Silvestre R,
Sastre B, Bernard JP, Lafontaine JS, Payan MJ, Dahan L, Pirrò N,
Seitz JF, et al: High histone deacetylase 7 (HDAC7) expression is
significantly associated with adenocarcinomas of the pancreas. Ann
Surg Oncol. 15:2318–2328. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang L, Bu L, Hu J, Xu Z, Ruan L, Fang Y
and Wang P: HDAC1 knockdown inhibits invasion and induces apoptosis
in non-small cell lung cancer cells. Biol Chem. 399:603–610. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jung KH, Noh JH, Kim JK, Eun JW, Bae HJ,
Xie HJ, Chang YG, Kim MG, Park H, Lee JY, et al: HDAC2
overexpression confers oncogenic potential to human lung cancer
cells by deregulating expression of apoptosis and cell cycle
proteins. J Cell Biochem. 113:2167–2177. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu C, Lv D, Li M, Zhang X, Sun G, Bai Y
and Chang D: Hypermethylation of miRNA-589 promoter leads to
upregulation of HDAC5 which promotes malignancy in non-small cell
lung cancer. Int J Oncol. 50:2079–2090. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang Y, Huang Y, Wang Z, Wang HT, Duan B,
Ye D, Wang C, Jing R, Leng Y, Xi J, et al: HDAC10 promotes lung
cancer proliferation via AKT phosphorylation. Oncotarget.
7:59388–59401. 2016.PubMed/NCBI
|
|
41
|
Lei Y, Liu L, Zhang S, Guo S, Li X, Wang
J, Su B, Fang Y, Chen X, Ke H, et al: Hdac7 promotes lung
tumorigenesis by inhibiting Stat3 activation. Mol Cancer.
16:1702017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gao S, Liu H, Hou S, Wu L, Yang Z, Shen J,
Zhou L, Zheng SS and Jiang B: MiR-489 suppresses tumor growth and
invasion by targeting HDAC7 in colorectal cancer. Clin Transl
Oncol. 20:703–712. 2018. View Article : Google Scholar
|
|
43
|
Wu MY, Fu J, Xiao X, Wu J and Wu RC:
MiR-34a regulates therapy resistance by targeting HDAC1 and HDAC7
in breast cancer. Cancer Lett. 354:311–319. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhu C, Chen Q, Xie Z, Ai J, Tong L, Ding J
and Geng M: The role of histone deacetylase 7 (HDAC7) in cancer
cell proliferation: Regulation on c-Myc. J Mol Med (Berl).
89:279–289. 2011. View Article : Google Scholar
|
|
45
|
Peixoto P, Blomme A, Costanza B, Ronca R,
Rezzola S, Palacios AP, Schoysman L, Boutry S, Goffart N, Peulen O,
et al: HDAC7 inhibition resets STAT3 tumorigenic activity in human
glioblastoma independently of EGFR and PTEN: New opportunities for
selected targeted therapies. Oncogene. 35:4481–4494. 2016.
View Article : Google Scholar : PubMed/NCBI
|